

Summary
Increasing evidence suggests that tRNA levels are dynamically and specifically regulated in response to internal and external cues to modulate the cellular translational program. However, the molecular players and the mechanisms regulating the gene-specific expression of tRNAs are still unknown. Using an inducible auxin-degron system to rapidly deplete RPB1 (the largest subunit of RNA Pol II) in living cells, we identified Pol II as a direct gene-specific regulator of tRNA transcription. Our data suggest that Pol II transcription robustly interferes with Pol III function at specific tRNA genes. This activity was further found to be essential for Maf1-mediated repression of a large set of tRNA genes during serum starvation, indicating that repression of tRNA genes by Pol II is dynamically regulated. Hence, Pol II plays a direct and central role in the gene-specific regulation of tRNA expression.
eTOC Blurb
Gerber et al. report that Pol II dynamically regulates tRNA transcription by Pol III via transcriptional interference, an activity also essential for Maf1-mediated repression of a large set of tRNA genes during serum starvation. The experimental system used also revealed that the Pol II CTD is not essential for productive elongation.
Graphical Abstract
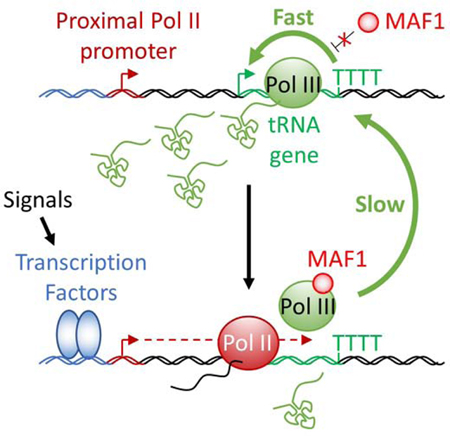
Introduction
Eukaryotic nuclear transcription is carried out by RNA polymerases I, II and III (Pols I, II and III). Pol I transcribes the single multi-copy gene that specifies the large ribosomal RNAs (28S rRNA, 18S rRNA, and 5.8S rRNA). Pol II transcribes genes that specify pre-messenger RNAs and long non-coding RNAs (the vast majority), as well as various small functional RNAs such as microRNAs and small nuclear RNAs (snRNAs). Finally, Pol III transcribes genes that specify small untranslated RNAs essential for protein synthesis, such as transfer RNAs (tRNAs) and the 5S ribosomal RNA, as well as a diverse group of short regulatory RNAs (Palazzo and Lee, 2015; Roeder and Rutter, 1969). In contrast to transcription by Pol II, which involves countless types of promoters and enhancers and many diverse transcriptional regulators, transcription of Pol III genes involves only three types of promoters that are regulated by a limited set of transcription factors (Dumay-Odelot et al., 2014; Schramm and Hernandez, 2002). Notably, transcription of all tRNA genes, other than those for selenocysteine tRNAs, is governed by type II internal promoters that require only the conserved multi-subunit transcription factors TFIIIB and TFIIIC in conjunction with Pol III. Initiation of tRNA transcription starts with the binding of TFIIIC to two intragenic boxes followed by the association of TFIIB with a region immediately upstream of the TSS. Finally, TFIIIB recruits Pol III to direct the first round of transcription. Upon reaching the terminator, a stretch of 4 or more thymidine residues, Pol III can be recycled to the same TFIIIB-bound promoter at much higher rates than the initial preinitiation complex formation, in a process known as facilitated recycling (Arimbasseri et al., 2014; Orioli et al., 2012).
tRNAs are stable molecules synthesized at very high rates. Their expression is co-regulated, primarily at the level of transcription initiation, in response to nutrient availability and cellular stresses via different signaling pathways (Grewal, 2015; Moir and Willis, 2013). Despite the apparent simplicity in the Pol III transcriptional machinery, the relative abundance of individual tRNAs varies considerably across different tissues and cell lines (Dittmar et al., 2006; Sagi et al., 2016) in response to stresses and changes in growth conditions (Cieśla et al., 2007; Orioli et al., 2016; Pang et al., 2014; Torrent et al., 2018), in support of cell proliferation or differentiation (Gingold et al., 2014) and in pathologies such as cancer (Goodarzi et al., 2016; Pavon-Eternod et al., 2009). Dynamic changes in the composition of the cellular tRNA repertoire in response to external and internal cues may in turn affect protein abundance, translation fidelity, protein folding and even mRNA stability (reviewed in Rak et al., 2018) to modify cellular metabolism or physiology.
The levels of a specific tRNA are determined by both its stability and its synthesis rate (Rak et al., 2018; Wichtowska et al., 2013). Surprisingly, however, there are no known tissue- or gene-specific transcription factors dedicated to tRNA gene transcription. Interestingly, in human and mouse cells, Pol II is often found in close proximity to tRNA genes that display high Pol III occupancy (Barski et al., 2010; Canella et al., 2012; Moqtaderi et al., 2010; Oler et al., 2010; Raha et al., 2010; Yeganeh et al., 2017). Different hypotheses have been proposed to explain these observations, and range from a spurious recruitment of Pol II at genes displaying high levels of Pol III (Canella et al., 2012; Yeganeh et al., 2017) to a requirement of Pol II activity to remodel the chromatin environment in order to facilitate Pol III recruitment. Treatment of cells with the transcriptional inhibitor α-amanitin at a concentration selective for Pol II was found to mildly reduce the expression of some Pol III targets (Barski et al., 2010; Listerman et al., 2007; Raha et al., 2010). However, and especially in light of the slow action of α-amanitin in vivo (Nguyen et al., 1996), these experiments did not establish that these effects were direct. In contrast, the expression levels of the Pol ll-transcribed AtNUDT22 gene in Arabidopsis negatively correlate with the expression of a set of overlapping Pol III-transcribed proline tRNA genes (Lukoszek et al., 2013). These results suggest transcriptional interference between the two types of polymerases rather than Pol II-assisted enhancement of Pol III activity. In order to unravel the primary function of Pol II at Pol III-transcribed genes in vivo, we established a human cell line that allows an inducible, rapid and selective depletion of RPB1 in living cells using an auxin- degron system.
Here, we report the identification of specific tRNA genes whose transcription by Pol III is directly under the control of elongating Pol II complexes. Unexpectedly, we found that the auxin-induced degradation of RPB1 in Pol II complexes was incomplete, generating elongation-competent but termination incompetent CTD-less (Pol IIB) complexes specifically at small nuclear RNA (snRNAs) and stably paused genes. By comparing the changes in the levels of nascent tRNAs in cells treated with the pause-release inhibitor DRB and in cells after auxin-induced RPB1 degradation, we further confirmed that Pol II passage through tRNA genes interferes robustly with their transcription. Pol II-mediated repression of Pol III, which appeared to be limited to a small number of loci in rich media conditions, was found to be essential for Maf1-mediated repression of a majority of tRNA genes during serum starvation. In addition, our experiments reveal that the downregulation of specific tRNA genes following Pol II inhibition or depletion is caused mainly by indirect effects such as loss of the MYC protein, which can explain the reported downregulation of particular tRNA genes in α-amanitin-treated cells (Barski et al., 2010; Raha et al., 2010). Hence, Pol II can regulate tRNA transcription by Pol III dynamically and in a gene-specific manner both directly, through transcriptional interference, and indirectly, by regulating the expression of MYC.
Results
Establishment of a cell line for rapid and inducible depletion of RPB1
To study the interplay between Pol II and Pol III, we established a HEK293 cell line expressing a full length RPB1 that is C-terminally tagged with a minimal auxin-sensitive degron (Natsume et al., 2016), and an orange fluorescent protein (Figure 1A) as the sole source of this essential subunit of Pol II as described in Figure S1A–C. In these cells, a rapid and auxin-dependent degradation of RPB1 was observed when the expression of the plant ubiquitin ligase OsTIRl was induced for 12 hrs with doxycycline (Dox) followed by addition of auxin for 2 hrs (Figure 1B). Three clones (#7, #12 and #19) displaying high responsiveness to the treatment were selected and used in all further experiments. A temporal analysis of these clones by flow cytometry revealed a rapid decay of fluorescence, indicative of RPB1, with a half-life of 33.2 min ± 1.8 min following auxin addition (Figure 1C–E). Microarray analysis of pools of RNA isolated from all three clones in untreated (Ctl), DOX-treated (Dox) or DOX treatment followed by auxin (DA) conditions revealed that steady-state levels of Pol II and Pol III transcripts were largely unchanged (Figure 1F–H). Surprisingly, about two third of the genes that were affected more than 2-fold (205/331 genes) were even upregulated in RPBl-depleted cells (Figure 1H). Pol II depletion on target genes was confirmed by RPB3 ChlP-seq for clones #7 and #19, which are, respectively, the most and least auxin-responsive clones and both of which showed an apparent complete loss of this endogenous subunit on all annotated genes (Figure 1I). Finally, sequencing of nascent RNAs isolated from elongating polymerases complexes (Werner and Ruthenburg, 2015; Wuarin and Schibler, 1994; Figure S1D) further revealed a clear reduction of transcription (Figure 1J). This approach is well suited to evaluate transcription by all three types of polymerase (Figure S1E) and efficiently separates nascent chains from mature tRNAs (Figure S1F). Hence, these results confirm that the degradation protocol is efficient and sufficiently rapid to prevent notable changes in steady-state RNA levels (except for highly unstable species) and should therefore allow for the evaluation of primary effects of RPB1 loss on Pol III transcription. To our surprise, the levels of RNAs in whole cell RNAs or in the polymerase-bound nascent RNA fraction were not dramatically affected by the DA-mediated depletion of Pol II. Pol II transcription is responsible for ~60% of total RNA synthesis in mammalian cells (Pombo et al., 1999; Wansink et al., 1993). Hence if Pol II activity was indeed fully lost, one would expect a substantial reduction (at least 50%) in the levels of nascent RNAs per cells. Yet our measurements revealed a mere 23% reduction (Figure 1K), indicating a significant residual level of Pol II transcription in DA-treated cells.
Figure 1. Establishment of a cell line for rapid and inducible depletion of RPB1.

(A) Schematic representation of the tagged RPB1 construct expressing a full length human RPB1 cDNA flanked with an N-terminal Flag-HA tag and a C-terminal Flag tag followed by a minimal auxin- sensitive degron, a V5 epitope and an mKO2 orange fluorescent protein. (B) Standard protocol for RPB1 depletion. (C) Time course flow cytometry experiment for clone #7 showing the progressive loss of fluorescence of DA-treated cells (in green). In red, untreated fluorescent cells. In blue, parental non-fluorescent cells. (D) Kinetics of fluorescence decay in all three clones. (E) % of remaining fluorescent cells at different time points during DA treatment for all 3 clones. (F) Correlation between gene expression levels in untreated (Ctl) and DOX-treated (Dox) pools of RNA prepared from all three clones. (G) Same as (F) but showing correlation between Ctl versus DA expression levels. (H) Number and type of genes affected >2-fold in the microarray analysis presented in (F) and (G). (I) Profile of RPB3 enrichment scores at all RefSeq genes for RPB1- expressing (Ctl) or RPBl-depleted (DA) clones #7 and 19#. TSS, transcription start site. TES, transcription end site. (J) Overlays of normalized nascent RNA and input-subtracted RPB3 ChlP-seq coverage at the MYC locus in RPBl-expressing (Ctl, blue) and RPBl-depleted (DA, red) cells for clones #7 and #19. (K) Quantification of RNA content in whole cell extract (Total) or nascent RNA fractions (Nascent) for clone #7 and #19 relative to Ctl. Data represent the average +/− SD.
Dox + auxin-treatment converts a fraction of initiated Pol II into termination-incompetent CTD-less complexes
In an attempt to understand these discrepancies, we evaluated nascent RNA expression for the upregulated genes in the microarray (Figure 2A). To our surprise, most of these genes were expressed only at low levels prior to RPB1 depletion (see also Figure 1G). Importantly, however, virtually all of them were located downstream of highly expressed genes and the increase in their expression appeared to originate from termination site read-through from neighboring genes (Figure 2A and 2C). Importantly, termination defects were not observed for most genes (Figure 1J and 2B). MST1L IncRNA, the most upregulated gene (21-fold in the microarray analysis), is located downstream of a Pol II-transcribed U1 snRNA gene that presented a marked transcription termination defect upon DA treatment (Figure 2C), a feature shared by all Pol II-transcribed snRNA genes (Figure 2D). In addition, termination site read-through was also particularly pronounced at genes, such as UBC (Figure 2E and 2F), previously reported to display stable Pol II pausing (Chen et al., 2015). These results prompted us to reevaluate whether the tagged RPB1 construct was indeed fully degraded.
Figure 2. DA-treatment converts a fraction of initiated Pol II into termination-incompetent Pol IIB.

(A) Profile of nascent RNA scores at Pol II genes > lkb upregulated in the microarray analysis in RPBl- expressing (Ctl) or RPBl-depleted (DA) cells for clones #7 and 19#. TSS, transcription start site. TES, transcription end site. (B) Same as (A) but genome-wide profile (all RefSeq RNA clusters) of nascent RNA scores downstream of TES. (C) Overlays of normalized nascent RNA and input-subtracted RPB3 coverage at the MST1L IncRNA locus (anti-sense of MST1L), showing termination site read-through from the upstream snRNA Ul-4 gene in DA treated clone #7 and #19. (D) Same as (B) but profile of nascent RNA scores downstream of all snRNA genes. (E) same as (C) at and downstream of the stably paused UBC gene showing termination site read-through for DA-treated clones #7 and #19 cells. (F) same as (B) but profiles of nascent RNA scores downstream of stably paused genes. (G) Schematic representation of tagged RPB1 protein and the different antibodies used to evaluate RPB1 degradation efficiency and immunoblot and corresponding Ponceau-stained membrane of chromatin pellets prepared from DA-treated and control cells. (H) Mirror plot presenting the summed extracted areas of peptides obtained from full length (Ctl, blue) and truncated RBP1 (DA, red) normalized to the signal at residue 365–382. Note that the peptides covering the perfect heptad repeats (1615–1804) are not included because two peptides could match multiple regions. (I) Quantification of the levels of Pol II subunit expression in nuclear extracts and chromatin pellets (see Figure S2C). Data represent the average +/− SD; *p<0.05 (T-test).
Taking advantage of the multiple tags and different antibodies available for RPB1 (Figure 2G), we probed the chromatin-bound fraction (containing only initiated polymerases) isolated from untreated and DA-treated cells for RPB1 expression. Immunoblot analyses revealed the formation of truncated RPB1 lacking the CTD entirely (Figure 2G, S2A–C). A mass spectrometric analysis of RPB1 isolated from control and DA-treated cells further confirmed that the RPB1 CTD was truncated up to the linker region in DA-treated cells and therefore did not contain any heptad repeats (Figure 2H). Immunoblotting for the endogenous Pol II subunits RPB2 and RPB3 revealed that about a third of the transcribing Pol II was still present on the chromatin after RPB1 depletion (Figure 2I and S2C). In contrast, Pol I and Pol III levels remained unaffected (Figure S2C and S2D). Since Pol II with a CTD containing only 5 heptad repeats (out of a total of 52) is deficient for initiation in the context of chromatin (Lux et al., 2005; Meininghaus et al., 2000), the absence of truncated RPB1 in both RIPA and NUN nuclear extracts (Figure S2A and S2C), containing nucleoplasmic proteins and non-initiated RNA polymerases (Figure S1D), suggests that the truncation occurred after initiation. Moreover, CTD-less Pol II (Pol IIB) is also deficient in termination (McCracken et al., 1997), and thus will remain associated with the chromatin. To confirm the absence of transcription initiation following DA treatment, serum-starved cells were stimulated with 20% fetal calf serum (FCS) following RPB1 depletion (Figure S2E). This treatment completely abolished induction of all serum-responsive genes (Figure S2G and S2H). Importantly, RPB1 depletion/truncation efficiencies were not affected by starvation (Figure S2F). Hence, the lack of RPB3 accumulation along target genes (Figure 1I) and the upregulation of non-expressed genes following RPB1 depletion (Figure 1H and 2A) are both explained by the conversion of a fraction of initiated Pol II into a form (CTD-free Pol IIB) that is competent for elongation but not for termination. However, the truncation was restricted to snRNA genes (Figure 2D), where the CTD is required for U snRNA 3’ processing and termination (Jacobs et al., 2004; Medlin et al., 2003), or genes displaying stable pausing (Figure 2F).
Pol II transcription interferes with RNA Pol III activity
To evaluate the impact of Pol II transcription on Pol III activity, we assessed the effects of Pol II depletion at a Pol-III transcribed MIR element located in the first intron of the Pol III subunit gene POLR3E (Yeganeh et al., 2017). ChlP-seq and nascent RNA-seq analyses revealed that loss of Pol II was accompanied by a clear upregulation of Pol III occupancy and activity at the MIR element (Figure 3A), indicating that Pol II negatively impacts Pol III activity via transcriptional interference. In addition, the same increase in MIR transcription was also observed in wild-type HEK293 cells treated for 1 hr with the CDK9 inhibitor DRB, as revealed by an analysis of published nascent RNA-seq data (Werner and Ruthenburg, 2015).
Figure 3. Pol II transcription interferes with RNA Pol III activity.

(A) Overlays of input-subtracted RPC62 and RPB3 ChlP-seq coverage and normalized nascent RNA in RPBl-expressing (Ctl, blue) or RPBl-depleted (DA, red) clone #7 and #19 cells at a MIR locus that is transcribed by Pol III in the anti-sense direction from the Pol ll-transcribed POLR3E gene. The nascent RNA coverage tracks of DRB-treated (yellow) and mock-treated (green) cells from Werner and Ruthenburg (2015) are also shown. The loss of elongating Pol II at this locus is correlated with an increase in Pol III occupancy and transcription of the MIR element in both DA-treated cells and DRB-treated cells. (B) tRNA genes affected >1.5-fold at the nascent RNA level with a FDR<0.05 in DRB-treated HEK293 cells (data ranked by Log2 fold change). tRNA genes affected significantly in DA-treated clones #7 and #19 cells are indicated by purple bars. Black rectangles identify the 3 model tRNA genes used in following experiments. (C) Profiles of nascent RNA, RPB3 and RPC62 enrichment scores at tRNA genes upregulated in both DA-treated and DRB-treated cells (1), upregulated in DRB only (2) or downregulated in DRB only (3) as defined in (B).
These results led us to postulate that Pol II recruited near tRNA loci could lead to similar repressions of Pol III activity if it transcribed through these loci. We then reasoned that a comparison of nascent-RNA levels in cells treated with DRB versus cells treated with DA, which generates a transcribing Pol IIB at both stably paused genes and snRNA genes, could provide a unique opportunity to gain mechanistic insights into potential functions of Pol II at tRNA loci. In this regard, DRB-treatment, which blocks Pol II pause-release, and thus Pol II transcription functions, is expected to reveal all Pol III-transcribed tRNA genes that are repressed by Pol II transcriptional interference. In contrast, the DA treatment should only enhance the Pol III-dependent transcription of a subset of DRB-induced tRNA genes. Thus, only tRNA genes that are directly repressed by Pol II and located away from any Pol II genes presenting DA-induced termination-site read-through are expected to be affected, since the resulting Pol IIB at the latter loci would generate read-though transcription that would compensate for the loss of local Pol II and maintain Pol III repression at these loci (Figure S3A and associated legend). This analysis revealed that out of a total of 371 expressed Pol III-dependent tRNA loci, 55 were affected by DRB (44 upregulated and 11 downregulated at least 1.5-fold with an FDR <0.05). In DA- treated HEK293 cells, only 8 of the 44 DRB-upregulated tRNA genes were also significantly increased by DA-mediated RPB1 depletion (Figure 3B, S3B and Table S2). tRNA genes that appeared enhanced or downregulated in DA-treated cells via Pol III-independent effects were excluded from further analysis. For these genes, the increase in transcription was caused solely by high level of Pol IIB read- though or loss of Pol II transcription at Pol II genes with overlapping Pol III genes (Figure S3C).
If Pol II indeed interferes with tRNA transcription, then the difference between the number of genes upregulated in DRB- versus DA-treated cells should lie in the presence of transcribing Pol IIB at the DRB-sensitive tRNA loci that were not affected by the DA-treatment. We therefore investigated nascent-RNA, Pol II and Pol III occupancy profiles at all affected tRNA loci, grouped by their response to DRB and DA, namely: (1) those upregulated both in DA- and DRB-treated cells at the nascent RNA level, (2) those upregulated only in DRB-treated cells and, finally, (3) those downregulated following DRB treatment (categories (1), (2) and (3) in Figure 3C). We did not observe Pol II densities at most tRNA loci (all categories) under control conditions (Figure 3C, 7Ctl and 19Ctl profiles), suggesting that Pol II does not accumulate at high levels near affected tRNA genes in HEK293. However, in DA-treated cells, Pol II densities were observed at tRNA loci upregulated under DRB only conditions and further confirmed by the presence of active transcription upstream of these loci following DA treatment (Figure 3C, (2) and 4A, tRNA-SeC-TCA-1–1). Hence, the difference between the 8 Pol III-dependent tRNA genes that are upregulated in both DA- and DRB-treated cells and the 36 Pol III-dependent tRNA genes upregulated only in DRB lies in the maintenance of active Pol II transcription in DA-treated cells at the Pol III-transcribed tRNA genes enhanced by DRB in wild-type HEK293 cells. Finally, genes downregulated by DRB were also negatively impacted by RPB1 depletion but presented clonal differences in their levels of expression (Figure 3C, (3)). Interestingly, Pol III occupancy was not as dramatically affected at these loci (Figure 3C). The lack of major changes in Pol III occupancy with changes in transcription is strikingly true at genes upregulated both by DRB and by DA-treatments. Since the density of a polymerase at a specific locus is determined by the ratio of the initiation frequency over the elongation rate (Ehrensberger et al., 2013), a higher tRNA synthesis rate without a change in the level of Pol III implies a concomitant increase in both initiation and elongation rates (see Discussion).
Figure 4. RPB1 depletion affects tRNA transcription rates.

(A) Overlays of input-subtracted RPC62 and RPB3 ChlP-seq coverage and normalized nascent RNA in RPBl-expressing (Ctl, blue) or RPBl-depleted (DA, red) clone #7 and #19 cells. The nascent RNA coverage tracks of DRB-treated (yellow) and mock-treated (green) cells are also shown. (B) Relative RNA expression levels in all three clones normalized to DMSO Ctl in RPBl-expressing (Ctl) or RPBl- depleted (DA) cells pre-treated with DRB or DMSO. (C) DA/Ctl ratios for data presented in (B). (D) Northern blot analysis of mature and pre-tRNAs in HEK293 cells treated with DRB or DMSO. Precursor sizes in HEK293 were obtained from SSB/La PAR-CLIP data from Gogakos et al. (2017). (E) Protocol used for the analysis of demethylated total and ethynyl-uridine (EU) labelled RNAs by RT-qPCR. (F) Relative expression levels of selected Pol I and Pol II RNAs in RPBl-expressing (Ctl) or RPBl-depleted (DA) cells for all 3 clones normalized to their levels under Ctl conditions. Data show the levels in total (Total) or metabolically labelled (EU-labelled) RNAs. (G) Same as (F) but for the three selected mature or precursor tRNAs. Data in (B), (C) and (F), (G) represent the average +/− SD; *p<0.05 (T-test)..
To further characterize the effects of Pol II inhibition/depletion on tRNA gene transcription in all three clones, we focused our attention on three model loci that are representative of the three categories of genes discussed above (Figure 4A): (1) tRNA-Thr-CGT-2–1 (pre-Thr), a DA-upregulated gene that is the second-most robustly overexpressed tRNA under DRB conditions; (2) tRNA-SeC-TCA-1–1 (pre-SeC), a gene that is regulated by type III external promoter and represents the third-most highly induced tRNA gene by DRB, but is unaffected by DA treatment. The lack of an effect of DA treatment at this locus correlates with the presence of read-through transcription originating from the stably paused FOSB gene following DA-mediated RPB1 truncation; and (3) tRNA-Ala-TGC-7–1 (pre-Ala), a tRNA gene downregulated by DRB and one of the four tRNA genes downregulated in our microarray analysis of DA-treated cells (Figure 1G and 1H).
First, we confirmed that DA and DRB treatments mediated their effects via the same pathway by incubating cells for 1 hr with DRB prior to RPB1 depletion. DRB treatment, like RPB1 depletion, efficiently reduced the mRNA levels of unstable (MYC, JUN) Pol II transcripts and, notably, there was no additive effect of RPB1 depletion and DRB treatment on their transcription (Figure 4B). Similarly, DRB and DA treatments each showed robust increases in pre-Thr tRNA levels and decreases in pre- Ala tRNA levels with combined treatments being essentially equivalent to the somewhat stronger DRB effects (Figure 4B and 4C, DA/Ctl ratio = 1). These results were further confirmed by Northern blots in DRB-treated cells (Figure 4D). Interestingly, in total RNA, pre-SeC tRNA levels remained unaffected following DA treatment but showed a statistically significant 1.7-fold increase with DRB. This latter observation confirmed that this tRNA is upregulated following DRB treatment but not by RPB1 depletion. Similar results could also be reproduced by pretreatment with the CDK9 inhibitor LDC00067 (LDC) and the initiation inhibitor Triptolide (Trip) pretreatment (Figure S4A–S4E). These treatments confirmed that the effects observed are caused by the loss of Pol II transcription itself since degrading or inhibiting pause-release or initiation of Pol II have all the same effects. Moreover, since the triptolide treatment that was used induces the complete loss of Pol II at the time of gene expression analysis (Figure S4A), it appears very clear that all the expression changes in DA-treated cells at these loci are directly caused by modulation of Pol III activity.
Taken together, these results indicate that Pol II activity rapidly modulates Pol III transcription both positively and negatively at specific subsets of tRNA genes. Moreover, it is likely that Pol II transcription per se, rather than pausing or recruitment mediates these effects. Indeed, the exact same outcomes were obtained with drugs (LDC and DRB) that stall polymerases at pause sites and with a drug (Trip) that blocks initiation and induces complete loss of Pol II in these cells. The absence of detectable Pol II densities near regulated tRNA loci is incompatible with the presence of stably paused Pol II but not with an infrequent passage of transcribing Pol II complexes through these genes and an associated interference with the Pol III transcription cycle.
RPB1 depletion affects tRNA transcription rates directly and indirectly
The fact that pre-Thr tRNA transcription can be enhanced without changes of Pol III occupancy or evidence of Pol II transcription prompted us to further evaluate whether these effects were indeed caused by modulation of Pol III activity or by changes in the stability or retention time of RNA precursors on chromatin. Thus, we monitored de novo synthesis of both stable tRNAs and their unstable precursors using metabolic labelling. Control or RPBl-depleted cells were incubated for 1 h with ethynyl-uridine (EU) (Figure 4E) and total RNA isolated from these cells was treated with an RNA demethylase (AlkB) to remove methyl groups that are known to interfere with RNA reverse transcription (Cozen et al., 2015; Zheng et al., 2015). Finally, EU-labelled RNAs were biotinylated, captured on streptavidin magnetic beads and analyzed by RT-qPCR. As expected, the synthesis of both stable and unstable mRNAs was substantially reduced (Figure 4F), but not abolished, by RPB1 degradation, consistent with the formation of elongating CTD-less complexes. The rapid loss of RPB1 did not alter EU incorporation either in Pol I transcripts or in most of the tested Pol III transcripts (Figure 4F and S4F). As expected, however, pre-Thr tRNA levels were increased in both total and EU-labelled RNA following DA treatment, consistent with an increase in transcriptional rates (Figure 4G). Furthermore, a significant increase of EU-labelled mature tRNA could also be measured for tRNA-Thr (EU-labelled pre-Thr tRNA, which is also detected by the primers used for the mature form, contributes only a small fraction (1/28) of the level measured; Figures S4G and S4H). In contrast, levels of both the pre- and mature Ala-tRNA were decreased in total RNA and EU-labelled fractions following DA treatment, suggesting that this tRNA is unstable in HEK293 cells (Figure 4G). Finally, increased levels of EU incorporation could be observed for pre-SeC tRNA but not for its mature form, consistent with the changes detected at the nascent RNA level. Hence, these changes mainly reflect higher EU incorporation into readthrough transcripts that are synthesized by CTD-less complexes and cannot be processed and remain associated with the chromatin.
To further confirm that these effects reflected changes in transcriptional rates rather than stability or processing of the tRNA precursors, control and DA-treated cells were incubated with actinomycin D (ActD) to fully block transcription by all RNA polymerases and pre-tRNA levels were measured at different time points. The decay rates of all three tested pre-tRNAs were not significantly affected by RPB1 depletion (Figure S4I and S4J), confirming that changes in expression levels mirrored alterations in Pol III transcriptional rates. In addition, changes in pre-tRNA synthesis rates following RPB1 depletion were also independent of protein synthesis, as revealed by pre-incubation with the translation inhibitor cycloheximide (CHX; Figure S4K). Hence, stimulation of pre- Thr tRNA transcription following RPBl-depletion is likely direct and not caused by the loss of unstable unknown repressor(s) requiring constant transcription-translation to maintain their levels. However, in the case of the downregulated pre-tRNAs, these experiments did not formally rule out an indirect effect such as loss of an unstable activator(s). Except for MYC, none of the genes encoding known Pol III factors or activators were among the 23 protein-coding genes affected in DA-treated cells (Figure 5A and Table S3). Indeed, MYC has been shown to exert a positive stimulatory role on Pol III transcription via recruitment of GCN5 and TRRAP (Kenneth et al., 2007). Since MYC levels rapidly decreased during the DA-treatment (Figure 5A and 5B), we decided to evaluate the MYC contribution to pre-tRNA expression by transfecting cells with a siRNA pool targeting MYC (siMyc) for 36 hrs prior to RPB1 depletion (Figure S5A). While MYC knock-down at the mRNA level was very efficient (Figure S5C), it also interfered with Dox-mediated induction of theTIRl ubiquitin ligase and, as a consequence, impaired RPB1 degradation in a large number of cells (Figure S5B and S5C). As a consequence, pre-Thr tRNA appeared less induced by the DA-treatment but its expression in RPB1- expressing cells still remained unaffected by MYC knock-down (Figure 5C). Similarly, pre-SeC tRNA levels were not significantly altered by siRNA-mediated depletion of MYC. In contrast, pre-Ala tRNA levels were reduced by MYC siRNAs to the same extent that they were reduced in DA-treated cells. Although the decrease in pre-Ala tRNA levels did not strictly parallel the extent of MYC mRNA reduction, this tRNA gene nonetheless appears to be sensitive to MYC levels. Hence, the downregulation of pre-Ala tRNA observed after RPB1 depletion seems to be caused, at least in part, by the loss of MYC and thus more likely reflects indirect effects. Interestingly, the three clones displayed different levels of expression of MYC protein (Figure 5A and 5B), which correlates with the difference in the levels of nascent RNAs observed in control versus treated cell for the tRNA loci downregulated by DRB (Figure 3C (3)).
Figure 5. Pol II activity regulates tRNA transcription directly and indirectly and is essential for the repression of most tRNA genes in serum-deprived cells.

(A) MYC immunoblot and corresponding Coomassie-stained membrane of nuclear extracts prepared from untreated (Ctl), Dox-treated and DA-treated cells for all three clones. (B) Quantification of data shown in (A). Histogram represents the average expression levels. (C) Relative pre-tRNA expression levels normalized to siCtl-treated cells for all 3 clones transfected with a non-targeting (siCtl) or MYC-targeting pool of siRNAs (siMyc) prior to RPB1 depletion. (D) MAF1 immunoblots and corresponding Coomassie-stained membrane of nuclear and cytoplasmic extracts. (E) MAF1 protein levels in cytoplasmic extracts prepared from RPBl-expressing or RPBl-depleted cells for all 3 clones. Histograms represent the average expression levels. (F) Relative pre-tRNA levels normalized to Ctl DMSO levels in RPBl-expressing (Ctl) or RPBl-depleted (DA) cells for all three clones treated with Rapa prior to or after auxin addition (see also Figure S5D and S5E). (G) Relative pre-tRNA levels normalized to Ctl cells kept in serum in RPBl-expressing (Ctl) or RPBl-depleted (DA) cells for all three clones cultured in the presence (black) or absence (white) of serum prior to and during RPB1- depletion. (H) DA/Ctl ratios for data shown in (G). (I) Microarray analysis of pools of RNA prepared from all three clones cultured in the presence (serum) or absence of serum (starved) for 20hrs. (J) Same as (I) but for cells treated with DA. Data in (C), (F), (G) and (H) represent the average +/− SD; *p<0.05, ns non-significant (T-test).
Pol II activity is essential for the repression of tRNA genes in serum-deprived cells
The fact that Pol II transcription loss did not affect the transcription of all tRNA genes suggests a direct participation of Pol II in the modulation of Pol III activity at specific loci rather than an alteration of the levels or activity of a general regulator. However, as shown above, changes in MYC levels specifically modulated pre-Ala expression. By analogy, mammalian and yeast tRNAs display different sensitivities to the global Pol III repressor MAF1 (Cieś et al., 2007; Orioli et al., 2016; Turowski et al., 2016). In human cells, MAF1 is essential for Pol III repression in response to unfavorable growth conditions, such as serum starvation (Michels et al., 2010). This Pol III repressor was also shown to exert a negative control under basal conditions, at least in IMR90 and HEK293 cells (Reina et al., 2006). First, we confirmed that MAF1 protein levels and subcellular localization were not affected by the DA treatment (Figure 5D and 5E). To investigate whether repression by Pol II transcription is achieved via the selective modulation of MAF1 activity or whether both exert independent negative effects on tRNA genes, we manipulated MAF1 activity using rapamycin (Rapa). This mTORCl inhibitor was shown to prevent MAF1 phosphorylation and inactivation in mammalian cells (Michels et al., 2010; Shor et al., 2010). Cells thus were treated either with DMSO or with Rapa prior to or after RPB1 depletion (Figure S5D). While Rapa could repress all three pre-tRNAs (Figure 5F), it had no impact on the stimulation of pre-Thr tRNA transcription or the repression of pre-Ala tRNA transcription caused by RPB1 depletion and did not change the response of pre-SeC tRNA transcription to RPB1 degradation. These effects were also not affected by inverting the order of RPB1 depletion and Rapa addition (Figure 5F and S5D–E). These results indicate that MAF1 and Pol II act via independent mechanisms to repress tRNA genes. Interestingly, the relative levels of pre-Thr tRNAs in Rapa plus DA-treated cells was similar to the level of pre-Thr tRNA in cells cultured in control conditions (Ctl, DMSO) (Figure 5F). Hence, loss of Pol II transcription cancelled the repression imposed by Rapa.
To evaluate the full extent of this effect of RPB1 depletion on Rapa-mediated activation of MAF1, which is normally activated under low growth conditions, we measured pre-tRNA expression in cells deprived of serum for 18 hrs prior to RPB1 depletion (Figure S2E). To our surprise, pre-Thr tRNA levels in serum-starved cells were up to 8-fold higher following RPB1 depletion, indicating that loss of Pol II activity for just 2 hrs was sufficient to fully reverse the normal low serum-mediated repression of this gene (Figure 5G and 5H). Similarly, RPB1 depletion in serum-starved cells restored pre-SeC tRNA expression to a level comparable to that observed in cells at high serum (Figure 5G and 5H). In contrast, the normal repression of pre-Ala tRNA transcription in serum-starved cells was enhanced by RPB1 depletion (Figure 5G and 5H, DA/Ctl <1). Strikingly, a microarray analysis performed with pools of RNAs prepared from all three clones treated or not with DA revealed that the transcriptional response of a large majority of tRNA genes affected by serum starvation in Ctl treated cells remain unaffected in serum-starved DA-treated cells (202 vs 50; compare Figure 5I to 5J). Finally, serum stimulation of deprived cells induced a rapid and robust induction of all three pre-tRNAs in the presence and absence of Pol II (Figure S5F). However, the kinetics of these inductions were different for the pre-Thr and pre-SeC tRNAs, which both displayed faster responses to serum stimulation in the absence of Pol II - as revealed by the slopes of the inductions in the first 30 min (Figure S5F, red triangles). Altogether these results indicate that RNA Pol II transcription plays an essential role in the regulation of the transcriptional response of most tRNA genes in response to environmental cues.
Pol II transcription can repress Pol III function directly from local promoters
The data accumulated so far are compatible with a model in which Pol II transcription can modulate Pol III activity directly via transcriptional interference or indirectly via loss of an unstable (co)activator(s). However, we were not able to show unambiguously that Pol II transcription occurs at tRNA genes that are upregulated by transcriptional inhibitors and by RPB1 depletion. Moreover, it also is not clear if these effects are restricted to HEK293 cells. Since Pol II was observed near 70% of all Pol III-bound tRNA loci in HeLa cells (Oler et al., 2010), we analyzed the presence of serine 2-phosphorylated (S2P) Pol II, a CTD modification associated with elongating complexes (Komarnitsky et al., 2000), at these loci in HeLa S3 cells using available ENCODE datasets. In this cell line, clear densities of Pol II and S2P-Pol II could be observed at both pre-Thr and pre-SeC tRNA loci but not at the pre-Ala tRNA locus (Figure 6A). If Pol II directly interferes with Pol III activity, one could expect that higher levels of Pol II should be correlated with greater repression. We tested this hypothesis using DRB, LDC and Trip treatments and measured pre-tRNA expression levels in HeLa S3 cells (Figure 6B and 6C). As expected, a 2-hr treatment with any of these inhibitors robustly stimulated expression of pre- Thr (up to 8-fold) and pre-SeC (up to 3-fold) tRNAs, but not pre-Ala tRNA (which was downregulated in all three cases as observed in HEK293 cells) (Figure 6C). In addition, similar results were also using IMR90 human primary fibroblasts treated with DRB, thereby indicating that these effects are not restricted to transformed cell lines. In these cells, a reduction of Pol II occupancy could also be observed at both pre-Thr and pre-SeC loci (Figure S6A and S6B). These data support the hypothesis that tRNA repression by transcribing Pol II complexes is both direct and not restricted to HEK293 cells. Next, we examined whether the tRNA-Thr locus displayed characteristics of a Pol II promoter by examining available ENCODE datasets for Pol II general transcription factor (GTFs) as well as gene-specific regulators in all available cell lines (Figure S6C). This analysis revealed the presence of GTFs (such as TFIIF and TFIID) as well as numerous transcriptional regulatory factors in the immediate vicinity of this Pol III gene. Among them, the Wnt-regulated factor TCF4 in HCT116 cells was further found to localize in close proximity of half of all the DRB-sensitive tRNA loci identified (Figure 6D). Hence, if our model is correct, one can make the prediction that TCF4 should be involved in the repression of the tRNA-Thr locus by stimulating Pol II transcription. To test this hypothesis, we first confirmed that the pre-Thr and pre-Ala tRNAs were indeed also regulated by Pol II in these cells (in contrast to Pol II-independent pre-GIu tRNAs) using transcriptional inhibitors (Figure 6E). These results further confirmed the generality of Pol II-mediated repression in yet another cell line. Next, we assessed the function of TCF4 by transfecting a plasmid expressing a dominant-negative TCF4 (AN-TCF4) (Korinek et al., 1997). Despite a relatively low transfection efficiency (Figure S6D), cells expressing the dominant-negative constructs (Figure S6E), contributed sufficiently to the analyzed pool of RNAs to reveal a highly specific upregulation of tRNA-Thr without any effects on the tRNA-Ala and tRNA-GIu loci (Figure 6F). Importantly, this upregulation was accompanied by a reduction of Pol II occupancy at the tRNA-Thr locus in cells expressing AN-TCF4 (Figure 6G). These results strongly indicate that Pol II, recruited specifically at local promoters by Pol II-specific transcriptional activators can regulate Pol III-dependent tRNA transcription via transcriptional interference.
Figure 6. Pol II repression of tRNA genes is not limited to HEK293 cells and likely occurs directly from local Pol II promoters.

(A) ChlP-seq coverage for total and Ser2-phosphorylated RPB1 and corresponding input (data from ENCODE) in HeLa S3 cells at three model pre-tRNA loci. (B) Relative mRNA levels in HeLa S3 cells treated for lhr with LDC, DRB and Trip normalized to levels in DMSO-treated cells. (C) Same as (B) but showing normalized relative pre-tRNA levels. (D) Venn diagram showing that half of the tRNA genes up-regulated by DRB in HEK293 cells are bound by TCF4 (within 500 bps from the tRNA genes) in HCT116 cells and Model of β-catenin/TCF4-mediated repression of tRNA genes sensitive to Pol II repression. (E) same as (B, C) but for HCT116 cells. Pre-GIu is used as a control (Pol ll-independent). (F) Relative mRNA and pre-tRNA levels in HCT116 cells transiently cotransfected with plasmids expressing a dominant-negative TCF4 construct (AN-TCF4) and an EGFP or just an EGFP (control). (G) Relative fold-change of Pol II occupancy at the tRNA-Thr, tRNA-Ala loci and MYC promoter in HCT116 cells cotransfected as in (F). Data in (B), (C) and (E)-(G) represent the average +/− SD; *p<0.05 (T- test).
Discussion
The relative simplicity in the Pol III transcriptional machinery allows an efficient global regulation of RNA species involved in protein biosynthesis to match cellular demands. However, several lines of evidence suggest that individual tRNAs are specifically modulated in response to internal and external cues to support specific roles in a large number of biological processes (see Rak et al., 2018). How gene-specific transcription of tRNAs genes is established remained largely unexplored in mammalian cells. During the past 10 years, numerous ChlP-seq studies revealed a surprising genomic colocalization of Pol II near Pol III genes (Barski et al., 2010; Canella et al., 2012; Moqtaderi et al., 2010; Oler et al., 2010; Raha et al., 2010; Yeganeh et al., 2017). However, the functional significance of these observations is difficult to address using conventional techniques since the mature forms of Pol III RNAs are very stable, highly abundant and difficult to measure.
To overcome these issues and explore functionally the connections between Pol II and Pol III transcription events, we designed cell lines in which the rapid depletion of RPB1 can be induced by the addition of auxin. Surprisingly, loss of Pol II resulted in rapid and severe consequences on the transcription of specific tRNA genes. Our approach unambiguously identified Pol II as a direct gene-specific regulator of tRNA transcription by Pol III. In addition, fortuitously, these cell lines also allowed us to assess the effects of Pol II transcription at tRNA loci as a result of a partial truncation of the CTD in Pol II complexes at snRNA and stably paused genes. Importantly, CTD-less polymerases are found exclusively as stable ternary complexes on the chromatin, suggesting that the processivity of Pol II is not controlled by the CTD. Although the CTD is not required for accurate transcription and elongation in DNA-templated cell-free transcription systems (Zehring et al., 1988), this RPB1 domain is vital for initiation in cells at endogenous genes (Lux et al., 2005; Meininghaus et al., 2000). Unexpectedly, our system also provides, for the first time, direct evidence that the CTD heptads (and their modifications) are not required for productive elongation at endogenous loci in living cells, which has broad implications for Pol II transcription in general.
Identification of Pol II as a gene-specific regulator of tRNA transcription by Pol III
Rapid depletion of RPB1 as well as pharmacological inhibition of Pol II revealed that Pol II transcription can modulate the synthesis of specific pre-tRNAs either positively or negatively. Our results suggest that the decrease in expression of Pol III targets observed following RPB1 depletion or in a-amanitin treated cells (Barski et al., 2010; Listerman et al., 2007; Raha et al., 2010) is most likely indirect and results from the loss of unstable Pol III regulators such as MYC, previously shown to be a positive regulator of Pol III transcription (Kenneth et al., 2007). In contrast, repression of certain tRNA loci by Pol II is direct and does not require de novo protein synthesis. In these cases, loss of Pol II transcription was accompanied by higher levels of nascent tRNAs associated with repressed loci and by increased incorporation of EU into corresponding precursors and mature tRNAs. Moreover, the stability of the precursors tested was not affected by RPB1 depletion, confirming modulation of the synthesis of these pre-tRNAs. Furthermore, the increase in tRNA transcription caused by Pol II inhibitors was not observed in RPBl-depleted cells when Pol II transcription was maintained by the termination-deficient, Pol IIB formed at neighboring Pol II genes. While the number of tRNA genes affected by Pol II is relatively small, we surprisingly also found that Pol II was essential to sustain the repression of a large number of tRNA genes during serum starvation. Altogether our results support a direct and essential role of Pol II in the repression of tRNA transcription not only in basal conditions but also in response to environmental cues and signaling pathways as diverse as the Wnt pathway.
A model for Pol III repression by transcriptional interference
Following the first round of transcription, terminating Pol III was found to be committed to reinitiation on the same template for multiple rounds without being released in solution in a process referred to as facilitated recycling (Dieci and Sentenac, 1996; Dieci et al., 2013). Polymerases engaged in such hyper-processive cycles transcribe their targets at much higher rates (between 5- to 10-fold) and are resistant to MAF1 (Cabart et al., 2008). Structural studies showed that MAF1 binds the same Pol III interface bound by TFIIIB, thereby preventing Pol III recruitment to cognate promoters (Vannini et al., 2010). Interestingly, reconstitution of Pol III elongating complexes in the presence of promoter-bound TFIIIB revealed that Pol III can interact with TFIIIB even in the gene body and likely during the entire transcription cycle (Han et al., 2018). This observation provides a mechanistic basis not only for facilitated recycling but also for the resistance of engaged polymerases to MAF1 in vitro. Although it still is not clear if facilitated recycling occurs in vivo (Arimbasseri et al., 2014), electron microscopic studies of 5S loci in yeast revealed an apparent interaction between the promoter and the terminator of some of these genes - an observation compatible with the existence of facilitated recycling in living cells (French et al., 2008). Hence, if recycling does occur in vivo, polymerases engaged in such hyper-processive loops would require an external player to break the cycling and allow MAF1 to establish repression. Based on our observations, transcribing Pol II passage through regulated tRNAs could assume this function by displacing, or at least interfering, even transiently, with TFIIIB-Pol III interactions and thus allowing MAF1 to interact with the engaged Pol III. Indeed, in the absence of TFIIIB, elongating Pol III complexes can interact with MAF1 (Vannini et al., 2010). However, upon reaching the end of a transcribed tRNA gene, the now Maf1-bound Pol III cannot be used for re-initiation - leading eventually to the repression of these genes (Figure 7). This model further explains our observation of an apparent reversion of tRNA repression following Pol II loss during serum starvation. Indeed, since MAF1 is 4- to 10-fold less abundant than Pol III in mammalian cells (Orioli et al., 2016), it is very likely that every tRNA gene will encounter at least one Maf1-free Pol III complex during the 2-hr period of RPB1 depletion in our analyses, thereby allowing facilitated recycling to be reestablished. In addition, since the dwell time of reinitiating polymerases at promoters and terminators has to be shorter than the dwell time of newly initiating Pol III, the proposed model also explains how higher rates of transcription can be achieved without increased Pol III occupancy.
Figure 7. A model for direct repression of Pol III activity at tRNA genes by Pol II transcription.

Model for the Pol II regulation of tRNA transcription rates. See Discussion for details
Hence, the presence of Pol II peaks close to tRNAs genes in some cell lines but not others could simply reflect cell line/tissue specific differences in nearby Pol II promoter activity. Higher local Pol II initiation rates would increase the chance of accumulation of elongating Pol II complexes at tRNA genes as they are slowed down by Pol III or its associated factors. Indeed, the strength of pre-Thr or pre-SeC tRNA stimulation following pharmacological inhibition of Pol II correlated with the higher levels of Pol II seen in HeLa or IMR90 cells. Our analysis also revealed that transcription factors such as T Cell factor 4 (TCF4) can indirectly repress the pre-Thr gene via Pol II recruitment in HCT116 cells. Hence, systematic identification of these promoters and the specific regulators involved in their regulation will provide insights into the regulation of particular tRNAs in response to environmental cues or pathologies. However, such a transcriptional control is meaningful only if a sufficient number of genes encoding a particular tRNA isodecoder (anticodon) are co-regulated. Interestingly, more than 50% of all expressed Ala-ACG, Arg-TCG, iMet, SeC and Ser-CGA and -GCT tRNAs appeared to be enhanced upon DRB treatment (Figure S7), which therefore could potentially affect the abundance of proteins relying on these tRNAs for their translation. This is particularly interesting in the case of initiator methionine (iMet) tRNA, which is essential for translation initiation and whose levels have been shown to control body size and developmental timing in Drosophila (Rideout et al., 2012). Moreover, an upregulation of this tRNA by only 1.4-fold was found to induce a global reprogramming of tRNA expression and to increase proliferation in human epithelial cells (Pavon-Eternod et al., 2013). However, the fact that most tRNA genes did not react to Pol II inhibition or depletion, or that some were even downregulated, suggests that the proposed mechanism is not general. It would therefore be of particular interest to evaluate whether these genes are capable of facilitated recycling in vivo and, if so, how these cycles are interrupted in adverse conditions. Alternatively, the dependency of pre-Ala tRNA levels on MYC levels may also indicate that high rates of transcription are perhaps achieved through stimulation of de novo initiation frequencies at this tRNA gene.
STAR Methods
Lead Contact and Materials Availability
Further information and requests should be directed to the Lead Contact, Robert G. Roeder (roeder(@)rockefeller.edu), and will be fulfilled after execution of a suitable Materials Transfer Agreement.
Experimental Model and Subject Details
Cell culture and cell line generation
HEK293 (RRID:CVCL_0045), HeLa-S3 (RRID:CVCL_0058), HCT116 (RRID:CVCL_0291) and IMR90 (RRID:CVCL_0347) cells were cultured in DMEM (high glucose, pyruvate; Thermofisher #11995040) supplemented with 10 % FCS and 100 U/ml PenStrep (Thermofisher #15140122). The parental HEK293 line expressing a doxycycline-inducible OsTIRl was established by cotransfection of the plasmid pMK243 and pSpCas9(BB)-2A-GFP expressing a chimeric guide RNA targeting the AAVS1 locus using X-tremeGENE 9 DNA Transfection Reagent (Sigma-Aldrich #6365779001) following the manufacturer’s instructions. Clones were obtained by limiting dilution of puromycin (2μg/ml; Thermofisher #A1113802) selected cells. After validation of the OsTIRl expression cassette integration into the AAVS1 locus and the ability to degrade a transfected AID-tagged H2B-YFP construct upon a 24-hr doxycycline (2μg/ml; Santa Cruz sc-337691) + auxin (500 μM; Santa Cruz sc-215171) treatment, one parental clone was stably transfected with a Pvul-linearized pcDNA-TagRPBl plasmid. After a one-week selection with 500 μg/ml Geneticin (Thermofisher #10131035), mKO2-positive cells were sorted by flow cytometry in bulk. Doxycycline + auxin (DA) sensitive cells were then selected by sorting in bulk mKO2 negative cells following a 24-hr DA treatment (see Figure S1A). After a recovery period of one week, cells were finally transfected with a pSpCas9(BB)-2A-GFP expressing a chimeric guide RNA targeting the POLR2A gene to inactivate specifically the endogenous RPB1. Two days after transfection, double positive (mKO2 and GFP) cells were sorted as single cells by flow cytometry and plated in four 96-well plates (384 clones), and 146 clones (38%) were finally obtained. GFP-expressing clones (stable integration of the CRISPR/Cas9 plasmid) were discarded, while other clones were screened for substantial cell death following a 12-hr DA treatment in the presence of puromycin. 47 clones (32%) passed this round of selection and complete loss of endogenous RPB1 was confirmed by immunoblotting of RIPA nuclear extracts. To establish the rapid degradation protocol, we first monitored the auxin response (loss of mKO2 fluorescence) over time (during 3hr) of cells incubated with increasing amounts of doxycycline (DOX) for 24 hrs prior to auxin addition. These experiments revealed that the dose of DOX only modulated the proportion of responsive cells but not the strength of the response. However, after a 24-hr period in the presence of a saturating dose of DOX (2 μg/ml), all mKO2 fluorescence was lost even in the absence of auxin. We therefore used a kinetic approach involving an induction of OsTIRl expression with 2 μg/ml DOX for different periods of time and monitoring the fluorescence response of mKO2 to auxin addition. The maximal response to 500 μM auxin with the minimal loss of fluorescence prior its addition was reached after a 12-hr induction with 2 ng/ml DOX followed by addition of auxin for at least 2 hrs. Three individual clones displaying high responsiveness to this protocol (#7, #12, and #19) were kept for all further experiments using the same basic protocol. For the control conditions, cells were incubated with medium supplemented with water instead of DOX for 12 hr prior to auxin addition. For pre-treatment with transcriptional inhibitors (100 μM DRB [Santa Cruz sc-200581], 10 μM LDC00067 [LDC; AdooQ Bioscience A14273–10mM-D], 10 μM triptolide [Trip; Santa Cruz sc-200122], 20 ng/ml actinomycin D [ActD; Santa Cruz sc-200906]) or 10 μM rapamycin (Rapa; EMD Millipore #553211), drugs were added prior to auxin addition (11 hrs after DOX) or 2 hrs after auxin addition in the case of rapamycin (after degradation) and for RNA stability measurement with ActD. For metabolic labelling, 0.5 mM ethynyl-uridine (EU) was added for 1 hr after a 2-hr treatment with auxin. For cycloheximide (CHX; Sigma Aldrich C4859) pre-treatment, the drug was added 15 min prior to auxin. For serum starvation/stimulation experiments, cells were incubated with DMEM without FCS for 6 hrs prior to OsTIRl induction by Dox and cells were starved for a total of 18 hrs prior to auxin addition. For serum stimulation experiments, cells were first starved and 2 hrs after auxin addition the culture medium was replaced with DMEM containing 20% FCS supplemented with 500 HM auxin. Finally, for experiments involving HeLa, HCT116 or IMR90 cells, transcriptional inhibitors were added 2 hrs prior to harvesting the cells. For all experiments involving treatments, cells were incubated with the corresponding vehicle only (water or DMSO) for control conditions (with or without RPB1 depletion) as indicated in the figure legends. For siRNA transfections, clones were transfected with siGENOME siRNA SMARTPools (Dharmacon non-targeting [D-001206–13-05] or human MYC-targeting [M-003282–07-0005] using Lipofectamine RNAiMAX Transfection Reagent (Thermofisher #13778100) following manufacturer’s instructions 22 hr prior to OsTIRl induction. siRNAs sequences are also indicated in Table S1. For the experiments using the TCF4 dominant- negative (AN-TCF4) expression plasmid, HCT116 cells cultured in 6-well dishes were transfected with either 1.5 ng of EGFP plasmid with 1.5 ng of carrier pBSKII plasmid (control condition) or 1.5 ng of EGFP plasmid and 1.5 ng of AN-TCF4 expressing plasmid per well using X-tremeGENE 9 DNA Transfection Reagent (Sigma-Aldrich #6365779001) following manufacturer’s instructions for 24hrs prior harvesting. For ChlP-qPCR experiments involving AN-TCF4 and GFP, transfected HCT116 cells were trypsinized and harvested for GFP-positive cell sorting 2 days after transfection. Cells were suspended in PBS/3%FBS solution containing TOPRO-3 (Thermofisher #T3605). GFP positive cells were sorted in PBS.
Method Details
Plasmids
CRISPR/Cas9 vectors were constructed according to (Ran et al., 2013) using the plasmid pSpCas9(BB)- 2A-GFP (PX458), a gift from Feng Zhang (Addgene plasmid # 48138; http://addgene.org/48138; RRID:Addgene_48138). The plasmid used to establish the parental HEK293 cell line expressing the OsTIRl ubiquitin ligase from the AAVS1 locus (Natsume et al., 2016), pMK243, was a gift a gift from Masato Kanemaki (Addgene plasmid # 72835; http://addgene.org/72835; RRID:Addgene_72835). Efficiencies of DA-mediated degradation of tagged proteins in parental clones were tested using the plasmid pcDNA5-H2B-AID-EYFP (Holland et al., 2012), a gift from Don Cleveland (Addgene plasmid # 47329; http://addgene.org/47329; RRID:Addgene_47329). The pcDNA-TagRPBl plasmid encoding the fluorescent degron-tagged full length RPB1 was constructed by In-Fusion cloning (Takara #639650) of a PCR amplified gene block (IDT) encoding the C-terminal tag (Flag-Degron-V5-mKO2) and a PCR amplified Flag-HA-Full length RPB1 cDNA (a gift from Dr. Sohail Malik) into a pcDNA3.1(+) plasmid (ThermoFisher #V79020) linearized with EcoRI. Cloning primers and gene block sequence are given in Table S1. The pcDNA/Myc DeltaN TCF4 was a gift from Bert Vogelstein (Addgene plasmid # 16513; http://addgene.org/16513; RRID:Addgene_16513).
RNA isolation, cDNA preparation and real-time qPCR analysis
Total RNA was prepared using TRIzol Reagent (Thermofisher #15596018) as described hereafter. Cells were harvested directly in TRIzol after removal of the culture medium. Following a 5 min incubation period, samples were centrifuged for 10 min at 12,500xg at 4°C to pellet high molecular weight genomic DNA. Phase separation was performed with 0.2 volume of chloroform and spun at 12,500xg for 15 min at 4°C after vigorous mixing for 30 sec. RNA in the recovered aqueous phase was precipitated with 0.8 volume of ispropanol at room temperature for 10 min and spun for 10 min at 12,500xg at 4°C. After complete removal of isopropanol, RNA pellets were washed twice with ice cold 80% Ethanol and resuspended in RNAse-free water and incubated for 5 min at 55°C. Isolated RNA was then treated with RQ1 RNase-Free DNase (Promega M6101) according to manufacturer’s instructions for 30 min at 37°C. Reactions were stopped using the provided stop solution, diluted to 200 nl with water and extracted with 1 volume of Phenol:Chloroform:lsoamyl Alcohol (Thermofisher #15593031). The aqueous phase was subsequently extracted with water-saturated chloroform prior to isopropanol precipitation as described before. RNA concentration was measured using a Nanodrop device. Quality and integrity were evaluated by A260/280 and A260/230 readings as well as electrophoresis on denaturing agarose or polyacrylamide gels. For nascent RNA preparation, nuclear pellets were prepared as described below (cell extract preparations) but with buffers supplemented with RNAsin Plus (Promega N2611). A fraction of the cell suspension was used for total RNA preparation (as described) before nuclei isolation and NUN buffer extraction. After the last wash with NUN buffer, chromatin pellets were thoroughly resuspended in TRIzol until complete decompaction of the pellets and then incubated 10 min at room temperate. RNA isolation was then performed as described before, but with addition of 30 μg UltraPure Glycogen (Thermofisher #10814010). cDNAs were synthesized from 3 ng RNA using a superscript III First-Strand Synthesis System (Thermofisher #18080051) with random hexamers following manufacturer’s instructions with a 10 min incubation step at room temperature prior to a 50-min incubation at 50°C. SYBR green RT-qPCRs were performed in 25 μl reactions with 2 μl of cDNAs diluted 1/10 in water using QuantiTect SYBR Green PCR Kit (QIAgen #204145) with primers used at a final concentration of 10 μM on a Applied Biosystems 7300 Real-Time PCR System. Primers for pre-tRNAs were designed using 3’ trailers sequences identified in (Gogakos et al., 2017). Efficiencies for each pair of primers were determined by serial dilution of cDNAs from total RNAs and taken into account for each RNA level measurements. Relative RNA levels were calculated using the geometric mean of two stable genes, PGK1 mRNA and PPIA mRNA, as reference. Primer sequences are given in Table S1.
RNA demethylation and EU-labelled RNA isolation
After isolation from cells incubated for 1 hr with 0.5 mM EU, 10 μg of total RNA was demethylated using the reagents included in the rtStar tRNA-optimized first-strand cDNA synthesis kit (Arraystar #AS-FS-004) following manufacturer’s instructions except first-strand cDNA synthesis was omitted and, instead, 5 μg of demethylated EU-labelled RNA was biotinylated in a click chemistry reaction performed with reagents provided in a Click-iT Nascent RNA Capture Kit (Thermofisher C10365) following manufacturer’s instructions. Metabolically labelled RNAs were finally captured on Dynabeads Streaptavidine T1 magnetic beads provided in the kit using 1 μg of biotinylated RNA as starting material following manufacturer’s instructions. Reverse transcription was finally performed on beads using superscript III First-Strand Synthesis System.
Cell fractionation, extracts preparation and immunoblotting
Cells were first trypsinized and washed with ice cold PBS. Cell pellets were then lysed in an ice-cold NP-40 homogenization buffer (10 mM Hepes-KOH [pH 7.6], 15 mM KCI, 2 mM EDTA, 0.15 mM spermine, 0.5 mM spermidine, 10% glycerol, 0.3M sucrose, 0.5% NP-40) and incubated 10 min on ice. A fraction of the lysate was used to prepare total RNA as described before for preparation of nascent RNA. NP-40 lysates were then layered on 30% cushion buffers (10 mM Hepes-KOH [pH 7.6], 15 mM KCI, 2 mM EDTA, 0.15 mM spermine, 0.5 mM spermidine, 10% glycerol, 0.87 M sucrose) in centrifugation tubes and spun 15 min at 3,500×g at 4°C. Cytoplasmic protein extracts were prepared from the upper phase by boiling with 1X final Laemmli protein sample buffer (4X Buffer: 240 mM Tris-HCI[pH 6.8], 40% glycerol, 8% SDS, 0.04% bromophenol blue, 5% β-mercaptoethanol). Isolated nuclei were resuspended in nuclear storage buffer (10 mM Hepes-KOH [pH 7.6], 100 mM KCI, 0.1 mM EDTA, 0.15 mM spermine, 0.5 mM spermidine, 10% glycerol). Nuclear RIPA extracts were obtained by pelleting nuclei and resuspending them in RIPA buffer (25 mM Tris-HCI [pH7.5], 150 mM NaCI, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS). After the RIPA extract preparation, an equivalent volume of lx Laemmli protein sample buffer was added to the RIPA pellet and sonicated until complete solubilization. The solubilized pellet and RIPA-nuclear extract were used for immunoblot analysis. NUN nuclear and pellet extracts were prepared my mixing 1 volume of nuclei in nuclear storage buffer with 1 volume of 2X NUN buffer (50 mM Hepes-KOH [pH 7.6] 0.6 M NaCI, 2% NP-40, 2M Urea) with a 30 min incubation on ice. After centrifugation for 30 min at 21,000×g at 4°C, NUN nuclear extracts were recovered (supernatant) and chromatin pellets were subsequently washed twice with lx NUN buffer and either processed to recover nascent RNAs (as described before) or incubated in urea buffer (8 M urea, 200 mM Tris-HCI, [pH 6.8], 1 mM EDTA, 5% SDS, 0.5M DTT) for 30 min at 37°C under agitation followed by sonication to prepare chromatin pellet protein extracts. All buffers (except SDS protein sample and urea buffer) were supplemented with 0.5 mM DTT (2 mM for 2x NUN buffer), 1/100 Protease Inhibitor Cocktail Set III (EMD Millipore #539134–1ML) and 1 tablet/lOml PhosSTOP Phosphatase Inhibitor Cocktail (Sigma Aldrich #04906837001) just before use. Antibodies used were polyclonal anti-RPBl (N-20, Santa Cruz sc-899), monoclonal anti-RPBl CTD (8WG16, Santa Cruz sc56767), polyclonal anti-RPB2 (H-201, Santa Cruz sc-67318), polyclonal anti-RPB3 (A303–771A, Bethyl), monoclonal anti-RPA194 (F-6, Santa Cruz sc-46699), polyclonal anti-RPC53 and anti-RPC62 (in house), monoclonal anti-Maf1 (H-2, Santa Cruz sc-515614), monoclonal anti-MYC (9E10, Santa Cruz sc-40), monoclonal anti-V5 (Thermofisher R960–25), anti-Flag (M2, Sigma Aldrich F1804). Secondary antibodies were anti-mouse and anti-rabbit IgG Hrp (respectively NA931 and NA934, GE health care).
Microscopy imaging
Fluorescence and phase contrast microscopy pictures of living cells were acquired on a Nikon eclipse Ti-S microscope with a 20X objective. Pictures of control and experimental conditions were acquired with the same settings and are presented with the same brightness/contrast adjustments.
Mass spectrometry
Samples for mass-spectrometric analysis of the full length and truncated RPB1 were prepared from 5 × 15cm plates of Clone #7. For both control (2 hrs auxin only) and DA-conditions (12 hrs DOX, 2 hrs auxin), cells were washed with lx PBS, trypsinized and harvested. Cell pellets were further washed with lx PBS, and nuclear extracts and pellets were prepared using a modified Dignam nuclear extraction protocol (Dignam et al., 1983). Briefly, cell pellets were swollen in hypotonic buffer (10 mM HEPES [pH7.9] 1.5 mM MgCI2, 10 mM KCI with protease inhibitors and 0.25 mM DTT), Dounce homogenized and spun down to recover the nuclei. The recovered nuclei were then resuspended in low salt buffer (20 mM HEPES [pH7.9], 20% Glycerol, 1.5mM MgCI2, 0.2mM EDTA, 20mM KCI supplemented with protease inhibitors and 0.25 mM DTT), Dounce homogenized again, and then adjusted to 420 mM KCI by adding high salt buffer (20 mM HEPES [pH7.9], 20% Glycerol, 1.5 mM MgCI2, 0.2 mM EDTA, 1.2 M KCI supplemented with protease inhibitors and 0.25 mM DTT) added in a drop-wise manner. Samples were recovered and centrifuged (13’000×g, 15min) and nuclear extract (supernatant) and nuclear pellets were separated. For the preparation of intact RPB1, nuclear extracts from control cells were used (as this fraction contains mainly unphosphorylated RPB1, which would otherwise complicate the peptide identification). Nuclear extracts were diluted to 300 mM KCI and 0.1% NP-40 was added. For CTD-less RPB1, nuclear pellets from DA-treated conditions were used since the CTD-truncated RPB1 is largely retained in the pellet in the Dignam extraction procedure as well. BC200 (1.2 V of the pellet, 300mM KCI final) was added to the pellet and inverted. Samples were sonicated 6 rounds with 3min intervals on ice. The sonicated samples were spun down and the solubilized pellets were used for downstream Flag (M2)-IP in presence of 0.1% NP-40. M2-agarose beads were added and incubated overnight in the presence of protease inhibitors. The M2 beads were washed 6 times in BC300 buffer (300 mM KCI, 0.1% NP-40), and bound proteins were eluted by adding 3xFLAG peptide (166 ng/ml, Sigma F4799) in BC300 buffer.
Eluted samples were loaded onto 4–20% gradient SDS/PAGE gel and stained with Colloidal Blue (ThermoFisher, #LC6025). Gel bands corresponding to the intact and truncated RPB1 were reduced with DTT and alkylated with iodoacetamide followed by digestion by a mix of trypsin and TLCK treated alpha chymotrypsin. Extracted peptides were analyzed by nano LC-MS/MS (EasyLC 1200 coupled to Fusion Lumos, Thermo Scientific)(Li et al., 2019), separated by reverse phase chromatography using a gradient increasing from 2% B/98%A to 35% B/65%A in 30 minutes (A: 0.1% formic acid, B: 80% acetonitrile/0.1% formic acid). MS data were queried against the UniProt Human database (2019) with semi-tryptic/semi-chymotryptic constraints using Proteome Discoverer 1.4 (Thermo Fischer) /Mascot 2.4 (Matrix Science). Oxidation of methionine and protein N-terminal acetylation was allowed as variable modifications. Matched peptides were filtered using <5% Percolator (Spivak et al., 2009) calculated False Discovery Rate and mass accuracy of 5 ppm or better. For each sample, extracted RBP1 peptide areas was summed per residue and plotted as a function of full length RBP1 (P24928) sequence using PepEx (Guerrero et al., 2014). Signals were normalized to the most abundant signal (residue 365–382). 17 peptides matched to the C-terminus of full-length RPB1 (~90% of the measured signal from 1590 to 1954) were unique to only one region of RPB1.
Northern Blot
tRNA-iMet Northern blots were performed using pre-hybridization with unfolder oligonucleotides as described in (Buvoli et al., 2000) with the following modifications. Total, nuclear or nascent RNAs were loaded on mini 6% urea-polyacrylamide gels. After electrophoresis, RNAs were transferred onto positively charged nylon membranes (Roche diagnostics #11 209 299 001) using a semi-dry electrophoretic transfer cell for 1 hr at 250 mA. RNAs were UV cross-linked (0.24J on the RNA side, 0.12V on the other side of the membrane) and membranes were further baked for 2 hr at 80°C. Pre-hybridization was performed in ULTRAhyb-Oligo buffer (Thermofisher AM8663) supplemented with 50 nM unfolder oligonucleotide for 12 hr at 42°C. Hybridization was then performed with 50 nM biotinylated oligo probes in the presence of 25 mM unfolder oligonucleotide overnight at 37°C. Membranes were subsequently washed twice for 10 min with 5X SSC, 0.5% SDS at room temperature then blocked for 15 min with 2X SSC, 0.5% SDS and 3% BSA at room temperature. Membranes were then incubated with Streptavidin-HRP (1:40,000, Thermofisher N100) in 2X SSC with 3% BSA and 0.5% SDS for 15 min, then washed quickly twice with 2X SSC. Membranes were subsequently washed with ABS buffer (10% BSA and 1% Triton X-100 in 2X SSC) for 5 min then rinsed twice with 2X SSC prior to film exposure using ECL-based detection.
For Pre-tRNA-Thr, tRNA-Thr, pre- tRNA-Ala and tRNA-Ala Northern blots, the same procedure was used except nylon membranes were pre-hybridized at 30°C in 2x diluted ULTRAhyb-Oligo buffer with 50 nM unfolder oligonucleotides for 12 hrs. Hybridization was performed at 30°C overnight in 2x diluted ULTRAhyb-Oligo buffer supplemented with 25 nM unfolder oligonucleotides and 5’ end 32P-labelled oligonucleotide probes (prepared by incubation of 30 pmol of oligonucleotide with 60 pmol γ−32P-ATP and T4 polynucleotide kinase for 2 hrs at 37°C and purified on Sephadex G25). Hybridized membranes were then washed twice for 10 min with 5X SSC, 0.5% SDS at room temperature followed by two 2 min washes with 2X SSC, 0.5%. Finally, membranes were exposed with radiographic films before development. Oligonucleotide sequences can be found in Table S1.
Cell Sorting
Cell sorting was performed using a FACS Ariall cell sorter (BD Biosciences). Time course analysis of DA-mediated RPB1 depletion was performed on FACS LSRII (BD Biosciences). For all flow cytometry procedures, cells were dissociated with 0.05% trypsin-EDTA, resuspended in cell culture medium, spun and resuspended in staining buffer (PBS supplemented with 3% FCS) containing TOPRO-3 at a 1:50,000 dilution (Thermofisher T3605). Analysis was performed using the Flowing Software 2 (http://flowingsoftware.btk.fi/).
Chromatin immunoprécipitation and library preparation
RPB3 and RPC62 ChlP-seq was performed as described in (Yu et al, Science, 2015) with the following modifications. Briefly, 2×l07 cells were fixed in 1% formaldehyde for 10 min at room temperature. For RPB3 and RPC62 ChlP-seq, fragmentation of fixed chromatin was performed by sonication of isolated nuclei (Branson Sonicator, Branson) to achieve enrichment of short (100–500 bp) chromatin fragments. 5 μg of RPB3 antibody (A303–771A, Bethyl) or 1 μl of RPC62 anti-serum was added to the chromatin lysate and incubated overnight at 4°C. The next day, Dynabeads Protein A (Thermo Fisher Scientific) was added and incubated with rotation at 4°C for 1.5 hours. Enriched DNA was isolated through extensive wash steps and subsequent reverse cross-linking and purification. To quantify ChlP-qPCR signals, real-time PCR was performed in 20 μl reactions using QuantiTect SYBR Green PCR Kit with primers used at a final concentration of 10 μM on an Applied Biosystems 7300 Real-Time PCR System. Primer sequences are given in Table S1. ChlP-seq libraries were prepared from 5–10 ng ChIP DNA following the instructions of Illumina TruSeq ChIP Library Preparation Kit (IP-202–1012). Libraries were then sequenced on Illumina HiSeq 2500 as 75-bp single-end runs at the Genomics Resource Center at the Rockefeller University.
Microarray analysis
Microarray analysis was performed at the Genomics Resource Center at the Rockefeller University using Affymetrix GeneChip Human Transcriptiome 2.0 Arrays (Thermofisher #902162) according to the manufacturer’s instructions. The data expressed as CEL files were normalized using the Signal Space Transformation-Robust Multi-Chip Analysis (SST-RMA) algorithm with Transcriptome Analysis Console (TAC) 4.0.1 (Thermofisher). Protein coding genes downregulated in DA-treated cells are listed in Table S3. piRNAs were excluded from the analysis.
Nascent RNA sequencing
Nascent RNA was converted to cDNA libraries using the TruSeq Stranded Total RNA Sample Prep kit with Ribo-zero Gold (lllumina) following manufacturer’s instructions and sequenced on lllumina HiSeq 2500 as 50-bp paired-end runs at the Genomics Resource Center at The Rockefeller University.
Sequencing Data analysis
Nascent RNA-seq data were aligned to the human hgl9 genome assembly using the lllumina RNA-Seq Alignment workflow version 1.1.1 with read mapping using TopHat 2 (Bowtie 2). Alignment files were then uploaded to the Galaxy web platform and analyzed using the public server at usegalaxy.org (Afgan et al., 2018). Coverage files were generated using bamCoverage tool (Galaxy Version 3.0.2.0) with bin sizes of 1 bp and RPKM normalization method. Pre-tRNA differential expression analysis was performed with edgeR tool (Galaxy Tool Version 3.20.7.2) using count files with standard parameters except that lowly expressed genes were filtered out if the total count number was inferior to less than 8. Count files were generated with featureCounts (Galaxy Tool Version vl.6.0) using the fragment counts option with a maximum fragment length of 200 bp to limit potential contribution of overlapping Pol II transcripts. Multi-mapping was allowed but assigned as fractional counts of the number of times mapped. A minimal mapping quality score of 3 per read was used. A pre-tRNA gene annotation file was used with the featureCounts tool containing all tRNAs genes (tRNA genes predicted by using tRNAscan-SE v.1.23 (Chan and Lowe, 2009)) including 40 bp of upstream and 275 bp of downstream genomic DNA to include leader and potential extended trailer sequences. Mitochondrial tRNA genes were excluded from the differential expression analysis. Profile plots for nascent RNA score distributions across genomic regions were generated using the computeMatrix tool (Galaxy Tool Version 2.5.0.0) followed by plotProfile (Galaxy Tool Version 2.5.0.0) using scale-regions as shown in presented plots. Regions plotted for tRNA gene profiles correspond to tRNA genes as defined by tRNAscan-SE v.1.23 or a subset of them. For ChlP-seq analysis, sequencing data were directly uploaded to the Galaxy web platform and data quality was verified with the FastQC tool (Galaxy tool version 0.69). Reads were mapped on the hgl9 genome assembly using Bowtie2 (Galaxy Tool Version 2.3.4.1) and input- subtracted coverage files were generated using bamCompare (Galaxy Tool Version 2.5.0.0) with signal extraction scaling (SES) option and normalization to lx coverage. Enrichment score profile plots were generated using computeMatrix (Galaxy Tool Version 2.5.0.0) followed by plotProfile (Galaxy Tool Version 2.5.0.0).
Quantification and Statistical Analysis
All quantitative analyses were performed with a minimum of three independent biological replicates. The Student’s t test was used to determine statistical significance. Values are reported as mean ± SD as indicated in figure and supplementary figure legends. Differential expression analysis of pre-tRNA levels using nascent RNA-seq data was performed using two biological replicates for each condition (clone #7 and #19, Ctl vs DA or two independent biological replicates each for Mock vs DRB treated HEK293 – data from Werner and Ruthenburg, 2015). Differential expression analysis and statistical significance were determined using edgeR (Robinson et al 2010, as described in the STAR methods section) with TMM normalization and quasi-likelihood F-test with robust settings. tRNA genes affected >1.5-fold at the nascent RNA level with a FDR<0.05 (Benjamini and Hochberg, 1995) were considered for analysis.
Data and Code Availability
See Key Resources Table.
Microarray, nascent RNA-seq and ChlP-seq data have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO superseries accession number GSE130878.
Nascent RNA-seq data from mock- and DRB-treated HEK293 were obtained from GEO accession GSE66478. ENCODE Tracks for Total/S2P-Pol II ChlP-seq and corresponding inputs in HeLa S3 cells were generated and analyzed by the lab of Michael Snyder and downloaded directly from the UCSC browser (Rosenbloom et al., 2013) (UCSC accession: wgEncodeEH000613, wgEncodeEH001838, wgEncodeEH000612 and wgEncodeEH000744 respectively).
Supplementary Material
Table S2. Nascent RNA fold-change and gene groups for DRB- and DA-sensitive tRNA genes. Related to Figure 3
Table S3. Protein coding genes downregulated following 2hrs DA-treatment. Related to Figure 1 and 5
Highlights.
Pol II plays a direct role in the gene-specific regulation of tRNA expression
Elongating Pol II represses Pol III activity via transcriptional interference
Pol II interference is essential for tRNA repression during serum starvation
Pol II CTD is not required for Pol II productive elongation
Acknowledgements
We thank Sohail Malik, Takashi Onikubo, Evelina Tutucci and Ueli Schibler for their comments on the manuscript, the Flow Cytometry Resource Center, Henrik Molina and the Proteomics core facility and the Genomics Core Facility of the Rockefeller University. This work was supported by NIH grants CA129325, DK071900 and CA202245 to R.G.R. A.G. was supported by a Swiss National Science Foundation Early Mobility Fellowship (P2GEP3_151952) and by a Human Frontier Science Program Long-Term Fellowship (LT001083/2014). K.I. was supported by a NCI T32 grant (CA009673) and a JSPS postdoctoral fellowship for research abroad.
Footnotes
Declaration of Interests
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Grüning BA, et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimbasseri GA (2018). Interactions between RNAP III transcription machinery and tRNA processing factors. Biochim. Biophys. Acta BBA - Gene Regul. Mech. 1861, 354–360. [DOI] [PubMed] [Google Scholar]
- Arimbasseri AG, Rijal K, and Maraia RJ (2014). Comparative overview of RNA polymerase II and III transcription cycles, with focus on RNA polymerase III termination and reinitiation. Transcription 5, e27369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Chepelev I, Liko D, Cuddapah S, Fleming AB, Birch J, Cui K, White RJ, and Zhao K (2010). Pol II and its associated epigenetic marks are present at Pol III-transcribed noncoding RNA genes. Nat. Struct. Mol. Biol. 17, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300. [Google Scholar]
- Buvoli A, Buvoli M, and Leinwand LA (2000). Enhanced detection of tRNA isoacceptors by combinatorial oligonucleotide hybridization. RNA 6, 912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabart P, Lee J, and Willis IM (2008). Facilitated recycling protects human RNA polymerase III from repression by Maf1 in vitro. J. Biol. Chem. 283, 36108–36117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canella D, Bernasconi D, Gilardi F, LeMartelot G, Migliavacca E, Praz V, Cousin P, Delorenzi M, Hernandez N, Hernandez N, et al. (2012). A multiplicity of factors contributes to selective RNA polymerase III occupancy of a subset of RNA polymerase III genes in mouse liver. Genome Res. 22, 666–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PP, and Lowe TM (2009). GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 37, D93–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Gao X, and Shilatifard A (2015). Stably paused genes revealed through inhibition of transcription initiation by the TFIIH inhibitor triptolide. Genes Dev. 29, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieśla M, Towpik J, Graczyk D, Oficjalska-Pham D, Harismendy O, Suleau A, Balicki K, Conesa C, Lefebvre O, and Boguta M (2007). Maf1 Is Involved in Coupling Carbon Metabolism to RNA Polymerase III Transcription. Mol. Cell. Biol. 27, 7693–7702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozen AE, Quartley E, Holmes AD, Robinson EH, Phizicky EM, and Lowe TM (2015). ARM-Seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods 12, 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieci G, and Sentenac A (1996). Facilitated Recycling Pathway for RNA Polymerase III. Cell 84, 245–252. [DOI] [PubMed] [Google Scholar]
- Dieci G, Bosio MC, Fermi B, and Ferrari R (2013). Transcription reinitiation by RNA polymerase III. Biochim. Biophys. Acta BBA - Gene Regul. Mech. 1829, 331–341. [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, and Roeder RG (1983). Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11,1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmar KA, Goodenbour JM, and Pan T (2006). Tissue-Specific Differences in Human Transfer RNA Expression. PLoS Genet 2, e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumay-Odelot H, Durrieu-Gaillard S, Ayoubi LE, Parrot C, and Teichmann M (2014). Contributions of in vitro transcription to the understanding of human RNA polymerase III transcription. Transcription 5, e27526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrensberger AH, Kelly GP, and Svejstrup JQ (2013). Mechanistic Interpretation of Promoter-Proximal Peaks and RNAPII Density Maps. Cell 154, 713–715. [DOI] [PubMed] [Google Scholar]
- French SL, Osheim YN, Schneider DA, Sikes ML, Fernandez CF, Copela LA, Misra VA, Nomura M, Wolin SL, and Beyer AL (2008). Visual Analysis of the Yeast 5S rRNA Gene Transcriptome: Regulation and Role of La Protein. Mol. Cell. Biol. 28, 4576–4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold H, Tehler D, Christoffersen NR, Nielsen MM, Asmar F, Kooistra SM, Christophersen NS, Christensen LL, Borre M, Sørensen KD, et al. (2014). A Dual Program for Translation Regulation in Cellular Proliferation and Differentiation. Cell 158, 1281–1292. [DOI] [PubMed] [Google Scholar]
- Gogakos T, Brown M, Garzia A, Meyer C, Hafner M, and Tuschl T (2017). Characterizing Expression and Processing of Precursor and Mature Human tRNAs by Hydro-tRNAseq and PAR-CLIP. Cell Rep. 20, 1463–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi H, Nguyen HCB, Zhang S, Dill BD, Molina H, and Tavazoie SF (2016). Modulated Expression of Specific tRNAs Drives Gene Expression and Cancer Progression. Cell 165, 1416–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SS (2015). Why should cancer biologists care about tRNAs? tRNA synthesis, mRNA translation and the control of growth. Biochim. Biophys. Acta BBA - Gene Regul. Mech. 1849, 898–907. [DOI] [PubMed] [Google Scholar]
- Guerrero A, Dallas DC, Contreras S, Chee S, Parker EA, Sun X, Dimapasoc L, Barile D, German JB, and Lebrilla CB (2014). Mechanistic Peptidomics: Factors That Dictate Specificity in the Formation of Endogenous Peptides in Human Milk. Mol. Cell. Proteomics MCP 13, 3343–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Yan C, Fishbain S, Ivanov I, and He Y (2018). Structural visualization of RNA polymerase transcription machineries. Cell Discov. 4, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, Fachinetti D, Han JS, and Cleveland DW (2012). Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc. Natl. Acad. Sei. U. S. A. 109, E3350–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs EY, Ogiwara I, and Weiner AM (2004). Role of the C-Terminal Domain of RNA Polymerase in U2 snRNA Transcription and 3′ Processing. Mol. Cell. Biol. 24, 846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenneth NS, Ramsbottom BA, Gomez-Roman N, Marshall L, Cole PA, and White RJ (2007). TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc. Natl. Acad. Sei. U. S. A. 104, 14917–14922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarnitsky P, Cho E-J, and Buratowski S (2000). Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 14, 2452–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, Wichen D. van, Weger R. de, Kinzler KW, Vogelstein B, and Clevers H. (1997). Constitutive Transcriptional Activation by a β-Catenin-Tcf Complex in APC−/− Colon Carcinoma. Science 275, 1784–1787. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Saucedo-Cuevas L, Yuan L, Ross D, Johansen A, Sands D, Stanley V, Guemez-Gamboa A, Gregor A, Evans T, et al. (2019). Zika Virus Protease Cleavage of Host Protein Septin-2 Mediates Mitotic Defects in Neural Progenitors. Neuron 101, 1089–1098.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Listerman I, Bledau AS, Grishina I, and Neugebauer KM (2007). Extragenic Accumulation of RNA Polymerase II Enhances Transcription by RNA Polymerase III. PLOS Genet. 3, e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukoszek R, Mueller-Roeber B, and Ignatova Z (2013). Interplay between polymerase II- and polymerase Ill-assisted expression of overlapping genes. FEBS Lett. 587, 3692–3695. [DOI] [PubMed] [Google Scholar]
- Lux C, Albiez H, Chapman RD, Heidinger M, Meininghaus M, Brack-Werner R, Lang A, Ziegler M, Cremer T, and Eick D (2005). Transition from initiation to promoter proximal pausing requires the CTD of RNA polymerase II. Nucleic Acids Res. 33, 5139–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, Patterson SD, Wickens M, and Bentley DL (1997). The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature 385, 357–361. [DOI] [PubMed] [Google Scholar]
- Medlin JE, Uguen P, Taylor A, Bentley DL, and Murphy S (2003). The C-terminal domain of pol II and a DRB-sensitive kinase are required for 3′ processing of U2 snRNA. EMBO J. 22, 925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meininghaus M, Chapman RD, Horndasch M, and Eick D (2000). Conditional Expression of RNA Polymerase II in Mammalian Cells DELETION OF THE CARBOXYL-TERMINAL DOMAIN OF THE LARGE SUBUNIT AFFECTS EARLY STEPS IN TRANSCRIPTION. J. Biol. Chem. 275, 24375–24382. [DOI] [PubMed] [Google Scholar]
- Michels AA, Robitaille AM, Buczynski-Ruchonnet D, Hodroj W, Reina JH, Hall MN, and Hernandez N (2010). mTORCl Directly Phosphorylates and Regulates Human MAF1. Mol. Cell. Biol. 30, 3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir RD, and Willis IM (2013). Regulation of pol III transcription by nutrient and stress signaling pathways. Biochim. Biophys. Acta BBA- Gene Regul. Mech. 1829, 361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moqtaderi Z, Wang J, Raha D, White RJ, Snyder M, Weng Z, and Struhl K (2010). Genomic Binding Profiles of Functionally Distinct RNA Polymerase III Transcription Complexes in Human Cells. Nat. Struct. Mol. Biol. 17, 635–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Kiyomitsu T, Saga Y, and Kanemaki MT (2016). Rapid Protein Depletion in Human Cells by Auxin-lnducible Degron Tagging with Short Homology Donors. Cell Rep. 15, 210–218. [DOI] [PubMed] [Google Scholar]
- Nguyen VT, Giannoni F, Dubois MF, Seo SJ, Vigneron M, Kédinger C, and Bensaude O (1996). In vivo degradation of RNA polymerase II largest subunit triggered by alpha-amanitin. Nucleic Acids Res. 24, 2924–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oler AJ, Alla RK, Roberts DN, Wong A, Hollenhorst PC, Chandler KJ, Cassiday PA, Nelson CA, Hagedorn CH, Graves BJ, et al. (2010). Human RNA Polymerase III transcriptomes and relationships to Pol II promoters, enhancer-binding factors and chromatin domains. Nat. Struct. Mol. Biol. 17, 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orioli A, Pascali C, Pagano A, Teichmann M, and Dieci G (2012). RNA polymerase III transcription control elements: Themes and variations. Gene 493, 185–194. [DOI] [PubMed] [Google Scholar]
- Orioli A, Praz V, Lhôte P, and Hernandez N (2016). Human MAF1 targets and represses active RNA polymerase III genes by preventing recruitment rather than inducing long-term transcriptional arrest. Genome Res. 26, 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo AF, and Lee ES (2015). Non-coding RNA: what is functional and what is junk? Front. Genet. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang YLJ, Abo R, Levine SS, and Dedon PC (2014). Diverse cell stresses induce unique patterns of tRNA up- and down-regulation: tRNA-seq for quantifying changes in tRNA copy number. Nucleic Acids Res. 42, el70–el70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavon-Eternod M, Gomes S, Geslain R, Dai Q, Rosner MR, and Pan T (2009). tRNA overexpression in breast cancer and functional consequences. Nucleic Acids Res. 37, 7268–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavon-Eternod M, Gomes S, Rosner MR, and Pan T (2013). Overexpression of initiator methionine tRNA leads to global reprogramming of tRNA expression and increased proliferation in human epithelial cells. RNA 19, 461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo A, Jackson DA, Hollinshead M, Wang Z, Roeder RG, and Cook PR (1999). Regional specialization in human nuclei: visualization of discrete sites of transcription by RNA polymerase III. EMBOJ. 18, 2241–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha D, Wang Z, Moqtaderi Z, Wu L, Zhong G, Gerstein M, Struhl K, and Snyder M (2010). Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc. Natl. Acad. Sci. U. S. A. 107, 3639–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rak R, Dahan O, and Pilpel Y (2018). Repertoires of tRNAs: The Couplers of Genomics and Proteomics. Annu. Rev. Cell Dev. Biol. 34, 239–264. [DOI] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Griining B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Diindar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina JH, Azzouz TN, and Hernandez N (2006). Maf1, a New Player in the Regulation of Human RNA Polymerase III Transcription. PLOS ONE 1, el34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout EJ, Marshall L, and Grewal SS (2012). Drosophila RNA polymerase III repressor Maf1 controls body size and developmental timing by modulating tRNAiMet synthesis and systemic insulin signaling. Proc. Natl. Acad. Sci. 109, 1139–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26,139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder RG, and Rutter WJ (1969). Multiple Forms of DNA-dependent RNA Polymerase in Eukaryotic Organisms. Nature 224, 234. [DOI] [PubMed] [Google Scholar]
- Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al. (2013). ENCODE Data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 41, D56–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagi D, Rak R, Gingold H, Adir I, Maayan G, Dahan O, Broday L, Pilpel Y, and Rechavi O (2016). Tissue- and Time-Specific Expression of Otherwise Identical tRNA Genes. PLOS Genet. 12, e1006264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm L, and Hernandez N (2002). Recruitment of RNA polymerase III to its target promoters. Genes Dev. 16, 2593–2620. [DOI] [PubMed] [Google Scholar]
- Shor B, Wu J, Shakey Q, Toral-Barza L, Shi C, Follettie M, and Yu K (2010). Requirement of the mTOR Kinase for the Regulation of Maf1 Phosphorylation and Control of RNA Polymerase Ill-dependent Transcription in Cancer Cells. J. Biol. Chem. 285, 15380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivak M, Weston J, Bottou L, Kail L, and Noble WS (2009). Improvements to the Percolator Algorithm for Peptide Identification from Shotgun Proteomics Data Sets. J. Proteome Res. 8, 3737–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrent M, Chalancon G, Groot N.S. de, Wuster A, and Babu MM (2018). Cells alter their tRNA abundance to selectively regulate protein synthesis during stress conditions. Sci Signal 11, eaat6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turowski TW, Lesniewska E, Delan-Forino C, Sayou C, Boguta M, and Tollervey D (2016). Global analysis of transcriptionally engaged yeast RNA polymerase III reveals extended tRNA transcripts. Genome Res. 26, 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini A, Ringel R, Kusser AG, Berninghausen O, Kassavetis GA, and Cramer P (2010). Molecular Basis of RNA Polymerase III Transcription Repression by Maf1. Cell 143, 59–70. [DOI] [PubMed] [Google Scholar]
- Wang Z, and Roeder RG (1997). Three human RNA polymerase Ill-specific subunits form a subcomplex with a selective function in specific transcription initiation. Genes Dev. 11, 1315–1326. [DOI] [PubMed] [Google Scholar]
- Wansink DG, Schul W, Kraan I. van der, Steensel B. van, Driel R. van, and Jong L. de (1993). Fluorescent labeling of nascent RNA reveals transcription by RNA polymerase II in domains scattered throughout the nucleus. J. Cell Biol. 122, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner MS, and Ruthenburg AJ (2015). Nuclear Fractionation Reveals Thousands of Chromatin-Tethered Noncoding RNAs Adjacent to Active Genes. Cell Rep. 12, 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichtowska D, Turowski TW, and Boguta M (2013). An interplay between transcription, processing, and degradation determines tRNA levels in yeast. Wiley Interdiscip. Rev. RNA 4, 709–722. [DOI] [PubMed] [Google Scholar]
- Wuarin J, and Schibler U (1994). Physical isolation of nascent RNA chains transcribed by RNA polymerase II: evidence for cotranscriptional splicing. Mol. Cell. Biol. 14, 7219–7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeganeh M, Praz V, Cousin P, and Hernandez N (2017). Transcriptional interference by RNA polymerase III affects expression of the Polr3e gene. Genes Dev. 31, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Yang W, Ni T, Tang Z, Nakadai T, Zhu J, and Roeder RG (2015). RNA polymerase II-associated factor 1 regulates the release and phosphorylation of paused RNA polymerase II. Science 350, 1383–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehring WA, Lee JM, Weeks JR, Jokerst RS, and Greenleaf AL (1988). The C-terminal repeat domain of RNA polymerase II largest subunit is essential in vivo but is not required for accurate transcription initiation in vitro. Proc. Natl. Acad. Sci. 85, 3698–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Qin Y, Clark WC, Dai Q, Yi C, He C, Lambowitz AM, and Pan T (2015). Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 12, 835–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Nascent RNA fold-change and gene groups for DRB- and DA-sensitive tRNA genes. Related to Figure 3
Table S3. Protein coding genes downregulated following 2hrs DA-treatment. Related to Figure 1 and 5
Data Availability Statement
See Key Resources Table.
Microarray, nascent RNA-seq and ChlP-seq data have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO superseries accession number GSE130878.
Nascent RNA-seq data from mock- and DRB-treated HEK293 were obtained from GEO accession GSE66478. ENCODE Tracks for Total/S2P-Pol II ChlP-seq and corresponding inputs in HeLa S3 cells were generated and analyzed by the lab of Michael Snyder and downloaded directly from the UCSC browser (Rosenbloom et al., 2013) (UCSC accession: wgEncodeEH000613, wgEncodeEH001838, wgEncodeEH000612 and wgEncodeEH000744 respectively).