
Figure 1.
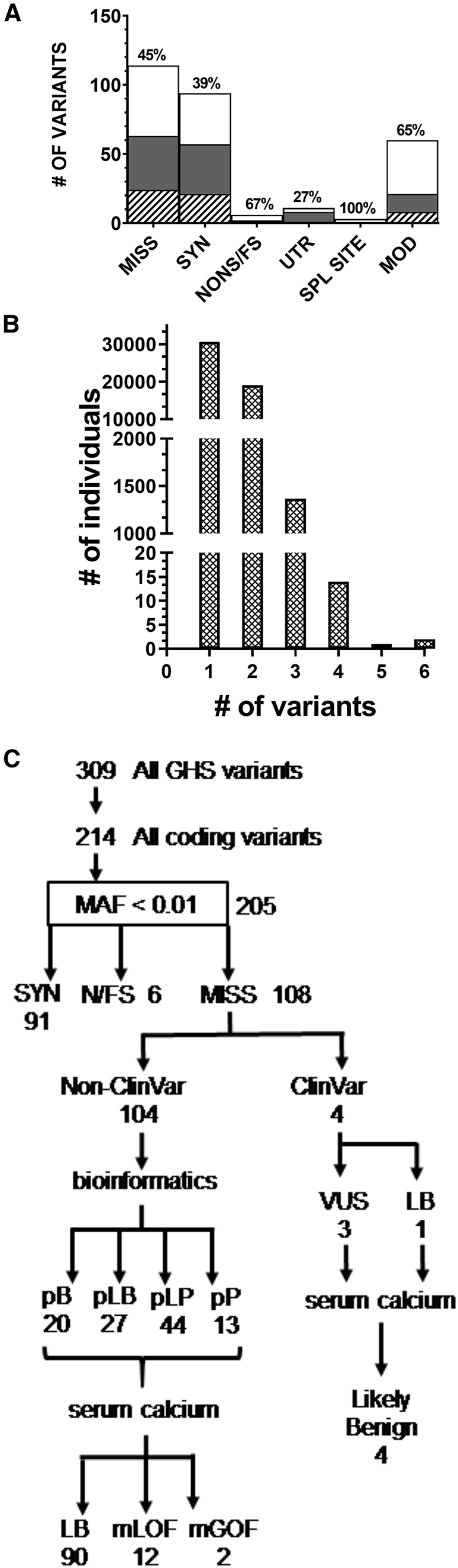
Characteristics and Bioinformatics Triage of CASR Variants in the 51,289 DiscovEHR Cohort
(A) Variants were classified into coding (missense [MISS], synonymous [SYN], and nonsense or frameshift [NONS/FS]) and non-coding (3′ or 5′ untranslated region [UTR], splice site [SPL SITE], or upstream/downstream modifier [MOD]) variants. Bar graph indicates numbers of variants in each class. The percent of variants not previously listed in variant databases is indicated above each bar. Filled or shaded portions of bars indicate the subset of variants new to this study (white), with RefSeq rsID number (gray) or without ID but found in population variant databases (crosshatched).
(B) Histogram of the common plus rare variant distribution in the 51,289 individual DiscovEHR cohort: 30,676 individuals had a single variant and 20,514 individuals had two or more variants (common + rare).
(C) Rare missense variants (n = 108) were sorted into those with (n = 4) and without (n = 104) ClinVar annotation. Variants were triaged by RMPath (see Supplemental Subjects and Methods) from variants of unknown significance into those predicted to be benign (pB), likely benign (pLB), likely pathogenic (pLP), and pathogenic (pP). Analysis of serum Ca concentrations allowed further sorting of all heterozygous missense variants into likely benign (LB), missense loss-of-function (mLoF), and missense gain-of-function (mGoF) categories. MAF, minor allele frequency; SYN, synonymous variant; N/FS, nonsense or frameshift variant; MISS, missense variant; VUS, variant of unknown significance.