

Abstract
The heart contains an abundant fibroblast population that may play a role in homeostasis, by maintaining the extracellular matrix (ECM) network, by regulating electrical impulse conduction, and by supporting survival and function of cardiomyocytes and vascular cells. Despite an explosion in our understanding of the role of fibroblasts in cardiac injury, the homeostatic functions of resident fibroblasts in adult hearts remain understudied. TGF-β-mediated signaling through the receptor-activated Smads, Smad2 and Smad3 critically regulates fibroblast function. We hypothesized that baseline expression of Smad2/3 in fibroblasts may play an important role in cardiac homeostasis. Smad2 and Smad3 were constitutively expressed in normal mouse hearts and in cardiac fibroblasts. In cultured cardiac fibroblasts, Smad2 and Smad3 played distinct roles in regulation of baseline ECM gene synthesis. Smad3 knockdown attenuated collagen I, collagen IV and fibronectin mRNA synthesis and reduced expression of the matricellular protein thrombospondin-1. Smad2 knockdown on the other hand attenuated expression of collagen V mRNA and reduced synthesis of fibronectin, periostin and versican. In vivo, inducible fibroblast-specific Smad2 knockout mice and fibroblast-specific Smad3 knockout mice had normal heart rate, preserved cardiac geometry, ventricular systolic and diastolic function, and normal myocardial structure. Fibroblast-specific Smad3, but not Smad2 loss modestly but significantly reduced collagen content. Our findings suggest that fibroblast-specific Smad3, but not Smad2, may play a role in regulation of baseline collagen synthesis in adult hearts. However, at least short term, these changes do not have any impact on homeostatic cardiac function.
Keywords: fibroblast, myocardium, extracellular matrix, TGF-β, Smad, collagen
Graphical Abstract
1. INTRODUCTION:
The adult mammalian heart contains a large population of fibroblasts (1),(2),(3) located in the cardiac interstitium. Resident cardiac fibroblasts can be activated in response to a wide range of injurious stimuli and play an important role in cardiac repair (4),(5), while contributing to adverse remodeling of the injured ventricle (6),(7),(8),(9),(10). A growing body of evidence challenges the notion that fibroblasts are unidimensional cells that simply serve to produce extracellular matrix (ECM) proteins. Fibroblasts exhibit remarkable transcriptomic heterogeneity (11), and can sense microenvironmental cues, undergoing phenotypic and functional changes (12),(13). In response to stimulation with neurohumoral signals, cytokines, growth factors, and components of the ECM, fibroblasts can acquire inflammatory, matrix-synthetic, matrix-degrading, or angiogenic phenotypes (14),(15),(4),(5),(16) serving as key orchestrators of reparative, fibrogenic and angiogenic cellular responses
The members of the Transforming Growth Factor (TGF)-β superfamily are crucial activators of fibroblasts in injured and remodeling hearts (17),(18). In vitro, stimulation of cardiac fibroblasts with TGF-βs triggers myofibroblast conversion, and promotes a matrix-preserving phenotype, inducing synthesis of structural collagens and upregulating expression of tissue inhibitors of metalloproteinases (TIMPs) (19),(20). Following binding to their receptors, TGF-βs act through signaling cascades involving intracellular effectors, the receptor-activated Smads (R-Smads) (4),(21),(22), or by transducing Smad-independent pathways (23),(24),(25). Although both Smad2 and Smad3 are activated in fibroblasts infiltrating infarcted and remodeling hearts, cell-specific loss-of-function studies suggested that Smad3 signaling plays a more important role in fibroblast activation in models of myocardial infarction and left ventricular pressure overload (26),(21),(4),(27). In contrast, myofibroblast-specific Smad2 loss had only transient effects on the reparative and fibrotic response to myocardial infarction (26).
Considering the evidence suggesting involvement of fibroblasts in cardiac repair and remodeling (9),(28), the focus on the role of fibroblasts in myocardial injury models is well-justified. However, cardiac fibroblasts may also be implicated in myocardial homeostasis, preserving structural integrity, facilitating impulse conduction, and supporting cardiomyocyte and microvascular function. The cardiac interstitial ECM, produced and maintained predominantly by fibroblast-like cells, provides essential structural support to the myocardium, preserving the mechanical properties of the ventricle. Moreover, the interstitial ECM proteins may transduce key signals to cardiomyocytes, promoting survival and supporting contractile function. Thus, baseline activity of cardiac fibroblasts may be important in cardiac homeostasis.
We hypothesized that fibroblast-specific Smad2 and/or Smad3 signaling may be important in regulation of baseline fibroblast function in adult mouse hearts. In order to test this hypothesis, we generated mice with inducible Smad2 and Smad3 disruption in quiescent collagen-expressing fibroblasts, and we performed in vitro experiments investigating the effects of Smad2/Smad3 loss on baseline ECM gene expression in cultured cardiac fibroblasts.
2. MATERIALS AND METHODS:
2.1. Smad2 and Smad3 knockdown experiments in cultured cardiac fibroblasts:
In order to examine the effects of Smad2 and Smad3 knockdown on cardiac fibroblast gene expression, fibroblasts were isolated and cultured from normal mouse (C57/BL6J) hearts as previously described (20),(29). Freshly dissected whole mouse hearts were minced and incubated in Liberase TM (Roche, 5401119001) supplemented with RNase I (Invitrogen™, AM2294) at recommended concentration for 30min at 37°C. Tissues were disrupted by pipetting up and down with a 1000μL tip. Cell suspensions were seeded in culture dishes (Corning, Cat#: 353003, Falcon® 100 mm X15 mm) with DMEM/F12 (Gibco™, 11320082) plus 10% FBS (Gibco™, 10100147), without antibiotic-antimycotic added. After 48h incubation at 37°C in 5% carbon dioxide, the medium and nonadherent cells were aspirated and fresh medium was added instead. Cells were ready to passage after another 48h incubation, when they grew to 80%−90% confluence. Mouse cardiac fibroblasts at passage 1 were seeded at 80% confluence (100 mm dishes) in DMEM/F12+10% FBS and were either transfected with 50 nM ON-TARGET plus mouse Smad2 siRNA, Smad3 siRNA, Smad2 siRNA+Smad3 siRNA, or transfected with a non-silencing control siRNA (Dharmacon) using Lipofectamine® 3000 Reagent (ThermoFisher Scientific). The ON-TARGET modification is shown to dramatically decrease the off-target effects of the siRNA (30). Fibroblasts cultured in plates were transfected with Smad2, Smad3, Smad2+Smad3, or control scrambled siRNA for 72h in DMEM/F12 with 10% FBS. Afterwards, cells were rinsed twice with DPBS, and then 1 mL Trizol solution was added in each dish for cell lysis and RNA extraction. Extracted RNA was used for PCR array analysis (n=4/group).
2.2. Generation of conditional fibroblast-specific Smad2 and Smad3 KO mice:
All animal studies were approved by the Institutional Animal Care and Use Committee at Albert Einstein College of Medicine. All animals were housed in the Albert Einstein College of Medicine animal institute (Ullman Building), a disease-free rodent facility. Animal care was in strict accordance with AAALAC and NIH guidelines. In order to study the role of fibroblast-specific Smad2 and Smad3 in homeostasis, Col1α2- CreERT mice (purchased from The Jackson Laboratory, Stock No: 029235) were bred with Smad2 fl/fl mice (purchased from The Jackson Laboratory, Stock No: 022074) or Smad3 fl/fl mice (from our colony) (4) to generate Col1α2-Cre;Smad2 fl/fl animals, Col1α2-Cre;Smad3 fl/fl animals and corresponding Smad2 fl/fl, Smad3 fl/fl controls. The Col1α2-CreERT line has been previously demonstrated to selectively target cardiac fibroblasts (31),(32),(33). Tamoxifen (Sigma, T5648) was dissolved in 5 % ethanol and 95 % corn oil to make a 20 mg/ml stock solution. Tamoxifen was administered intraperitoneally every 24 hours for 5 consecutive days (75mg/kg/day) to generate mice with inducible Smad2 loss (FS2KO, Col1α2-Cre;Smad2 fl/fl animals treated with tamoxifen), animals with inducible Smad3 loss (FS3KO, Col1α2-Cre;Smad3 fl/fl animals treated with tamoxifen), and corresponding tamoxifen-treated Smad2 fl/fl and Smad3 fl/fl controls. Mice were 4 weeks old during initiation of tamoxifen treatment and were followed up for 8 weeks. Both male (M) and female (F) mice were studied. A total of 27 Smad2 fl/fl (M, n=13, F, n=14), 25 FS2KO (M, n=12, F, n=13), 20 Smad3 fl/fl (M, n=11, F, n=9), and 21 FS3KO mice (M, n=11, F, n=10) were studied.
2.3. Isolation and culture of cardiac fibroblasts from FS2KO, FS3KO and control mice, protein extraction and western blotting:
In order to confirm loss of Smad2 and Smad3 in cardiac fibroblasts, cells were isolated from Smad2 fl/fl, FS2KO, Smad3 fl/fl, and FS3KO mice at 4 weeks after tamoxifen injection and cultured in DMEM/F12 (Gibco™, 11320082) with 10% FBS (Gibco™, 10100147) as described above. Briefly, medium was changed every 48h and experiments were performed on fibroblasts at passage 1, when cells grew to 80%−90% confluence (100mm dish). Protein was extracted from cardiac fibroblasts as previously described (34). Western blotting was performed using antibodies to Smad2 (Cell Signaling Technology, 5339), Smad3 (Cell Signaling Technology, 9523), and GAPDH (Santa Cruz, 25778). The gels were imaged by ChemiDoc™ MP System (Bio Rad) and analyzed by Image Lab 3.0 software (Bio Rad).
2.4. Echocardiography:
Echocardiographic studies were performed in FS2KO, FS3KO and corresponding Smad2 fl/fl and Smad3 fl/fl control animals at baseline, and 4 weeks and 8 weeks after tamoxifen administration, using the Vevo 2100 system (VisualSonics. Toronto ON), as previously described (35). Parasternal long-axis (PSLAX) M-mode was used for measurement of systolic and diastolic ventricular and wall diameters. PSLAX IVS tool was used to measure the end-diastolic thickness of the left ventricular anterior wall (LVAWd), and the end-diastolic thickness of the left ventricular posterior wall (LVPWd), The left ventricular end-diastolic volume (LVEDV) and the left ventricular end-systolic volume (LVESV) were measured as indicators of dilative remodeling. The left ventricular ejection fraction (LVEF= [(LVEDV-LVESV) / LVEDV] ×100%) was measured for assessment of systolic ventricular function. Dimensions of the ascending aorta were measured using PSLAX-B mode images. Electrocardiography was used to calculate heart rate from more than 10 cardiac cycles. For assessment of diastolic function, we performed Doppler echocardiography and tissue Doppler imaging (TDI), as previously described (36). Transmitral LV inflow velocities were measured by pulsed-wave Doppler. Peak early E wave (E) and late A wave (A) filling velocities were measured. TDI was obtained by placing a 1.0-mm sample volume at the medial annulus of the mitral valve. Analysis was performed for the early (e′) and late (a′) diastolic velocity. The mitral inflow E velocity-to-tissue Doppler e′ wave velocity ratio (E/e′) was calculated to assess diastolic function. All Doppler spectra were recorded for 3–5 cardiac cycles at a sweep speed of 100 mm/s. The echocardiographic offline analysis was performed by a sonographer blinded to the study groups.
2.5. Assessment of Smad2 and Smad3 expression and phosphorylation in normal mouse tissues and in cultured cardiac fibroblasts:
In order to assess baseline expression and phosphorylation lavels of Smad2 and Smad3, organs (heart, lung, spleen, liver, kidney, skin, pancreas and thymus) were harvested from 8 week-old WT C57Bl6J mice. Protein was extracted as previously described (34). Halt™ Phosphatase Inhibitor Cocktail (Thermo Scientific™, 78420) was used to preserve the phosphorylation state of proteins during and after tissue lysis or protein extraction. Fibroblasts isolated from WT C57/BL6J and Smad3 KO mouse hearts (19) were cultured in 100 mm dishes with DMEM/F12 plus 10% FBS. When cells at passage 1 grew to 80%–90% confluence, medium was changed to DMEM/F12 without serum overnight. TGF-β1 (10 ng/ml, 30 min) was added as positive control for phosphorylation of Smad2 and Smad3. Western blotting was performed using antibodies to p-Smad2 (Cell Signaling Technology, 3108), Smad2 (Cell Signaling Technology, 5339), pSmad3 (Cell Signaling Technology, 9520), Smad3 (Cell Signaling Technology, 9523), and GAPDH (Santa Cruz, 25778).
2.6. Histology and machine learning-based quantitative analysis of myocardial collagen content:
For histopathological analysis murine tissues were fixed in zinc-formalin (Z-fix; Anatech, Battle Creek, MI), and embedded in paraffin. In order to assess collagen content, sections were stained with picrosirius red to label the collagen fibers. Picrosirius red-stained slides were scanned using the bright field settings of Zen 2.6 Pro software and the Zeiss Imager M2 microscope (Carl Zeiss Microscopy, New York NY). Using default algorithms of the Intellesis Trainable Segmentation module of Zen 2.6 Pro software (Carl Zeiss Microscopy, New York NY), a model was trained on multiple fields of different regions of the baseline myocardium to segment SR stained collagen fibers. Red fibrillar staining representing interstitial and perivascular collagen was considered the object of interest, while the rest of myocardium was considered as background. Automatic analysis of 10 fields from 3 different sections from each mouse heart was performed using the trained model. Collagen content was expressed as the percentage of the picrosirius red-stained area to the total myocardial area.
2.7. RNA extraction, qPCR and qPCR array analysis:
TRIzol (Invitrogen™) based method was used to extract total RNA from cultured fibroblasts at passage 1 according to the manufacturer’s instructions. RNA concentration was measured by a spectrophometer at 260nm and 280nm. For each reverse transcription reaction, 1 μg of total RNA was converted to cDNA using iScript ™ cDNA synthesis kit (Bio-Rad, 1708890). Quantitative PCR (qPCR) was performed using SsoFast EvaGreen Supermix reagent (Bio-Rad) on the CFX384™ Real-Time PCR Detection System (Bio-Rad) and the thermal cycler apparatus from Bio-Rad follow the manufacturer’s recommendations: enzyme was activated at 95°C for 30 sec, followed by 40 cycles of 5 sec at 95°C, 5 sec at 60°C. In order to confirm loss of Smad3 and Smad2 in vivo, qPCR was performed with the following primer pairs: GAPDH forward 5’-AGGTCGGTGTGAACGGATTTG-3’, GAPDH reverse 5’-TGTAGACCATGTAGTTGAGGTCA-3’, Smad2 forward 5’-ATCTTGCCATTCACTCCGCC-3’, Smad2 reverse 5’-TCTGAGTGGTGATGGCTTTCTC −3’, Smad3 forward 5’-CACGCAGAACGTGAACACC-3’, and Smad3 reverse 5’-GGCAGTAGATAACGTGAGGGA-3’, The housekeeping gene GAPDH was used as internal control. The qPCR procedure was repeated three times in independent runs; gene expression levels were calculated using the ΔCT method.
In order to assess gene expression of extracellular matrix-related genes in cultured fibroblasts following Smad2 or Smad3 knockdown, we used the RT2 Profiler™ PCR Array Mouse Extracellular Matrix & Adhesion Molecules (QIAGEN, PAMM-013ZE) according to manufacturer’s protocol. RNA extraction was performed using Trizol. A total of 400ng RNA was reverse-transcribed into cDNA using the RT2 first strand kit. The same thermal profile conditions were used for all primers sets: 95°C for 10 minutes, 40 cycles at 95°C for 15 seconds followed at 60°C for 1 minute. The data obtained were exported to SABiosciences PCR array web-based template where it was analyzed using the ΔCT method.
2.8. Statistical analysis:
For comparisons of two groups unpaired, 2-tailed Student’s t-test using (when appropriate) Welch’s correction for unequal variances was performed. The Shapiro-Wilk test was used to assess normality of the distributions. The Mann-Whitney test was used for comparisons between 2 groups that did not show Gaussian distribution. For comparisons of multiple groups, 1-way ANOVA was performed followed by Tukey’s multiple comparison test. For comparisons of several groups with control Dunnett’s test was used. The Kruskall-Wallis test, followed by Dunn’s multiple comparison post-test was used when one or more groups did not show Gaussian distribution.
3. RESULTS:
3.1. Constitutive expression of Smad2 and Smad3 in normal mouse tissues and in cultured cardiac fibroblasts.
We used western blotting to study the expression of Smad2 and Smad3 in normal mouse tissues and in cultured cardiac fibroblasts. All tissues studied showed significant constitutive expression of Smad2 (Figure 1A). The thymus, pancreas, skin, spleen and lung had the highest levels of Smad2 expression (Figure 1A, C). Constitutive expression of p-Smad2 was low in all organs studied. The skin, lung and heart exhibited identifiable bands of p–Smad2 (Figure 1A). Smad3 was also ubiquitously expressed in mouse tissues (Figure 1B, D). The liver, kidney and heart had the highest levels of Smad3 expression (Figure 1B, D). Constitutive p-Smad3 expression was low in all organs studied. Cultured cardiac fibroblasts had high levels of baseline Smad2 and Smad3 expression, but low levels of Smad2 and Smad3 phosphorylation (Figure 1A–B). As expected, TGF-β1 stimulation triggered Smad2 and Smad3 phosphorylation in cardiac fibroblasts. Specificity of the antibodies was confirmed using Smad3 KO cells (Figure 1A–B).
Figure 1. Constitutive expression of Smad2 and Smad3 in mouse tissues and in cultured cardiac fibroblasts.
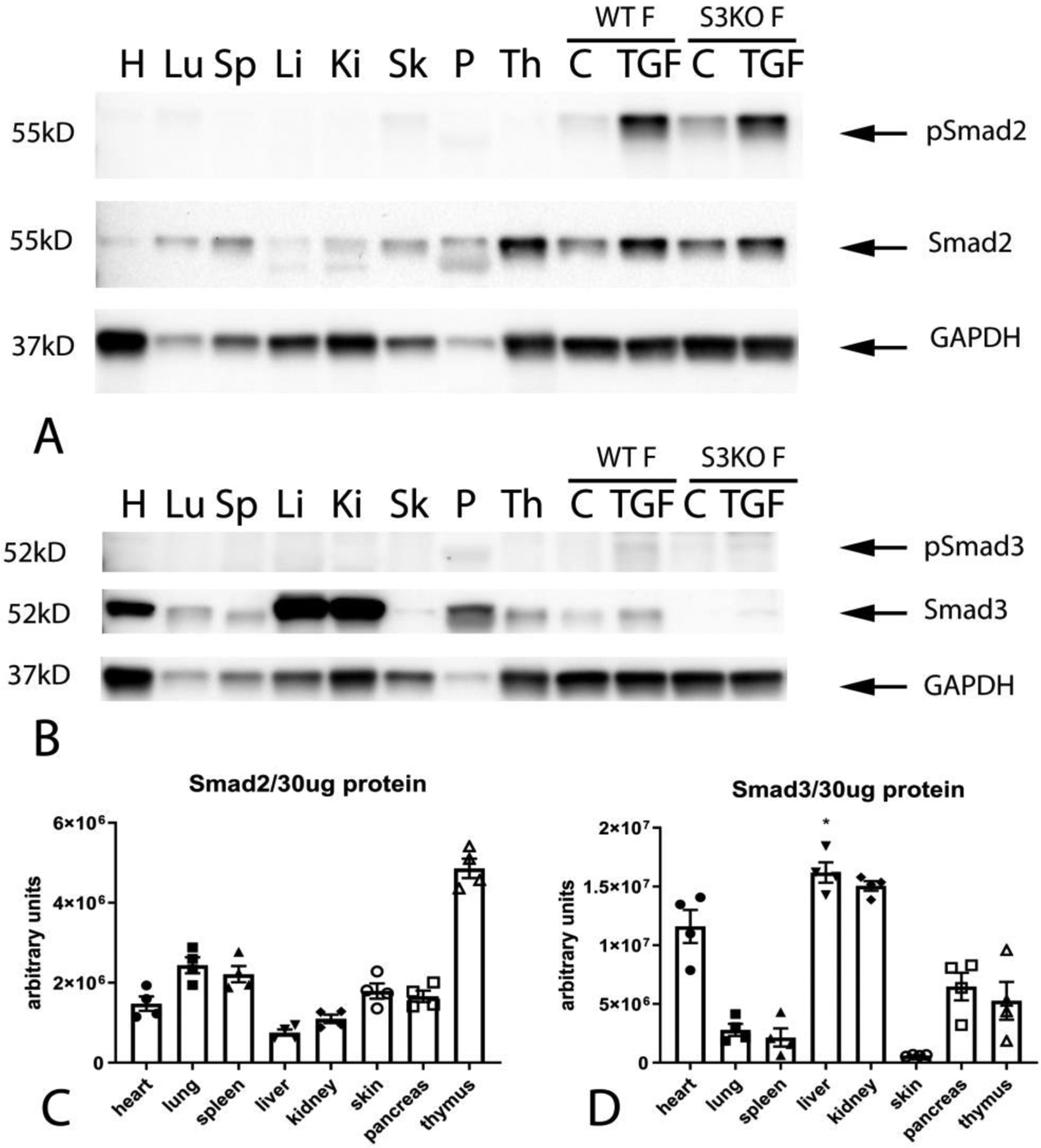
All mouse tissues had significant constitutive expression of Smad2 (A). The thymus (Th), pancreas (P), skin (Sk), spleen (Sp) and lung (Lu) had the highest levels of Smad2 expression (A, C). Constitutive expression of p-Smad2 was low in all organs studied (A). The skin (Sk), lung (Lu) and heart (H) had identifiable bands of p–Smad2 (A). Smad3 was also ubiquitously expressed in mouse tissues (B, D). The liver (Li), kidney (Ki) and heart (H) had the highest levels of Smad3 expression (B, D). Constitutive p-Smad3 expression was low in all organs studied. Relative expression of Smad2 (C) and Smad3 (D) was normalized to the total amount of protein, due to marked differences in baseline expression of the housekeeping protein GAPDH between tissues. Cultured cardiac fibroblasts (WT F) had high levels of baseline Smad2 and Smad3 expression, but low levels of Smad2 and Smad3 phosphorylation (A–B). TGF-β1 stimulation (TGF) triggered Smad2 and Smad3 phosphorylation in cardiac fibroblasts. Specificity of the antibodies was confirmed using Smad3 KO cells (S3KO F) (A–B). n=4.
3.2. Smad3 mediates collagen I transcription, whereas Smad2 stimulates collagen V synthesis in cultured cardiac fibroblasts.
qPCR demonstrated that Smad2 siRNA KD (for 72h) in cultured serum-stimulated cardiac fibroblasts specifically reduced Smad2 levels without affecting Smad3 expression (Figure 2A). A PCR array was used to examine the effects of Smad2 and Smad3 KD on matrix gene synthesis (Table 1) Smad3 KD markedly attenuated Smad3 synthesis without affecting Smad2 levels (Figure 2B). Combined Smad2 and Smad3 knockdown markedly reduced levels of both Smad2 and Smad3 (Figure 2A–B). Smad3, but not Smad2 KD significantly attenuated collagen I a1 expression (Figure 2C) mRNA expression. Neither Smad2 nor Smad3 KD significantly affected transcription of collagen II a1 and collagen III a1 (Figure 2D–E). Combined Smad2 and Smad3 KD attenuated synthesis of collagen I a1 and collagen II a1, but did not affect collagen III a1 levels (Figure 2C–E). Smad2, but not Smad3 KD significantly reduced collagen V a1 expression (Figure 2F), whereas collagen VI levels were not affected by Smad2, Smad3 or combined Smad2 and Smad3 KD (Figure 2G).
Figure 2: Effects of Smad2 and Smad3 on baseline collagen gene transcription in cardiac fibroblasts.
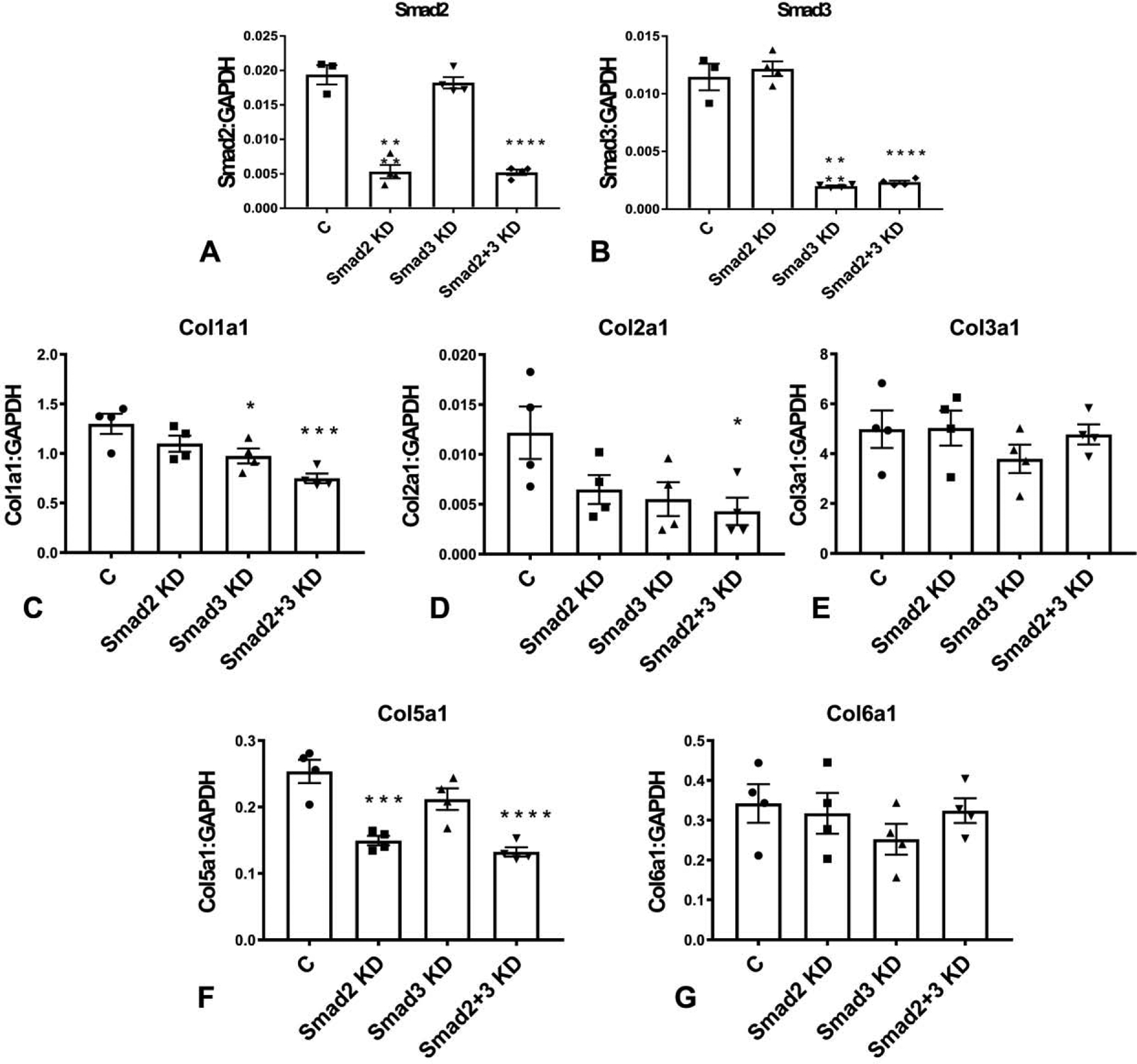
The effectiveness and specificity of Smad2 and Smad3 KD was demonstrated using qPCR (A–B). Full PCR array data on the effects of Smad2 and Smad3 KD on ECM and adhesion molecules gene synthesis are provided in Table 1. Smad3, but not Smad2 KD significantly attenuated collagen I a1 mRNA expression (C). Neither Smad2 nor Smad3 KD affected transcription of collagen II a1 and collagen III a1 (D–E). Combined Smad2 and Smad3 KD attenuated synthesis of collagen I a1 and collagen II a1, but did not affect collagen III a1 levels (C–E). Smad2, but not Smad3 KD significantly reduced collagen V a1 expression (F), whereas collagen VI levels were not affected by Smad2, Smad3 or combined Smad2 and Smad3 KD (G). (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 vs control (C), n=4/group, ANOVA followed by Dunnett’s multiple comparisons test).
Table 1:
Effects of Smad2, Smad3 and combined Smad2/Smad3 loss on extracellular matrix gene synthesis by cardiac fibroblasts
| Gene name | Gene symbol | Smad2 KD | Smad3 KD | Smad2+Smad3 KD | |||
|---|---|---|---|---|---|---|---|
| Fold | p-value | Fold | p-value | Fold | p-value | ||
| A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1motif, 1 | Adamts1 | 1.20 | 0.1026 | 0.57 | 0.0004† | 1.07 | 0.9612 |
| A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1motif, 2 | Adamts2 | 1.00 | >0.9999 | 0.79 | 0.4414 | 0.95 | 0.9259 |
| A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1motif, 5 | Adamts5 | 1.79 | 0.0011† | 0.46 | 0.0172† | 1.21 | 0.5691 |
| A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1motif, 8 | Adamts8 | 0.71 | 0.3369 | 1.99 | 0.0009† | 0.91 | 0.9268 |
| CD44 antigen | Cd44 | 0.85 | 0.0811 | 0.57 | 0.0006† | 0.64 | 0.002† |
| Cadherin 1 | Cdh1 | ND | - | ND | - | ND | - |
| Cadherin 2 | Cdh2 | 0.78 | 0.0617 | 0.77 | 0.0421† | 0.83 | 0.1401 |
| Cadherin 3 | Cdh3 | 0.44 | 0.0342† | 1.40 | 0.2144 | 0.62 | 0.0955 |
| Cadherin 4 | Cdh4 | ND | - | ND | - | ND | - |
| Contactin 1 | Cntn1 | 0.82 | 0.9399 | 0.63 | 0.8804 | 1.31 | 0.9942 |
| collagen type I, alpha1 | Col1a1 | 0.86 | 0.2259 | 0.75 | 0.034† | 0.58 | 0.001† |
| collagen type II, alpha1 | Col2a1 | 0.53 | 0.1216 | 0.42 | 0.0645 | 0.34 | 0.028† |
| collagen type III, alpha1 | Col3a1 | 1.02 | >0.9999 | 0.76 | 0.4218 | 0.99 | 0.99 |
| collagen type IV, alpha1 | Col4a1 | 0.89 | 0.2537 | 0.57 | 0.0001† | 0.79 | 0.0168† |
| collagen type IV, alpha2 | Col4a2 | 0.97 | 0.8428 | 0.86 | 0.2308 | 0.85 | 0.0984 |
| collagen type IV, alpha3 | Col4a3 | 1.23 | 0.9918 | 0.66 | 0.5862 | 0.81 | 0.7044 |
| collagen type V, alpha1 | Col5a1 | 0.59 | 0.0003† | 0.83 | 0.1002 | 0.53 | <0.0001† |
| collagen type VI, alpha1 | Col6a1 | 0.93 | 0.9558 | 0.73 | 0.3592 | 0.98 | 0.9818 |
| Connective tissue growth factor | Ctgf | 0.96 | 0.9705 | 0.73 | 0.1052 | 0.58 | 0.0115† |
| Catenin (Cadherin associated protein), alpha1 | Ctnna1 | 0.96 | 0.8678 | 0.90 | 0.3221 | 1.00 | 0.9981 |
| Catenin (Cadherin associated protein), alpha2 | Ctnna2 | 1.19 | 0.9997 | 0.58 | 0.3288 | 1.03 | 0.8801 |
| Catenin (Cadherin associated protein), beta1 | Ctnnb1 | 1.00 | >0.9999 | 0.95 | 0.8839 | 1.20 | 0.0875 |
| Extracellular matrix protein 1 | Ecm1 | 0.92 | 0.6037 | 1.20 | 0.0914 | 1.26 | 0.0351† |
| Elastin microfibril interfacer 1 | Emilin1 | 1.13 | 0.8245 | 0.75 | 0.2225 | 0.86 | 0.4733 |
| Ectonucleoside triphosphate diphosphohydrolase 1 | Entpd1 | 0.88 | 0.7972 | 0.66 | 0.1511 | 0.90 | 0.9441 |
| Fibulin 1 | Fbln1 | 0.63 | 0.2275 | 0.93 | 0.9068 | 0.90 | 0.8722 |
| Fibronectin 1 | Fn1 | 0.71 | 0.0011† | 0.76 | 0.0061† | 0.63 | 0.0002† |
| Hyaluronan and proteoglycan link protein 1 | Hapln1 | 4.11 | 0.0337† | 3.29 | 0.4624 | 1.20 | >0.9999 |
| Hemolytic complement | Hc | 1.32 | 0.7713 | 1.30 | 0.79 | 0.81 | 0.8101 |
| Intercellular adhesion molecule 1 | Icam1 | 0.66 | 0.0003† | 0.81 | 0.0217† | 0.71 | 0.0011† |
| Integrin alpha2 | Itga2 | 0.72 | 0.1937 | 0.63 | 0.0375† | 0.77 | 0.2425 |
| Integrin alpha3 | Itga3 | 1.23 | 0.5389 | 1.27 | 0.419 | 1.07 | 0.9919 |
| Integrin alpha4 | Itga4 | 0.85 | 0.0618 | 0.54 | <0.0001† | 0.59 | <0.0001† |
| Integrin alpha5 | Itga5 | 0.84 | 0.1638 | 1.07 | 0.689 | 0.90 | 0.5138 |
| Integrin alpha1, epithelial-associated | Itgae | ND | - | ND | - | ND | - |
| Integrin alpha L | Itgal | 2.13 | 0.0003† | 0.39 | 0.0185† | 0.95 | 0.977 |
| Integrin alpha M | Itgam | 1.27 | 0.4943 | 1.33 | 0.3749 | 1.18 | 0.7086 |
| Integrin alpha V | Itgav | 0.86 | 0.1827 | 1.03 | 0.964 | 0.95 | 0.8274 |
| Integrin alpha X | Igtax | 1.42 | 0.278 | 1.30 | 0.5194 | 1.30 | 0.4876 |
| Integrin beta1 | Itgb1 | 0.69 | <0.0001† | 0.96 | 0.7791 | 0.78 | 0.0017† |
| Integrin beta2 | Itgb2 | 1.45 | 0.1434 | 1.06 | 0.983 | 1.12 | 0.8774 |
| Integrin beta3 | Itgb3 | 0.75 | 0.2061 | 0.92 | 0.7783 | 0.85 | 0.3933 |
| Integrin beta4 | Itgb4 | 0.88 | 0.3398 | 0.93 | 0.553 | 0.92 | 0.4494 |
| Laminin,alpha1 | Lama1 | 0.69 | 0.9534 | 0.94 | >0.9999 | 0.59 | 0.6767 |
| Laminin,alpha2 | Lama2 | 1.91 | 0.003† | 1.11 | 0.9619 | 1.36 | 0.2962 |
| Laminin,alpha3 | Lama3 | 0.86 | 0.7634 | 0.37 | 0.2565 | 0.78 | 0.6245 |
| Laminin, beta2 | Lamb2 | 1.36 | 0.2233 | 1.13 | 0.8831 | 1.46 | 0.1447 |
| Laminin, beta3 | Lamb3 | 0.94 | >0.9999 | 0.55 | 0.4203 | 0.65 | 0.5299 |
| Laminin, gama1 | Lamc1 | 1.14 | 0.1251 | 1.06 | 0.6642 | 0.96 | 0.8442 |
| Matrix metallopeptidase 10 | Mmp10 | 0.77 | 0.3023 | 1.21 | 0.9662 | 0.57 | 0.0399† |
| Matrix metallopeptidase 11 | Mmp11 | 1.34 | 0.1113 | 1.42 | 0.0496† | 1.46 | 0.0414† |
| Matrix metallopeptidase 12 | Mmp12 | 1.37 | 0.8689 | 1.90 | 0.2164 | 1.60 | 0.4346 |
| Matrix metallopeptidase 13 | Mmp13 | 1.56 | 0.0317† | 0.54 | 0.0739 | 0.99 | >0.9999 |
| Matrix metallopeptidase 14 | Mmp14 | 1.27 | 0.0205† | 1.31 | 0.0108† | 1.44 | 0.001† |
| Matrix metallopeptidase 15 | Mmp15 | 0.51 | 0.0092† | 1.03 | 0.9927 | 0.72 | 0.1573 |
| Matrix metallopeptidase I, alpha | Mmp1a | 2.23 | <0.0001† | 0.88 | 0.7969 | 1.94 | 0.0004† |
| Matrix metallopeptidase 2 | Mmp2 | 1.42 | 0.0098† | 0.65 | 0.0085† | 0.96 | 0.7422 |
| Matrix metallopeptidase 3 | Mmp3 | 1.52 | 0.5465 | 1.08 | 0.9911 | 2.01 | 0.1364 |
| Matrix metallopeptidase 7 | Mmp7 | ND | - | ND | - | ND | - |
| Matrix metallopeptidase 8 | Mmp8 | 1.90 | 0.1009 | 1.26 | 0.9574 | 1.13 | 0.9999 |
| Matrix metallopeptidase 9 | Mmp9 | 1.91 | 0.1663 | 1.52 | 0.3911 | 1.24 | 0.9028 |
| Neural cell adhesion molecule 1 | Ncam1 | 0.56 | 0.0071† | 0.85 | 0.4708 | 0.33 | 0.0003† |
| Neural cell adhesion molecule 2 | Ncam2 | ND | - | ND | - | ND | - |
| Platelet/endothelial cell adhesion molecule 1 | Pecam1 | 1.01 | 0.9991 | 1.11 | 0.6413 | 1.20 | 0.1953 |
| Periostin, osteoblast specific factor | Postn | 0.62 | 0.0017† | 0.86 | 0.2503 | 0.38 | <0.0001† |
| Selectin, endothelial cell | Sele | 0.83 | 0.6299 | 1.22 | 0.5334 | 1.71 | 0.0076† |
| Selectin, lymphocyte | Sell | ND | - | ND | - | ND | - |
| Selectin, platelet | Selp | 0.99 | 0.9978 | 0.69 | 0.0193† | 1.02 | >0.9999 |
| Sarcoglycan, epsilon | Sgce | 1.57 | 0.0127† | 0.98 | 0.9993 | 1.27 | 0.357 |
| Secreted acidic cysteine rich glycoprotein | Sparc | 1.19 | 0.1279 | 0.92 | 0.6975 | 1.05 | 0.9429 |
| Sparc/osteonectin, cwcv and kazal-like domains proteoglycan 1 | Spock1 | ND | - | ND | - | ND | - |
| Secreted phosphoprotein 1 | Spp1 | 1.59 | 0.6204 | 2.31 | 0.028† | 1.39 | 0.7043 |
| synaptotagmin 1 | Syt1 | 1.72 | 0.8436 | 1.12 | 0.9061 | 0.92 | 0.9725 |
| Transforming growth factor, beta induced | Tgfbi | 0.68 | 0.0226† | 1.27 | 0.0538† | 0.72 | 0.0414† |
| Thrombospondin 1 | Thbs1 | 0.90 | 0.4343 | 0.76 | 0.0224† | 0.70 | 0.0045† |
| Thrombospondin 2 | Thbs2 | 0.52 | 0.0658 | 0.89 | 0.8188 | 0.56 | 0.0745 |
| Thrombospondin 3 | Thbs3 | 1.66 | 0.0525† | 1.30 | 0.8955 | 0.97 | >0.9999 |
| Tissue inhibitor of metalloproteinase 1 | Timp1 | 1.10 | 0.8511 | 0.81 | 0.5181 | 0.99 | >0.9999 |
| Tissue inhibitor of metalloproteinase 2 | Timp2 | 1.12 | 0.7962 | 1.51 | 0.0167† | 1.59 | 0.0067† |
| Tissue inhibitor of metalloproteinase 3 | Timp3 | 0.77 | 0.3058 | 0.43 | 0.0078† | 0.30 | 0.0016† |
| Tenascin C | Tnc | 0.64 | 0.0067† | 0.80 | 0.1059 | 0.72 | 0.0196† |
| Vascular cell adhesion molecule 1 | Vcam1 | 1.87 | 0.0008† | 1.00 | 0.9944 | 1.62 | 0.0221† |
| Versican | Vcan | 0.61 | 0.0092† | 1.06 | 0.9172 | 0.84 | 0.3146 |
| Vitronectin | Vtn | ND | - | ND | - | ND | - |
P value <0.05.
ND-Not Detected
3.3. Smad3, but not Smad2 mediates collagen IV a1 mRNA synthesis in cardiac fibroblasts.
Next, we examined the effects of Smad2 and Smad3 on expression of basement membrane genes by cardiac fibroblasts. Smad3 KD, but not Smad2 KD attenuated collagen IV a1 synthesis; however, effects on collagen IV a2 and collagen IV a3 gene expression did not reach statistical significance (Figure 3A–C).
Figure 3: Effects of Smad2 and Smad3 KD on fibroblast expression of basement membrane genes.
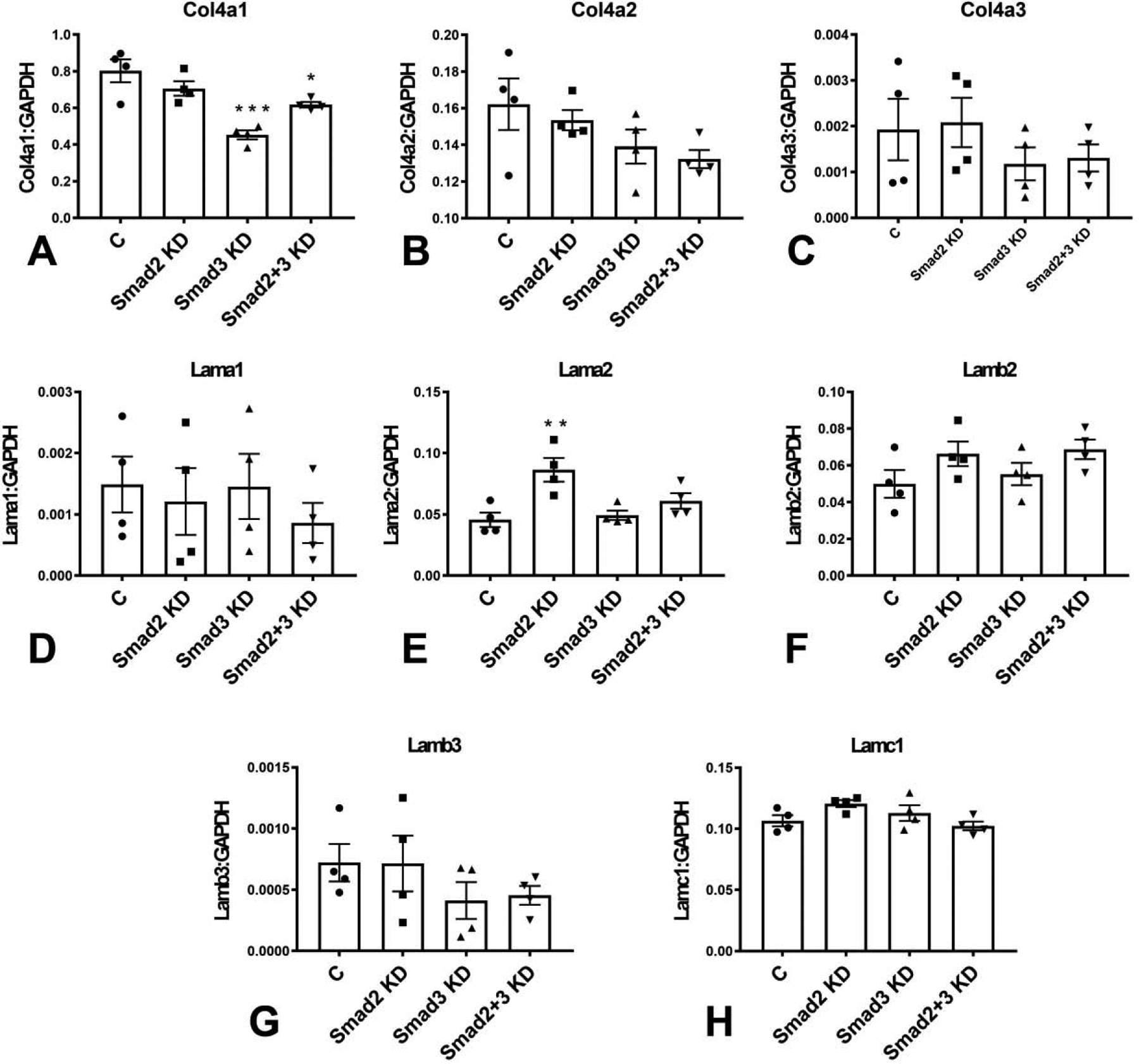
Smad3 KD, but not Smad2 KD attenuated collagen IV a1 synthesis (A); however, effects on collagen IV a2 and collagen IV a3 gene expression did not reach statistical significance (B–C). Smad3 KD had no significant effects on laminin α1, α2 and β2 mRNA expression (D–F). In contrast, Smad2 KD significantly increased laminin α2 transcription (E) without affecting laminin α1 and β2 levels (D, F). (*p<0.05, **p<0.01, ***p<0.001 vs control (C), n=4/group, ANOVA followed by Dunnett’s multiple comparisons test).
3.4. Smad2 restrains laminin α2 chain mRNA synthesis.
Smad3 KD had no significant effects on laminin α1, α2 and β2 expression (Figure 3D–F). In contrast, Smad2 KD significantly increased laminin α2 transcription (Figure 3E) without affecting laminin α1 and β2 levels (Figure 3D, 3F).
3.5. Role of Smad2 and Smad3 in expression of specialized matrix proteins by cultured cardiac fibroblasts.
Next, we examined the role of Smad2 and Smad3 in expression of fibronectin and matricellular genes by cardiac fibroblasts. Smad2, Smad3 and combined Smad2/Smad3 KD attenuated expression of fibronectin in cardiac fibroblasts (Figure 4A), suggesting that both Smad2 and Smad3 are involved in mediating fibronectin transcription in cardiac fibroblasts. In contrast, Smad2 and Smad3 had distinct effects on synthesis of matricellular genes. Tenascin-C expression was significantly reduced in Smad2 and Smad2/Smad3 KD cells, but not in Smad3 KD fibroblasts (Figure 4B). Effects of Smad2 and Smad3 KD on SPARC expression were not statistically significant (Figure 4C). Smad3 KD significantly increased osteopontin/Spp1 mRNA expression (Figure 4D). Smad2KD and combined Smad2/Smad3 KD, but not Smad3 KD attenuated expression of periostin (Figure 4E). Versican expression was attenuated in Smad2, but not in Smad3 KD cells (Figure 4F). Thrombospondin (TSP)-1/Thbs1 was the highest expressed TSP in cultured cardiac fibroblasts. Smad3 KD and combined Smad2/Smad3 KD, but not Smad2 KD significantly reduced TSP-1 synthesis (Figure 4G). In contrast, effects of Smad2 and Smad3 KD on TSP-2/Thbs2 expression did not reach statistical significance (Figure 4H). Low levels of TSP-3/Thbs3 expression were noted in cardiac fibroblasts; Smad2 KD, but not Smad3 KD increased TSP-3 expression levels (Figure 4I).
Figure 4: Effects of Smad2 and Smad3 on fibroblast expression of fibronectin and matricellular genes.
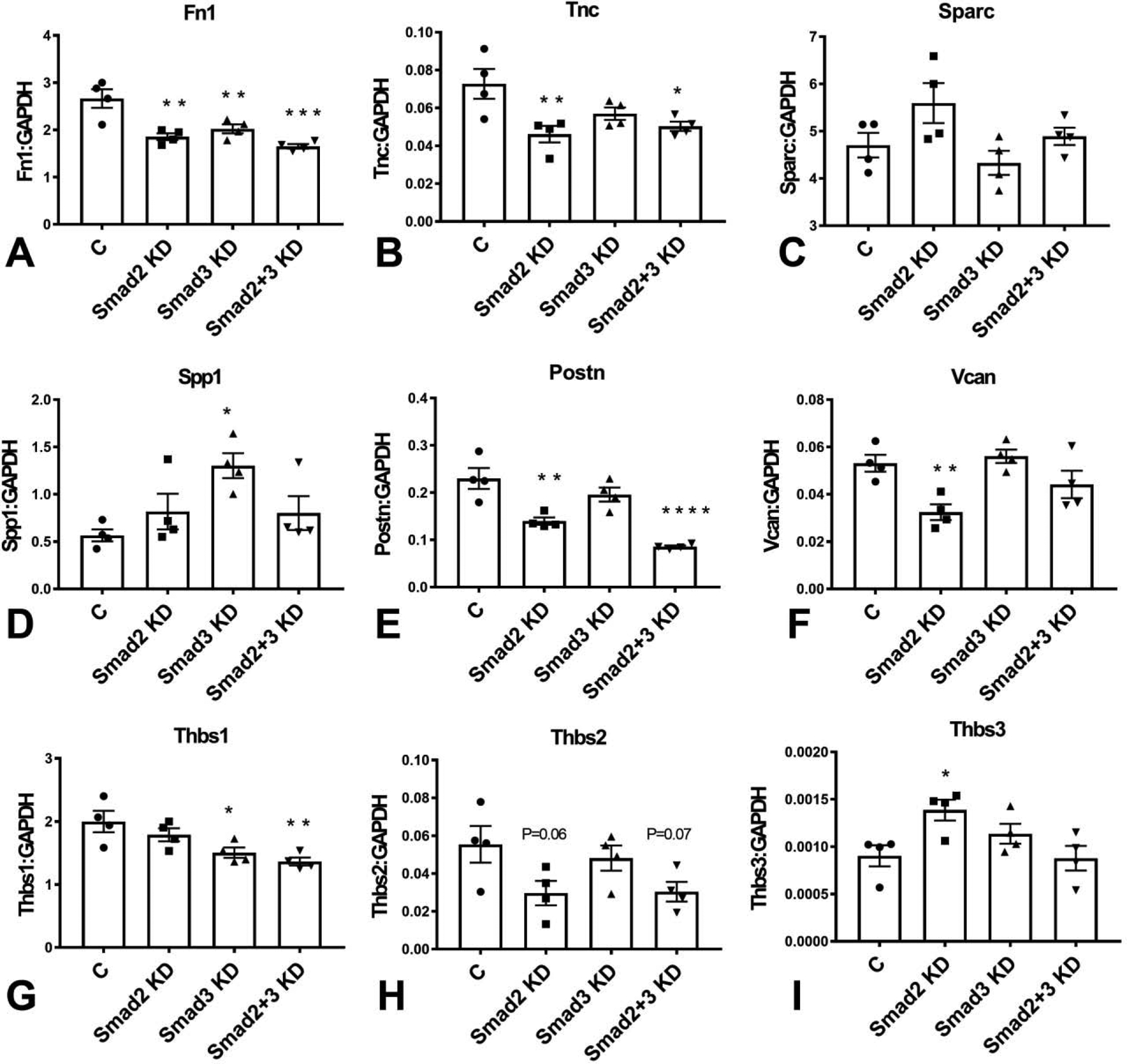
Smad2, Smad3 and combined Smad2/Smad3 KD attenuated expression of fibronectin mRNA (Fn1) in cardiac fibroblasts (A). Tenascin-C (TnC) expression was significantly reduced in Smad2 and Smad2/Smad3 KD cells, but not in Smad3 KD fibroblasts (B). Effects of Smad2 and Smad3 KD on SPARC expression were not statistically significant (C). Smad3 KD significantly increased osteopontin/Spp1 mRNA expression (D). Smad2KD and combined Smad2/Smad3 KD, but not Smad3 KD attenuated expression of periostin (Postn, E). Versican (Vcan) expression was attenuated in Smad2, but not in Smad3 KD cells (F). Smad3 KD and combined Smad2/Smad3 KD, but not Smad2 KD significantly reduced TSP-1 synthesis (G). In contrast, effects of Smad2 and Smad3 KD on TSP-2/Thbs2 expression did not reach statistical significance (H). Smad2 KD, but not Smad3 KD increased TSP-3/Thbs3 expression levels (I). (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 vs. control (C), n=4/group, ANOVA followed by Dunnett’s multiple comparisons test).
3.6. Effects of Smad2 and Smad3 on fibroblast expression of proteases.
Cardiac fibroblasts expressed high levels of several proteases, including members of the ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) and matrix metalloproteinase (MMP) families. Smad3 loss significantly reduced baseline mRNA expression of ADAMTS1 and ADAMTS5. In contrast, Smad2 KD did not affect ADAMTS1 levels, but significantly increased ADAMTS5 expression (Figure 5A–B). Smad2 and Smad3 had distinct effects in regulation of MMP and TIMP mRNA synthesis. Smad2 KD increased MMP1a, MMP2 and MMP14 mRNA expression, suggesting that Smad2-mediated actions restrain MMP synthesis (Figure 5C–E). In contrast, Smad3 KD had no effects on MMP1a, but reduced MMP2 expression and increased MMP14 synthesis (Figure 5C–E). Smad3, but not Smad2 disruption significantly reduced TIMP3 expression (Figure 5F)
Figure 5: Effects of Smad2 and Smad3 on fibroblast expression of ADAMTS and MMP family members.
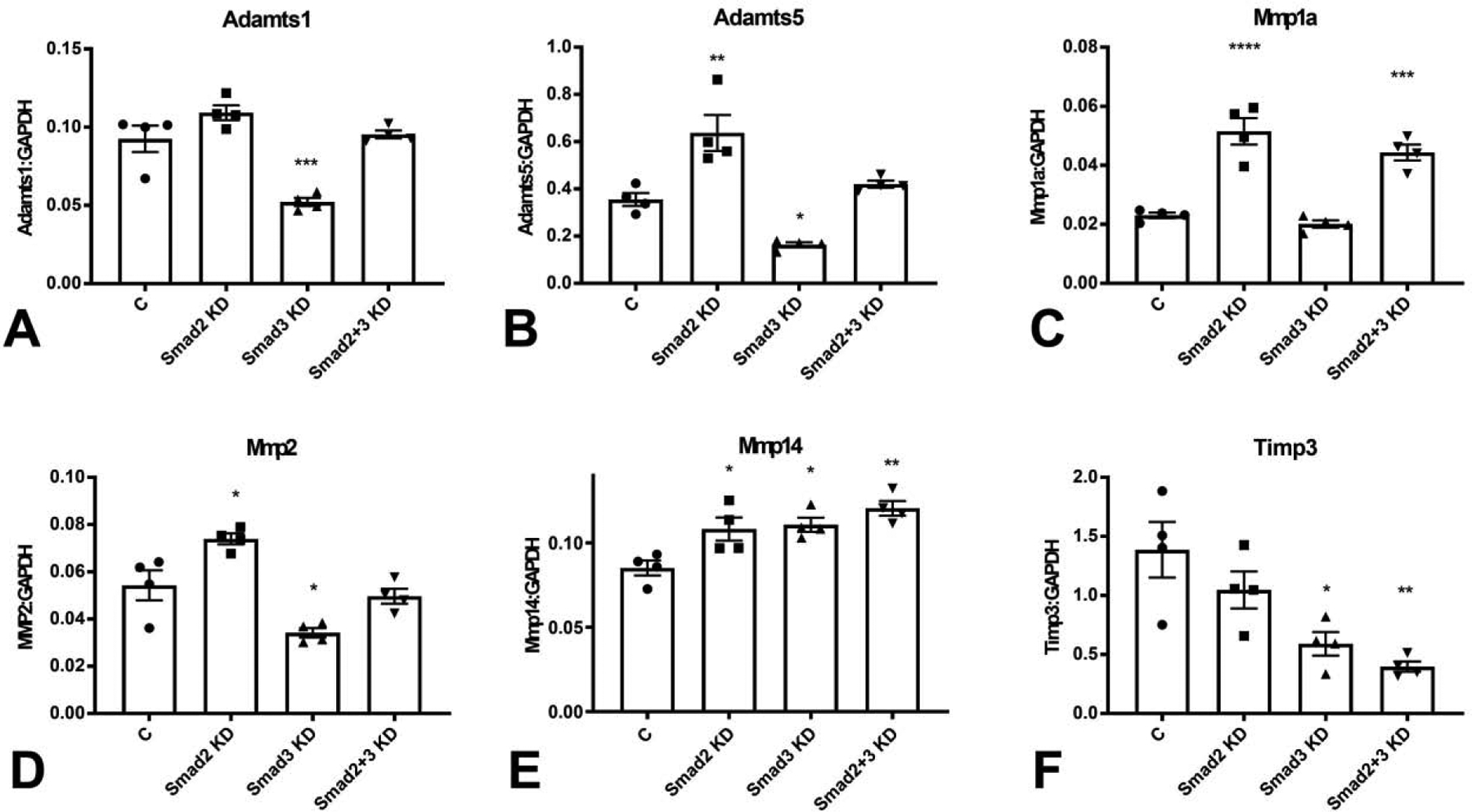
Smad3 KD significantly reduced Adamts1 (A) and Adamts5 (B) mRNA expression levels. In contrast, Smad2 KD had no effects on Adamts1 expression and accentuated Adamts5 synthesis (A–B). Smad2 KD increased expression of MMP1a (C), and MMP2 (D). In contrast, Smad3 KD had no effects on MMP1a expression, and reduced MMP2 levels (C–D). Smad2 and Smad3 KD significantly increased MMP14 expression (E). TIMP3 levels were markedly reduced following Smad3 KD (F). (*p<0.05, **p<0.01, ***p<0.001, vs. control (C), n=4/group, ANOVA followed by Dunnett’s multiple comparisons test).
3.7. Effects of Smad2 and Smad3 on fibroblast adhesion molecule expression.
Expression of adhesion molecules by fibroblasts modulates their matrix-synthetic and matrix remodeling properties (37). Both Smad2 and Smad3 mediated ICAM-1 synthesis by cardiac fibroblasts. Smad2 KD, Smad3 KD and cells with combined Smad2 and Smad3 KD had significantly attenuated expression of ICAM-1 (Figure 6A). Fibroblasts also exhibited significant expression of NCAM-1 (Figure 6B) and VCAM-1 (Figure 6C). Smad2 KD, but not Smad3 KD reduced NCAM-1 mRNA expression. In contrast, Smad2 KD increased VCAM-1 mRNA levels, suggesting that SMad2 signaling may restrain VCAM-1 synthesis. CD44 has been critically implicated in fibroblast activation (38), serving as a ligand for hyaluronan (39) and osteopontin (40), and accentuating TGF-β signaling. Smad3 KD, but not Smad2 KD significantly attenuated CD44 expression in cardiac fibroblasts (Figure 6D).
Figure 6: Effects of Smad2 and Smad3 loss on fibroblast expression of adhesion molecules.
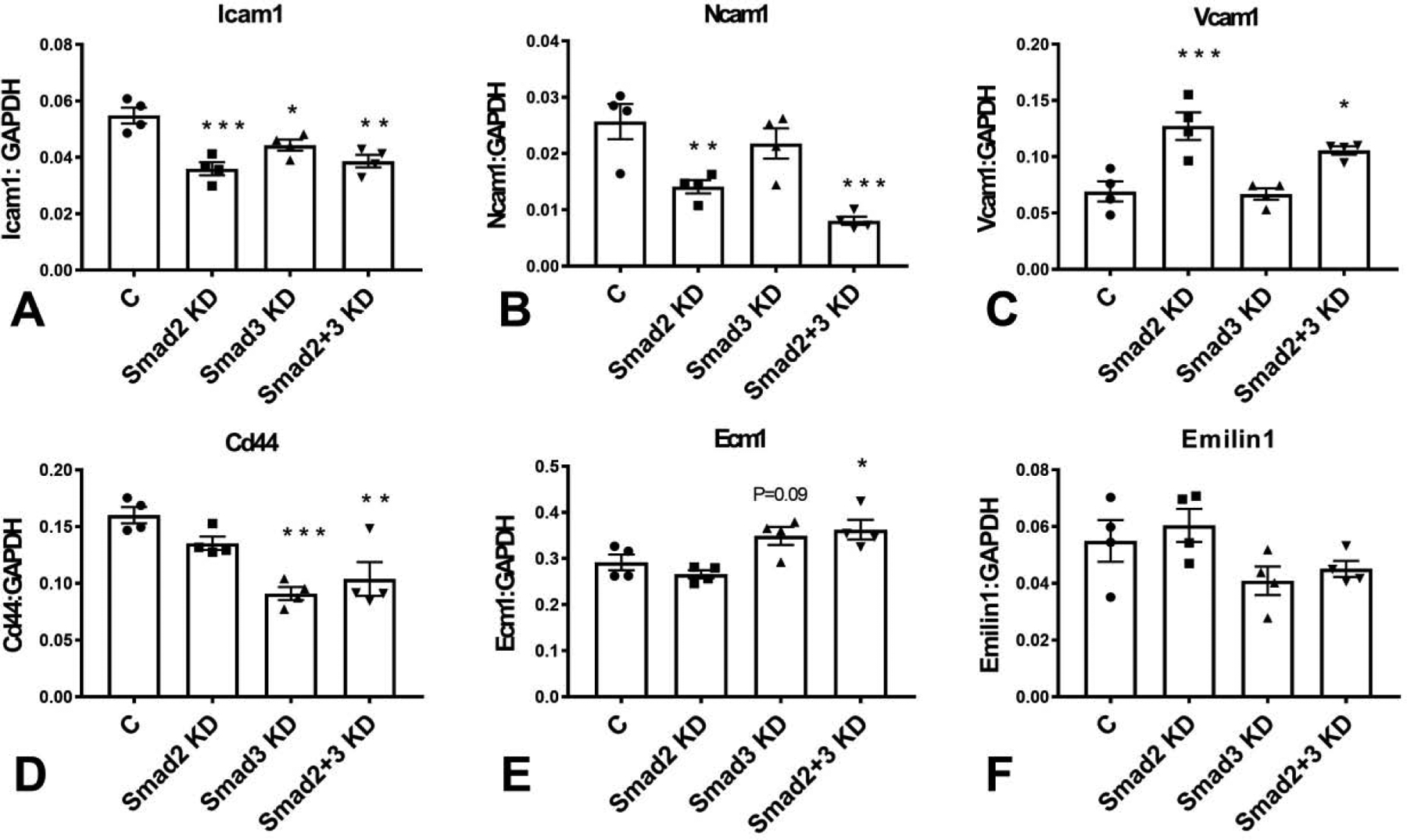
Both Smad2 and Smad3 mediated ICAM-1 synthesis by cardiac fibroblasts. Smad2 KD, Smad3 KD and cells with combined Smad2 and Smad3 KD had significantly attenuated expression of ICAM-1 (A). Fibroblasts also exhibited significant expression of NCAM-1 (B) and VCAM-1 (C). Smad2 KD, but not Smad3 KD reduced NCAM-1 mRNA expression. In contrast, Smad2 KD increased VCAM-1 mRNA levels, suggesting that Smad2 signaling may restrain VCAM-1 synthesis. Smad3 KD, but not Smad2 KD significantly attenuated CD44 expression in cardiac fibroblasts (D). Cardiac fibroblasts synthesized significant amounts of ECM-1 mRNA. Neither Smad2 nor Smad3 KD had significant effects on ECM1 expression; however, combined KD of Smad2 and Smad3 increased ECM1 mRNA levels (E). Cardiac fibroblasts expressed significant amounts of EMILIN-1; however, Smad2 and Smad3 KD did not affect EMILIN-1 expression (F). (*p<0.05, **p<0.01, ***p<0.001 vs control (C), n=4/group, ANOVA followed by Dunnett’s multiple comparisons test).
3.8. Effects of Smad2 and Smad3 on Ecm1 and emilin1 expression.
The extracellular matrix protein ECM1 is upregulated in aging hearts and has been suggested to act as a fibroblast activator(41). Cardiac fibroblasts synthesized significant amounts of ECM-1 mRNA. Neither Smad2 nor Smad3 KD had significant effects on ECM1 expression; however, combined KD of Smad2 and Smad3 increased ECM1 mRNA levels (Figure 6E). Emilin-1 is a member of the emilin family of extracellular matrix proteins, involved in elastogenesis and in regulation of fibroblast proliferation. Cardiac fibroblasts expressed significant amounts of EMILIN-1; however, Smad2 and Smad3 KD did not affect EMILIN-1 expression (Figure 6F).
3.9. Generation of mice with fibroblast-specific Smad2 and Smad3 loss.
Our in vitro studies suggested an important role of Smad2 and Smad3 in regulating matrix gene transcription in cardiac fibroblasts cultured in serum. In order to test the in vivo role of Smad2 and Smad3 in regulating structure and function of the adult heart, we generated mice with inducible fibroblast-specific loss of Smad2 and Smad3 (FS2KO and FS3KO mice respectively) using the Collagen I α2-CreERT driver. In order to document cell-specific loss of Smad2 and Smad3, cardiac fibroblasts were harvested from the hearts of FS2KO, Smad2 fl/fl, FS3KO and Smad3 fl/fl mice, 4 weeks after tamoxifen injection. Subsequently, hearts were digested with Liberase and cell suspensions were seeded in dishes with DMEM/F12 plus 10% FBS. Medium was changed every 48h until fibroblasts grew to 80%−90% confluence. Documentation of Smad2 and Smad3 loss were performed on cells at passage 1. FS2KO cardiac fibroblasts had a significant 50% reduction in Smad2 mRNA and protein expression, when compared with cardiac fibroblasts harvested from Smad2 fl/fl hearts (Figure 7A–C). In contrast, fibroblasts from FS3KO and Smad3 fl/fl mice had comparable Smad2 expression levels (Figure 7A, D–E). FS3KO mice had a statistically significant 60% reduction in Smad3 mRNA and protein levels (Figure 7F, I–J). Smad3 expression was comparable between FS2KO and Smad2 fl/fl cardiac fibroblasts (Figure 7F, G–H).
Figure 7: Documentation of fibroblast-specific Smad2 and Smad3 loss in FS2KO and FS3KO animals.
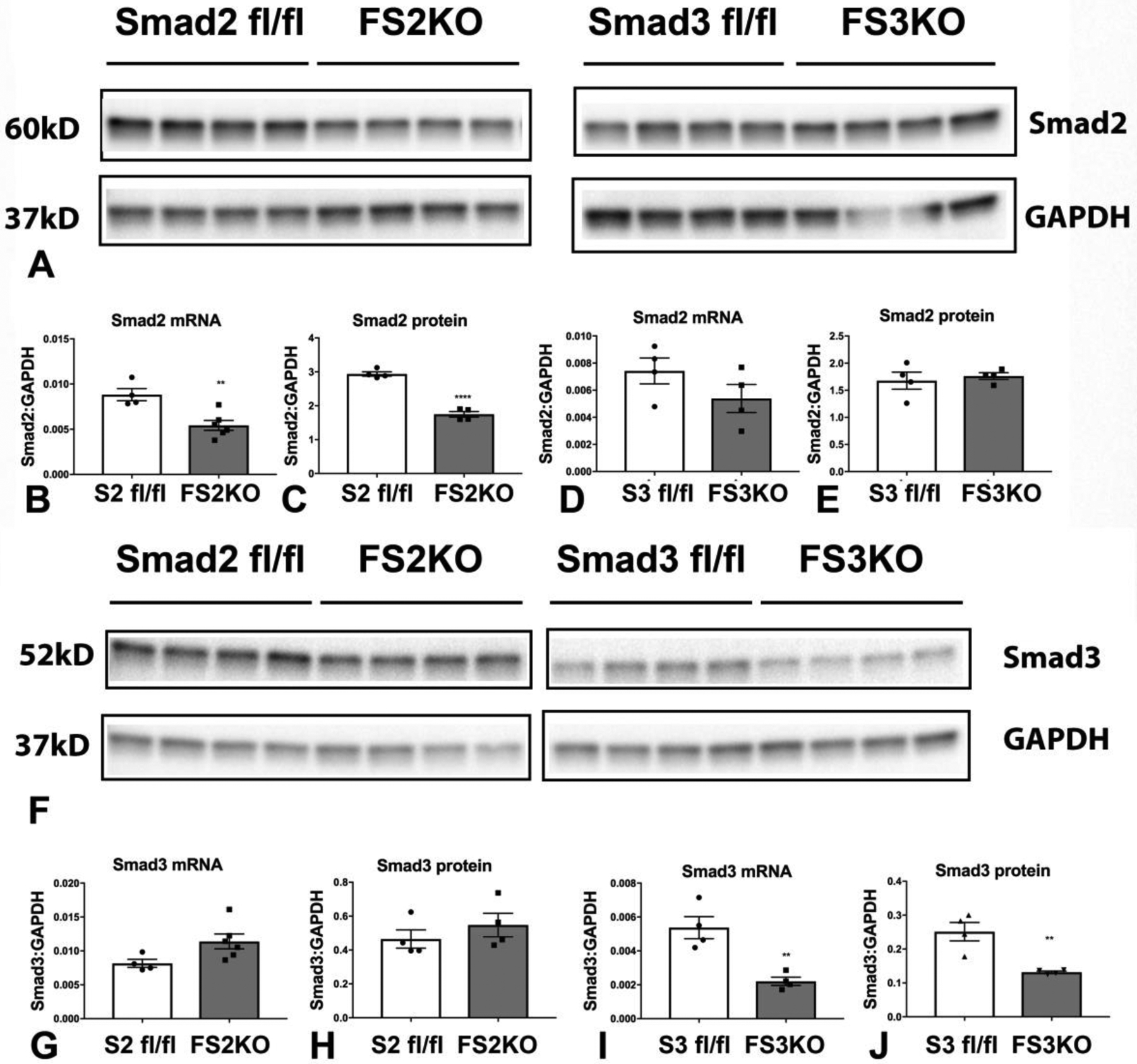
FS2KO cardiac fibroblasts had a significant 50% reduction in Smad2 mRNA and protein expression, when compared with cardiac fibroblasts harvested from Smad2 fl/fl hearts (A–C). In contrast, FS3KO fibroblasts and Smad3 fl/fl fibroblasts had comparable Smad2 expression levels (A, D–E). FS3KO mice had a statistically significant 60% reduction in Smad3 mRNA and protein levels (F, I–J). Smad3 expression was comparable between FS2KO and Smad2 fl/fl cardiac fibroblasts (F, G–H) (**p<0.01, ****p<0.0001, n=4/group, unpaired t test).
3.10. FS2KO mice have normal baseline cardiac geometry, and preserved systolic and diastolic function.
In order to examine whether fibroblast-specific Smad2 disruption affects baseline cardiac geometry and function, we performed echocardiographic analysis in FS2KO and Smad2 fl/fl mice at baseline and 4–8 weeks after tamoxifen injection. Male and female FS2KO mice had comparable left ventricular ejection fraction, LVEDV and LVESV with corresponding Smad2 fl/fl controls (Figure 8A–F). End-diastolic left ventricular anterior wall thickness was comparable between groups in both male and female mice (Figure 8G–H). Female FS2KO mice had a modest but statistically significant reduction in left ventricular posterior wall thickness (*p<0.05), when compared with age-and sex-matched Smad2 fl/fl controls (Figure 8J). Dimensions of the ascending aorta were comparable between groups (Figure 8K–L). No significant differences in heart rate were noted (Figure 8M–N). Tissue Doppler imaging showed that FS2KO mice and corresponding Smad2 fl/fl controls had comparable E/E’ ratio, suggesting that Smad2 loss in cardiac fibroblasts does not significantly affect baseline diastolic function (Figure 8O–P).
Figure 8: Fibroblast-specific Smad2 loss does not affect baseline cardiac geometry, systolic and diastolic function.
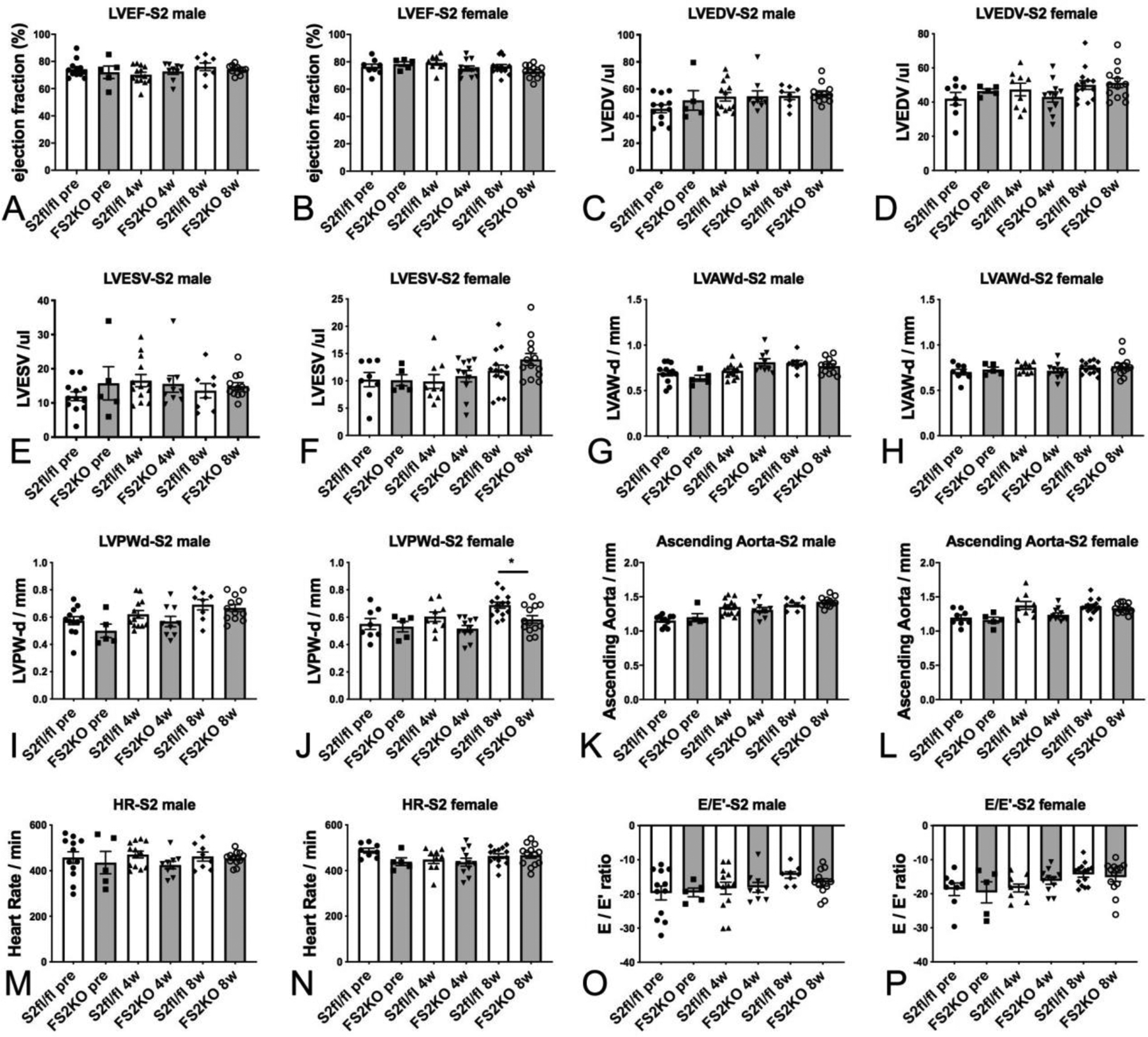
Echocardiographic analysis was performed in FS2KO and Smad2 fl/fl mice at baseline and 4–8 weeks after tamoxifen injection. Male and female FS2KO mice had comparable LVEF, LVEDV and LVESV with corresponding Smad2 fl/fl controls (A–F). End-diastolic left ventricular anterior wall thickness was comparable between groups in both male and female mice (G–H). Female FS2KO mice had a modest but statistically significant reduction in left ventricular posterior wall thickness (*p<0.05), when compared with Smad2 fl/fl controls (J). Dimensions of the ascending aorta were comparable between groups (K–L). No significant differences in heart rate were noted (M–N). Tissue Doppler imaging showed that FS2KO mice and corresponding Smad2 fl/fl controls had comparable E/E’ ratio, suggesting that Smad2 loss in cardiac fibroblasts does not significantly affect baseline diastolic function (O–P). Sample size: Smad2 fl/fl (males, n=13; females, n=14), FS2KO (males, n=12; females, n=13). ANOVA followed by Sidak’s multiple comparisons test.
3.11. FS3KO mice have normal left ventricular geometry and preserved systolic and diastolic function.
In order to examine the effects of fibroblast-specific Smad3 disruption on cardiac geometry and function, we compared echocardiographic endpoints between FS3KO and Smad3 fl/fl mice at baseline and 4–8 weeks after tamoxifen injection. Male and female FS3KO mice had comparable left ventricular ejection fraction, LVEDV, LVESV, and end-diastolic left ventricular anterior and posterior wall thickness with corresponding Smad3 fl/fl controls (Figure 9A–J). Dimensions of the ascending aorta (Figure 9K–L) and heart rate (Figure 9M–N) were also comparable between groups. Tissue Doppler imaging showed that FS3KO mice and corresponding Smad3 fl/fl controls had comparable E/E’ ratio, suggesting that Smad3 loss in cardiac fibroblasts does not significantly affect baseline diastolic function (Figure 9O–P).
Figure 9: Fibroblast-specific Smad3 loss does not affect baseline cardiac geometry, systolic and diastolic function.
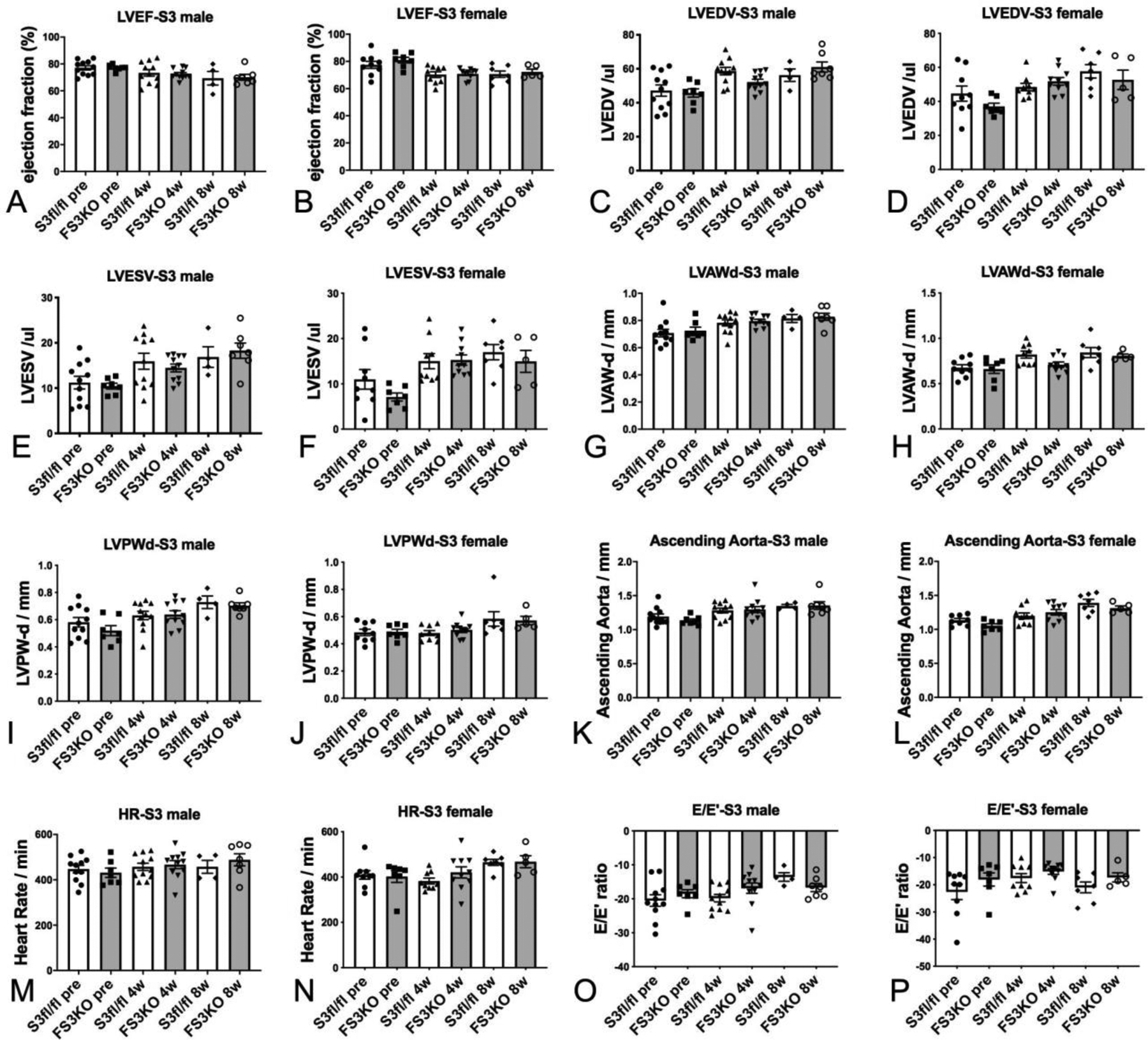
Echocardiographic endpoints were compared between FS3KO and Smad3 fl/fl mice at baseline and 4–8 weeks after tamoxifen injection. Male and female FS3KO mice had comparable LVEF, LVEDV, LVESV, and end-diastolic left ventricular anterior and posterior wall thickness with corresponding Smad3 fl/fl controls (A–J). Dimensions of the ascending aorta (K–L) and heart rate (M–N) were also comparable between groups. Tissue Doppler imaging showed that FS3KO mice and corresponding Smad3 fl/fl controls had comparable E/E’ ratio, suggesting that fibroblast-specific Smad3 loss does not significantly affect baseline diastolic function (O–P). Sample size: Smad3 fl/fl (male, n=11; female, n=9), FS3KO mice (male, n=11; female, n=10). ANOVA followed by Sidak’s multiple comparisons test.
3.12. Fibroblast-specific Smad3, but not Smad2, loss reduces collagen content in uninjured myocardium.
In order to examine the effects of fibroblast-specific Smad2 and Smad3 on the extracellular matrix network, we stained myocardial sections with Sirius red to label collagen fibers. 8 weeks after tamoxifen injection, FS2KO and FS3KO mice appeared to have normal patterns of endomysial, perimysial and perivascular collagen (Figure 10A–H). In order to perform unbiased quantitative analysis of collagen content with validated a machine learning approach using Zen intellesis software (Figure 10I–K). Quantitation of collagen content showed that FS2KO and Smad2 fl/fl controls had comparable myocardial collagen content (Figure 10L). In contrast, FS3KO mice had a modest, but significant reduction in myocardial collagen content when compared with Smad3 fl/fl animals (Figure 10M), suggesting that Smad3 may play a role in regulation of tissue collagen content under homeostatic conditions. In order to examine whether these changes are associated with alterations in fibroblast expression of matrix genes, we compared matrix gene expression between fibroblasts harvested from FS3KO and FS2KO mice and corresponding controls (Figure 11, Tables 2 and 3). Cardiac fibroblasts from FS3KO mice had significantly reduced baseline expression of Col2a1 (Figure 11C), Col4a1 (Figure 11G), Col4a2 (Figure 11I), thrombospondin-1 (Figure 11M) and TIMP3 (Figure 11O), and trends towards decreased expression levels of Col1a1, Col3a1 and Col5a1 that did not reach statistical significance (Figure 11A, E, K). These findings supported the notion that fibroblast Smad3 plays a role in regulation of baseline matrix synthesis in normal adult hearts. In contrast, fibroblasts from FS2KO only exhibited a modest but significant reduction in TSP-1 expression levels (Figure 11N), without any significant changes in expression levels of any other matrix genes (Figure 11). Thus, the findings of the fibroblast-specific analysis of gene expression were consistent with the histological quantification of collagen, suggesting that Smad3, but not Smad2, may play a role in regulation of baseline matrix gene expression in adult mice.
Figure 10: Inducible fibroblast-specific Smad3 loss reduces myocardial collagen content.
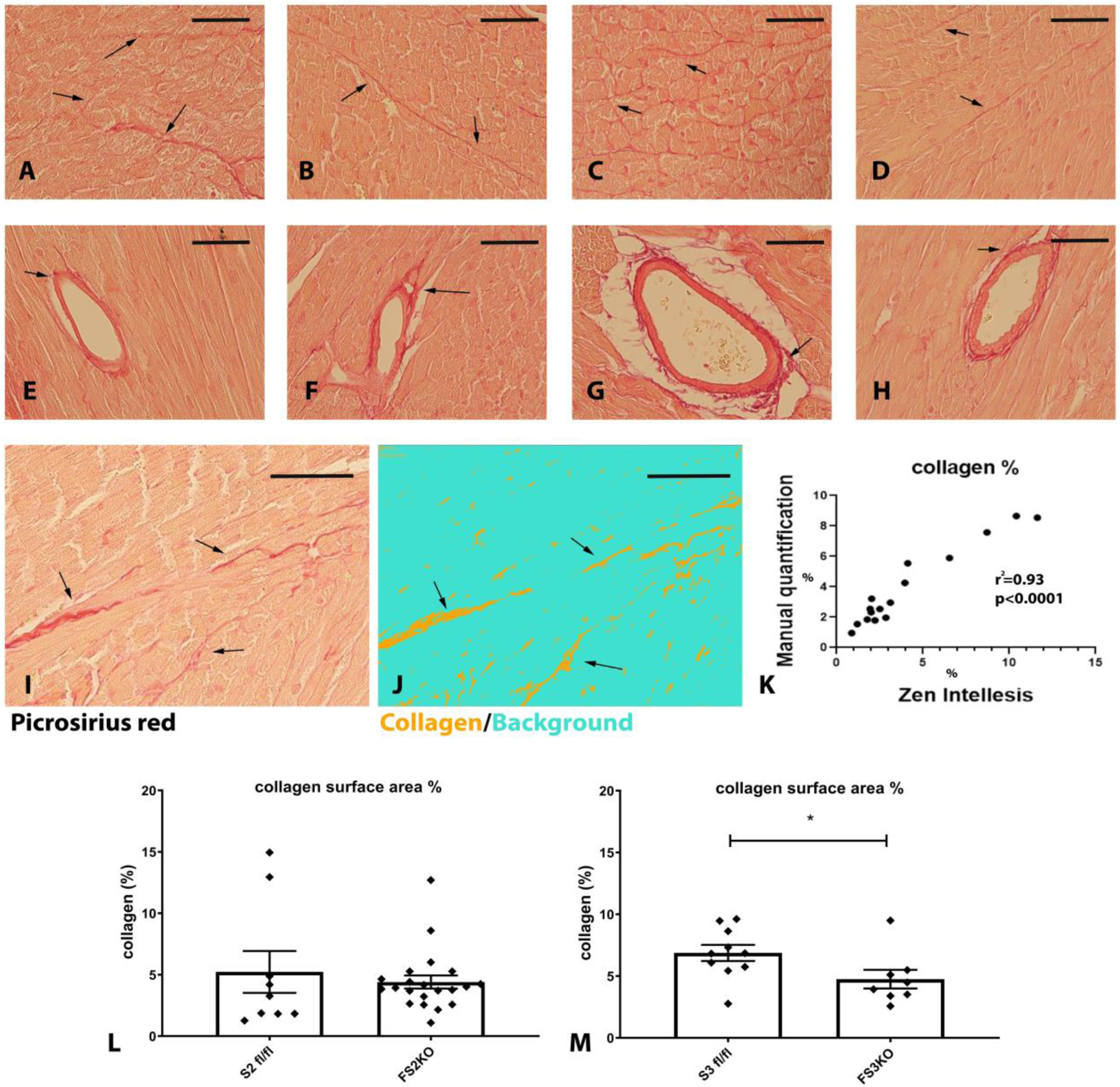
Myocardial sections from Smad2 fl/fl (A, E), FS2KO (B, F), Smad3 fl/fl (C, G) and FS3KO (D, H) mice were stained with picrosirius red to label interstitial (A–D) and perivascular (E–H) collagen fibers (arrows). Mice were studied 8 weeks after tamoxifen injection, FS2KO and FS3KO mice appeared to have preserved myocardial structure. Quantitative analysis of collagen content was performed using a machine learning-based system for objective unbiased analysis. I–K: The artificial intelligence-guided model was tested using 16 different field from 4 mice and showed excellent correlation (r2=0.93, p<0.0001, n=16) with manual measurements. L: FS2KO mice and Smad2 fl/fl controls had comparable myocardial collagen content (p=NS, n=9–21/group, Mann-Whitney test). M: When compared with Smad3 fl/fl animals, FS3KO mice had a modest, but significant reduction in myocardial collagen content (*p<0.05, n=8–10/group, Mann-Whitney test). Scalebar= 50μm.
Figure 11: Fibroblasts harvested from FS3KO hearts exhibit attenuated synthesis of ECM genes.
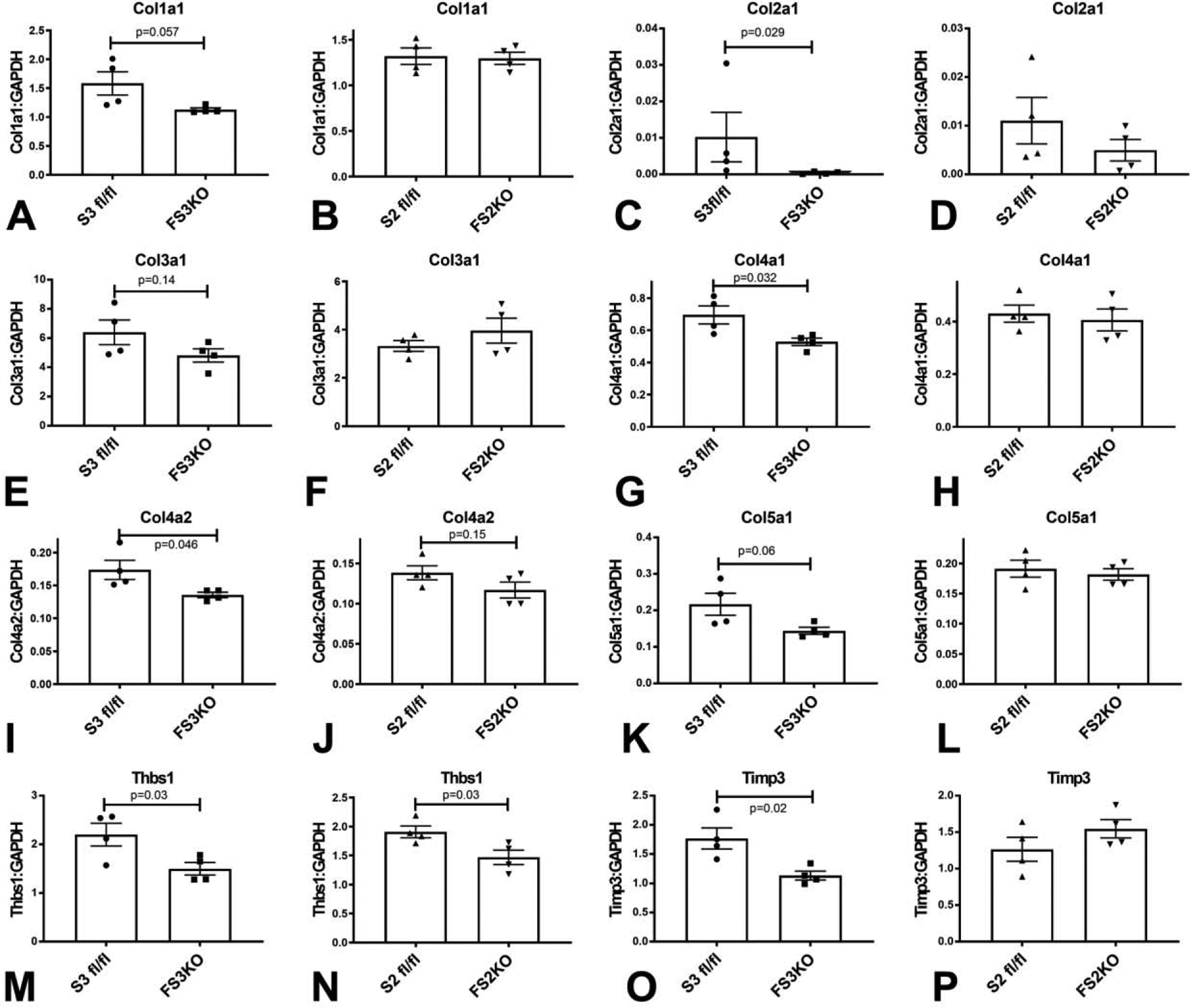
When compared with Smad3 fl/fl cardiac fibroblasts, FS3KO fibroblasts had significantly lower mRNA expression of Col2a1(C), Col4a1 (G), Col4a2 (I), Thrombospondin-1/Thbs1 (M) and TIMP3 (O), and exhibited trends towards reduced expression of Col1a1 (A), Col3a1 (E) and Col5a1 (K). In contrast, when compared with corresponding Smad2 fl/fl controls, fibroblasts harvested from FS2KO hearts had comparable expression of Col1a1 (B), Col2a1 (D), Col3a1 (F), Col4a1 (H), Col4a2 (J), Col5a1 (L) and TIMP3 (P). Thbs1 levels were significantly reduced in the absence of Smad2 (N). n=4/group. Unpaired t-test.
Table 2:
Baseline extracellular matrix gene expression levels in cardiac fibroblasts from fibroblast-specific Smad2 KO (FS2KO) mice.
| Gene name | Gene symbol | Smad2 fl/fl (mean±SEM) | FS2KO (mean±SEM) | p-value |
|---|---|---|---|---|
| A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1motif, 1 | Adamts1 | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1motif, 2 | Adamts2 | 0.1242±0.008133 | 0.1495±0.01546 | 0.1978 |
| A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1motif, 5 | Adamts5 | 0.07718±0.01298 | 0.1003±0.01372 | 0.2665 |
| A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1motif, 8 | Adamts8 | 4.250e-005±4.787e-006 | 6.750e-005±1.702e-005 | 0.2070 |
| CD44 antigen | Cd44 | 0.1194±0.003434 | 0.1327±0.007079 | 0.1422 |
| Cadherin 1 | Cdh1 | ND | ND | - |
| Cadherin 2 | Cdh2 | 0.01873±0.003922 | 0.02185±0.002256 | 0.5166 |
| Cadherin 3 | Cdh3 | 0.0001700±3.162e-005 | 0.0001100±2.799e-005 | 0.2052 |
| Cadherin 4 | Cdh4 | ND | ND | - |
| Contactin 1 | Cntn1 | ND | ND | - |
| collagen type I, alpha1 | Col1a1 | 1.322±0.09085 | 1.298±0.06676 | 0.8358 |
| collagen type II, alpha1 | Col2a1 | 0.01105±0.004770 | 0.004953±0.002213 | 0.2907 |
| collagen type III, alpha1 | Col3a1 | 3.325±0.2212 | 3.958±0.5178 | 0.3038 |
| collagen type IV, alpha1 | Col4a1 | 0.4297±0.03268 | 0.4059±0.04170 | 0.6693 |
| collagen type IV, alpha2 | Col4a2 | 0.1384±0.008707 | 0.1169±0.009966 | 0.1554 |
| collagen type IV, alpha3 | Col4a3 | 0.0001200±2.614e-005 | 0.0001700±4.163e-005 | 0.3484 |
| collagen type V, alpha1 | Col5a1 | 0.1914±0.01411 | 0.1818±0.009520 | 0.5945 |
| collagen type VI, alpha1 | Col6a1 | 0.3600±0.04680 | 0.3853±0.03039 | 0.6657 |
| Connective tissue growth factor | Ctgf | 2.578±0.2733 | 2.827±0.1393 | 0.4485 |
| Catenin (Cadherin associated protein), alpha1 | Ctnna1 | 0.08657±0.008150 | 0.1025±0.007268 | 0.1950 |
| Catenin (Cadherin associated protein), alpha2 | Ctnna2 | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| Catenin (Cadherin associated protein), beta1 | Ctnnb1 | 0.1322±0.005469 | 0.1472±0.006247 | 0.1213 |
| Extracellular matrix protein 1 | Ecm1 | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| Elastin microfibril interfacer 1 | Emilin1 | 0.08706±0.006891 | 0.09406±0.004824 | 0.4369 |
| Ectonucleoside triphosphate diphosphohydrolase 1 | Entpd1 | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| Fibulin 1 | Fbln1 | 0.008356±0.001318 | 0.007695±0.001472 | 0.7491 |
| Fibronectin 1 | Fn1 | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| Hyaluronan and proteoglycan link protein 1 | Hapln1 | ND | ND | - |
| Hemolytic complement | Hc | ND | ND | - |
| Intercellular adhesion molecule 1 | Icam1 | 0.04526±0.003715 | 0.08703±0.008140 | 0.0034† |
| Integrin alpha2 | Itga2 | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| Integrin alpha3 | Itga3 | 0.006343±0.0004632 | 0.006505±0.0006678 | 0.8489 |
| Integrin alpha4 | Itga4 | 0.004250±0.0006292 | 0.008163±0.001345 | 0.0388† |
| Integrin alpha5 | Itga5 | 0.07631±0.01056 | 0.06432±0.003967 | 0.3287 |
| Integrin alpha1, epithelial-associated | Itgae | ND | ND | - |
| Integrin alpha L | Itgal | 0.0002500±8.165e-006 | 0.0002975±3.705e-005 | 0.2922 |
| Integrin alpha M | Igam | 2.250e-005±9.465e-006 | 4.250e-005±1.887e-005 | 0.3801 |
| Integrin alpha V | Itgav | 0.07016±0.01001 | 0.07201±0.007132 | 0.8854 |
| Integrin alpha X | Igtax | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| Integrin beta1 | Itgb1 | 0.3718±0.03817 | 0.4283±0.02015 | 0.2390 |
| Integrin beta2 | Itgb2 | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| Integrin beta3 | Itgb3 | 0.007356±0.0006389 | 0.007835±0.0005224 | 0.6857 |
| Integrin beta4 | Itgb4 | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| Laminin,alpha1 | Lama1 | 0.0007726±7.470e-005 | 0.0006245±9.608e-005 | 0.6857 |
| Laminin,alpha2 | Lama2 | 0.008000±0.001080 | 0.008758±0.001548 | 0.7020 |
| Laminin,alpha3 | Lama3 | ND | ND | - |
| Laminin, beta2 | Lamb2 | 0.1242±0.008133 | 0.1495±0.01546 | 0.1978 |
| Laminin, beta3 | Lamb3 | 0.0001549±2.649e-005 | 0.0002531±9.019e-005 | 0.3365 |
| Laminin, gama1 | Lamc1 | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| Matrix metallopeptidase 10 | Mmp10 | 0.0009981±0.0002581 | 0.0009728±5.866e-005 | 0.9291 |
| Matrix metallopeptidase 11 | Mmp11 | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| Matrix metallopeptidase 12 | Mmp12 | 0.0001067±2.493e-005 | 0.0001167±4.918e-005 | 0.8617 |
| Matrix metallopeptidase 13 | Mmp13 | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| Matrix metallopeptidase 14 | Mmp14 | 0.07840±0.007007 | 0.07942±0.004432 | 0.9059 |
| Matrix metallopeptidase 15 | Mmp15 | 0.0007500±0.0002500 | 0.001201±0.0001180 | 0.2857 |
| Matrix metallopeptidase I, alpha | Mmp1a | 0.0005438±0.0001033 | 0.0004808±7.519e-005 | 0.6394 |
| Matrix metallopeptidase 2 | Mmp2 | 0.04275±0.006897 | 0.04616±0.003666 | 0.6781 |
| Matrix metallopeptidase 3 | Mmp3 | 0.1488±0.03419 | 0.1464±0.03906 | 0.9641 |
| Matrix metallopeptidase 7 | Mmp7 | ND | ND | - |
| Matrix metallopeptidase 8 | Mmp8 | 0.001260±0.0001659 | 0.001073±0.0002067 | 0.5064 |
| Matrix metallopeptidase 9 | Mmp9 | ND | ND | - |
| Neural cell adhesion molecule 1 | Ncam1 | 0.01511±0.001621 | 0.01639±0.002848 | 0.7091 |
| Neural cell adhesion molecule 2 | Ncam2 | ND | ND | - |
| Platelet/endothelial cell adhesion molecule 1 | Pecam1 | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| Periostin, osteoblast specific factor | Postn | 0.01174±0.002937 | 0.008773±0.001060 | 0.3781 |
| Selectin, endothelial cell | Sele | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| Selectin, lymphocyte | Sell | ND | ND | - |
| Selectin, platelet | Selp | 0.02509±0.007001 | 0.04889±0.01727 | 0.3429 |
| Sarcoglycan, epsilon | Sgce | 0.03075±0.003301 | 0.03869±0.003668 | 0.1588 |
| Secreted acidic cysteine rich glycoprotein | Sparc | 3.999±0.08188 | 4.519±0.4423 | 0.3261 |
| Sparc/osteonectin, cwcv and kazal-like domains proteoglycan 1 | Spock1 | ND | ND | - |
| Secreted phosphoprotein 1 | Spp1 | 0.1475±0.004519 | 0.1012±0.03215 | 0.2461 |
| synaptotagmin 1 | Syt1 | ND | ND | - |
| Transforming growth factor, beta induced | Tgfbi | 0.002250±0.0002500 | 0.002053±0.0004777 | 0.8000 |
| Thrombospondin 1 | Thbs1 | 1.907±0.1020 | 1.467±0.1256 | 0.0349† |
| Thrombospondin 2 | Thbs2 | 0.05250±0.008005 | 0.03626±0.002433 | 0.2000 |
| Thrombospondin 3 | Thbs3 | 0.002192±0.0001814 | 0.001634±0.0002342 | 0.1087 |
| Tissue inhibitor of metalloproteinase 1 | Timp1 | 0.05011±0.004495 | 0.05468±0.002875 | 0.4241 |
| Tissue inhibitor of metalloproteinase 2 | Timp2 | 1.399±0.05310 | 1.565±0.1505 | 0.3375 |
| Tissue inhibitor of metalloproteinase 3 | Timp3 | 1.265±0.1647 | 1.547±0.1258 | 0.2228 |
| Tenascin C | Tnc | 0.1975±0.02316 | 0.1565±0.006735 | 0.1399 |
| Vascular cell adhesion molecule 1 | Vcam1 | 0.1053±0.01070 | 0.1123±0.008587 | 0.6282 |
| Versican | Vcan | 0.01832±0.005616 | 0.01193±0.0009556 | 0.3399 |
| Vitronectin | Vtn | ND | ND | - |
P value <0.05.
ND-Not Detected
Table 3:
Baseline extracellular matrix gene expression levels in cardiac fibroblasts from fibroblast-specific Smad3 KO (FS3KO) mice
| Gene name | Gene symbol | Smad3 fl/fl (mean±SEM) | FS3KO (mean±SEM) | p-value |
|---|---|---|---|---|
| A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1motif, 1 | Adamts1 | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1motif, 2 | Adamts2 | 0.2270±0.04090 | 0.1796±0.01951 | 0.3359 |
| A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1motif, 5 | Adamts5 | 0.1784±0.02840 | 0.1528±0.01668 | 0.4669 |
| A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1motif, 8 | Adamts8 | 5.750e-005±1.109e-005 | 5.750e-005±1.315e-005 | >0.9999 |
| CD44 antigen | Cd44 | 0.1203±0.008070 | 0.1126±0.007737 | 0.5179 |
| Cadherin 1 | Cdh1 | ND | ND | - |
| Cadherin 2 | Cdh2 | 0.03067±0.004226 | 0.02008±0.001042 | 0.0510 |
| Cadherin 3 | Cdh3 | 0.0001100±2.273e-005 | 8.250e-005±3.065e-005 | 0.4982 |
| Cadherin 4 | Cdh4 | ND | ND | - |
| Contactin 1 | Cntn1 | ND | ND | - |
| collagen type I, alpha1 | Col1a1 | 1.584±0.2012 | 1.129±0.03113 | 0.0571 |
| collagen type II, alpha1 | Col2a1 | 0.01022±0.006811 | 0.0003875±0.0001616 | 0.0286† |
| collagen type III, alpha1 | Col3a1 | 6.392±0.8426 | 4.811±0.4546 | 0.1496 |
| collagen type IV, alpha1 | Col4a1 | 0.6953±0.05561 | 0.5286±0.02332 | 0.0327† |
| collagen type IV, alpha2 | Col4a2 | 0.1737±0.01462 | 0.1355±0.004078 | 0.0457† |
| collagen type IV, alpha3 | Col4a3 | 0.0001325±1.493e-005 | 7.000e-005±8.165e-006 | 0.0104† |
| collagen type V, alpha1 | Col5a1 | 0.2166±0.03011 | 0.1440±0.009342 | 0.0609 |
| collagen type VI, alpha1 | Col6a1 | 0.4923±0.03000 | 0.4311±0.03351 | 0.2223 |
| Connective tissue growth factor | Ctgf | 2.001±0.05805 | 1.694±0.2973 | 0.3795 |
| Catenin (Cadherin associated protein), alpha1 | Ctnna1 | 0.1082±0.008349 | 0.07824±0.004927 | 0.0215† |
| Catenin (Cadherin associated protein), alpha2 | Ctnna2 | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| Catenin (Cadherin associated protein), beta1 | Ctnnb1 | 0.1350±0.006237 | 0.1192±0.005547 | 0.1065 |
| Extracellular matrix protein 1 | Ecm1 | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| Elastin microfibril interfacer 1 | Emilin1 | 0.1401±0.01728 | 0.1160±0.005231 | 0.2288 |
| Ectonucleoside triphosphate diphosphohydrolase 1 | Entpd1 | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| Fibulin 1 | Fbln1 | 0.006493±0.001556 | 0.004165±0.0005900 | 0.2114 |
| Fibronectin 1 | Fn1 | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| Hyaluronan and proteoglycan link protein 1 | Hapln1 | ND | ND | - |
| Hemolytic complement | Hc | ND | ND | - |
| Intercellular adhesion molecule 1 | Icam1 | 0.05568±0.004796 | 0.05175±0.002562 | 0.4975 |
| Integrin alpha2 | Itga2 | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| Integrin alpha3 | Itga3 | 0.003108±0.0003279 | 0.002097±0.0005673 | 0.1739 |
| Integrin alpha4 | Itga4 | 0.008000±0.001954 | 0.006000±0.0009129 | 0.3895 |
| Integrin alpha5 | Itga5 | 0.04811±0.007806 | 0.03566±0.001261 | 0.2090 |
| Integrin alpha1, epithelial-associated | Itgae | ND | ND | - |
| Integrin alpha L | Itgal | 0.0005550±0.0001202 | 0.002135±0.0004533 | 0.0151† |
| Integrin alpha M | Itgam | 0.007605±0.0006380 | 0.02065±0.006379 | 0.1330 |
| Integrin alpha V | Itgav | 0.07837±0.008046 | 0.06176±0.002823 | 0.0994 |
| Integrin alpha X | Igtax | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| Integrin beta1 | Itgb1 | 0.4998±0.04867 | 0.3983±0.03534 | 0.1425 |
| Integrin beta2 | Itgb2 | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| Integrin beta3 | Itgb3 | 0.006515±0.0007208 | 0.005433±0.0005200 | 0.2691 |
| Integrin beta4 | Itgb4 | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| Laminin,alpha1 | Lama1 | 0.0006850±0.0004440 | 0.0003373±0.0001388 | 0.8857 |
| Laminin,alpha2 | Lama2 | 0.01510±0.002387 | 0.01050±0.001041 | 0.1276 |
| Laminin,alpha3 | Lama3 | ND | ND | - |
| Laminin, beta2 | Lamb2 | 0.2270±0.04090 | 0.1796±0.01951 | 0.3359 |
| Laminin, beta3 | Lamb3 | 0.0003100±5.642e-005 | 0.0003403±4.560e-005 | 0.6912 |
| Laminin, gama1 | Lamc1 | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| Matrix metallopeptidase 10 | Mmp10 | 0.0009200±0.0002996 | 0.0007273±7.612e-005 | 0.5724 |
| Matrix metallopeptidase 11 | Mmp11 | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| Matrix metallopeptidase 12 | Mmp12 | 0.02601±0.0005370 | 0.02907±0.007396 | 0.7067 |
| Matrix metallopeptidase 13 | Mmp13 | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| Matrix metallopeptidase 14 | Mmp14 | 0.08783±0.006574 | 0.08574±0.006208 | 0.8857 |
| Matrix metallopeptidase 15 | Mmp15 | 0.0005925±7.157e-005 | 0.0005000±0.0002887 | >0.9999 |
| Matrix metallopeptidase I, alpha | Mmp1a | 0.007983±0.002195 | 0.006278±0.001565 | 0.5505 |
| Matrix metallopeptidase 2 | Mmp2 | 0.04454±0.006701 | 0.04175±0.001250 | 0.7081 |
| Matrix metallopeptidase 3 | Mmp3 | 0.3447±0.08269 | 0.3010±0.07823 | 0.7144 |
| Matrix metallopeptidase 7 | Mmp7 | ND | ND | - |
| Matrix metallopeptidase 8 | Mmp8 | 0.004628±0.001785 | 0.002308±0.0005025 | 0.2574 |
| Matrix metallopeptidase 9 | Mmp9 | ND | ND | - |
| Neural cell adhesion molecule 1 | Ncam1 | 0.01641±0.003805 | 0.008610±0.0006935 | 0.1313 |
| Neural cell adhesion molecule 2 | Ncam2 | ND | ND | - |
| Platelet/endothelial cell adhesion molecule 1 | Pecam1 | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| Periostin, osteoblast specific factor | Postn | 0.01017±0.001744 | 0.005487±0.001245 | 0.0714 |
| Selectin, endothelial cell | Sele | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| Selectin, lymphocyte | Sell | ND | ND | - |
| Selectin, platelet | Selp | 0.01438±0.001676 | 0.008881±0.0005781 | 0.0211† |
| Sarcoglycan, epsilon | Sgce | 0.03166±0.002014 | 0.03925±0.002428 | 0.0571 |
| Secreted acidic cysteine rich glycoprotein | Sparc | 4.222±0.3581 | 3.930±0.2464 | 0.5262 |
| Sparc/osteonectin, cwcv and kazal-like domains proteoglycan 1 | Spock1 | ND | ND | - |
| Secreted phosphoprotein 1 | Spp1 | 0.2215±0.04578 | 0.08450±0.03218 | 0.0499† |
| synaptotagmin 1 | Syt1 | ND | ND | - |
| Transforming growth factor, beta induced | Tgfbi | 0.002168±0.0003201 | 0.004750±0.001031 | 0.0538 |
| Thrombospondin 1 | Thbs1 | 2.199±0.2340 | 1.497±0.1276 | 0.0389† |
| Thrombospondin 2 | Thbs2 | 0.05461±0.01732 | 0.03425±0.005618 | 0.6857 |
| Thrombospondin 3 | Thbs3 | 0.0009700±0.0001891 | 0.0006808±0.0001137 | 0.2378 |
| Tissue inhibitor of metalloproteinase 1 | Timp1 | 0.08552±0.008394 | 0.08235±0.004471 | 0.7504 |
| Tissue inhibitor of metalloproteinase 2 | Timp2 | 1.001±0.04551 | 1.126±0.1519 | 0.4593 |
| Tissue inhibitor of metalloproteinase 3 | Timp3 | 1.766±0.1795 | 1.133±0.07485 | 0.0173† |
| Tenascin C | Tnc | 0.1372±0.01384 | 0.1166±0.006884 | 0.2324 |
| Vascular cell adhesion molecule 1 | Vcam1 | 0.05594±0.009035 | 0.04700±0.002160 | 0.4002 |
| Versican | Vcan | 0.008155±0.001039 | 0.005431±0.0008986 | 0.0946 |
| Vitronectin | Vtn | ND | ND | - |
P value <0.05.
ND-Not Detected
4. DISCUSSION:
Our study reports for the first time that Smad2 and Smad3 play important and distinct roles in regulation of baseline ECM gene synthesis in cultured cardiac fibroblasts. Moreover, in adult mouse hearts fibroblast-specific Smad3, but not Smad2 is involved in regulation of collagen content. Although mice with fibroblast-specific Smad3 disruption had reduced myocardial collagen content and exhibited attenuated expression of several ECM genes in cardiac fibroblasts, these perturbations had no short-term consequences on left ventricular function.
4.1. Smad2 and Smad3 signaling activate distinct aspects of the ECM transcriptome.
Both Smad2 and Smad3 have been suggested to transduce critical TGF-β-induced activating signals in fibroblasts. We have previously demonstrated that in cultured cardiac fibroblasts, Smad3 signaling stimulates TGF-β-driven fibroblast to myofibroblast conversion (19), and mediates, at least in part, the matrix-synthetic and matrix-preserving fibroblast response triggered by TGF-β (19),(21). Pro-fibrotic effects of Smad3 have also been reported in renal (42), lung (43) and dermal fibroblasts (44). In contrast, the effects of Smad2 on fibroblast phenotype have been less consistent. Some studies have reported a role for Smad2 in mediating TGF-β-driven ECM gene synthesis by renal fibroblasts (45). Other investigations suggested that Smad2 is not involved in regulation of ECM gene synthesis by activated myofibroblasts (46), and have suggested that Smad2 actions may oppose the fibrogenic effects of Smad3 (47). Differences in the strategies used to disrupt Smad2 and Smad3, study of distinct fibrosis-associated genes that may be differentially regulated by Smad2 and Smad3, and the heterogeneity of fibroblasts harvested from various tissues may account for the conflicting findings. Our study suggests that in isolated cardiac fibroblasts cultured in the presence of serum, both Smad2 and Smad3 are implicated in baseline ECM gene expression. Smad2 mediated Col5a1, Lama2, TnC, Postn and Vcan synthesis, whereas Smad3 stimulated Col1a1, Col4a1 and Thbs1 expression (Figures 2–4). Fibronectin (Fn1) gene synthesis was mediated through both Smad2 and Smad3 (Figure 4A). Our findings do not support the notion that Smad2 may oppose actions of Smad3 on ECM gene synthesis. The distinct targets of Smad2 and Smad3 may explain conflicting conclusions between various in vitro studies that may have assessed effects on the fibrogenic profile of fibroblasts on the basis of expression of selected ECM genes.
4.2. The role of fibroblasts in cardiac homeostasis.
In developing hearts, fibroblasts have been suggested to play a role in formation of myocardial tissue (48),(49). However, whether fibroblasts in adult hearts play an important role in regulation of cardiac structure and function remains unknown. The adult mammalian heart contains a large population of fibroblasts (1), predominantly derived from epicardial sources (7),(8). In the adult myocardium, cardiac fibroblasts may regulate homeostatic function through several different pathways. First, continuous secretion of ECM proteins by fibroblasts may be important in preservation of the matrix network, providing mechanical support to the myocardium. Second, fibroblasts express gap junction proteins (such as Cx43 and Cx45) (50), and may be involved in regulation of cardiac electrical activity. Third, through their close spatial association with cardiomyocytes and vascular cells, fibroblasts may support cardiomyocyte survival and contractile function, and may control the density and function of the microvasculature. The signals involved in baseline regulation of fibroblast function are poorly understood. Fibroblast-specific loss-of-function approaches suggest that both activating and inhibitory signals may be involved in regulation of fibroblast populations in adult hearts. Platelet-derived growth factor receptor (PDGFR)-α signaling is critically involved in maintenance of the fibroblast population in adult mouse hearts (51). On the other hand, constitutive activity of the Hippo pathway kinases large tumor suppressor kinase (LATS)1 and LATS2 promotes fibroblast quiescence in the myocardium (52), suggesting that maintaining the resting fibroblast state is an active process.
4.3. Fibroblast-specific Smad3 regulates collagen content in the adult mouse heart
In vivo, inducible fibroblast-specific disruption of Smad3 significantly reduced myocardial collagen content (Figure 10). In contrast, fibroblast-specific Smad2 loss had no significant effects on myocardial collagen levels. Moreover cardiac fibroblasts harvested from FS3KO mice exhibited lower levels of expression of several ECM genes (Figure 11). The effects of Smad3 loss on ECM gene synthesis were less impressive in cells harvested from FS3KO animals (Figure 11), when compared to the effects of Smad3 siRNA KD (Figures 2–7). This may be due to the higher efficiency of siRNA KD, or may reflect compensatory mechanisms that may preserve ECM synthesis in vivo. Taken together, our findings suggest that Smad3, but not Smad2 plays a role in maintaining ECM synthesis in adult hearts.
4.4. Attenuated collagen deposition in FS3KO mice has no short-term impact on cardiac systolic and diastolic function
Despite the reduction in myocardial collagen content in FS3KO hearts, fibroblast-specific Smad3 loss had no effects on cardiac systolic and diastolic function (Figure 9). Our observations and the recently reported absence of systolic dysfunction in fibroblast-specific PDGFR-α KO mice (despite a prolonged reduction in fibroblast density) (51) are consistent with a limited role for baseline fibroblast actions in function of the adult heart. Moreover, global loss of the transcription factor scleraxis, an important regulator of fibroblast function had no effects on echocardiographically-assessed left ventricular function (53),(54), despite a marked reduction in fibroblast density and activity (53). Thus, low level baseline activity of cardiac fibroblasts may be sufficient to preserve the structural integrity and function of the adult mammalian heart. Although this is a plausible interpretation of the data, several limitations of our study preclude definitive conclusions regarding the absence of a homeostatic role of fibroblast-specific R-Smads. First, using the Col1a2-CreERT driver, we achieved only 50–60% loss of Smad2 and Smad3 in cardiac fibroblasts. Residual Smad2/3 expression may be sufficient to support fibroblast function and preserve myocardial homeostasis. Second, functional and histological analyses were performed 8 weeks after tamoxifen administration for cell-specific Smad2 and Smad3 deletion. The relatively long half-life of many ECM proteins in normal adult hearts (55) may limit the consequences of reduced ECM gene transcription on cardiac function. Longer intervals of fibroblast-specific R-Smad deletion may be associated with significant changes in ECM structure and cardiac function. Third, we did not examine the effects of combined fibroblast-specific Smad2 and Smad3 loss in vivo. Disruption of both pathways may cause more pronounced perturbations in the ECM network that could be associated with dysfunction. Fourth, in young animals, a relatively modest attenuation of collagen content may not significantly affect myocardial mechanisms. However, the effects of fibroblast-specific Smad3 loss may become functionally significant in aging hearts, attenuating senescence-associated collagen deposition that may contribute to diastolic dysfunction (56). Unfortunately, due to the practical challenges of cell-specific loss-of-function studies in senescent mice, the hypothesis that disruption of fibroblast activation may limit aging-associated diastolic dysfunction has not been tested.
4.5. What is the basis for the contrasting in vivo and in vitro observations?
The impressive effects of Smad2 and Smad3 KD on baseline gene expression of cultured cardiac fibroblasts seem to contrast the unimpressive in vivo consequences of fibroblast-specific Smad2 and Smad3 loss. The contrasting findings of in vitro and in vivo experiments reported in our study reflect, at least in part, the inherent problems in studying resting fibroblasts using in vitro models. Fibroblasts are highly dynamic cells; their phenotype is dependent on the culture environment. When cultured in plates, a significant proportion of cardiac fibroblasts undergoes myofibroblast conversion, thus exhibiting characteristics of injury-associated cells (57). The use of serum during the culture process may have further accentuated fibroblast activation in our experiments assessing ECM gene synthesis. Thus, the significant effects of Smad2 and Smad3 on baseline gene expression in cultured cardiac fibroblasts may reflect activation responses, rather than a role for R-Smads in fibroblast homeostasis.
4.6. Conclusions
In recent years, the cardiovascular community has focused on the role of fibroblasts in pathologic conditions, such as myocardial infarction and heart failure. This is very well-justified, considering the important role of fibroblasts in reparative, fibrotic and remodeling responses. However, the potential role of fibroblasts in cardiac homeostasis has been neglected. Our findings suggest that fibroblast-specific Smad3 signaling may play a role in regulating collagen content in normal adult hearts. Although attenuated collagen deposition in FS3KO hearts had no impact on left ventricular function in young adult mice, chronic modulation of baseline fibroblast activity may have significant effects on age-associated fibrosis and diastolic dysfunction.
HIGHLIGHTS.
Smad2 and Smad3 regulate extracellular matrix genes in cardiac fibroblasts.
Fibroblast-specific Smad3 knockout (FS3KO) mice have reduced myocardial collagen.
Decreased myocardial collagen in FS3KO mice does not affect cardiac function.
Fibroblast-specific Smad2 loss does not affect myocardial collagen content.
Fibroblast-specific Smad2 knockout mice have preserved ventricular function.
5. SOURCES OF FUNDING:
This work was supported by NIH R01 grants HL76246 and HL85440, and by Department of Defense grants PR151029, PR151134, and PR181464. Dr. Shuaibo Huang was supported by China Scholarship Council grant 201603170222. Dr. Bijun Chen and Dr. Claudio Humeres are supported by American Heart Association post-doctoral grants.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES:
- 1.Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, et al. Revisiting Cardiac Cellular Composition. Circ Res. 2016;118(3):400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Souders CA, Bowers SL, and Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105(12):1164–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tallquist MD, and Molkentin JD. Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol. 2017;14(8):484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A, Conway SJ, Graff JM, and Frangogiannis NG. Opposing Actions of Fibroblast and Cardiomyocyte Smad3 Signaling in the Infarcted Myocardium. Circulation. 2018;137(7):707–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maruyama S, Nakamura K, Papanicolaou KN, Sano S, Shimizu I, Asaumi Y, van den Hoff MJ, Ouchi N, Recchia FA, and Walsh K. Follistatin-like 1 promotes cardiac fibroblast activation and protects the heart from rupture. EMBO Mol Med. 2016;8(8):949–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bageghni SA, Hemmings KE, Yuldasheva NY, Maqbool A, Gamboa-Esteves FO, Humphreys NE, Jackson MS, Denton CP, Francis S, Porter KE, et al. Fibroblast-specific deletion of interleukin-1 receptor-1 reduces adverse cardiac remodeling following myocardial infarction. JCI Insight. 2019;5( [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, Kamran P, Muller AM, Volz KS, Tang Z, et al. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ Res. 2014;115(7):625–35. [DOI] [PubMed] [Google Scholar]
- 8.Moore-Morris T, Guimaraes-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;124(7):2921–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC JL, Aronow BJ, Tallquist MD, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nature communications. 2016;7(12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruiz-Villalba A, Simon AM, Pogontke C, Castillo MI, Abizanda G, Pelacho B, Sanchez-Dominguez R, Segovia JC, Prosper F, and Perez-Pomares JM. Interacting resident epicardium-derived fibroblasts and recruited bone marrow cells form myocardial infarction scar. J Am Coll Cardiol. 2015;65(19):2057–66. [DOI] [PubMed] [Google Scholar]
- 11.Vidal R, Wagner JUG, Braeuning C, Fischer C, Patrick R, Tombor L, Muhly-Reinholz M, John D, Kliem M, Conrad T, et al. Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight. 2019;4(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Humeres C, and Frangogiannis NG. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl Sci. 2019;4(3):449–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roche PL, Filomeno KL, Bagchi RA, and Czubryt MP. Intracellular signaling of cardiac fibroblasts. Compr Physiol. 2015;5(2):721–60. [DOI] [PubMed] [Google Scholar]
- 14.Mouton AJ, Ma Y, Rivera Gonzalez OJ, Daseke MJ 2nd, Flynn ER, Freeman TC, Garrett MR, DeLeon-Pennell KY, and Lindsey ML. Fibroblast polarization over the myocardial infarction time ontinuum shifts roles from inflammation to angiogenesis. Basic Res Cardiol. 2019;114(2):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anzai A, Choi JL, He S, Fenn AM, Nairz M, Rattik S, McAlpine CS, Mindur JE, Chan CT, Iwamoto Y, et al. The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J Exp Med. 2017;214(11):3293–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Travers JG, Kamal FA, Robbins J, Yutzey KE, and Blaxall BC. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res. 2016;118(6):1021–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanna A, and Frangogiannis NG. The Role of the TGF-beta Superfamily in Myocardial Infarction. Front Cardiovasc Med. 2019;6(140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frangogiannis NG. Transforming Growth Factor (TGF)-beta in tissue fibrosis. J Exp Med.2020;217((3)):e20190103 10.1084/jem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, and Frangogiannis NG. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. 2010;107(3):418–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, Wang XF, and Frangogiannis NG. Essential Role of Smad3 in Infarct Healing and in the Pathogenesis of Cardiac Remodeling. Circulation. 2007;116(2127–38. [DOI] [PubMed] [Google Scholar]
- 21.Russo I, Cavalera M, Huang S, Su Y, Hanna A, Chen B, Shinde AV, Conway SJ, Graff J, and Frangogiannis NG. Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program. Circ Res. 2019;124(8):1214–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biernacka A, Dobaczewski M, and Frangogiannis NG. TGF-beta signaling in fibrosis. Growth Factors. 2011;29(5):196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, Gunaje J, Otsu K, and Davis J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation. 2017;136(6):549–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bageghni SA, Hemmings KE, Zava N, Denton CP, Porter KE, Ainscough JFX, Drinkhill MJ, and Turner NA. Cardiac fibroblast-specific p38alpha MAP kinase promotes cardiac hypertrophy via a putative paracrine interleukin-6 signaling mechanism. FASEB J. 2018:fj201701455RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeglinski MR, Roche P, Hnatowich M, Jassal DS, Wigle JT, Czubryt MP, and Dixon IM. TGFbeta1 regulates Scleraxis expression in primary cardiac myofibroblasts by a Smad-independent mechanism. Am J Physiol Heart Circ Physiol. 2016;310(2):H239–49. [DOI] [PubMed] [Google Scholar]
- 26.Huang S, Chen B, Su Y, Alex L, Humeres C, Shinde AV, Conway SJ, and Frangogiannis NG. Distinct roles of myofibroblast-specific Smad2 and Smad3 signaling in repair and remodeling of the infarcted heart. J Mol Cell Cardiol. 2019;132(84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127(10):3770–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Mollmann H, et al. Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice. Circ Res. 2016;118(12):1906–17. [DOI] [PubMed] [Google Scholar]
- 29.Bujak M, Dobaczewski M, Gonzalez-Quesada C, Xia Y, Leucker T, Zymek P, Veeranna V, Tager AM, Luster AD, and Frangogiannis NG. Induction of the CXC chemokine interferon-gamma-inducible protein 10 regulates the reparative response following myocardial infarction. Circ Res. 2009;105(10):973–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackson AL, Burchard J, Leake D, Reynolds A, Schelter J, Guo J, Johnson JM, Lim L, Karpilow J, Nichols K, et al. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA. 2006;12(7):1197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lal H, Ahmad F, Zhou J, Yu JE, Vagnozzi RJ, Guo Y, Yu D, Tsai EJ, Woodgett J, Gao E, et al. Cardiac fibroblast glycogen synthase kinase-3beta regulates ventricular remodeling and dysfunction in ischemic heart. Circulation. 2014;130(5):419–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ubil E, Duan J, Pillai IC, Rosa-Garrido M, Wu Y, Bargiacchi F, Lu Y, Stanbouly S, Huang J, Rojas M, et al. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature. 2014;514(7524):585–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swonger JM, Liu JS, Ivey MJ, and Tallquist MD. Genetic tools for identifying and manipulating fibroblasts in the mouse. Differentiation. 2016;92(3):66–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biernacka A, Cavalera M, Wang J, Russo I, Shinde A, Kong P, Gonzalez-Quesada C, Rai V, Dobaczewski M, Lee DW, et al. Smad3 Signaling Promotes Fibrosis While Preserving Cardiac and Aortic Geometry in Obese Diabetic Mice. Circ Heart Fail. 2015;8(4):788–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, and Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. 2010;176(5):2177–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alex L, Russo I, Holoborodko V, and Frangogiannis NG. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol. 2018;315(4):H934–H49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsushita Y, Hasegawa M, Matsushita T, Fujimoto M, Horikawa M, Fujita T, Kawasuji A, Ogawa F, Steeber DA, Tedder TF, et al. Intercellular adhesion molecule-1 deficiency attenuates the development of skin fibrosis in tight-skin mice. J Immunol. 2007;179(1):698–707. [DOI] [PubMed] [Google Scholar]
- 38.Huebener P, Abou-Khamis T, Zymek P, Bujak M, Ying X, Chatila K, Haudek S, Thakker G, and Frangogiannis NG. CD44 Is Critically Involved in Infarct Healing by Regulating the Inflammatory and Fibrotic Response. J Immunol. 2008;180(4):2625–33. [DOI] [PubMed] [Google Scholar]
- 39.Midgley AC, Rogers M, Hallett MB, Clayton A, Bowen T, Phillips AO, and Steadman R. Transforming growth factor-beta1 (TGF-beta1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J Biol Chem. 2013;288(21):14824–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anwar A, Li M, Frid MG, Kumar B, Gerasimovskaya EV, Riddle SR, McKeon BA, Thukaram R, Meyrick BO, Fini MA, et al. Osteopontin is an endogenous modulator of the constitutively activated phenotype of pulmonary adventitial fibroblasts in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2012;303(1):L1–L11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hardy SA, Mabotuwana NS, Murtha LA, Coulter B, Sanchez-Bezanilla S, Al-Omary MS, Senanayake T, Loering S, Starkey M, Lee RJ, et al. Novel role of extracellular matrix protein 1 (ECM1) in cardiac aging and myocardial infarction. PLoS One. 2019;14(2):e0212230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qu XL, Li XL, Zheng YW, Ren Y, Puelles VG, Caruana G, Nikolic-Paterson DJ, and Li JH. Regulation of Renal Fibrosis by Smad3 Thr388 Phosphorylation. American Journal of Pathology. 2014;184(4):944–52. [DOI] [PubMed] [Google Scholar]
- 43.Ramirez AM, Takagawa S, Sekosan M, Jaffe HA, Varga J, and Roman J. Smad3 deficiency ameliorates experimental obliterative bronchiolitis in a heterotopic tracheal transplantation model. American Journal of Pathology. 2004;165(4):1223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verrecchia F, Chu ML, and Mauviel A. Identification of novel TGF-beta /Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem. 2001;276(20):17058–62. [DOI] [PubMed] [Google Scholar]
- 45.Heeg MHJ, Koziolek MJ, Vasko R, Schaefer L, Sharma K, Muller GA, and Strutz F. The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kidney International. 2005;68(1):96–109. [DOI] [PubMed] [Google Scholar]
- 46.Duan WJ, Yu X, Huang XR, Yu JW, and Lan HY. Opposing roles for Smad2 and Smad3 in peritoneal fibrosis in vivo and in vitro. Am J Pathol. 2014;184(8):2275–84. [DOI] [PubMed] [Google Scholar]
- 47.Meng XM, Huang XR, Chung ACK, Qin W, Shao XL, Igarashi P, Ju WJ, Bottinger EP, and Lan HY. Smad2 Protects against TGF-beta/Smad3-Mediated Renal Fibrosis. Journal of the American Society of Nephrology. 2010;21(9):1477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Furtado MB, Costa MW, and Rosenthal NA. The cardiac fibroblast: Origin, identity and role in homeostasis and disease. Differentiation. 2016;92(3):93–101. [DOI] [PubMed] [Google Scholar]
- 49.Furtado MB, Costa MW, Pranoto EA, Salimova E, Pinto AR, Lam NT, Park A, Snider P, Chandran A, Harvey RP, et al. Cardiogenic genes expressed in cardiac fibroblasts contribute to heart development and repair. Circ Res. 2014;114(9):1422–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Camelliti P, Borg TK, and Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res. 2005;65(1):40–51. [DOI] [PubMed] [Google Scholar]
- 51.Ivey MJ, Kuwabara JT, Riggsbee KL, and Tallquist MD. Platelet-derived growth factor receptor-alpha is essential for cardiac fibroblast survival. Am J Physiol Heart Circ Physiol. 2019;317(2):H330–H44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao Y, Hill MC, Li L, Deshmukh V, Martin TJ, Wang J, and Martin JF. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 2019;33(21–22):1491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bagchi RA, Roche P, Aroutiounova N, Espira L, Abrenica B, Schweitzer R, and Czubryt MP. The transcription factor scleraxis is a critical regulator of cardiac fibroblast phenotype. BMC Biol. 2016;14(21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Levay AK, Peacock JD, Lu Y, Koch M, Hinton RB Jr., Kadler KE, and Lincoln J. Scleraxis is required for cell lineage differentiation and extracellular matrix remodeling during murine heart valve formation in vivo. Circ Res. 2008;103(9):948–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jugdutt BI. Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation. 2003;108(11):1395–403. [DOI] [PubMed] [Google Scholar]
- 56.Biernacka A, and Frangogiannis NG. Aging and Cardiac Fibrosis. Aging Dis. 2011;2(2):158–73. [PMC free article] [PubMed] [Google Scholar]
- 57.Shinde AV, Humeres C, and Frangogiannis NG. The role of alpha-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim Biophys Acta. 2017;1863(1):298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]