

Abstract
Disturbed iron (Fe) ion homeostasis and mitochondrial dysfunction have been implicated in neurodegeneration. Both processes are related, because central Fe ion consuming biogenetic pathways take place in mitochondria and affect their oxidative energy metabolism. Iron is imported into mitochondria by the two homologous Fe ion importers mitoferrin-1 and mitoferrin-2. To elucidate more specifically the role of mitochondrial Fe ions for brain energy metabolism and for proper neuronal function, we generated mice with a neuron-specific knockout of mitoferrin-1 (Slc25a37−/− or mfrn-1−/−) and compared them with corresponding control littermates (mfrn-1flox/flox). Mice lacking neuronal mfrn-1 exhibited no obvious anatomical or behavioral abnormalities as neonates, young or adult animals. However, they exhibited a moderate decrease in brain mitochondrial O2-consumption with complex-I substrates of the electron transport chain (p < 0.05), indicating a moderate suppression of electron transport. While these mice did not exhibit altered basal fear levels, inquisitiveness or motor skills in specific neurobiological test batteries, they clearly exhibited decreased spatial learning skills and missing establishment of stable spatial memory in Morris water maze, as compared to floxed controls (p < 0.05). We thus conclude that mitochondrial Fe ion supply is an important player in neuronal energy metabolism and proper brain function and that the carrier mitoferrin-1 cannot be completely replaced by mitoferrin-2 or other as yet unknown Fe ion carriers.
Keywords: Brain, Mitoferrin-1, Mitoferrin-2
1. Introduction
Both mitochondrial impairment and impaired iron homeostasis have been associated with neurodegenerative diseases, such as Morbus Parkinson (PD), Alzheimeŕs disease (AD) and amyotrophic lateral sclerosis (ALS) [4,6,13,14,22]. Despite a long lasting debate, it remains unclear, if mitochondria and/or iron (Fe) play an active role in the disease process. At least the rare disorder NBIA (neurodegeneration with brain iron accumulation) clearly underpins the role of brain iron overload in neurodegeneration [24].
Interestingly, two major iron consuming biochemical pathways, i.e. heme synthesis and biogenesis of iron-sulfur clusters (ISC) largely take place within mitochondria, thus connecting the two potential pathogenic pathways [2]. For ISC biogenesis, an iron binding protein of the mitochondrial matrix space, frataxin (FXN) is essentially required [19]. FXN also plays a role in heme synthesis [26]. In both biosynthetic pathways FXN accepts iron, imported through mitoferrins into the mitochondrial matrix space, and delivers it to acceptors involved in ISC or heme genesis. In the inherited neurodegenerative disorder, Friedreich’s Ataxia (FA), a GAA trinucleotide expansion in the first intron leads to a dramatic loss of FXN, together with a suppression of ISC biogenesis [5]. This deadly disease is thus the most clear-cut example that shows that a functional impairment of mitochondrial iron utilization can act as the sole cause of neurodegeneration in the brain.
It has been established, that two homologous solute carriers of the inner mitochondrial membrane, the mitoferrins 1 and 2, are involved in mitochondrial iron uptake as a prerequisite of its utilization within the organelles [18,25,28]. While iron-loaded heme and ISCs are largely exported from the mitochondria for incorporation into cytosolic, nuclear or other proteins, at least a fraction of these prosthetic groups remains within mitochondria, e.g. as electron carriers within the large protein complexes of the electron transport chain (ETC) of the inner mitochondrial membrane [27]. Correct mitochondrial iron import is therefore essential for the function of the ETC and, as a consequence, for oxidative phosphorylation (OXPHOS), i.e. the most important route of ATP production in neurons. Mitochondrial iron import and energy metabolism are thus interconnected. Since deficits in ETC/OXPHOS have been extensively debated as a co-factor in neurodegeneration [31], a better understanding of this interconnection is desired.
In the past it had been shown that mitoferrin-1 (mfrn-1) is unequivocally necessary for correct maturation of erythrocytes in mice, while it is critical in the liver only under high heme demand [28]. These results may eventually be interpreted in terms of an unequivocal importance of mfrn-1 solely in cells with an extreme mitochondrial Fedemand (erythropoiesis, liver cells during aminolevulenic acid-rich diet). It may be suggested that mfrn-2 can largely compensate for losses of mfrn-1 in most cell types, where Fe transport capability is far from being saturated.
Also Paradkar and colleagues [18] important results, regarding impact and partial, but incomplete exchangeability of both mitoferrins in various cell types. They demonstrated by siRNA-mediated knockdown that either variant participates in mitochondrial heme synthesis in non-erythroid cells. E.g. in mouse 3T3 fibroblasts a 27% (58%) decline of heme synthesis occurred with siRNA-oligos directed to mfrn-1 (mfrn-2), while synchromous targeting of both iron importers reduced heme synthesis by 84%, all measured under conditions, where the availability of protoporphyrin IX was not limiting. In addition, synthesis levels in fibroblasts with mfrn-1 knockdown could be rescued by enforced overexpression of mfrn-2 and vice versa, further underpinning exchangeability of both importers for mitochondrial iron import in non-erythroid cells. In contrast, differentiated murine erythroleukemia cells responded solely to mfrn-1 knockdown with a severe decrease of globin accumulation, in accordance with a predominant role of this carrier in hemoglobinization and maturation of erythroid cells. Taken together, a general participation of both iron importers in non-erythroid cells could be assumed, but no analyses were so far available for neuronal cultures or mouse brain.
Interestingly, some hints in the literature suggest a pronounced relation between Fe ion availability, mitochondria and hippocampal dysfunction. For instance, it is known that Fe-deficiency in neonatal rats with IDA (neonatal Fe-deficiency anemia) leads to deficits of hippocampus-based learning and memory, which are reflected by shorter hippocampal CA1 apical dendrites with less organized branching. More recently, it has been shown in embryonic hippocampal neuron cultures that Fe-deficiency caused a downregulation of mRNA levels of genes associated with neuronal development and synaptic function, as well as of genes coding mitochondrial proteins [3]. In addition, mitochondrial maximal respiration and ATP production were decreased, as well as number and length of primary dendrites and length of branches [3]. A larger number of reports were dedicated to the opposite phenomenon, i.e. hampering of hippocampal function by iron overload, including the potential benefit of Fe-chelators for improvement of hippocampal function [7,10,23,30]. Effects of iron overload may also involve mitochondrial mechanisms [20]. A specific role of the mitochondrial iron pool has not been analyzed.
We thus decided to create neuronal mfrn-1 knockout (KO) mice in order to measure the role of mfrn-1 for proper function of the mitochondrial electron transport chain (ETC), i.e. energy metabolism, in the brain. Since an altered ETC function was observed as a primary indicator for a role of mfrn-1 in the brain, we proceeded to behavioral studies, in order to seek a neuro-biologic phenotype.
2. Methods
2.1. Animals and genotyping
Mice were housed in groups of 3–4 animals with free access to food and water at 20–21 °C, 50–60% air humidity and 12 h day/night cycle (lights on 6 a.m.). All experiments were performed during the light period of the animals. Newly developed offspring were monitored according to the guidelines to exclude any suffering. A total of 60 animals were used for experiments. Animal maintenance and all experiments were performed in accordance to the EU directive 2010/63 and were approved by the local animal care committee (Landesverwaltungsamt Sachsen-Anhalt: 42502-2-1364 UniMD).
Mice of the strain C57BL6/J, which had been floxed to allow credependent removal of exon 2) of mfrn-1 had been generated in the laboratory of Diane Ward and had been used earlier for successful knockout of mfrn-1 in erythrocyte precursors or liver cells [28]. The functionality of the exon excision had thus already been proven earlier in other mouse tissues. Homozygous mice (mfrn-1flox/flox) were crossed with a cre-driver mouse [29], in which cre expression was controlled by the neuron-specific nestin (nes) promotor. These mice were hemizygous for nes-cre (cre+/−). All offspring in F1 thus were heterozygous for the floxed allele of mfrn-1 and about 50% contained a nes-cre allele. Crossing of these cre-positive mice with mfrn-1flox/flox yielded a fraction of mice which were homozygous mfrn-1flox/flox and hemizygous for cre. These mice thus harbored a knockout of mfrn-1 in neurons and are further designated as mfrn-1−/−. Littermates without cre, further designated mfrn-1flox/flox were used as controls in all biochemical measurements and behavioral experiments.
A fraction of the newly generated knockout mice of both sexes was carefully examined as neonates, at an age of 4 weeks and periodically as adult mice until they had reached the maximal age to be used in experiments. The development of knockout mice was normal and no obvious anatomical or behavioral deviations occurred. No significant differences in mean body weight (for males or females, respectively) occurred between the two genotypes between the ages of 1 month and 6 months (latest experiments). This observation excluded a severe diseased phenotype of the mice and allowed all behavioral experiments according to permission 42502-2-1364 UniMD of the local animal care committee (Landesverwaltungsamt Sachsen-Anhalt). Moreover, this observation already suggested some compensation of a neuronal loss of mfrn-1 by mfrn-2 and/or unknown Fe carriers.
Genotyping from tail tips was performed with mfrn-1 primers as described earlier [28]. Tail tips from non-transgenic mice were used as controls to indicate the wild-type allele (wt) of mfrn-1. Amplicon length was 173 bp in wild-type mice, which could be distinguished by fragment size from the floxed allele (flox, 273 bp). Presence of the nestincre construct [29] in tail tips distinguished the knockout mice from their cre-negative littermates, used as controls. In some cases the PCR system for mfrn-1 was also applied to explanted brain tissue of 2 week-old neonates, in order to verify the excision of exon 2 in brain tissue by the occurrence of the corresponding KO allele (428 bp). A liver specimen was used to assess specificity (KO allele missing, flox allele present). PCR fragments were separated on 8% polyacrylamide gels on a Multiphor-II device (Pharmacia, Uppsala, Sweden) and gels were silver-stained. Hae-III digested Phi-X-174-DNA served as size marker.
2.2. Oxygraphic measurements
Brains of ten knockout mice and corresponding cre-negative littermates (controls) of both sexes (6 months old) were used for biochemical analyses. To assess the function of the electron transport chain (ETC) of isolated brain mitochondria of a single mouse, mitochondria were prepared immediately after killing of the mouse and rapid brain preparation. The mitochondrial fraction of whole brain extracts was prepared by homogenization with a Potter-Elvejhem homogenizer in the cold, followed by centrifugation in cold mannitol-medium, as described earlier [12]. Mitochondrial preparations were then immediately measured in an oxygraphic device (Oroboros), basically as described earlier [12], without any time delay or storage. Concentrations of mitochondrial protein were adjusted to 0.06 mg/ml for oxygraphy. The measurements assessed the O2 consumption of freshly isolated brain mitochondria in the presence of 2 mM ADP and those ETC substrates, which allow a maximal flow of electrons fed into NADH:ubichinon-oxidorectuctase (complex I), i.e. 10 mM malat +10 mM pyruvate). After this record, the typically two times lower O2-consumption, which can be reached by feeding electrons from 10 mM succinate into succinate dehydrogenase (SDH, complex II) was measured in the same run in addition. To exclude any participation of complex I, this enzyme complex was completely poisoned by the classical complex-I inhibitor rotenone (1.5 μM) in this period. Furthermore, every experiment started with an oxygraphic record without ADP, which was taken as the baseline of unspecific O2-consumption to calculate the true ETC-specific rates, as depicted in Fig. 2 comparing the two genotypes. Subsequent ADP addition also served as quality control of mitochondria. As expected, ADP dramatically increased the O2-consumption in every single experiment, thus demonstrating undamaged mitochondria. At the end of every measurement electron flow was completely blocked by poisoning of cytochrome c oxidase (COX) with azide, in order to test, if the resulting low levels of unspecific O2 consumption were similar to the initial levels without ADP. This was always the case, thus demonstrating stability of the system over the time period of oxygraphic measurement.
Fig. 2.
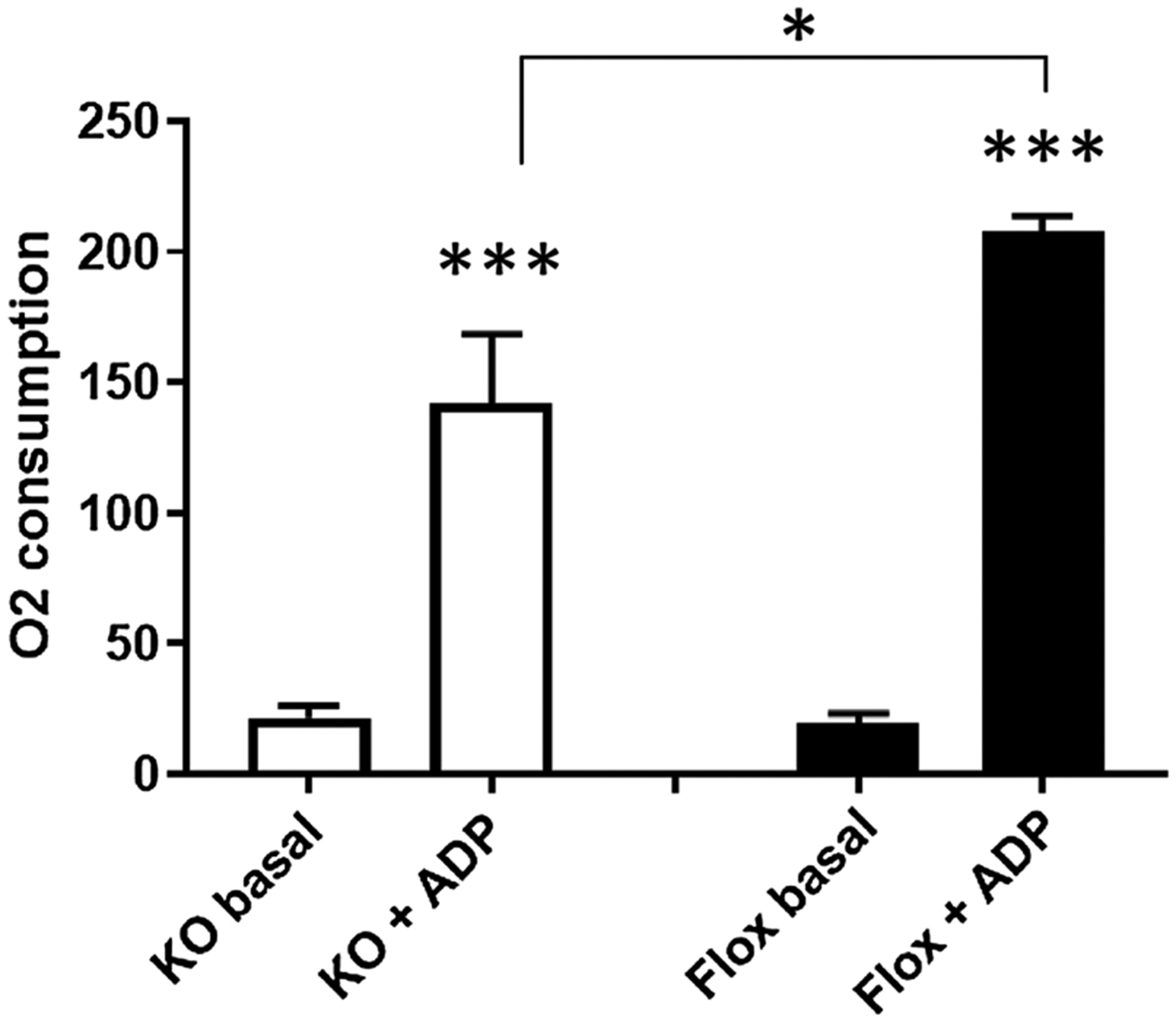
Induction of maximal respiration by ADP in isolated brain mitochondria. The basal O2-consumption with complex I substrates in the absence of ADP is strongly increased by the addition of 2 mM ADP in brain mitochondria isolated from neuronal Mfrn-1 KO mice, as well as in organelles derived from corresponding flox controls, indicating intact brain mitochondria in both animal lines.
2.3. Iron measurement
Hippocampus, cerebellum and frontal cortex of 7 neuronal Mfrn-1 knockout mice and 5 flox control mice (6 months old) were separately homogenized in the buffer used for oxygraphy to allow measurement of total tissue iron, normalized to total tissue protein, in order to assess a potential gross disturbance of iron homeostasis. Iron and protein levels, were measured with routine photometric procedures established at the Institute of Clinical Chemistry and Pathobiochemistry, based on the Roche Cobas platform (Roche Diagnostics, Mannheim, Germany) in consideration of quality assurance. Fe2+ was assessed as Ferrozine complex after reduction of Fe3+ to Fe2+ via ascorbate, thereby allowing measurement of total tissue iron.
2.4. Brain histology
The brains of two Mfrn-1 knockout mice and two flox control mice (6 months old) were fixed in 4% formalin, embedded in paraffin and 4 μm thick histologic slices were stained with H&E to assess gross brain alterations and with Nissl stain to asses more in detail neuronal alterations, especially in the hippocampal region. Microscopic examinations of the slices were performed by an experienced neuropathologist (C.M.).
2.5. Behavioral experiments
All behavioral tests were performed in adult, i.e. 3–4 months old, male mice. All behavioral experiments were performed during the light period of the animals. The light/dark cycle was 12 h/12 h and light was switched on at 7 a.m. To assess different aspects of the phenotype that might affect the result of the learning and memory task, all animals went through a battery of different tests in the following sequence: (1) elevated plus maze, (2) open field (3) novel object recognition and (4) Morris water maze. To check whether the genetic modification alters anxiety behavior or spontaneous motor activity which might then interfere with the subsequent performance in the learning tasks, we conducted an elevated plus maze and an open field test. For a detailed description of these tests see e.g. [9,21]. In brief, the plus maze was elevated 40 cm above the floor and the arm length was 30 cm. The animals got 5 min to explore the maze and the time the animals spent in the different zones of the maze was scored by a video tracking system (AnyMaze, Stoelting Co., Wood Dale, IL). The open field consisted of a quadratic arena with a side length of 50 cm. The animals got 10 min to explore the arena and their behavior was automatically scored by a video tracking system (AnyMaze, Stoelting Co., Wood Dale, IL). Both experiments were performed with 14 floxed and 11 KO animals.
To assess the learning and memory capacities of these animals, we first performed a novel object recognition task. For this we used a rectangular arena (30 × 45 cm2) and placed 2 identical objects in it. The animals were given 10 min to freely explore the arena and the objects (object presentation trial), and afterwards were put back into their home cages. 24 h later, the animals were re-exposed to the arena, but this time one of the two objects was replaced by a novel object (novel object trial). The experiments were recorded by the AnyMaze software, which analyzed the general activity of the animals. In addition, the software was used to manually score the exploration times of the objects by an experimenter blind to the genotype of the animals. A minimum total exploration time for both objects (familiar + novel object) of less than 10 s was set as exclusion criterion. By applying this criterion 12 floxed and 9 KO animals were included in the final analysis of this experiment.
To gain further insights into the learning and memory capacities of the animals, we performed a Morris water maze task. A circular pool with 150 cm diameter filled with white colored water (22 °C ± 1 °C) served as water maze. The maze was virtually devided into 4 quadrants and the submerged hidden target platform was placed in the center of one of the 4 quadrants. The experiments were recorded and analyzed by a video tracking system (AnyMaze, Stoelting Co., Wood Dale, IL). For 4 consecutive days, the animals were exposed to 4 training trials per day (20 min inter-trial interval). In each training trial the animals were inserted in the maze at pseudorandomized starting points and the time they needed to reach the platform (latency) was scored. If an animal didn’t find the platform within 60 s, it was gently guided by the experimenter to the platform. On the 5th day we performed a probe trial in which we removed the hidden platform and let the animals swim for 60 s. The memory performance was assessed by the time the animals spent in the previous target quadrant. The Morris water maze was performed with 22 animals (11 floxed, 11 KO mice)
2.6. Statistics
All oxygraphic data were compared between KO mice and cre-negative littermates by the Mann-Whitney U test The same test was applied to compare complex-I dependent O2 consumption with and without ADP in each mouse strain as a proof of ADP-dependent induction, i.e. quality of the mitochondrial preparation, and was used to compare regional tissue iron content between the two genotypes. The behavioral data were analyzed by analysis of variances (ANOVA), if applicable repeated measure ANOVAs were performed. Significant main effects in the ANOVAs were followed by Fisher-LSD post hoc tests, if applicable. All calculations were performed with SPSS, release 24. Significance was assumed for p < 0.05.
3. Results
3.1. Generating of mfrn-1 knockout mice
The structure of the mfrn-1 wild type locus, the targeting strategy to create a lox-P flanked exon 2, which becomes excised in the presence of cre, had already been published [28]. Fig. 1(A–D) shows a modified version of the figure presented in the original publication for overview. Specificity for removal of exon 2 in neurons was reached in our breeding program by usage of a neuron-specific nestin-cre driver mouse. The aspect of a potential role of additional nestin-promoter activity in radial glia is illuminated in the discussion section.
Fig. 1.
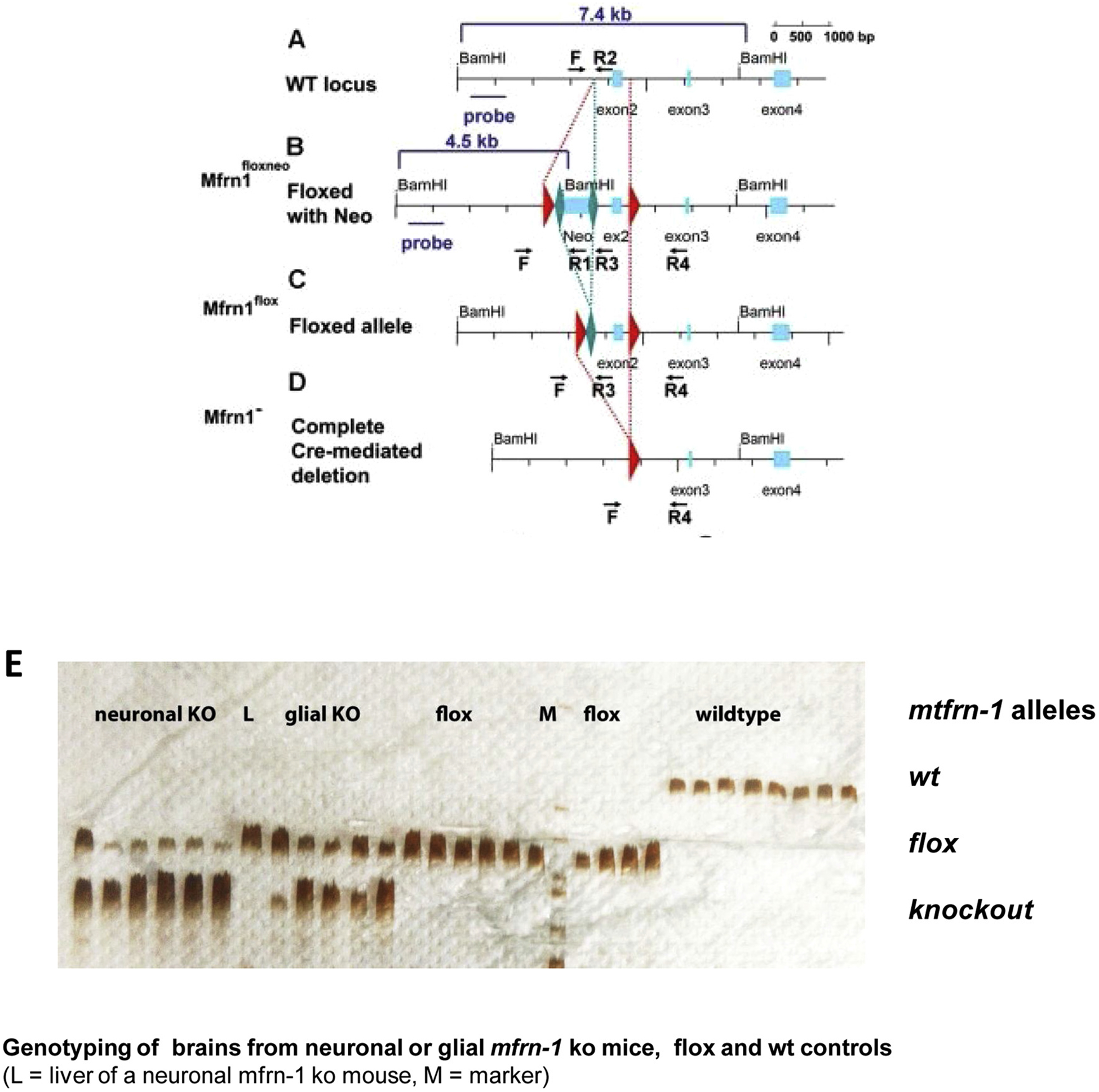
(A–D) Overview about the genomic structure of the wild type mouse mfrn-1 locus (A), construction of floxed allele with selectable neo-cassette (B), removal of neo-cassette to obtain a pure floxed allele (C) and graphical representation of a cre-mediated deletion of exon 2 (D), modified according to Troadec et al. [28]. (E) Analysis of neuronal knockout mice: Knockout occurs in the brains of 2 week-old neonates. Silver-stained polyacrylamide gel of a PCR showing the general presence of the conditional Mfrn-1 allele (Flox, intermediate band) and the absence of the wildtype allele (upper band, visible in non-transgenic animals only) in all mice of the breeding program, i.e. homozygosity for Flox. Those mice, which were positive for NES-driven Cre (neuronal KO) exhibited the corresponding KO allele (lowest bands) in brain tissue, which was absent in liver samples (L). Cre-negative mice contained only Flox, but no KO allele in the brain. ‘Glial KO’ refers to mice containing a GFAP-driven Cre, resulting in a glial Mfrn-1 KO. These mice were not subjected to closer examination as adults and are thus not subject of the present paper. However, they also show the expected KO band in the brain of neonates. M = molecular weight marker, low molecular weights on the upper edge.
At the final stage of our breeding program all mice were homozygous for the floxed allele of mfrn-1, which could be distinguished from the alleles of non-transgenic mice (wt) by the longer length of the PCR fragments derived from tail tips. An additional PCR for cre distinguished neuronal mfrn-1 knockout (KO) mice (further designated mfrn-1−/−) from the cre-negative control littermates (designated mfrn-1flox/flox). It could be observed that often even the PCR reaction from the tail tips of mfrn-1−/− mice exhibited an additional heavier band (weak), corresponding in molecular weight to the allele with excised exon 2. This could be explained by the presence of at least small amounts of neurons in the tail tips. However, in brains of mfrn-1−/− mice we expected a strong knockout-indicating band, which should never occur in the brains of mfrn-1flox/flox controls. This was precisely the case, as exemplified in Fig. 1(E). It can be clearly seen that the knockout-indicating band in brain tissue of two-week old neonates was restricted to the KO mice. Brains of control littermates solely contained the original floxed allele, in which no cre-mediated excision had taken place. The persisting (weaker) signals of the flox-allele in the KO mice were expected, because the second dominating cell species of the brain, i.e. glial cells, and other non-neuronal cells still contained the original floxed allele due to the neuron-specificity of cre expression, driven by the nestin promoter. This specificity was further confirmed by the absence of the KO band in liver samples of neuronal KO mice.
3.2. Significant decrease of mitochondrial O2-consumption in mfrn-1−/−
We next isolated brain mitochondria from a total of ten mfrn-1−/− mice of both sexes and mfrn-1flox/flox controls of both sexes (6 months old). Up to this age mfrn-1−/− mice had not shown any visible anatomic or behavioral deviation as compared to age-matched mfrn-1flox/flox controls or non-transgenic mice of the same strain and did not develop any observable peculiarities thereafter. Preparation of fresh brain mitochondria of every single mouse was started within a time interval of 10 min after scarifying the animal and the preparations were immediately subjected to oxygraphy without time delay. Only one or two mice could be measured per day and mice of both genotypes were measured in mixed order in the same Oroboros oxygraph by the same person (L.B.) in order to exclude a bias by unknown variations of experimental conditions. Moreover, the basal buffer for oxygraphy was only once prepared and kept in frozen aliquots.
The buffer in the oxygraphic cell of the Oroboros machine initially contained only the complex-I substrates malate and pyruvate without ADP. As expected, only low rates of O2-consumption were observed, i.e. 21.6 ± 4.48 (19.4 ± 3.86) nmol sec−1 ml−1 in mfrn-1−/− and mfrn-1flox/flox, respectively. These rates should be low in intact mitochondria due to the rapid depletion of endogenous ADP, leading to an inhibition of proton reflux and subsequently to blockade of electron flow. In mitochondria with an intact inner membrane and intact coupling between electron flow and oxidative phosphorylation (OXPHOS) addition of 2 mM ADP is expected to dramatically increase electron flow and thus O2-consumption, as it was the case in both mouse genotypes with significant increases (p < 0.001) of 8.81 ± 2.26 (12.7 ± 2.95) –fold in mfrn-1−/− and mfrn-1flox/flox, respectively (Fig. 2). While the low basal and unspecific O2-consumption did not differ significantly between the two genotypes, the ADP-induced maximal respiration was about one third lower in the knockout mice (p < 0.05) as compared to control littermates (Fig. 3). It should be kept in mind that nestin-cre mediated knockout was chosen in order to restrict the knockout to neurons, especially for later behavioral analyses searching for neurobiological deficits. Since glial cells remain unaltered, the moderate decrease of maximal respiration (one third) shown in Fig. 3 is expected to represent a stronger decay in the neuronal mitochondria, which cannot be measured separately by biochemical approaches.
Fig. 3.
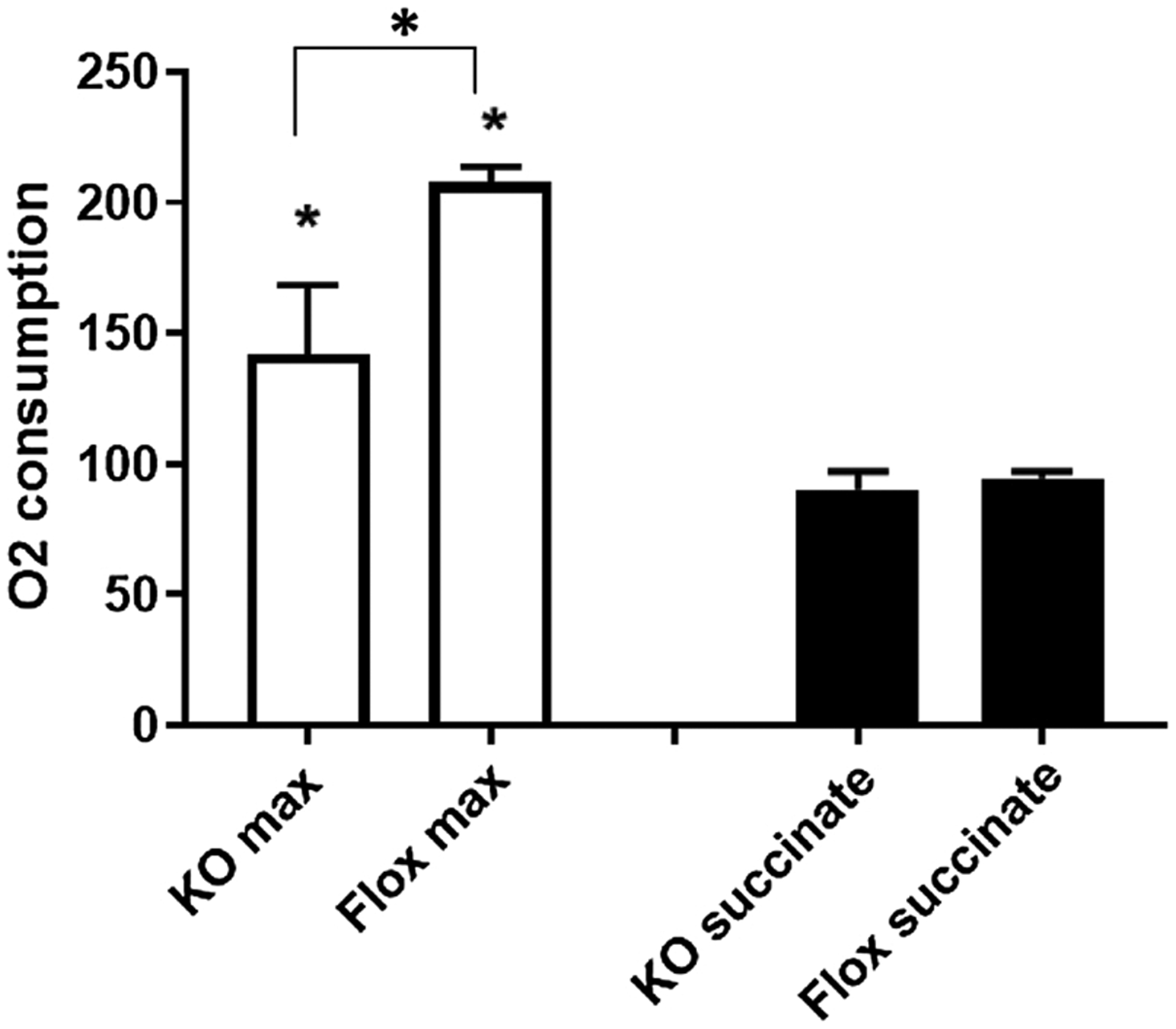
Decreased maximal respiration in KO mice. The achievable maximal respiration rate (O2-consumption) with complex I substrates was significantly lower in neuronal Mfrn-1 KO mice as compared to corresponding flox controls, while the lower rates of complex II driven O2-consumption (substrate succinate in the presence of rotenone) were not significantly lower in the KO mice.
As generally expected in oxygraphic measurements, respiration by the complex-II (SDH) substrate succinate - in the presence of 1.5 μM rotenone to exclude any influence of complex-I - reached only about half of the maximal values obtained with complex-I substrates. This lower comoplex-II respiration did not differ significantly between the two genotypes (Fig. 3). It was only 5% lower in the KO mice (n.s.).
Taken together, these biochemical results demonstrated for the first time that mfrn-1 plays a role for neuronal mitochondria and seemingly cannot be completely substituted by other Fe-transporters. However, at this point it could not be concluded with certainty that the moderate decrease of complex-I dependent electron flow in the KO mice (Fig. 3) resembles a true defect of aerobic energy metabolism leading to a functional impairment of neurons. A severe defect of brain energy metabolism seemed unlikely, because the mice showed normal behavior at first sight. Moreover, the degree of suppression of the maximal electron flow seemed too low to suggest an energy (ATP) deficit in the brain, since maximal electron flow may usually not be required to fulfill the ATP demand. On the other hand, one argument was of interest at this point, i.e. that the true ETC suppression of neurons should be higher, since glial cells still contain mitoferrin-1, thus reducing effects seen in total brain mitochondria. It was thus meaningful to further look for limited alterations of brain functions in behavioral assays.
Although global or regional alterations of brain iron content were not expected to occur after knocking out a mitochondrial iron importer, total tissue iron normalized to tissue protein, was determined separately in hippocampus, cerebellum and frontal cortex of 7 knockout mice and 5 flox controls (6 months-old). No significant differences between knockout mice and controls occurred in any region (data not shown), thereby excluding that gross alterations of total iron may explain the observed moderate mitochondrial dysfunction.
3.3. Basic behavioral characterization of mfrn-1−/−
The elevated plus maze test revealed no difference in anxiety behavior between the two genotypes. So there was no difference in the time the animals spent in the open arms (57.7 ± 10 s (flox), 63.9 ± 7.2 s (ko)), closed arms (146.9 ± 10.2 s (flox), 149.1 ± 10.7 s (ko)), or the center region (93.4 ± 5.3 s (flox), 87 ± 7.9 s (ko)). This finding was supported by an ANOVA revealing only a significant effect for the time the animals spent in the different zones of the maze (F3,69 = 48.2, p > 0.0001), but there was neither a significant effect of the genotype nor a significant interaction of these two factors (F’s ≤ 0.2, p’s ≥ 0.82).
Similarly, as anxiety behavior, the spontaneous motor behavior was not altered between the two genotypes. We observed no differences between the two genotypes in the traveled distance (41.8 ± 3.6 m (flox), 37.2 ± 3.8 m (KO)) or the average speed (6.9 ± 0.6 cm/s (flox), 6.2 ± 0.6 cm/s (ko)). Statistical analysis by ANOVAs revealed no significant differences in these parameters between the two geno types (F’s ≤ 0.7, p’s ≥ 0.4). Thus overall it can be concluded that basal behavioral parameters in respect to anxiety and spontaneous motor activity are unchanged in mfrn1-ko mice.
3.4. Learning and memory capacities of mfrn-1−/−
In the novel object recognition task, the animals were first exposed to two identical objects. In this first trial the animals showed no difference in exploration time (flox: 12.3 ± 1 s vs. 13.5 ± 1.9 s; ko:10.3 ± 1.5 s vs. 10.9 ± 1.9 s; F1,19 = 1.2, p = 0.3) as well as no preference for one of the two objects (F1,19 = 1.1, p = 0.3). There was also no significant interaction of these two factors (genotype x object: F1,19 = 0.1, p = 0.7), further indicating similar explorative behavior in both genotypes in the object presentation trial.
24 h later, we re-exposed the animals to the arena but replaced now one object by a novel object. Here, we observed a clear and significant preference for the novel object (Fig. 4; F1,19 = 61.9, p < 0.0001). Again, we observed no significant differences between the two genotypes (F1,19 = 1.3, p = 0.3) and no interaction of the factors object and genotype (F1,19 = 0.1, p = 0.8). Thus, overall, these data clearly show that the neuronal deletion of mfrn1 does not result in an impaired memory for objects, since neuronal mfrn1-ko exhibited an unaltered preference for the novel object.
Fig. 4.
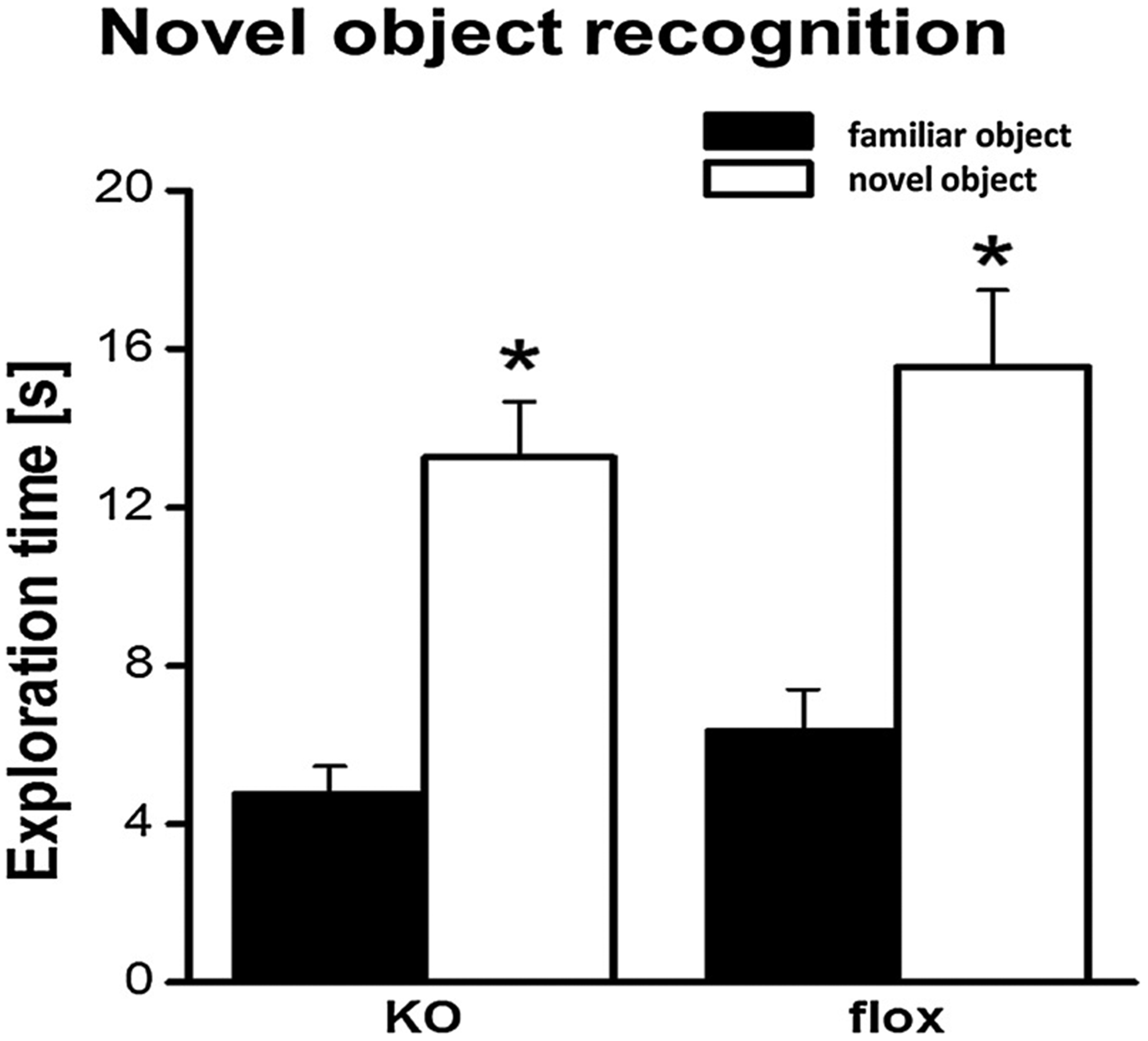
Novel object recognition: Both genotypes showed a clear preference for the novel object, indicated by significantly longer exploration times of the novel compared to the familiar object (* p < 0.05).
In the Morris water maze test, we first trained the animals for four days to find the hidden platform. Across the training days we observed a slight reduction in swimming speed of mfrn1-ko mice (Fig. 5A), which reached statistical significance (genotype: F1,19 = 4.27, p = 0.05, training day: F3,19 = 12.1, p = 0.002, genotype x training day: F3,19 = 6.5, p = 0.02). A Fisher-LSD post hoc comparison revealed significant differences between ko and flox mice on training day three and four. However, since we didn’t observe any differences in swimming speed on day 1, 2 and 5 (probe trial) as well as in all other performed behavioral tasks, this might be rather a random than a systematic effect. Nevertheless, this slightly reduced swimming speed might affect the analysis of the latencies to reach the platform. Therefore, we decided to analyze the mean distance to the platform instead, as this parameter is independent of the swimming speed. Here we observed a general improvement across the training days (F3,19 = 35.8, p < 0.0001). In addition, we observed a significant effect of the gen ning day (F3,19 = 6.4, p = 0.02). Post-hoc Fisher-LSD comparisons revealed significant differences on training day three and four (Fig. 5B). Thus, mfrn1-ko mice exhibited an impaired learning performance in the spatial water maze task.
Fig. 5.

Morris water maze test: A: During the training trials we observed a slightly reduced swimming speed over time and significantly different swimming speeds between the two genotypes. Therefore, we analyzed the mean distance to the platform instead of the latency to see the learning progress of the animals. (B). Here we observed that KO mice were significantly further away from the platform than their flox littermates. On the fifth day, we performed a probe trial (C,D), in which we removed the target platform and let the animal swim for one minute. Here, only the flox mice showed a clear and significant (*) preference for the previous target quadrant, as compared to the opposite, the adjacent right or adjacent left quadrants. The KO mice did not spend significantly different times in the four quadrants (C). The total distance the mice swam as well as the average speed did not differ between the genotypes during the 60 s of the probe trial (D). * indicates a significant difference (p < 0.05).
On the fifth day, we removed the hidden platform from the maze and let the animals swim for 60 s (probe trial) and analyzed the time the animals spent in previous target quadrant. Here we observed that flox animals spent most of the time in the previous target quadrant, while mfrn1-ko mice showed no preference for a quadrant (Fig. 5C). This finding is further corroborated by an ANOVA revealing a tendency for the factor genotype (F1,19 = 3.0, p = 0.1) and significant effects for the factor zone (F3,19 = 19.1, p < 0.0001) and the interaction of these two factors (F3,19 = 5.2, p = 0.03). In addition, the mean distance from the platform was significantly (F1,19 = 9.7, p = 0.006) larger in ko mice (0.65 ± 0.4 m) than in flox mice (0.48 ± 0.5 m). In the probe trial neither the speed nor the total distance swam (Fig. 5D) during the 60 s differed between the genotypes (F’s ≤ 2.8, p’s ≥ 0.1). Thus, overall mfrn1 ko mice exhibited an impaired spatial learning during the water maze training as well as an impaired memory performance in the probe trial. Microscopic examination of two brains of 6-month old naïve knockout and control mice did neither reveal gross alterations, such as regional hypotrophy visible in H&E staining (not shown), nor specific neuronal alterations in Nissl staining in the hippocampus or other brain regions (Fig. 6). In the course of this work it was not possible to analyze quantitatively dendritic branching or synapse density in the hippocampus of trained animals, which may be worth to be analyzed in follow-up studies.
Fig. 6.

Brain histology from mfrn-1−/− (KO) and control (FLOX) mice.
Examples of Nissl-stained hippocampal tissue from KO and FLOX mice do not reveal structural alterations or neuronal damage.
4. Discussion
4.1. Mitoferrin-1 is necessary for brain mitochondria and influences hippocampal function
In the present work, we have elucidated the role of mitochondrial mitoferrin-1 (mfrn-1) in neurons. We found that neuronal knockout of mfrn-1 leads to a moderate, but observable decline of complex-I dependent electron transport of isolated brain mitochondria, indicating that mfrn-2 or other transporters cannot completely compensate for the loss of mfrn-1. We further demonstrated that hippocampus-dependent spatial learning and memory show a surprisingly pronounced decline in the knockout mice, emphasizing the role of mfrn-1 and mitochondrial iron for the establishment of a stable memory.
Since the exact timing of the onset of neuronal cre-activity in mouse embryos is not known for the cre-driver mice used in the present study, it cannot be excluded that neuronal mfrn-1 may be essential for fetal development. However, it can be concluded that no lethal or gross deficits occur in neonatal mfrn-1−/− mice (knockout proven) or during later maturation of the nervous system and during adulthood. These results suggest that the transport function of mfrn-1 in neuronal mitochondria can largely be replaced by mfrn-2 as shown for other non-erythroid cell types [18] and/or unknown Fe ion-carriers. On the other hand, the observed moderate, but certainly underestimated ETC suppression in neuronal mitochondria of KO mice could explain the observed deficit of hippocampus-dependent spatial learning and memory, as probed by the ‘Morris water maze’. A moderate decline of OXPHOS and other Fe-dependent mitochondrial processes (ISC and heme biogenesis) may moderately hamper the development of correctly organized neuronal circuits during brain maturation and eventually adult neuronal function selectively in some regions of pronounced sensitivity. The hippocampus, whose dysfunction is of eminent interest for the most common neurodegenerative diseases in humans, Alzheimeŕs disease, may be among such sensitive regions.
To the best of our knowledge this is the first report, which clearly connects the deficiency of a mitochondrial iron importer in neurons, and thus their mitochondrial iron pool, to learning and memory. Only a general association between iron and hippocampus had so far been described in the literature. The majority of these reports were dedicated to hippocampal damage in situations of iron overload [7,10,23,30], which may occur in the setting of rare diseases, but does not shed much light on the role of physiological iron levels in brain function. More relevant with regard to a reduction of the metal were a few studies of IDA rats (neonatal Fe-deficiency anemia) exhibiting memory deficits and corresponding CA1 micromorphological alternations. In hippocampal neuron cultures it had been shown that a general Fe-deficiency, elicited by an Fe-chelator, alters the expression of genes associated with neuronal development and synaptic function, but also of mitochondrial genes resulting in a down-regulation of maximal mitochondrial respiration and ATP synthesis [3]. In the present study, we used a more specific approach, which selectively attacks mitochondrial iron import in neurons and found also a moderate reduction of maximal respiration (O2 consumption with ETC complex I substrates) in hippocampal tissue and observed deficits in the hippocampus-dependent
Morris water maze task. Because the starting point of our model is specifically the induction of a mitochondrial iron defect, we conclude that a disturbed function of this organelle potentially leads to the observed hippocampal dysfunction. Further studies are required to elucidate, if a moderate disturbance of early development or synaptic dysfunction of the adult hippocampus are more likely mediators of the observed neurologic phenotype. Tamoxifen-inducible nestin-cre driver mice may be helpful for this purpose.
4.2. Mitoferrin-1 and electron transport chain (ETC)
In the present study, we applied the analysis of mitochondrial O2 consumption as a sensitive measure to detect a metabolic suppression of these organelles by mitoferrin-1 knockout, because the ETC contains Fe-dependent reactive centers.
Complex-I, NADH:ubiquinone-oxidoreductase, is a large L-shaped electron transport complex of the inner mitochondrial membrane [8,16]. It is positioned at the beginning of the electron transport chain (ETC) and transfers electrons from reduced NADH, the main electron donor generated during citric acid cycle, to the mobile electron carrier ubiquinone (coenzyme Q), which further delivers the electrons to ETC complex-III. Within complex-I the electrons pass through a series of fixed redox carriers, initially reducing flavin mononucleotide in a twoelectron step, but then being further channeled to Fe-S clusters (ISC) within complex-I, which finally reduce the flexible small molecule electron carrier coenzyme Q [16]. Fe is thus required for the function of complex-I. While short-term fluctuations of Fe may not necessarily hamper the ETC, a partial, but life-long depletion of Fe in the mitochondrial matrix of neurons by knockout of a relevant carrier is expected to hamper ISC biogenesis and as a consequence ETC function. This could be measured by lower reduction of O2 to H2O, i.e. lower O2- consumption of brain mitochondria in our oxygraphic experiments. Since complex-II (SDH) and further downstream complexes also contain Fe-dependent groups (ISC and heme), it may have been expected that long-term Fe-depletion may have also decreased total electron flow to O2, if electrons from succinate were fed into complex-II of the ETC, while poisoning complex-I. Complex-II also reduces coenzyme Q and thus uses the same terminal redox pathway within the ETC [15]. However, no significant alteration of O2-consumption with succinate had been observed in mfrn-1−/− brains, in contrast to the usage of complex-I substrates. It may be that only the highest possible fluxes through the ETC, which are generally achievable with complex-I substrates, allowed the detection of a minor ETC deficit present in mfrn-1−/−neurons. Moreover, only respiration with complex I substrates utilizes all possible Fe-centers and may thus be expected to be most sensitive.
4.3. Potential role of radial glia in mfrn-1−/− mice
Although the nestin promoter has been commonly used for neuronal gene expression, it has been shown to exhibit activity in radial glia, too. In the context of the present work, it may be of special interest that radial glia like stem cells were shown to participate in adult hippocampal neurogenesis [1,17]. It can thus not be completely excluded that mfrn-1 KO in radial glia may contribute to the neuro-biologic phenotype in our mice. However, a dominant role of mfrn-1 KO in these cells for the observed deficits in Morris Water Maze seems unlikely, because the chosen classical settings of Morris Water Maze should be quite insensitive to detect an eventual participation of hippocampal neurogenesis. In contrast, more specialized precautions would have been necessary to modify this assay, in order to measure the influence of neurogenesis sensitively [11].
In conclusion, the present study demonstrates for the first time memory impairment in mice by moderate manipulation of mitochondrial Fe, the mediating pathways of which have to be further elucidated.
Acknowledgements
We highly appreciate the help of PD. Dr. Frank Norbert Gellerich, Neurology, Otto-von-Guericke University, Magdeburg, Germany, who carefully introduced Lisa Baldauf into the technique of oxygraphic measurements of isolated mouse brain mitochondria. Furthermore, we thank him for the possibility to use his oxygraphic devices for the project. We further thank Colette Obst and David Scharnetzki for their technical support as well as Astrid Froebe and Mathias Wolff (Central Animal Facility, Otto-von-Guericke University, Magdeburg) for their assistance in mouse care and for initial characterization of the health status of the new mouse genotype as prescribed by law. All animal experiments were performed according to law (permission 42502-2-1364, UniMD, Landesverwaltungsamt Sachsen-Anhalt). The project was partially financed by a grant‚ Mitoferrin-1 im ZNS und bei ALS’, (Stiftung für medizinische Wissenschaft) as well as NIDDK grant DK052380 to D.M.W. We thank Prof. G. Schütz (DKFZ, Heidelberg) for kind help regarding cre driver mice.
References
- [1].Aelvoet SA, Pascual-Brazo J, Libbrecht S, Reumers V, Gijsbers R, Van den Haute C, Baekelandt V, Long-term fate mapping using conditional lentiviral vectors reveals a continuous contribution of radial glia-like cells to adult hippocampal neurogenesis in mice, PLoS One 10 (2015) e0143772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Barupala DP, Dzul SP, Riggs-Gelasco PJ, Stemmler TL, Synthesis, delivery and regulation of eukaryotic heme and Fe-S cluster cofactors, Arch. Biochem. Biophys 592 (2016) 60–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bastian TW, von Hohenberg WC, Mickelson DJ, Lanier LM, Georgieff MK, Iron deficiency impairs developing hippocampal neuron gene expression, energy metabolism, and dendrite complexity, Dev. Neurosci 38 (2016) 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bozzo F, Mirra A, Carri MT, Oxidative stress and mitochondrial damage in the pathogenesis of ALS: new perspectives, Neurosci. Lett 636 (2017) 3–8. [DOI] [PubMed] [Google Scholar]
- [5].Burk K, Friedreich Ataxia: current status and future prospects, Cerebellum Ataxias 4 (2017) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cardoso S, Carvalho C, Correia SC, Seica RM, Moreira PI, Alzheimer’s Disease, From mitochondrial perturbations to mitochondrial medicine, Brain Pathol. (Zurich, Switzerland) 26 (2016) 632–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chtourou Y, Slima AB, Gdoura R, Fetoui H, Naringenin mitigates iron-induced anxiety-like behavioral impairment, mitochondrial dysfunctions, ectonucleotidases and acetylcholinesterase alteration activities in rat Hippocampus, Neurochem. Res 40 (2015) 1563–1575. [DOI] [PubMed] [Google Scholar]
- [8].Clason T, Ruiz T, Schagger H, Peng G, Zickermann V, Brandt U, Michel H,Radermacher M, The structure of eukaryotic and prokaryotic complex I, J. Struct. Biol 169 (2010) 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Endres T, Lessmann V, Age-dependent deficits in fear learning in heterozygous BDNF knock-out mice, Learn. Mem 19 (2012) 561–570. [DOI] [PubMed] [Google Scholar]
- [10].Figueiredo LS, de Freitas BS, Garcia VA, Dargel VA, Kobe LM, Kist LW, Bogo MR, Schroder N, Iron loading selectively increases hippocampal levels of ubiquitinated proteins and impairs hippocampus-dependent memory, Mol. Neurobiol 53 (2016) 6228–6239. [DOI] [PubMed] [Google Scholar]
- [11].Garthe A, Kempermann G, An old test for new neurons: refining the Morris water maze to study the functional relevance of adult hippocampal neurogenesis, Front. Neurosci 7 (2013) 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gellerich Frank N., Gizatullina Z, Trumbekaite S, Korzeniewski B, Gaynutdinov T, Seppet E, Vielhaber S, Heinze H-J, Striggow F, Cytosolic Ca2+ regulates the energization of isolated brain mitochondria by formation of pyruvate through the malate–aspartate shuttle, Biochem. J 443 (2012) 747–755. [DOI] [PubMed] [Google Scholar]
- [13].Hadzhieva M, Kirches E, Mawrin C, Review: iron metabolism and the role of iron in neurodegenerative disorders, Neuropathol. Appl. Neurobiol 40 (2014) 240–257. [DOI] [PubMed] [Google Scholar]
- [14].Hare DJ, Double KL, Iron and dopamine: a toxic couple, Brain 139 (2016) 1026–1035. [DOI] [PubMed] [Google Scholar]
- [15].Hatefi Y, The mitochondrial electron transport and oxidative phosphorylation system, Annu. Rev. Biochem 54 (1985) 1015–1069. [DOI] [PubMed] [Google Scholar]
- [16].Lenaz G, Fato R, Genova ML, Bergamini C, Bianchi C, Biondi A, Mitochondrial Complex I: structural and functional aspects, Biochim. Biophys. Acta 1757 (2006) 1406–1420. [DOI] [PubMed] [Google Scholar]
- [17].Moss J, Gebara E, Bushong EA, Sanchez-Pascual I, O’Laoi R, El M’Ghari I, Kocher-Braissant J, Ellisman MH, Toni N, Fine processes of Nestin-GFP-positive radial glia-like stem cells in the adult dentate gyrus ensheathe local synapses and vasculature, Proc. Natl. Acad. Sci. U. S. A 113 (2016) E2536–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Paradkar PN, Zumbrennen KB, Paw BH, Ward DM, Kaplan J, Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2, Mol. Cell. Biol 29 (2009) 1007–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Parent A, Elduque X, Cornu D, Belot L, Le Caer JP, Grandas A, Toledano MB, D’Autreaux B, Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols, Nat. Commun 6 (2015) 5686. [DOI] [PubMed] [Google Scholar]
- [20].Park J, Lee DG, Kim B, Park SJ, Kim JH, Lee SR, Chang KT, Lee HS, Lee DS, Iron overload triggers mitochondrial fragmentation via calcineurin-sensitive signals in HT-22 hippocampal neuron cells, Toxicology 337 (2015) 39–46. [DOI] [PubMed] [Google Scholar]
- [21].Psotta L, Rockahr C, Gruss M, Kirches E, Braun K, Lessmann V, Bock J, Endres T, Impact of an additional chronic BDNF reduction on learning performance in an Alzheimer mouse model, Front. Behav. Neurosci 9 (2015) 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Requejo-Aguilar R, Bolanos JP, Mitochondrial control of cell bioenergetics in Parkinson’s disease, Free Radic. Biol. Med 100 (2016) 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Salkovic-Petrisic M, Knezovic A, Osmanovic-Barilar J, Smailovic U, Trkulja V, Riederer P, Amit T, Mandel S, Youdim MB, Multi-target iron-chelators improve memory loss in a rat model of sporadic Alzheimer’s disease, Life Sci. 136 (2015) 108–119. [DOI] [PubMed] [Google Scholar]
- [24].Schneider SA, Neurodegenerations with brain Iron accumulation, Parkinsonism Relat. Disord 22 (Suppl 1) (2016) S21–25. [DOI] [PubMed] [Google Scholar]
- [25].Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, Gwynn B, Lambert AJ, Wingert RA, Traver D, Trede NS, Barut BA, Zhou Y, Minet E, Donovan A, Brownlie A, Balzan R, Weiss MJ, Peters LL, Kaplan J, Zon LI, Paw BH, Mitoferrin is essential for erythroid iron assimilation, Nature 440 (2006) 96–100. [DOI] [PubMed] [Google Scholar]
- [26].Soderberg C, Gillam ME, Ahlgren EC, Hunter GA, Gakh O, Isaya G, Ferreira GC, Al-Karadaghi S, The structure of the complex between Yeast Frataxin and ferrochelatase: Characterization and pre-steady state reaction of ferrous iron delivery and heme synthesis, J. Biol. Chem 291 (2016) 11887–11898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Stiban J, So M, Kaguni LS, Iron-sulfur clusters in mitochondrial metabolism: multifaceted roles of a simple cofactor, Biochemistry. Biokhimiia 81 (2016) 1066–1080. [DOI] [PubMed] [Google Scholar]
- [28].Troadec MB, Warner D, Wallace J, Thomas K, Spangrude GJ, Phillips J, Khalimonchuk O, Paw BH, Ward DM, Kaplan J, Targeted deletion of the mouse Mitoferrin1 gene: from anemia to protoporphyria, Blood 117 (2011) 5494–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G, Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety, Nat. Genet 23 (1999) 99–103. [DOI] [PubMed] [Google Scholar]
- [30].Ward RJ, Dexter DT, Crichton RR, Neurodegenerative diseases and therapeutic strategies using iron chelators, J. Trace Elem. Med. Biol 31 (2015) 267–273. [DOI] [PubMed] [Google Scholar]
- [31].Zsurka G, Kunz WS, Mitochondrial involvement in neurodegenerative diseases, IUBMB Life 65 (2013) 263–272. [DOI] [PubMed] [Google Scholar]