

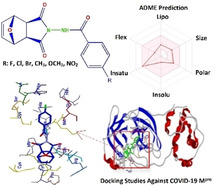
Abstract
A series of novel Norcantharimide derivatives were synthesized and their structures were characterized by FTIR, 1H and 13C NMR spectroscopy as well as elemental analyses. The absorption, distribution, metabolism and excretion (ADME) properties of the synthesized molecules were investigated. The results obtained in silico demonstrated that these molecules can be considered as orally active drug candidates due to their physicochemical properties. Also, docking studies demonstrated that all derivatives exhibit a good theoretical affinity with MolDock Score in between 124–138 against the main protease of Coronavirus Disease 2019 (COVID‐19 Mpr°) that caused worldwide epidemics. We believe that newly synthesized norcantharimide derivatives can guide many future studies in organic synthesis, medicine and pharmaceutical applications.
Keywords: ADME, Cantharidin, COVID-19 Mpr°, Drug design, Medicinal chemistry, Molecular docking studies, Norcantharidin, Norcantharimide
A series of new novel Norcantharimides have been synthesized and their absorption, distribution, metabolism and excretion (ADME) properties were investigated. The results obtained in silico showed that these molecules can be considered orally active drug candidates, taking into account their physicochemical properties. In addition, placement studies have shown that all derivatives exhibit good theoretical affinity with the 124–138 MolDock Score against the main protease of Coronavirus Disease 2019 (COVID‐19 Mpr°), which causes worldwide outbreaks.
1. Introduction
Cantharidin, which is by blister beetles as a chemical defense against predators, is a toxic monoterpene. It is one of the first pharmacological natural products used by humans. The dried body of Mylabris (Mylabris phalerata and M. cichorii), the Chinese blister beetle, has been used in Chinese medicine for thousands of years to treat malignant tumors such as hepatoma, breast cancer and colorectal cancer.
Cantharidin (1), a natural compound, has been used by the Chinese people as an anticancer agent for many years in traditional treatment methods.1, 2, 3 However, since it is a natural toxin, it is not used in chemotherapeutic studies today.4 Norcantharidin and norcantharimide, which are derivatives of cantharidin, have gained a great interest due to their less toxic effect.5 Norcantharidin (2) is the demethylated form of cantharidin. Demethylation reduces not only liver toxicity of cantharidine, but also the biological activity of the obtained norcantharidin. Cantharidin and its analogs are potent inhibitor of serine / threonine protein phosphatase 1 and 2 A (PP1 and PP2 A).6, 7, 8, 9 It is found that compounds containing norcantharidin ring inhibited cell proliferation in vitro and showed anticancer activity against various cancer cells. Also, current research results show that norcantharimides or isoindole‐1,3‐dion derivatives have a much higher anti‐cancer activity on many other types of cancer. For example, Kose et al. synthesized new norcantharimide derivatives and investigated the cytotoxic activities of these compounds. They reported that the new norcantharimide derivatives show high anticancer activity in lung and breast cancer.10
In recent years, many researchs have been carried out about the anti‐cancer and anti‐viral effects of cantharidine derivatives. In one of these studies, Cantharidin was used in chemical treatment of viral infection of Molluscum contagiosum (MC) in the skin.11 Meanwhile, in another study, it was proposed to use Norcantharidin to inhibit HIV in vitro.12 Yet in the literature, no study can be found on the anti‐viral activity of Norcantharimide derivatives. Also, there is no study on the effect of these compounds on COVID‐19 Mpr° has been conducted so far.
Computer models provide information about the possible effects of the compounds on metabolism and whether they are suitable for begin used as medicine without performing experimental studies. Cheminformatics allows us to understand its pharmacokinetic, physicochemical, solubility, absorption, and similar properties from the chemical structure of a molecule. Many new molecules are synthesized every year in the world. Testing the bioactivity of these molecules as in vitro and in vivo results manifest very high costs. Therefore, ADME and predicting the targets of the molecule have become essential for those sectors in which these molecules can be used.13
In this study, novel norcantharimide analog compounds were synthesized. The structures of the synthesized compounds were characterized using spectrometric methods (1H, 13C‐NMR, IR and Elemental Analysis). Besides, investigation of the physicochemical, pharmacokinetics and Drug‐likeness properties of the synthesized molecules for oral administration was aimed.
The COVID‐19 has infected more than to million people and is spreading at a rapidly speed. Many efforts have been performed by the scientists to develop treatment methods. With this study, we have aimed to contribute these efforts by investigating the interactions of the new norcantharimide derivatives with the potential target protein COVID‐19 Mpr°. COVID‐19 Mpr° has the main role in the replication and transcription of the Corona virus in the cell. Inhibition of this enzyme may stop the replication and transcription of the corona virus. This development is an important step in the battle against the virus.14
2. Results and discussion
2.1. Chemistry
For the synthesis of norcantharimide derivatives (6 a–f), 2‐amino‐1H‐4,7‐epoxyisoindole‐1,3(2H)‐dione (4) were used as the key compound. This key compound was obtained by reaction of 3a, 4,7,7a‐tetrahydro‐4,7‐epoxyisobenzofuran‐1,3‐dione (3) with hydrazine hydrate in the presence of methyl alcohol. 3a,4,7,7a‐tetrahydro‐4,7‐epoxyisobenzofuran‐1,3‐dione (3) was prepared via an exo‐selective cycloaddition of furan and maleic anhydride (Scheme 1).
Scheme 1.
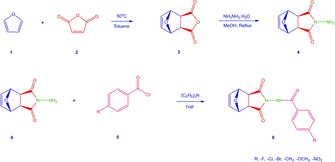
Synthesis of norcantharimide analog compounds.
The reaction of the appropriate benzoyl chlorides with 2‐amino‐1H‐4,7‐epoxyisoindole‐1,3(2H)‐dione (4) in the presence of THF gave the norcantharimide derivatives in 57–65 % yield 6 a–f (Scheme 1).
2.2. The Characterization of compounds
The structures of the norcantharimide derivatives that we synthesized in our study are similar. Among these molecules, 6 b was chosen as the model molecule. When the IR spectra of the synthesized compounds were investigated, characteristic bands of the expected functional groups were seen. When the FTIR spectrum of the compound 6 b given the structure formula in Figure 2 is examined, the presence of a single and strong N−H band (∼3330 cm−1) confirms the presence of the amide. The aromatic C−H band was observed at about 3010 cm−1. There are two carbonyl stretches in the spectra of cyclic imides corresponding to in‐phase and out of phase stretching of two C=O groups. These usually appear from 1730 to 1680 cm−1. In the phthalimide molecule, the splitting between the symmetric and anti‐symmetric stretching modes is 42 cm−1, with the υs(C=O) and υas(C=O) appearing as absorptions of medium and very high intensity at 1769 and 1727 cm−1, respectively.15, 16
Figure 2.
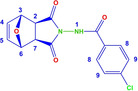
Molecular structure of compound 6 b
Figure 1.
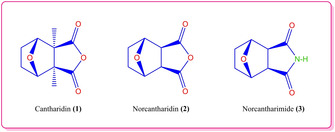
Chemical structures of cantharidin (1), norcantharidin (2) and norcantharimide (3).
The band in the FTIR spectrum at 1650 cm−1 belongs to the C=O stretching mode in the amide group. The C=C aromatic stretching was observed at 1580 cm−1. The C−O group was observed at 1100 cm−1. The 1H‐NMR spectrum of the compound 6 b in DMSO‐d6 is shown in Figure 3.
Figure 3.
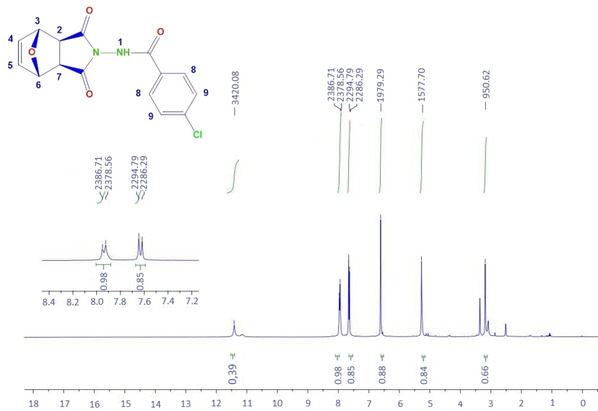
The 1H‐NMR spectrum of compound 6 b
The single peak at δ 11.40 ppm corresponds to hydrogen (N‐H) (1) in the amide group. The double peaks appearing at δ 7.93 and 7.64 ppm belong to the hydrogens bound to the C8 and C9 carbons in the aromatic ring. Since the aromatic ring is para‐substituted, the peaks are seen as doublet of doublet. The single peaks at δ 6.43 and δ 5.25 ppm belong to the hydrogens on the C4‐C5 and C3‐C6 carbons, respectively. The peaks of the hydrogens on the C2 and C7 carbons are observed singlet at about δ 3.17 ppm.
The 13C‐APT NMR spectrum of the compound 6 b in DMSO‐d6 is shown in Figure 4.
Figure 4.
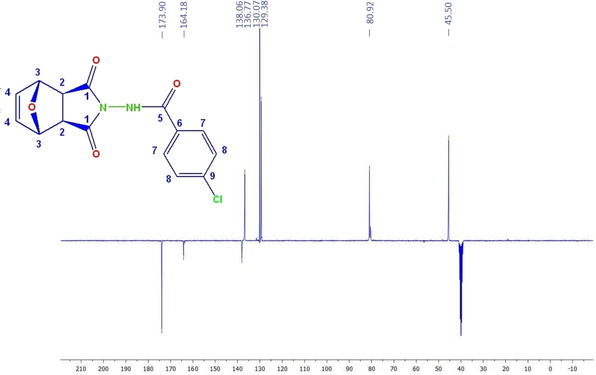
The 13C‐APT NMR spectrum of compound 6 b
When the 13C‐APT NMR spectrum of compound 6 b is investigated, the peak of the carbons in the C=O group in the imide ring (C1 and C8) is observed at δ 173.9 ppm. The peak of the carbonyl carbon (C=O) in the amide group (C9) is observed at about δ 164.2 ppm. The peaks in the range of δ 129.4‐138.1 ppm region belong to aromatic Ar−C (C10, C11, C12 and C13) carbons. The peak of carbons C4 and C5 is observed at δ 136.8 ppm. The peaks of carbons C3‐C6 and C2‐C7 are observed at δ 80.9 and δ 45.5 ppm, respectively.
2.3. ADME prediction
In silico ADME studies were performed to detect some physicochemical properties, such as lipophilicity, water solubility, and pharmacokinetic properties through the login‐free website http://www.swissadme.ch.17 Absorption (% ABS) of compounds from intestinal was figured out by: % ABS=109×(0.345xTopological Polar Surface Area (TPSA)).18
ADME prediction study of the designed compounds demonstrated Table 1.
Table 1.
ADME results.
|
ADME properties |
6 a |
6 b |
6 c |
6 d |
6 e |
6 f |
6 |
4 |
Nelfinavir |
|---|---|---|---|---|---|---|---|---|---|
|
Molecular weight (g/mol) |
302.26 |
318.71 |
363.16 |
298.29 |
314.29 |
329.26 |
284.27 |
180.16 |
567.78 |
|
Num. rotatable bonds |
3 |
3 |
3 |
3 |
4 |
4 |
3 |
0 |
12 |
|
Num. H‐bond acceptors |
5 |
4 |
4 |
4 |
5 |
6 |
4 |
4 |
5 |
|
Num. H‐bond donors |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
4 |
|
Log S (ESOL) |
−1.98 |
−2.41 |
−2.73 |
−2.12 |
−1.89 |
−1.89 |
−1.81 |
0.06 |
−6.36 |
|
WLOGP |
0.45 |
0.54 |
0.65 |
0.20 |
−0.10 |
−0.20 |
−0.11 |
−1.58 |
4.37 |
|
MLOGP |
1.52 |
1.64 |
−2.73 |
1.37 |
0.84 |
1.04 |
1.12 |
−0.39 |
3.20 |
|
TPSA (Å2) |
75.71 |
75.21 |
75.71 |
75.21 |
84.94 |
121.53 |
75.71 |
72.63 |
127.20 |
|
Absorption ( % ABS) |
82 |
83 |
82 |
83 |
80 |
67 |
83 |
84 |
65.115 |
|
Log K p (skin permeation) (cm/s) |
−7.85 |
−7.58 |
−7.81 |
−7.64 |
−8.02 |
−8.21 |
−7.81 |
−8.54 |
−5.74 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
SwissADME section gives a physicochemical property of possible oral drug candidates according to five different rules determined by the Lipinski, Ghose, Veber, Egan, and Muegge.18, 19, 20, 21 Our compounds have an acceptable pharmacokinetics profile given these drug‐likeness rules than norcantharidin. Quantitative classes of solubility in the water is defined as insoluble<−10<poorly<−6<moderately<−4<soluble<−2<very<0<highly. According to the results, molecule 6 e and 6 f with the result of −1.89 is the most soluble in water and is very soluble. Other molecules were classified as soluble with results in the −2, −3 range. Adding benzene ring to norcantharidin reduced the water solubility of the synthesized compounds. Substitution −Cl, −Br, and −OCH3 decreased the solubility of the compound 6. The −F and −NO2 groups did not affect the water solubility of this compound.
The online program displays whether the compounds are suitable for oral administration with the bioavailability radar panel containing physicochemical properties such as flexibility, insolubility, lipophilicity, size, and polarity. Molecules with oral bioavailability have shown in the colored region (Figure 5). Results obtained from computational methods showed that our molecules are suitable for oral application like norcantharidin. Except for NO2 groups, other groups slightly decreased the absorption rate of the modified compounds in the small intestine. The absorption compounds from the small intestine are above 80 %, except for the 6 a compound. Therefore, the absorption potential of the compounds has very high.
Figure 5.
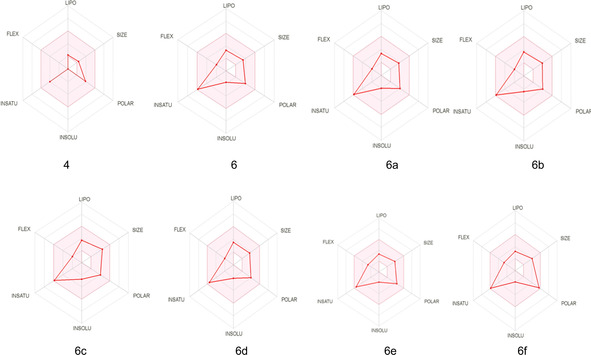
Radar related to physicochemical properties and structure of molecules (Criterias: Lipophilicity:‐0.7 <XLOGP3< +5.0, Size: 150 MW 500 g/mol, Polarity: 20<TPSA<130 Å2, Insolubility: 0<log S<6, Insaturation, Flexibility: 0.25<rotatable bonds<9)
Blood‐Brain Barrier (BBB) diffusion of CNS‐inactive compounds due to toxicity is a crucial parameter. The fact that newly synthesized compounds do not cross the blood brain‐barrier makes them a good drug candidate. Cytochromes P450 (CYP) are the major enzymes involved in the metabolic drug elimination.22 Their inhibition leads to toxic or other undesirable adverse effects because of the lower clean up and collection of the drug or its metabolites.23 Five major isoforms (CYP1 A2, CYP2 C19, CYP2 C9, CYP2D6, CYP3 A4) are responsible for the elimination of the majority of therapeutic molecules.24 Our derivatives have not demonstrated like compound 4 any inhibition effect on major CYP 450s.
2.4. Docking studies
Possible docking modes between compounds and the COVID‐19 Mpr° were studied using the Molegro Virtual Docker 7.25 The score function used is MolDock score with the coordinates of the position are X: −10.85 Y: 15.32, and Z: 68.39 at 120.832 Å3 volume, 417.28 Å2 surface, and 0.30 grid resolution. The target the COVID‐19 Mpr° (PDB id: 6LU7; Resolution 2.16 Å were downloaded in pdb format from protein data bank (http://www.rcsb.org/pdb).26 Nelfinavir used as positive control.
Coronavirus caused worldwide epidemics. We tested our compounds against the protease protein of 2019‐nCoV as In silico. The scores of the steric and hydrogen bond interactions between derivatives and COVID‐19 Mpr° presented in Table 2 and 3.
Table 2.
MolDock score, steric interaction and the hydrogen bond energy of the docked compounds.
|
Energy overview: Descriptors |
Total Energy |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
Nelfinavir |
4 |
6 |
6 a |
6 b |
6 c |
6 d |
6 e |
6 f |
|
|
Protein‐Ligand interactions |
Protein Ligand Steric (by PLP) |
−169.050 |
−77.745 |
−118.8 |
−116.168 |
−123.423 |
−116.037 |
−116.025 |
−123.686 |
−112.436 |
|
Protein Ligand Hydrogen bonds |
−6.261 |
−1.049 |
−0.523 |
−1.164 |
−1.942 |
−1.207 |
−1.616 |
−2.291 |
−1.091 |
|
|
Internal Ligand interactions |
22.284 |
−6.532 |
−5.215 |
−10.939 |
−11.977 |
−10.788 |
−8.614 |
−11.039 |
−10.979 |
|
|
MolDock Score |
−146.766 |
−85.325 |
−124.5 |
−128.271 |
−137.342 |
−128.032 |
−126.255 |
−137.016 |
−124.506 |
|
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Table 3.
H‐bonds interacting with COVID‐19 Mpr°
|
No |
The number of hydrogen bond |
Energy |
Length |
|---|---|---|---|
|
Ref. Mol. |
1 2 3 4 |
−0.984251 −2.5 −2.5 −1.68321 |
3.18633 2.86281 3.05449 2.80543 |
|
4 |
1 |
−1.04876 |
2.60146 |
|
6 |
1 |
−0.522533 |
3.12828 |
|
6 a |
1 |
−1.16392 |
3.10061 |
|
6 b |
1 |
−1.94159 |
2.79785 |
|
6 c |
1 |
−1.20656 |
3.07903 |
|
6 d |
1 |
−1.61612 |
3.10632 |
|
6 e |
1 2 |
−0.656968 −1.63358 |
2.4622 2.84309 |
|
6 f |
1 |
−1.09128 |
2.60589 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
The 2D animation of hydrogen bond and some steric interactions is shown in Figure 6.
Figure 6.
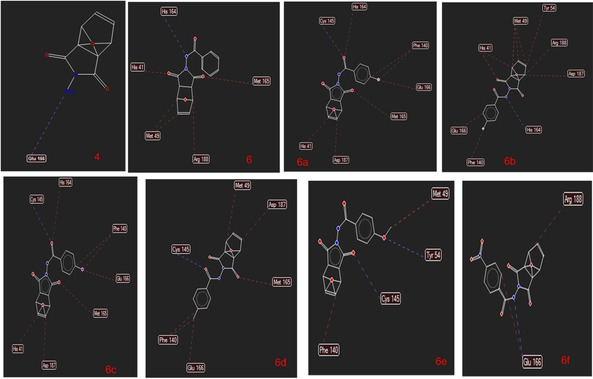
The 2D animation of hydrogen bond and some steric interactions.
Our docking analysis indicates that molecules interact with the COVID‐19 Mpr° with values between the 124–138 MolDock score.
Derivatives the best binding to COVID‐19 Mpr° among the synthesized compounds are 6 b and 6 e (Figure 7). Compound 6 e makes two hydrogen bonds with Cys 145, and Tyr 54 and steric interactions with Met 49, and Phe 140. It can be deducted from Figure 6 that 6 a, 6 c, and 6 d (with Cys 145), 6 and 6 b (with His 164), 6 f (with Glu 166) are formed hydrogen bonds in the ligands‐ COVID‐19 Mpr° complex formation. The new analogs significantly enhanced the interaction of cantharidin compounds with COVID‐19 Mpr° when compared to cantharidin. Our analogs exhibited more interaction with COVID‐19 Mpr° than some drugs such as lopinavir (ABT‐378), caspofungin acetate, atazanavir, indinavir.27 They exhibited a slightly lower affinity compared to the drug nelfinavir. But, as Table 1 is analyzed, our derivatives have better physicochemical properties than it, and can be modified to more active molecules. These results indicated that newly synthesized cantharidin derivatives can use to design and discover new compounds interacting with COVID‐19 Mpr°.
Figure 7.
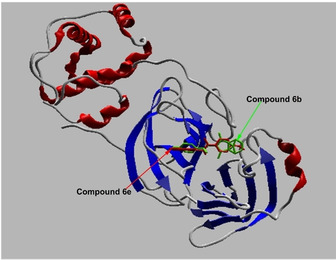
The binding of compounds 6 b and 6 e to COVID‐19 Mpr°.
3. Conclusion
In this study, 6 new compounds, which are Norcantharimide analogs were synthesized. The structures of the synthesized compounds were elucidated in detail by the spectroscopic methods (FTIR, 1H‐ and 13C‐APT NMR) and elemental analysis. According to the results of ADME, new molecules have the necessary properties for oral administration. Docking analyses demonstrated that these compounds can interact with COVID‐19 Mpr°. Interactions between the COVID‐19 Mpr° and the molecule are changed with the exchange of substituted groups. Therefore, we believe that they can be used as leading molecules in further research to develop new molecules that affect COVID‐19 Mpr° activity.
Supporting Information Summary
Experimental procedures for synthesis, of compounds 3, 4 and 6 (6 a–f) are available in supporting information. Also FT‐IR, 1H/ 13C ‐NMR spectra and elemental analysis results of compounds 6 a–f are available in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We would like to thank Prof. Dr. Yılmaz Yıldırır, Prof. Dr. Ali Dişli, Assoc. Prof. Dr. Serkan Yavuz, Research Assistant Doğukan Doyduk and Öykü Göktunalı for their valuable opinions and advice.
H. Özkan, Ş. Adem, ChemistrySelect 2020, 5, 5422.
References
- 1. Wang G. S., J. Ethnopharmacol. 1989, 26, 147–162. [DOI] [PubMed] [Google Scholar]
- 2. Lin P. Y., Shi S. J., Hsu F. L., Chen C. F., J. Chin. Chem. Soc. 1998, 45, 323–326. [Google Scholar]
- 3. Nickolls L., Teare D., Br. Med. J. 1954, 2, 1384–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu D., Chen Z., Anti-Cancer Agent. Me. 2009, 9, 392–396. [DOI] [PubMed] [Google Scholar]
- 5. Hill T. A., Stewart S. G., Ackland S. P., Gilbert J., Sauer B., Sakoff J. A., McCluskey A., Bioorgan. Med. Chem. 2007, 15, 6126–6134. [DOI] [PubMed] [Google Scholar]
- 6. Honkanen R. E., FEBS Letters. 1993, 330, 283–286. [DOI] [PubMed] [Google Scholar]
- 7. McCluskey A., Walkom C., Bowyer M. C., Ackland S. P., Gardiner E., Sakoff J. A., Bioorg. Med. Chem. Lett. 2001, 11, 2941–2946. [DOI] [PubMed] [Google Scholar]
- 8. Massicot F., Dutertre-Catella H., Pham-Huy C., Liu X. H., Duc H. T., Warnet J. M., Basıc. Clın. Pharmacol. 2005, 96, 26–32. [DOI] [PubMed] [Google Scholar]
- 9. Baba Y., Hirukawa N., Sodeoka M., Bioorgan. Med. Chem. 2005, 13, 5164–5170. [DOI] [PubMed] [Google Scholar]
- 10. Kose A., Kaya M., Kishalı N. H., Akdemir A., Şahin E., Kara Y., Mohamed G. Ş., Bioorg. Chem. 2020, 94, 103421. [DOI] [PubMed] [Google Scholar]
- 11. Robinson G., Townsend S., Jahnke M. N., Curr. Dermatol. Rep. 2020, 9, 83–92. [Google Scholar]
- 12. Peng Z. G., Jiang J. D., Wu D. Z., Chen H. S., Acta Pharm. Sin. B. 2010, 45, 224–227. [Google Scholar]
- 13. Ferreira L. L., Andricopulo A. D., Drug Discov. Today. 2019, 24, 1157–1165. [DOI] [PubMed] [Google Scholar]
- 14. Jin J., Du Z., Xu X., Nature, DOI 10.1038/s415860202223. [Google Scholar]
- 15. Torrico-Vallejos S., Erben M. F., Piro O. E., Castellano E. E., Della Védova C. O., J. Mol. Struct. 2010, 975, 227–233. [Google Scholar]
- 16. Binev I. G., Stamboliyska B. A., Binev Y. I., Velcheva E. A., Tsenov J. A., J. Mol. Struct. 1999, 513, 231–243. [Google Scholar]
- 17. Daina A., Michielin O., Zoete V., SwissADME Sci. Rep-Uk. 2017, 7, 42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lipinski C. A., Lombardo F., Dominy B. W., Feeney P. J., Adv. Drug Deliver. Rev. 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- 19. Muegge I., Heald S. L., Brittelli D., J. Med. Chem. 2001, 44, 1841–1846. [DOI] [PubMed] [Google Scholar]
- 20. Egan W. J., Merz K. M., Baldwin J. J., J. Med. Chem. 2000, 43(21), 3867–3877. [DOI] [PubMed] [Google Scholar]
- 21. Ghose A. K., Viswanadhan V. N., Wendoloski J. J., J. Comb. Chem. 1999, 1, 55–68. [DOI] [PubMed] [Google Scholar]
- 22. Veber D. F., Johnson S. R., Cheng H. Y., Smith B. R., Ward K. W., Kopple K. D., J. Med. Chem. 2002, 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- 23. Hollenberg P. F., Drug Met. Rev. 2002, 34, 17–35. [DOI] [PubMed] [Google Scholar]
- 24. Kirchmair J., Göller A. H., Lang D., Kunze J., Testa B., Wilson I. D., Glen R. C., Schneider G., Nat. Rev. Drug Dıscov. 2015, 14, 387–404. [DOI] [PubMed] [Google Scholar]
- 25.A. Molegro, (Aarhus C, Denmark), MVD 7.0 Molegro Virtual Docker, DK-8000, 2019.
- 26. Liu X., Zhang B., Jin Z., Yang H., Rao Z., Biorxiv. DOI 10–1101/20200226964882. [Google Scholar]
- 27. Contini A., Chemrxiv. DOI 10–26434/chemrxiv.11847381. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary