

Abstract
Generation of pancreatic β cells from human pluripotent stem cells (hPSCs) holds promise as a cell replacement therapy for diabetes. Here, we establish a link between the state of the actin cytoskeleton and the expression of pancreatic transcription factors that drive pancreatic lineage specification. Bulk and single-cell RNA sequencing demonstrated that different degrees of actin polymerization biased cells toward various endodermal lineages, and that conditions favoring a polymerized cytoskeleton strongly inhibited NEUROG3-induced endocrine differentiation. Using latrunculin A to depolymerize the cytoskeleton during endocrine induction, we developed a two-dimensional differentiation protocol for generating human pluripotent stem cell-derived β (SC-β) cells with improved in vitro and in vivo function. SC-β cells differentiated from four hPSC lines exhibited first and second phase dynamic glucose-stimulated insulin secretion. Transplantation of islet-sized aggregates of these cells rapidly reversed severe pre-existing diabetes in mice at a rate close to that of human islets and maintained normoglycemia for at least 9 months.
Introduction
Methods for differentiating hPSCs toward pancreatic β cells offer the promise of a cell therapy for the treatment of diabetes.1,2 These differentiation strategies rely on the precise activation and repression of specific developmental pathways with soluble growth factors and small molecules,3 guiding hPSCs in a stepwise fashion through intermediate endodermal and pancreatic progenitor stages. 4–15 While endodermal progenitors can also produce non-pancreatic lineages, such as intestine16 or hepatocytes,17 differentiation toward a SC-β cell fate requires expression of certain pancreatic transcription factors in the proper order. Specifically, NKX6–1+ pancreatic progenitors must be generated before certain endocrine genes turn on, as premature expression of these genes, such as NEUROG3,18 results in non-functional polyhormonal cells.11,19,20 NKX6–1+ pancreatic progenitors can be generated in both two-dimensional6,14 and three-dimensional cell culture,4,21 but further robust differentiation to a SC-β cell fate has been achieved only in three-dimensional culture, with cells grown either as suspension clusters4 or as aggregates on an air-liquid interface.6 The reason for this requirement and, in particular, the effects of the insoluble microenvironment on pancreatic fate choice, are poorly understood.
Cells can sense their microenvironment through transmembrane proteins called integrins. Different combinations of the α and β integrin subunits dictate the extracellular matrix (ECM) proteins to which a cell can adhere. Integrins bound to ECM proteins form complexes with other adhesion proteins that anchor and promote assembly of the actin cytoskeleton, providing a means for cells to generate mechanical forces. While these forces allow cells to migrate and change shape, they can also be transduced into intracellular biochemical signaling. In particular, material properties of the ECM, such as matrix stiffness,22,23 geometry,24,25 and adhesion density26–28, have been shown to guide stem cell differentiation by modulating the degree of cytoskeletal polymerization and the recruitment of adhesion proteins, altering downstream signaling pathways.29–32
Here, we demonstrate that the state of the actin cytoskeleton strongly influences NEUROG3-dependent endocrine induction and subsequent SC-β cell specification. We found that manipulating cell-biomaterial interactions and the state of the actin cytoskeleton alters the timing of endocrine transcription factor expression and the ability of pancreatic progenitors to differentiate into SC-β cells. This result allowed us to overcome the requirement for three-dimensional culture in SC-β cell differentiation and create a fully planar protocol. SC-β cells differentiated from four hPSC lines with our planar protocol exhibited better or similar dynamic glucose-stimulated insulin secretion (GSIS) and reversed diabetes in mice faster than SC-β cells generated with a suspension protocol. We also found that the benefits of modulating actin polymerization extend to other endodermal lineages, enhancing directed differentiation to pancreatic exocrine, hepatocytes, and intestinal cells.
Results
The actin cytoskeleton regulates NEUROG3 levels in PDX1-expressing pancreatic progenitors
Differentiation of hPSCs into SC-β cells typically proceeds through several stages that try to mimic in vivo development (Fig. 1a). To better understand the role of the microenvironment in this process, we generated stage 3 PDX1+ pancreatic progenitor cells with our suspension-based differentiation protocol,5 created a single-cell dispersion from these clusters, and seeded cells onto tissue culture polystyrene (TCP) plates coated with a wide variety of ECM proteins (Fig. 1a-b, Supplementary Fig. 1a). Cells at the beginning of stage 4 do not express NKX6–1 or NEUROG3 (Supplementary Fig. 1c). If NEUROG3 turns on before NKX6–1, the cells become polyhormonal cells that ultimately resolve into α-cells.33,34 Therefore, stage 4 is designed to generate NKX6–1+ pancreatic progenitors, and stage 5 initiates endocrine induction of these progenitors by inducing NEUROG3.4 The most striking observation from these experiments was that plating the cells down for the duration of stage 4 on most ECM proteins prevented premature expression of NEUROG3, in contrast to normal suspension clusters, while reaggregating the cells back into clusters after single-cell dispersion greatly increased NEUROG3 expression (Fig. 1c, Supplementary Fig. 1d). Two downstream NEUROG3 targets, NKX2.2 and NEUROD1, followed the same decreasing trend, while SOX9 expression increased (Fig. 1c, Supplementary Fig. 1d). The ECM protein that induced the highest NEUROG3 expression was laminin 211, which correlated with visual observations of poorer cell attachment compared to the other ECM conditions (Supplementary Fig. 1a,d). A colorimetric antibody-based integrin adhesion assay at the beginning and end of stage 4 confirmed high expression of integrin subunits that bind to collagens I and IV (α1, α2, β1), fibronectin (αV, β1, α5β1), vitronectin (αV, β1, αVβ5) and some, but not all, laminin isoforms (α3, β1) (Supplementary Fig. 1b). Thus, strong attachment to the culture surface, rather than the composition of a particular ECM coating, seemed to prevent premature endocrine induction during stage 4.
Figure 1. The state of the cytoskeleton controls expression of the transcription factors NEUROG3 and NKX6–1 in pancreatic progenitors.

(a) Schematic of the differentiation protocol5 used for suspension differentiations and plate down studies. (b) Images of clusters at the beginning of stage 4 dispersed and plated onto ECM-coated TCP for the remainder of the protocol. Scale bar = 100 μm. (c) qRT-PCR of pancreatic genes at the end of stage 4 of cells plated on collagen I at the beginning of stage 4 compared to regular suspension clusters or clusters reaggregated after dispersion (Tukey’s HSD test, n = 4). (d) qRT-PCR of pancreatic genes at the end of stage 4 of cells plated on collagen I gels of varying heights at the beginning of stage 4. Increasing the height of collagen I gels fixed to TCP correlates with decreasing the effective stiffness experienced by cells (ANOVA, n = 4). (e) qRT-PCR of plated stage 4 cells treated with some commonly used cytoskeletal-modulating compounds to identify latrunculin A as a potent endocrine inducer (Dunnett’s multiple comparisons test, n = 4). (f) Immunostaining of plated cells at the end of stage 4 demonstrating that a 1 μM latrunculin A treatment increased NEUROG3+ cells and decreased NKX6–1+ cells when administered during stage 4. Scale bar = 50 μm. (g) Latrunculin A dose response of pancreatic gene expression added during stage 4 measured with qRT-PCR (ANOVA, n = 4). (h) Immunostaining of plated stage 4 cells treated for 24 hours with 1 μM latrunculin A, demonstrating depolymerization of F-actin but maintenance of PDX1 expression. (i) Western blot quantification of the G/F actin ratio within cells under different culture formats and treated with latrunculin A (n = 3). All data was generated with HUES8. All data is represented as the mean, and all error bars represent SEM. Individual data points are shown for all bar graphs. ns = not significant, * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
Cells plated on TCP plates experience greater substrate stiffness than cells cultured in suspension. The high stiffness of TCP resists pulling forces generated by the actin cytoskeleton, providing mechanical feedback that can alter intracellular signaling.29–32 To test the influence of substrate stiffness on endocrine induction, we plated PDX1-expressing pancreatic progenitors on TCP plates overlaid with type 1 collagen gels of various heights, as increasing gel height lowers the effective stiffness experienced by the cell.35 Increasing gel height led to increases in NEUROG3, NKX2.2, and NEUROD1 and decreases in SOX9, consistent with endocrine induction (Fig. 1d). NKX6–1 expression followed the reverse trend as NEUROG3, indicating that premature NEUROG3 expression during stage 4 induced by a soft substrate is detrimental to NKX6–1 induction in pancreatic progenitors.
To further probe how cell adhesion affects endocrine induction throughout stage 4, we tested several compounds that influence cytoskeletal dynamics36: latrunculin A, latrunculin B, cytochalasin D, jasplakinolide, blebbistatin, nocodazole, Y-27632, Y15, and GDC-0994. Clusters at the beginning of stage 4 were single-cell dispersed, seeded onto TCP plates coated with dilute collagen I, and then treated with compounds for the duration of stage 4. This screen revealed that latrunculin A, which binds and sequesters monomeric actin,36,37 greatly increased expression of NEUROG3 as well as its downstream targets NKX2.2 and NEUROD1 (Fig. 1e-f). NEUROG3 expression in response to latrunculin A treatment was dose-dependent for HUES8 (Fig. 1g) and two induced pluripotent stem cell (iPSC) lines (Supplementary Fig. 2a). Latrunculin B, a less potent form of the compound, increased NEUROG3 expression in a dose-dependent manner as well but required more than a 10x higher concentration to achieve a similar effect (Supplementary Fig. 2b). NKX6–1 expression followed the reverse trend as NEUROG3 (Fig. 1f-g), again highlighting the need to prevent NEUROG3 expression in order to turn on NKX6–1 during stage 4.
Treatment of plated stage 4 cells with 1 μM latrunculin A for 24 hours resulted in almost complete depolymerization of F-actin (Fig. 1h), an increased G/F-actin ratio (Fig. 1i, Supplementary 2d), and high NEUROG3 expression. The G/F-actin ratio correlated with NEUROG3 expression for all conditions (Fig. 1c), with the plated cells having the lowest levels, followed by the suspension culture, then reaggregated clusters, and finally the plated cells treated with latrunculin A. In contrast, adding the actin polymerizer jasplakinolide to pancreatic progenitors during reaggregation after dispersion attenuated NEUROG3 expression (Supplementary Fig. 2c). These data show that the polymerization state of the actin cytoskeleton correlates with expression of the important pancreatic transcription factors NEUROG3 and NKX6–1.
The cytoskeletal state of pancreatic progenitors guides their fate selection
Next, we performed single-cell RNA sequencing on plated pancreatic progenitors treated for all five days of stage 4 with either latrunculin A or nocodazole. Latrunculin A depolymerizes F-actin of these plated progenitors, whereas nocodazole depolymerizes microtubules, leading to increased polymerization and contraction of F-actin (Supplementary Fig. 3a).24,38 Unsupervised clustering of treated and untreated cells distinguished five populations (Fig. 2a-b). By examining genes important to different stages of pancreatic development, we identified these five populations (Fig. 2c, Supplementary Fig. 3b). As expected, there was a pancreatic progenitor population that had high expression of PDX1 and SOX9 but lacked expression of endocrine genes. There were two separate endocrine populations: early endocrine cells with high expression of NEUROG3 and late endocrine cells with lower NEUROG3 levels but increased expression of downstream markers such as NKX2–2, CHGA, and ISL1. Some of the late endocrine cells also expressed insulin. However, both endocrine populations lacked NKX6–1 expression, indicating premature endocrine induction. A fourth population was characterized by high expression of the ductal marker KRT7 and the acinar marker PRSS1 that encodes cationic trypsinogen, indicating an exocrine progenitor phenotype. A final population of undefined endoderm lacked pancreatic markers and had high expression of SOX2 and SOX3 (Supplementary Fig. 3b).
Figure 2. Single-cell RNA sequencing demonstrates that cytoskeletal state directs pancreatic progenitor fate.

(a-b) tSNE projections of single-cell RNA sequencing performed on plated stage 4 cells either untreated (n = 286 cells), treated with 0.5 μM latrunculin A (n = 579 cells), or treated with 5 μM nocodazole (n = 197 cells). Unsupervised clustering of the combined cell population from all three conditions revealed five separate clusters. (c) Violin plots indicating the relative expression of important pancreatic genes in each cluster. (d) The percentage of cells from the indicated treatment condition that were within each of the five clusters. (e) Violin plot of NKX6–1 expression within the pancreatic progenitor cluster. All data was generated with HUES8.
Cytoskeletal state during stage 4 strongly affected the distribution of cells into these five populations (Fig. 2d). In untreated plated control cells, the largest population was comprised of pancreatic progenitors (93.0%), with very few of the cells expressing endocrine (2.8%) or exocrine (4.2%) genes. Latrunculin A treatment sharply decreased the pancreatic progenitor population (19.9%) and increased both early (18.8%) and late (33.9%) endocrine cells. These results correspond to the preceding qRT-PCR data, demonstrating that plating pancreatic progenitors prevents NEUROG3 from turning on and that latrunculin A is a potent endocrine inducer. Additionally, 26.3% of latrunculin A treated cells were characterized as undefined endoderm, comprising almost all of this population. Treatment with nocodazole led to the greatest fraction of exocrine-like progenitors (66.5%) and few endocrine cells (3.6%). Although all conditions had some pancreatic progenitors, only the plated control cells expressed much NKX6–1 (Fig. 2e). These data suggest that an optimal cytoskeletal state is needed for NKX6–1 expression during stage 4. Specifically, a depolymerized cytoskeleton during stage 4 leads to endocrine induction before NKX6–1 can turn on, while a hyper-activated cytoskeleton induced by nocodazole also prevents NXK6–1 expression and instead promotes an exocrine progenitor–like fate. Thus, the polymerization state of the actin cytoskeleton in pancreatic progenitors is a crucial regulator of pancreatic cell fate.
Differentiation to SC-β cells is temporally regulated by the actin cytoskeleton
To further explore how cytoskeletal state modulates expression of pancreatic transcription factors, we added latrunculin A throughout different stages of differentiation after pancreatic progenitors were plated on type 1 collagen-coated TCP. Without latrunculin A treatment, plated pancreatic progenitors had poor differentiation efficiency (Fig. 3a), and the resulting cells secreted little insulin (Fig. 3b). Adding 0.5 μM latrunculin A throughout either stage 4 (pancreatic progenitors) or stage 6 (SC-β cell maturation) increased both general endocrine induction (CHGA+) and β cell specification (NKX6–1+/c-peptide+) (Fig. 3a). However, addition of 0.5 μM latrunculin A during stage 5, which is designed to induce endocrine differentiation, led to the greatest increase in endocrine induction, SC-β cell specification, and GSIS (Fig. 3a-b). These data demonstrate that attachment of pancreatic progenitors onto TCP inhibits SC-β cell differentiation, which is overcome by stage-dependent depolymerization of the actin cytoskeleton with latrunculin A.
Figure 3. Latrunculin A treatment during stage 5 increases the efficiency of SC-β cell specification of plated pancreatic progenitors.

(a) Flow cytometry two weeks into stage 6 for NKX6–1, CHGA, and C-peptide of plated cells as per Fig. 1a, untreated or treated with 0.5 μM latrunculin A throughout stage 4, 5, or 6 (Dunnett’s multiple comparisons test, n = 4). (b) Static GSIS two weeks into stage 6 of plated cells, untreated or treated with 0.5 μM latrunculin A throughout stage 4, 5, or 6 (paired two-sided t-test compares between low and high glucose for a particular sample, Dunnett’s test compares insulin secretion at high glucose to the control, n = 4). (c) Optimization of latrunculin A concentration and timing during stage 5 for plated cells. Static GSIS was performed after 2 weeks of stage 6 (paired two-sided t-test compares low and high glucose, unpaired two-sided t-test compares high glucose of plated control and 24 hour 1 μM latrunculin A treatment, n = 4). (d) Insulin content of plated cells two weeks into stage 6, untreated or treated 24 hours with 1μM latrunculin A on the first day of stage 5 (unpaired two-sided t-tests, n = 3). (e) Proinsulin/insulin ratio of plated cells two weeks into stage 6, untreated or treated for 24 hours on the first day of stage 5 with 1 μM latrunculin A (unpaired two-sided t-tests, n = 3). (f) qRT-PCR measuring endocrine (left) and non-endocrine (right) gene expression of plated cells two weeks into stage 6, untreated or treated for 24 hours on the first day of stage 5 with 1 μM latrunculin A (unpaired two-sided t-tests, n = 3). (g) Immunostaining for AFP and c-peptide of plated cells two weeks into stage 6, untreated or treated for 24 hours on the first day of stage 5 with 1 μM latrunculin A. Scale bar = 100 μm. (h) Images of aggregating plated cells after one week in stage 6. Scale bar = 250 μm. (i) Dynamic glucose-stimulated insulin secretion of stage 6 cells exhibiting first and second phase insulin release (n = 3). All data was generated with HUES8. All data is represented as the mean, and all error bars represent SEM. Individual data points are shown for all bar graphs. ns = not significant, * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
To optimize the benefits of latrunculin A for differentiation of SC-β cells, we tested a range of durations and concentrations during stage 5 (Fig. 3c). Both duration and concentration influenced GSIS, with a 1 μM treatment during the first 24 hours of stage 5 having the most benefit at the shortest and lowest dose. This 24-hour treatment seemed sufficient to rescue SC-β cell specification, while extended culture with latrunculin A in stage 5 hampered this effect. A 24-hour 1 μM latrunculin A treatment increased total insulin content (Fig. 3d), improved pro-insulin/insulin ratio (Fig. 3e), and increased expression of endocrine genes (Fig. 3f). Expression of markers associated with other endodermal lineages was reduced (Fig. 3f), as were regions of off-target cell types that were easily distinguished visually by differences in cell morphology and that stained for non-pancreatic markers, such as alpha-fetoprotein (AFP) (Fig. 3g). Latrunculin A treated cells that remained on the plate in stage 6 were functional in a static GSIS assay (Fig. 3c) but could also be aggregated into clusters in 6-well plates on an orbital shaker (Fig. 3h) for assessment of dynamic GSIS. These clusters exhibited both first and second phase insulin secretion (Fig. 3i).
Collectively, these data demonstrate that adequate cytoskeletal polymerization is important for the pancreatic progenitor program during stage 4, and that actin depolymerization during stage 5 endocrine induction is required for differentiation into SC-β cells. Specifically, the high stiffness of TCP induces actin polymerization that prevents premature NEUROG3 expression and promotes NKX6–1 expression during stage 4, but it inhibits NEUROG3 expression and subsequent SC-β cell specification during stage 5. Treatment with latrunculin A during stage 5 depolymerizes the cytoskeleton and enables efficient generation of SC-β cells on TCP without a three-dimensional cell arrangement.
A planar SC-β cell differentiation method
The experiments described above used our suspension differentiation protocol5 for the first 3 stages to produce pancreatic progenitors followed by plating of the cells on ECM-coated TCP for continued differentiation (Fig. 1a). To demonstrate a fully planar protocol for generating SC-β cells (Fig. 4a), hPSCs were propagated on Matrigel-coated TCP until confluency, at which point the medium was switched from mTeSR1 to differentiation medium. Similar to earlier experiments, adding latrunculin A during stage 4 sharply increased expression of NEUROG3 and its downstream targets and decreased NKX6–1 expression (Supplementary Fig. 4a). In the fully planar protocol, almost no SC-β cells could be generated without latrunculin A (Fig. 4b), consistent with the requirement for three-dimensional culture in prior reports.4–10 However, a 24-hour 1 μM latrunculin A treatment at the beginning of stage 5 greatly increased endocrine induction and SC-β cell specification while decreasing the generation of off-target lineages (Fig. 4b and Supplementary Fig. 4b-d).
Figure 4. SC-β cells differentiated with the new planar protocol express β cell markers and function in vitro.

(a) Schematic of the new planar protocol for making SC-β cells incorporating a 1 μM latrunculin A treatment for the first 24 hour of stage 5. The cells were plated onto Matrigel-coated TCP throughout this differentiation process. (b) Flow cytometry after one week in stage 6 of planar cells from HUES8 with and without stage 5 latrunculin A treatment measuring endocrine induction (CHGA+) and SC-β cell specification (C-peptide+/NKX6–1+) (unpaired two-sided t-tests, n = 4). (c) Flow cytometry of islet and SC-β cells markers for stage 6 cells differentiated from HUES8, 1013–4FA, and 1016SeVA hPSC lines after being aggregated into clusters for one week (n = 4). (d) qRT-PCR of islet and disallowed (LDHA, SLC16A1) genes for stage 6 cells and human islets (Dunnett’s multiple comparisons test, n = 4 for SC-β cells, n = 3 for human islets). (e) Immunostaining of aggregated planar stage 6 cells from HUES8. (f) Insulin content of planar stage 6 cells (n = 4). (g) Proinsulin/insulin content ratio for planar stage 6 cells (n = 4). (h) Static GSIS for planar stage 6 cells (paired two-sided t-tests, n = 11 for HUES8, n = 4 for 1013–4FA and 1016SeVA). (i) Dynamic GSIS for planar stage 6 cells generated from HUES8 (n = 7), 1013–4FA (n = 3), and 1016SeVA (n = 4) compared with stage 6 cells generated with the suspension protocol (HUES8, n = 4; 1013–4FA, n = 4; 1016SeVA, n = 3) and with human islets (n = 4). Insulin units are shown in both μIU and ng. All data shown in this figure is of cells generated with the planar differentiation protocol unless otherwise noted. All data is represented as the mean, and all error bars represent SEM. Individual data points are shown for all bar graphs. ns = not significant, * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
To further characterize our planar protocol, we applied it to three hPSC lines that we had studied previously5—HUES8, 1013–4FA, and 1016SeVA. After one week in stage 6, the cells were aggregated into clusters on an orbital shaker for testing in the same in vitro and in vivo assays used for cells from the suspension protocol. This yielded aggregates with up to ~40% SC-β cells (NKX6–1+/c-peptide+) and low percentages of polyhormonal cells (c-peptide+/GCG+ or c-peptide+/SST+) (Fig. 4c). Expression of many β cell and islet genes was similar to that in human islets, but MAFA and UCN3 expression remained low (Fig. 4d), as reported with our suspension protocol.5 MAFB expression, however, was similar to that in human islets, and MAFB protein co-stained with c-peptide (Supplementary Fig. 4g). Most cells in the clusters showed c-peptide staining in the cytoplasm and were co-positive with other important β-cell markers (Fig. 4e, Supplementary Fig. 4e-f). Specifically, PDX1, NKX6–1, NKX2–2, NEUROD1, and ISL1 were present in the nucleus of the majority of c-peptide-positive cells. Most cells also expressed CHGA, a general marker of endocrine cells. Somatostatin and glucagon-expressing cells were present in smaller fractions than c-peptide-expressing cells. The protein immunostaining corresponded to the flow cytometry quantification, which showed that a majority of the cells expressed c-peptide and CHGA (Fig. 4b), while a much smaller fraction expressed glucagon or somatostatin (Fig. 4c).
SC-β cells generated with the planar protocol responded to multiple glucose challenges (Supplementary Fig. 5a), increased insulin secretion with exposure to various secretagogues (Supplementary Fig. 5b), produced insulin granules as observed with electron microscopy (Supplementary Fig. 5c), and exhibited glucose-responsive calcium signaling (Supplementary Fig. 5d). SC-β cells derived from all three lines had similar insulin content (Fig. 4f), pro-insulin/insulin ratio (Fig. 4g), and static GSIS (Fig. 4h), providing evidence of reproducibility across these three lines. Gene expression, insulin content, proinsulin/insulin ratio, and static GSIS of planar SC-β cells generated from the HUES8 line were similar to those produced with the suspension protocol (Supplementary Figs. 6a-d).
We previously reported much weaker dynamic GSIS with SC-β cells generated from the 1013–4FA and 1016SeVA lines compared to HUES8 using our suspension protocol5. The planar protocol, however, improves dynamic GSIS for the 1013–4FA and 1016SeVA lines, with results for 1013–4FA approaching those of human islets, including second phase insulin secretion (Fig. 4i). Notably, elevated insulin secretion at high glucose appropriately diminished in ~10 minutes when the cells were switched to low glucose, similar to the response of human islets. Furthermore, application of the planar protocol to an additional iPSC line, BJFF.6,39 that did not propagate and differentiate with our suspension protocol (Supplementary Fig. 5e) successfully generated SC-β cells that exhibited dynamic GSIS function (Supplementary Fig. 5f-g), suggesting that the planar protocol enables generation of SC-β cells from cell lines not compatible with other protocols. Finally, we scaled up the planar protocol in standard T-75 cell culture flasks and generated 0.81 million cells per cm2, which showed equivalent robust GSIS (Supplementary Fig. 5h-i).
For in vivo evaluation of SC-β cell function, we transplanted HUES8 stage 6 clusters underneath the kidney capsule of streptozotocin (STZ)-induced diabetic mice (Supplementary Fig. 5j) in accordance with the regulations of the Washington University Institutional Animal Care and Use Committee. Fasting glucose levels approached those of untreated controls within two weeks after transplantation, which was similar to, but slightly slower than, human islet transplants, and remained below 200 mg/dL until the grafts were removed at 36 weeks post-transplant (Fig. 5a). Diabetes reversal in mice receiving SC-β cells generated with the suspension protocol, however, took approximately three weeks longer (Fig. 5a). STZ-treated mice receiving the planar SC-β cell transplants had similar glucose tolerance as untreated control mice at both 3 and 10 weeks (Fig. 5a) and high levels of serum human insulin that were regulated by glucose (Fig. 5b, Supplementary Fig. 4k). A nephrectomy at week 36 to remove the human grafts resulted in rapid loss of glycemic control and confirmed that restoration of glucose homeostasis arose from the transplants (Fig. 5a). Immunostaining of excised kidneys revealed large regions of c-peptide, NKX6–1, and PDX1 positive cells, while few glucagon or somatostatin positive cells were present (Fig. 5c). Furthermore, no overgrowths were observed (Fig. 5c). These data demonstrate that our planar protocol generates functional SC-β cells capable of rapidly reversing severe pre-existing diabetes in mice.
Figure 5. SC-β cells generated with the new planar protocol can rapidly cure pre-existing diabetes in mice.
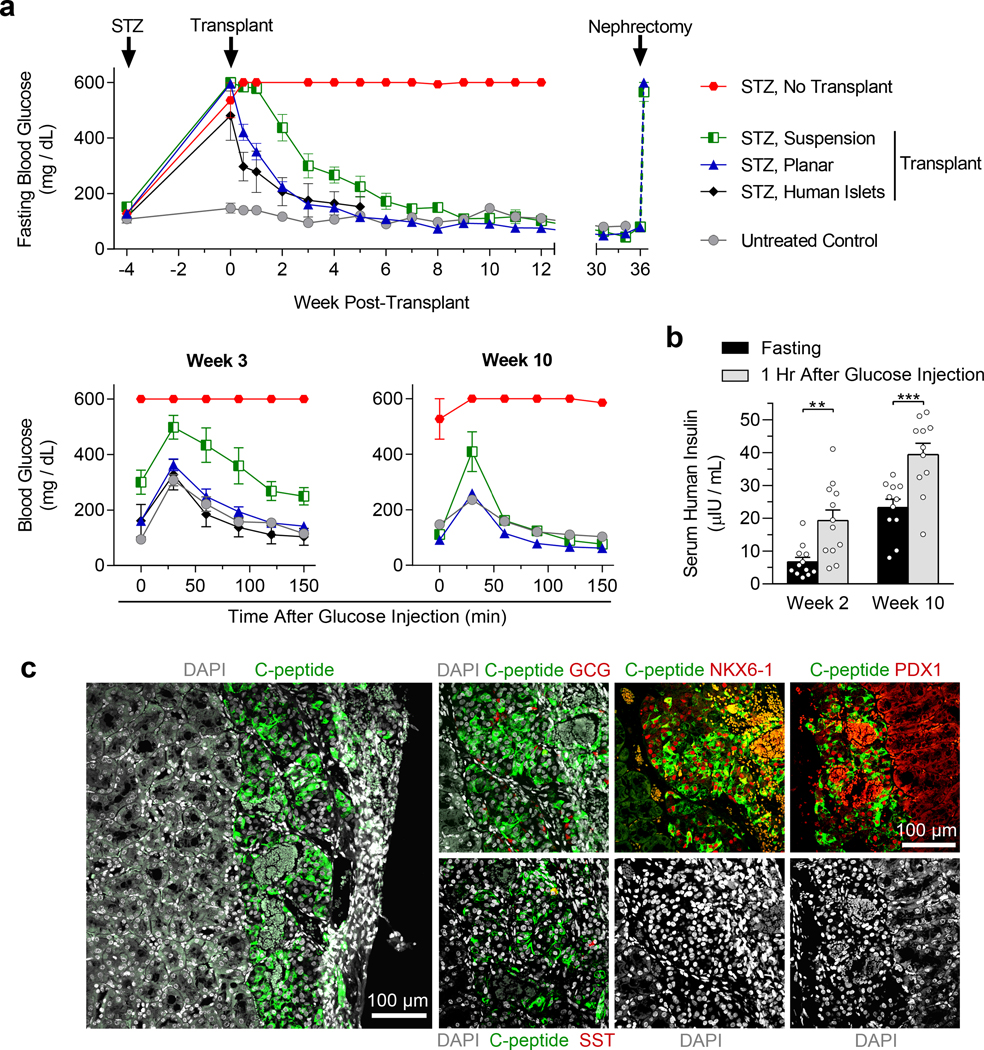
(a) Diabetes was induced with STZ in a total of 30 mice. 4 weeks after injection, SC-β cells were transplanted underneath the kidney capsule of the mice (planar transplant, n = 12; suspension transplant, n = 6; human islet transplant, n = 5, no transplant, n = 7). Additionally, 5 non-STZ mice served as non-diabetic controls. Glucose tolerance tests were performed 3 and 10 weeks after transplantation. A nephrectomy was performed 36 weeks after transplantation on mice receiving planar and suspension SC-β cells. All STZ mice that did not receive a transplant died by week 29. (b) In vivo GSIS of mice receiving the planar SC-β cell transplants 2 and 10 weeks after transplantation measuring human insulin (paired two-sided t-tests, n = 12 for week 2, n = 11 for week 10). (c) Immunostaining of sectioned kidneys transplanted with SC-β cells generated with the planar protocol 3 weeks after transplantation showing endocrine cell markers. Scale bars = 100 μm. All data was generated with HUES8 using either the planar protocol outlined in Fig. 4a or the suspension protocol.5 All data is represented as the mean, and all error bars represent SEM. Individual data points are shown for all bar graphs. ns = not significant, * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
Cytoskeletal modulation influences endodermal cell fate
To further investigate the effects of the cytoskeleton on endodermal cell fate choice, we performed bulk RNA sequencing at stage 6 of the SC-β cell protocol on cells that had been plated during stage 4 and which were treated with latrunculin A during either the pancreatic progenitor stage (stage 4) or during endocrine induction (stage 5). These cells were also compared with untreated plated and suspension differentiations. A heat map of the 1,000 most differentially expressed genes illustrates that the timing of latrunculin A treatment had a drastic effect on the expression profile of the resulting cells (Fig 6a). Specifically, the optimal stage 5 latrunculin A treatment shifted the gene expression profile of plated cells toward that of the suspension SC-β cell differentiation (Fig. 6a), increasing expression of β cell and islet genes (Fig. 6b). When directly compared with the suspension differentiation, however, the plated stage 5 treated cells actually had lower levels of GCG and SST, similar levels of INS, and higher levels of genes associated with insulin processing and secretion (Fig. 6b, Supplementary Fig. 5l). Many other differentially expressed genes among all conditions were associated with non-endocrine lineages (Fig. 6b-d): stage 4 latrunculin A treatment increased intestine and stomach marker expression and the plated control showed increased expression of genes associated with the liver and esophagus. Thus, the timing of cytoskeletal modulation affected endodermal lineage specification.
Figure 6. The state of the cytoskeleton influences endodermal cell fate.

(a) Suspension (n = 5) and plated pancreatic progenitors differentiated to stage 6 as per Fig. 1a either untreated (n = 6), treated with 0.5 μM latrunculin A throughout stage 4 (n = 6), or treated with 1 μM latrunculin A for the first 24 hours of stage 5 (n = 6). Bulk RNA sequencing at two weeks into stage 6 was used to generate a heat map of the 1,000 most differentially expressed genes between the stage 5 latrunculin A treatment and plated control. (b) A heat map from bulk RNA sequencing of select genes from multiple endodermal lineages. (c) A volcano plot from bulk RNA sequencing data showing expression differences of select genes between untreated plated cells and stage 5 latrunculin A treated cells. (d) Gene enrichment analysis from bulk RNA sequencing of select gene sets from multiple endodermal lineages. All data was generated with HUES8.
In light of this finding, we tested incorporation of latrunculin A and nocodazole into established differentiation protocols for generating exocrine pancreas,40 intestine,16,41 and liver.42 In an exocrine differentiation, nocodazole treatment drastically increased gene expression of two trypsin isoforms (PRSS1 by 26x and PRSS2 by 52x) as well as immunostaining of these proteins (Supplementary Fig 7a). Nocodazole in the intestinal differentiation, on other hand, increased CDX2 immunostaining as well as gene expression by 17 fold (Supplementary Fig. 7b). (Supplementary Fig. 7a-c). In contrast, latrunculin A during an intestinal differentiation led to a 12 fold increase in the expression of the intestinal stem cell marker LGR5 (Supplementary Fig. 7b). Furthermore, gene markers for Paneth cells, which are known to be important for LGR5+ intestinal stem cell viability,43 were also elevated with latrunculin A treatment. Finally, although both nocodazole and latrunculin A increased hepatocyte gene expression in a liver differentiation, immunostaining for albumin was more abundant with nocodazole treatment while AFP was more prevalent with latrunculin A treatment, suggesting differences in hepatic phenotype (Supplementary Fig. 7c). While the dosing and timing of latrunculin A and nocodazole could benefit from further optimization, these data indicate that cytoskeletal modulation may increase differentiation efficiency to a range of endodermal lineages.
Discussion
We have demonstrated that the actin cytoskeleton strongly influences human pancreatic cell fate specification. By controlling the state of the cytoskeleton with cell arrangement (two- vs. three-dimensional, Fig. 1b-c), substrate stiffness (Fig. 1d), or directly with chemical treatment (Fig. 1e-i), we found that a polymerized cytoskeleton prevents premature induction of NEUROG3 expression in pancreatic progenitors but also inhibits subsequent differentiation to SC-β cells. Appropriately timed cytoskeletal depolymerization with latrunculin A overcomes this inhibition to enable robust generation of SC-β cells in planar culture. We used these findings to adapt a suspension differentiation protocol to traditional two-dimensional culture (Fig. 4a), generating SC-β cells from four cell lines that exhibited both first- and second-phase dynamic insulin secretion (Fig. 4i). Moreover, these SC-β cells rapidly reversed severe pre-existing diabetes in mice nearly as fast as human islets and about three weeks faster than cells generated with the suspension protocol (Fig. 5a).
Our planar protocol appears generalizable to a larger number of cell lines than previous protocols, as it led to large improvements in dynamic GSIS of SC-β cells generated from the 1013–4FA and 1016SeVA lines, as compared with the suspension protocol (Fig. 4i). Dynamic GSIS for SC-β cells generated from the 1013–4FA line even approached that of human islets (Fig. 4i). Translatability of differentiation strategies is a longstanding challenge in the field and has been particularly problematic when studying patient-derived iPSCs, which often have weak SC-β cell phenotypes.9,44–46 Furthermore, many hPSC lines can be difficult to adapt to suspension culture. For example, although the BJFF.6 iPSC line did not propagate in our normal suspension bioreactors (Supplementary Fig. 5e), we produced SC-β cells with dynamic function from this cell line with the planar protocol (Supplementary Fig. 5f-g). This improved reproducibility across cell lines may better facilitate the study of diabetic phenotypes (e.g., loss of GSIS, particularly first phase insulin secretion), drug screening, and autologous cell replacement therapy for diabetes.1
This planar protocol generates approximately the same number of cells on a per media volume basis as the suspension protocol (~1 million cells/mL). Importantly, it can produce cell numbers sufficient for drug screening or for a small number of transplantation doses for autologous therapy. For example, a 68 kg diabetic patient would require a therapeutic dose of 340–750 million SC-β cells,4 requiring six to thirteen standard T-75 culture flasks. While not demonstrated here, production could potentially be scaled up further with larger-format flasks, such as multilayer hyperflasks with a surface area of 1720 cm2, generating up to 1.4 billion cells per batch, or with microcarriers in suspension culture.47
Our findings appear to parallel actin dynamics during islet development in vivo, whereby the cytoskeleton is reorganized in cells of the developing pancreatic ducts to induce delamination and subsequent islet formation.48 The brief, 24-hour time requirement for latrunculin A treatment in our method is likely due to a positive feedback loop that maintains NEUROG3 expression after it has been turned on.49 We found that only latrunculin A and B could induce endocrine differentiation, whereas other compounds that decrease F-actin polymerization and cytoskeletal contractility, such as cytochalasin D and blebbistatin, did not. This discrepancy is likely due to the different mechanistic effects of these compounds on the cytoskeleton.36 Further investigation of the signaling mechanisms by which the cytoskeleton and cell adhesion regulate endocrine cell fate50 could reveal additional cytoskeleton-modifying compounds that direct pancreatic cell fate.
Another observation from this work is that cytoskeletal state influences not only SC-β cell differentiation but endodermal lineage specification more broadly. Depending on the timing of latrunculin A treatment during SC-β cell differentiation, gene signatures of exocrine, liver, esophagus, stomach, and intestine were detected in stage 6. We applied these findings by adding cytoskeletal-modulating compounds during directed differentiation protocols for exocrine pancreas, intestine, and liver, improving differentiation outcomes in each protocol. Our study as a whole emphasizes that cytoskeletal dynamics work synergistically with soluble biochemical factors to regulate endodermal cell fate, opening new opportunities to improve differentiation outcomes.
Online Methods
Stem Cell Culture.
Three stem cell lines previously used in SC-β cell differentiation protocols were utilized in this study, including the HUES8 hESC line and two non-diabetic human iPSC lines (1013–4FA and 1016SeVA).5 A fourth iPSC line (BJFF.6)39 not previously used for SC-β cell differentiations was obtained from the iPSC core at Washington University in St. Louis. Human islets were purchased from Prodo Labs for comparison. Experiments were performed with the HUES8 line unless indicated otherwise. The experiments using human stem cells were approved by the Washington University ESCRO committee (approval #15–002) with appropriate consent and conditions. Undifferentiated cells were propagated with mTeSR1 (StemCell Technologies, 05850) in a humidified incubator at 5% CO2 at 37 °C. For suspension culture, cells were passaged every 3 days with Accutase (StemCell Technologies, 07920) and seeded at 0.6 × 106 cells/mL in 30 mL spinner flasks (Reprocell, ABBWVS03A) at 60 RPM on a magnetic stir plate (Chemglass). For planar culture, cells were passaged every 4 days with TrypLE (Life Technologies, 12–604-039) and seeded onto Matrigel (Corning, 356230) coated 6-well plates at 3–5 million cells/well, with density dependent on cell line. All cells were seeded in mTeSR1 supplemented with 10 μM Y-27632 (Abcam, ab120129).
SC-β Cell Differentiation.
Suspension Protocol.
72 hours after passaging, cells in 30 mL spinner flasks were differentiated in a 6 stage protocol as previously described, using the following formulations.5 Stage 1 (3 days): S1 medium + 100 ng/ml Activin A (R&D Systems, 338-AC) + 3 μM CHIR99021 (Stemgent, 04–0004-10) for 1 day. S1 medium + 100 ng/ml Activin A for the next 2 days. Stage 2 (3 days): S2 medium + 50 ng/ml KGF (Peprotech, AF-100–19). Stage 3 (1 day): S3 medium + 50 ng/ml KGF + 200 nM LDN193189 (Reprocell, 040074) + 500 nM PdBU (MilliporeSigma, 524390) + 2 μM retinoic acid (MilliporeSigma, R2625) + 0.25 μM SANT1 (MilliporeSigma, S4572) + 10 μM Y27632. Stage 4 (5 days): S3 medium + 5 ng/mL Activin A + 50 ng/mL KGF + 0.1 μM retinoic acid + 0.25 μM SANT1 + 10 μM Y27632. Stage 5 (7 days): S5 medium + 10 μM ALK5i II (Enzo Life Sciences, ALX-270–445-M005) + 20 ng/mL Betacellulin (R&D Systems, 261-CE-050) + 0.1 μM retinoic acid + 0.25 μM SANT1 + 1 μM T3 (Biosciences, 64245) + 1 μM XXI (MilliporeSigma, 595790). Stage 6 (7–25 days): Enriched serum-free medium (ESFM). On the first day of stage 6, clusters were resized by single-cell dispersing with TrypLE and reaggregating in a 6-well plate on an orbital shaker (Benchmark Scientific, OrbiShaker) at 100 RPM in ESFM.
The base differentiation media formulations used in each stage were as follows. S1 medium: 500 mL MCDB 131 (Cellgro, 15–100-CV) supplemented with 0.22 g glucose (MilliporeSigma, G7528), 1.23 g sodium bicarbonate (MilliporeSigma, S3817), 10 g bovine serum albumin (BSA) (Proliant, 68700), 10 μL ITS-X (Invitrogen, 51500056), 5 mL GlutaMAX (Invitrogen, 35050079), 22 mg vitamin C (MilliporeSigma, A4544), and 5 mL penicillin/streptomycin (P/S) solution (Cellgro, 30–002-CI). S2 medium: 500 mL MCDB 131 supplemented with 0.22 g glucose, 0.615 g sodium bicarbonate, 10 g BSA, 10 μL ITS-X, 5 mL GlutaMAX, 22 mg vitamin C, and 5 mL P/S. S3 medium: 500 mL MCDB 131 supplemented with 0.22 g glucose, 0.615 g sodium bicarbonate, 10 g BSA, 2.5 mL ITS-X, 5 mL GlutaMAX, 22 mg vitamin C, and 5 mL P/S. S5 medium: 500 mL MCDB 131 supplemented with 1.8 g glucose, 0.877 g sodium bicarbonate, 10 g BSA, 2.5 mL ITS-X, 5 mL GlutaMAX, 22 mg vitamin C, 5 mL P/S, and 5 mg heparin (MilliporeSigma, H3149). ESFM: 500 mL MCDB 131 supplemented with 0.23 g glucose, 10.5 g BSA, 5.2 mL GlutaMAX, 5.2 mL P/S, 5 mg heparin, 5.2 mL MEM nonessential amino acids (Corning, 20–025-CI), 84 μg ZnSO4 (MilliporeSigma, 10883), 523 μL Trace Elements A (Corning, 25–021-CI), and 523 μL Trace Elements B (Corning, 25–022-CI).
For experiments investigating the effects of plating pancreatic progenitors, cells were differentiated with the suspension protocol for stages 1–3. At the end of stage 3, clusters were single cell dispersed with TrypLE and plated onto tissue culture plates coated with various ECM proteins at 0.625 ×106 cells/cm2. Differentiation media for the remainder of this hybrid protocol were the same as for the suspension protocol with the exception that Y-27632 and Activin A, which were originally included to maintain cluster integrity for suspension clusters5 but were not necessary when cells were plated, were omitted on days 2–5 of stage 4. Additional compounds were added as indicated in each experiment: 1 μM latrunculin A (Cayman Chemical, 10010630), 1 μM latrunculin B (Cayman Chemical, 10010631), 1 μM cytochalasin D (MilliporeSigma, C2618), 1 μM jasplakinolide (Cayman Chemical, 11705), 10 μM blebbistatin (MilliporeSigma, 203389), 1 μM nocodazole (Cayman Chemical, 13857), 1 μM Y-15 (Cayman Chemical, 14485), 10 μM Y-27632, and 10 μM GDC-0994 (Selleckchem, S7554). A variety of ECM coatings were initially tested with this plating methodology, including collagen I (Corning, 354249), collagen IV (Corning, 354245), fibronectin (Gibco, 33016–015), vitronectin (Gibco, A14700), Matrigel (Corning, 356230), gelatin (Fisher, G7–500), and laminins 111, 121, 211, 221, 411, 421, 511, and 521 (Biolamina, LNKT-0201). All subsequent experiments with this hybrid protocol were performed on collagen I.
Planar Protocol.
24 hours after passaging, cells seeded onto 6 or 24-well plates coated with diluted Matrigel at 0.313–0.521 × 106 cells/cm2 were differentiated with a new 6 stage protocol using the following formulations, with media changes every day. Stage 1 (4 days): BE1 + 100 ng/mL Activin A + 3 μM CHIR99021 for the first 24 hours, followed with 3 days of BE1 containing 100 ng/mL Activin A only. Stage 2 (2 days): BE2 + 50 ng/mL KGF. Stage 3 (2 days): BE3 + 50 ng/mL KGF, 200 nM LDN193189, 500 nM TPPB (Tocris, 53431), 2 μM retinoic acid, and 0.25 μM SANT1. Stage 4 (4 days): BE3 + 50 ng/mL KGF, 200 nM LDN193189, 500 nM TPPB, 0.1 μM retinoic acid, and 0.25 μM SANT1. Stage 5 (7 days): S5 + 10 μM ALK5i II + 20 ng/mL Betacellulin + 0.1 μM retinoic acid + 0.25 μM SANT1 + 1 μM T3 + 1 μM XXI. 1 μM Latrunculin A was added to this media for the first 24 hours only. Stage 6 (7–25 days): Cultures were kept on the plate with ESFM for the first 7 days. To move to suspension culture, cells could be single cell dispersed with TrypLE and placed in 6 mL ESFM within a 6-well plate at a concentration of 4–5 million cells/well on an orbital shaker at 100 RPM. Assessments were performed 5–8 days after cluster aggregation.
The base differentiation media formulations that differed from the suspension protocol were as follows. BE1: 500 mL MCDB 131 supplemented with 0.8 g glucose, 0.587 g sodium bicarbonate, 0.5 g BSA, and 5 mL GlutaMAX. BE2: 500 mL MCDB 131 supplemented with 0.4 g glucose, 0.587 g sodium bicarbonate, 0.5 g BSA, 5 mL GlutaMAX, and 22 mg vitamin C. BE3: 500 mL MCDB 131 supplemented with 0.22 g glucose, 0.877 g sodium bicarbonate, 10 g BSA, 2.5 mL ITS-X, 5 mL GlutaMAX, and 22 mg vitamin C.
Microscopy and Immunocytochemistry.
Bright field images were taken with a Leica DMi1 inverted light microscope, and fluorescence images were captured with a Nikon A1Rsi confocal microscope. For immunostaining, cells were fixed with 4% paraformaldehyde (PFA) at room temperature for 30 minutes. They were then blocked and permeabilized for 45 minutes at room temperature with an immunocytochemistry (ICC) solution consisting of 0.1% triton X (Acros Organics, 327371000) and 5% donkey serum (Jackson Immunoresearch, 017000–121) in phosphate-buffered saline (PBS, Corning, 21–040-CV). Samples were then incubated with primary antibodies diluted in ICC solution overnight at 4 ºC, washed with ICC, incubated with secondary antibodies diluted in ICC for 2 hours at room temperature, and stained with DAPI for 15 minutes at room temperature.
For histological sectioning, whole SC-β cell clusters generated with the planar protocol and mouse kidneys containing transplanted cells were fixed overnight with 4% PFA at 4 ºC. The in vitro clusters were also embedded in Histogel (Thermo Scientific, hg-4000–012). These samples were then paraffin-embedded and sectioned by the Division of Comparative Medicine (DCM) Research Animal Diagnostic Laboratory Core at Washington University in St. Louis. Paraffin was removed from sectioned samples with Histoclear (Thermo Scientific, C78–2-G), and antigen retrieval was carried out in a pressure cooker (Proteogenix, 2100 Retriever) with 0.05 M EDTA (Ambion, AM9261). Slides were blocked and permeabilized with ICC solution for 45 minutes, incubated with primary antibodies in ICC solution overnight at 4 ºC, and incubated with secondary antibodies for 2 hours at room temperature. Slides were then sealed with DAPI Fluoromount-G (Southern Biotech, 0100–20).
Primary antibodies were diluted in ICC solution at 1:300 unless indicated otherwise: rat anti-C-peptide (DSHB, GN-ID4-S), 1:100 mouse anti-NKX6–1 (DSHB, F55A12-S), goat anti-PDX1(R&D Systems, AF2419), sheep anti-NEUROG3 (R&D Systems, AF3444), rabbit anti-somatostatin (ABCAM, ab64053), mouse anti-glucagon (ABCAM, ab82270), mouse anti-NKX2–2 (DSHB, 74.5A5-S), goat anti-NEUROD1 (R&D Systems, AF2746), mouse anti-ISL1 (DSHB, 40.2d6-s), rabbit anti-CHGA (ABCAM, ab15160), rabbit anti-MAFB (MilliporeSigma, HPA005653), 1:100 sheep anti-PRSS1/2/3 (R&D Systems, AF3586), 1:100 mouse-anti-KRT19 (Dako, MO888), goat anti-KLF5 (R&D Systems, AF3758), rabbit anti-CDX2 (Abcam, ab76541), mouse anti-AFP (Abcam, ab3980), and rabbit anti-albumin (Abcam, ab207327). F-actin was stained directly with 1:200 TRITC-conjugated phalloidin (MilliporeSigma, FAK 100).
Secondary antibodies were diluted in ICC solution at 1:300. All secondary antibodies were raised in donkey: anti-goat alexa fluor 594 (Invitrogen, A11058), anti-goat alexa fluor 647 (Invitrogen, A21447), anti-mouse alexa fluor 488 (Invitrogen, A21202), anti-mouse alexa fluor 594 (Invitrogen, A21203), anti-mouse alexa fluor 647 (Invitrogen, A31571), anti-rabbit alexa fluor 488 (Invitrogen, A21206), anti-rabbit alexa fluor 594 (Invitrogen, A21207), anti-rabbit alexa fluor 647 (Invitrogen, A31573), anti-rat alexa fluor 488 (Invitrogen, A21208), anti-sheep alexa fluor 594 (Invitrogen, A11016).
qRT-PCR.
RNA was extracted from either whole clusters or cells directly on the plate with the RNeasy Mini Kit (Qiagen, 74016). Samples were treated with a DNAse kit (Qiagen, 79254) during extraction. The High Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems, 4368814) was used to synthesize cDNA on a thermocycler (Applied Biosystems, A37028). The PowerUp SYBR Green Master Mix (Applied Biosystems, A25741) was used on a StepOnePlus (Applied Biosystems), and real time PCR results were analyzed using a ΔΔCt methodology. TBP and GUSB were both used as housekeeping genes. Primer sequences were as follows.
Collagen Gels.
Type 1 collagen (Corning, 354249) gels were created at a concentration of 5 mg/mL using 10x PBS, sterile deionized water, and 1M NaOH according to the manufacturer’s instructions. Various volumes of this collagen solution were pipetted into the center of wells of a 24-well plate and briefly centrifuged to obtain a uniform coating. Collagen gel heights were calculated based on the volume of collagen gel solution, the radius of the 24-well plate, and the equation for the height of a cylinder.
G/F actin ratio.
The G/F actin ratio was determined by western blot following the instructions of the G-actin/F-actin In Vivo Assay Kit (Cytoskeleton, Inc, BK037). The Precision Plus Protein Kaleidoscope Protein Standard (Biorad, 161–0375) was used to indicate molecular weights. Western blots were visualized using SuperSignal West Pico PLUS Chemiluminescent substrate (ThermoScientific, 34577) and the Odyssey FC (LI-COR) imager.
Integrin Assay.
To quantify which integrins were expressed on the surface of pancreatic progenitors, cells generated in suspension culture were dispersed with TrypLE at either the end of stage 3 or stage 4 and plated onto wells coated with monoclonal antibodies for different α and β integrin subunits using the Alpha/Beta Integrin-Mediated Cell Adhesion Array Combo Kit (MilliporeSigma, ECM532). Integrin expression was quantified according to the manufacturer’s instructions.
Single-Cell RNA Sequencing.
Cells generated with the suspension protocol were single-cell dispersed with TrypLE from clusters at the end of stage 3 and seeded onto collagen 1 coated 24-well plates at 0.625 ×106 cells/ cm2. Either 0.5 μM latrunculin A or 5 μM nocodazole were added throughout the entirety of stage 4. At the end of stage 4, cells were single-cell dispersed, suspended in Dulbecco’s Modified Eagle Medium, and submitted to the Washington University Genome Technology Access Center. Library preparation was done using the Chromium Single Cell 3’ Library and Gel Bead Kit v2 (10x Genomics, 120237). Briefly, single cells were isolated in emulsions using a microfluidic platform, and each single cell emulsion was barcoded with a unique set of oligonucleotides. The GemCode Platform was used to carry out reverse transcription within each single cell emulsion, which was amplified to construct a library. The libraries were sequenced with paired-end reads of 26×98bp using the Illumina HiSeq2500.
We used Seurat v2.0 to perform single cell RNA analyses.51 Duplicate cells and cells with high mitochondrial gene expression were filtered out using FilterCells (>9000 total genes, <5000 total genes and >6% mitochondrial genes for Untreated Control, >6000 genes, <2500 total genes and >5% mitochondrial genes for latrunculin A, >12000 genes, <2200 total genes and >4% mitochondrial genes for nocodazole). Each data set was normalized using global-scaling normalization. FindVariableGenes identified and removed outlier genes using scaled z-score dispersion. We then combined datasets and performed a canonical correlation analysis (CCA) with RunMultiCCA (num.ccs = 20). AlignSubspace was used to align the CCA subspaces and generated a new dimension reduction for integrated analysis. Unsupervised TSNE plots were generated using RunTSNE (dim.use = 1:17), and the resulting clusters were defined and labeled using FindMarkers. We used VlnPlot (Violin plots) and FeaturePlot (tsne plots) to visualize differences in gene expressions across each cluster and across conditions.
Flow Cytometry.
Cells were single-cell dispersed with TrypLE and fixed with 4% PFA for 30 minutes. Cells were then washed with PBS and incubated in ICC solution for 45 minutes at room temperature, incubated with primary antibodies overnight at 4 ºC, and incubated with secondary antibodies for 2 hours at room temperature. Cells were then washed twice with ICC solution and filtered before running on the LSRII flow cytometer (BD Biosciences). Analysis was completed with FlowJo.
Glucose Stimulated Insulin Secretion.
Static GSIS.
To assess the function of cells produced by the hybrid protocol, static GSIS was performed with cells still attached to 96 or 24-well tissue culture plates. To assess function of clusters generated with the planar protocol, approximately 30 clusters were collected and placed in tissue culture transwell inserts (MilliporeSigma, PIXP01250) in a 24-well plate. All cells were first washed twice with KRB buffer (128 mM NaCl, 5 mM KCl, 2.7 mM CaCl2, 1.2 mM MgSO4, 1 mM Na2HPO4, 1.2 mM KH2PO4, 5 mM NaHCO3, 10 mM HEPES (Gibco, 15630–080), and 0.1% BSA). Cells were first incubated in KRB containing 2 mM glucose at 37ºC for one hour, after which this solution was discarded and replaced with fresh 2 mM glucose KRB solution. After an additional hour, the supernatant was collected. 20 mM glucose KRB was added for the next hour, after which the supernatant was again collected. Cells were washed with fresh KRB during each solution change. Cells were then single-cell dispersed with TrypLE and counted with the Vi-Cell XR (Beckman Coulter). Supernatants from the low and high glucose challenges were quantified with a human insulin ELISA (ALPCO, 80-INSHU-E10.1), and cell counts were used to normalize insulin secretion.
In a subset of experiments, one of the following secretagogues was added to the high glucose challenge: 300 μM Tolbutamide (MilliporeSigma, T0891), 10 nM Exedin-4 (MilliporeSigma, E7144), 100 μM IBMX (MilliporeSigma, I5879), or 30 mM KCL (Thermo Fisher, BP366500). For measuring insulin secretion in response to multiple glucose challenges, cells were treated sequentially with two cycles of 1 hour incubations as follows: low (2mM), high (20mM), low (2mM), then high (20mM). The supernatant was collected after the 1 hour incubation of each challenge.
Dynamic GSIS.
Dynamic function of SC-β cells was assessed with a perifusion setup as we previously reported.5 0.015 inch inlet and outlet tubing (ISMATEC, 070602–04i-ND) was connected with 0.04” connection tubing (BioRep, Peri-TUB-040) to 275-μl cell chambers (BioRep, Peri-Chamber) and dispensing nozzles (BioRep, PERI-NOZZLE). Approximately 30 SC-β cell clusters were washed twice with KRB buffer and loaded into the chambers, sandwiched between two layers of hydrated Bio-Gel P-4 polyacrylamide beads (Bio-Rad, 150–4124). These chambers were connected to a high precision 8-channel dispenser pump (ISMATEC, ISM931C) and immersed in a 37 °C water bath for the remainder of the assay. A 2 mM glucose KRB solution was perfused through the chambers for the first 90 minutes at a flow rate of 100 μL/min. After this equilibration period, effluent was collected in 2 minute time intervals, switching glucose solutions as follows: 2 mM glucose KRB for 12 minutes, 20 mM glucose KRB for 24 minutes, and 2 mM glucose KRB for 16 minutes. The SC-β cell clusters were then lysed with a solution of 10 mM Tris (MilliporeSigma, T6066), 1 mM EDTA (Ambion, AM9261), and 0.2% Triton-X (Acros Organics, 327371000). DNA was quantified using the Quant-iT Picogreen dsDNA assay kit (Invitrogen, P7589) and was used to normalize insulin values quantified with a human insulin ELISA.
Electron Microscopy.
Cell clusters were fixed with 2.5% glutaraldehyde, 1.25% PFA, and 0.03% picric acid in a 0.1 M sodium cacodylate buffer at 7.4 pH, similar as previously reported4 and processed at the Harvard Medical School Electron Microscopy Facility. Samples were imaged using a JEM-1400+ transmission electron microscope.
Calcium Flux Analysis.
Calcium imaging was performed similar as previously reported. 4 Clusters were attached to Matrigel, incubated with 20 μM Fluo4-AM (Life Technologies, F14217) in KRB buffer with 2 mM glucose for 45 minutes, washed, and incubated at sequential treatments of 2 mM glucose, 20 mM glucose, 2 mM glucose, and 20 mM glucose plus 30 mM KCl in KRB buffer. Fluo4-AM fluorescence was imaged using a Leica DMI4000 fluorescence microscope and quantified with ImageJ.
Insulin and Proinsulin Content.
Whole SC-β cell clusters or cells attached to culture plates were washed twice thoroughly with PBS. Half of the clusters or an equivalent well of plated cells were immersed in TrypLE for cell counts on the Vi-Cell XR. For the other half of the samples, a solution of 1.5% HCl and 70% ethanol was added to either the clusters in eppendorf tubes or directly onto plated cells. After 15 minutes, the plated cells were pipetted vigorously and transferred to eppendorf tubes. The eppendorf tubes from both clusters and plated cells were kept at −20°C for 72 hours, vortexing vigorously every 24 hours. Samples were then centrifuged at 2100 RCF for 15 minutes. The supernatant of each sample was collected, neutralized with an equal volume of 1 M TRIS (pH 7.5), and quantified using a proinsulin ELISA kit (Mercodia, 10–1118-01) and a human insulin ELISA kit. Proinsulin and insulin secretion were normalized to the viable cell counts.
Transplantation Studies.
In vivo studies were carried out in accordance to the Washington University Institutional Animal Care and Use Committee regulations (protocol 20180203). 7-week-old male immunodeficient mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) were purchased from Jackson Laboratories. Randomly selected mice were induced with diabetes by administering 45 mg/kg STZ (R&D Systems, 1621500) in PBS for 5 consecutive days via intraperitoneal injection. Mice became diabetic approximately one week after STZ treatment. After 2 more weeks, we performed transplant surgeries by injecting either 5 million SC-β cells generated with the planar protocol, 5 million SC-β cells generated with the suspension protocol, or 5 million human islet cells under the kidney capsule of diabetic mice anaesthetized with isoflurane. The human islet transplants were done in a later cohort of mice from the SC-β cell transplantations. All mice were monitored weekly after transplant surgeries. Removal of the kidneys containing SC-β cells were performed during week 36 after transplant.
We performed fasting blood glucose measurements, glucose tolerance tests, and in vivo GSIS for our in vivo assessments. Mice were fasted 4–6 hours for all studies. For fasting measurements, blood glucose levels were obtained from a tail bleed using a handheld glucometer (Bayer, 9545C). For glucose tolerance tests, we injected 2 g/kg glucose in 0.9% saline (Moltox, 51–405022.052) and measured blood glucose every 30 minutes for 150 minutes. For in vivo GSIS, we collected approximately 30 μL of blood via tail bleed using microvettes (Sarstedt, 16.443.100) before and 60 minutes post glucose injection. The blood samples were centrifuged at 2500 rpm for 15 minutes at 4°C and the serum was collected to be quantified with the Human Ultrasensitive Insulin ELISA kit (ALPCO, 80-ENSHUU-E01.1) and Mouse C-peptide ELISA kit (ALPCO, 80-CPTMS-E01).
Bulk RNA Sequencing.
Cells generated with the suspension protocol were single-cell dispersed from clusters with TrypLE at the end of stage 3 and seeded onto collagen 1 coated 24-well plates at 0.625 ×106 cells/ cm2. Either 0.5 μM latrunculin A was added throughout the entirety of stage 4 or 1 μM latrunculin A was added for the first 24 hours of stage 5. Cells generated from the planar protocol without latrunculin A treatment as well as clusters generated with the suspension protocol were used as controls. After two weeks in stage 6, RNA was extracted with the RNeasy Mini Kit (Qiagen, 74016), including a DNase treatment (Qiagen, 79254) during extraction. Samples were delivered to Washington University in St. Louis Genome Technology Access Center for library preparation and sequencing. Samples were prepared by RNA depletion using Ribo-Zero according to library kit manufacturer’s protocol, indexed, pooled, and sequenced on an Illumina HiSeq2500.
Differential gene expression analysis was performed using EdgeR. 52,53 DGEList was used to create the count object and normalize the data using the trimmed mean M-values (TMM) method with calcNormFactors. Pairwise comparisons were performed using exactTest and used topTags to obtain differentially expressed genes and their respective log fold change (logFC) and adjusted p-value (FDR). These values were used to generate volcano plots using ggplot2. Hierarchical clustering and heatmaps were performed and generated with heatmap.2 (gplots) using logCPM calculated expression levels. Gene set analyses were performed with gene set enrichment analysis (GSEA).54,55 Lineage specific gene sets including Exocrine (GO: 0035272,M13401), Pancreas Beta cells (Hallmark, M5957) and Intestinal epithelial (GO: 0060576, M12973) were obtained from the Molecular Signatures Database (MdigDB).54–57 Gene sets for liver, esophagus and stomach were customized using the Human Protein Atlas58 and literature.16,59–64
Differentiation to Other Endodermal Lineages.
For differentiation to other endodermal lineages, HUES8 stem cells were cultured and passaged normally. Differentiations were initiated 24 hours after seeding 24-well plates at 0.521 × 106 cells/cm2. Protocols for exocrine pancreas,40 intestine,16,41 and liver42 were adapted from literature. Either latrunculin A or nocodazole were added as indicated in each protocol. All three differentiation protocols used the same stage 1 to induce endoderm. Stage 1 (4 days): BE1 + 100ng/mL Activin A + 3μM CHIR99021 for the first 24 hours, followed with 3 days of BE1 containing 100ng/mL Activin A only.
Exocrine Pancreas.
Stage 2 (2 days): BE2 + 50 ng/mL KGF. Stage 3 (2 days): BE3 + 50 ng/mL KGF, 200 nM LDN193189, 500 nM TPPB, 2 μM retinoic acid, and 0.25 μM SANT1. Stage 4 (4 days): BE3 + 50 ng/mL KGF, 200 nM LDN193189, 500 nM TPPB, 0.1 μM retinoic acid, and 0.25 μM SANT1. Either 1 μM latrunculin A was added for the first 24 hours of this stage, or 1 μM nocodazole was added for the entirety of stage 4. Stage 5 (6 days): S5 + 10 ng/mL bFGF. 10 mM nicotinamide (MilliporeSigma, 72340) was added for the last two days.
Intestine Differentiation.
Stage 2 (4 days): BE2 + 3 μM CHIR99021 + 500 ng/mL FGF4 (R&D Systems, 235-F4). Either 1 μM latrunculin A was added for the first 24 hours of this stage, or 1 μM nocodazole was added for the entirety of stage 2. Stage 3 (7 days): BE3 + 500 ng/mL R-spondin1 (R&D Systems, 4645-RS) + 100 ng/mL EGF (R&D Systems, 236-EG) + 200 nM LDN193189.
Liver Differentiation.
Stage 2 (2 days): BE2 + 50 ng/mL KGF. Stage 3 (4 days): BE3 + 10 ng/mL bFGF + 30 ng/mL BMP4 (R&D Systems, 314-BP). For the first 24 hours only, 2 μM retinoic acid and either 1 μM latrunculin A or 1 μM nocodazole were added. Stage 4 (5 days): BE3 + 20 ng/mL OSM (R&D Systems, 295-OM) + 20 ng/mL HGF (R&D Systems, 294-HG) + 100 nM dexamethasone (MilliporeSigma, D4902).
Statistics and Reproducibility.
Data analysis was performed in GraphPad Prism, version 7. Analyzed data was evaluated by either two-sided t-tests or ANOVA followed by either Dunnett’s multiple comparison test or Tukey’s HSD test. The following convention is used for indicating p-values: ns = not significant, * = p < 0.05, ** = p < 0.01, *** = p < 0.001. All data is presented as the mean, and all error bars represent SEM. The sample size (n) indicates the total number of independent biological samples. During in vitro experiments, each replicate for planar cells was a completely separate well of a tissue culture plate, and each of these wells was processed individually (e.g., separate RNA extractions and PCR reactions). For suspension clusters, each replicate was a group of clusters that was collected and processed together. During in vivo experiments, each mouse was considered to be a replicate.
Throughout the manuscript, representative microscopy images are shown, but these were replicated by multiple independent experiments as follows. Figure 1: (b) This plating method was reproduced numerous times (>20) as it was the basis of all experiments in Figures 1–3. (f) This staining result was reproduced by three independent differentiations. (h) This staining result was reproduced by three independent differentiations. Figure 3: (g) This staining result was reproduced by two independent differentiations. (h) This aggregation method was successfully reproduced in all subsequent differentiations (>20) and is incorporated into our protocol methodology. Figure 4: (e) This staining result was reproduced by many clusters from two independent differentiations. Figure 5: (c) Staining results were obtained from three transplanted mice. Supplemental Figure 1: (a) Plating on the different ECM proteins was reproduced by three independent experiments. Subsequent experiments that plated cells at the beginning of stage 4 used collagen 1. Supplemental Figure 3: (a) This staining result was reproduced by two independent differentiations. Supplemental Figure 4: (e-f) These staining results were reproduced by many clusters from a single differentiation of each line. (g) This staining result was reproduced by three independent differentiations. Supplemental Figure 5: (c) Electron microscopy for insulin granules was performed on multiple clusters from two independent differentiations. (f-g) SC-β cell clusters were generated from the BJFF.6 cell line from three independent differentiations.
Data availability
All single-cell and bulk RNA sequencing data have been deposited to the Gene Expression Omnibus under accession number GSE137659. Any other data and protocol information used in the present the manuscript are available from the corresponding author upon reasonable request.
Supplementary Material
| Gene name | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|
| TBP | GCCATAAGGCATCATTGGAC | AACAACAGCCTGCCACCTTA |
| GUSB | CGTCCCACCTAGAATCTGCT | TTGCTCACAAAGGTCACAGG |
| INS | CAATGCCACGCTTCTGC | TTCTACACACCCAAGACCCG |
| CHGA | TGACCTCAACGATGCATTTC | CTGTCCTGGCTCTTCTGCTC |
| NEUROD1 | ATCAGCCCACTCTCGCTGTA | GCCCCAGGGTTATGAGACTAT |
| SST | TGGGTTCAGACAGCAGCTC | CCCAGACTCCGTCAGTTTCT |
| GCG | AGCTGCCTTGTACCAGCATT | TGCTCTCTCTTCACCTGCTCT |
| PDX1 | CGTCCGCTTGTTCTCCTC | CCTTTCCCATGGATGAAGTC |
| NKX2–2 | GGAGCTTGAGTCCTGAGGG | TCTACGACAGCAGCGACAAC |
| NKX6–1 | CCGAGTCCTGCTTCTTCTTG | ATTCGTTGGGGATGACAGAG |
| ISL1 | TCACGAAGTCGTTCTTGCTG | CATGCTTTGTTAGGGATGGG |
| GCK | ATGCTGGACGACAGAGCC | CCTTCTTCAGGTCCTCCTCC |
| MAFB | CATAGAGAACGTGGCAGCAA | ATGCCCGGAACTTTTTCTTT |
| AFP | TGTACTGCAGAGATAAGTTTAGCTGAC | CCTTGTAAGTGGCTTCTTGAACA |
| PRSS1 | TATCAGCAGGCCACTGCTAC | CCTCCAGGACTTCGATGTTG |
| CDX2 | GAACCTGTGCGAGTGGATG | GGTGATGTAGCGACTGTAGTGAA |
| SOX2 | TTGCTGCCTCTTTAAGACTAGGA | TAAGCCTGGGGCTCAAACT |
| KRT19 | AGGATGCTGAAGCCTGGTT | GGTCAGTAACCTCGGACCTG |
| SERPINA1 | CCCTGTTTGCTCCTCCGATAA | GATGCCCCACGAGACAGAAG |
| FAH | GCCAGTGTGCTGGAAAAGTG | CTGGCAGGGAGGCTTTACAC |
| HNF4A | GGACATGGCCGACTACAGTG | CTCGAGGCACCGTAGTGTTT |
| CEBPA | TATAGGCTGGGCTTCCCCTT | AGCTTTCTGGTGTGACTCGG |
| CYP3A4 | CACCCCCAGTTAGCACCATT | CCACGCCAACAGTGATTACA |
| FABP1 | TCTCCGGCAAGTACCAACTG | GATTTCCGACACCCCCTTGA |
| LGR5 | CTTGGTGCCCAAAGCTCA | TCTTTTCCAGGTATGTTCATTGC |
| ASCL2 | CACTGGGGATCTGTGGACTG | TTCTGTAAGGCCCAAAGCGT |
| FABP2 | GCCCAAGGACAGACCTGAAT | CAAGTGCTGTCAAACGCCAT |
| MUC2 | CAGCTCATCTCGTCCGTCTC | GTGTAGGTGTGTGTCAGCGA |
| MMP7 | CATGATTGGCTTTGCGCGAG | CTACCATCCGTCCAGCGTTC |
| LYZ | TCAGCCTAGCACTCTGACCT | GCCCTGGACCGTAACAGAAA |
| PRSS2 | GCTACAAGTCGGCAATTAACTCA | CGATGTTGTGCTCTCCCAGT |
| AMY2B | GGAGCCTCTGTGTTTCTTTGTT | GCACTTGAAGGACACGGGA |
| NR5A2 | CCGACAAGTGGTACATGGAA | TCCGGCTTGTGATGCTATTA |
| ALDH1 | ATCAAAGAAGCTGCCGGGAA | GCATTGTCCAAGTCGGCATC |
| TAT | CAGTCCCCGAGGTGATGATG | CTGAGTGTGGGTGTGGTTGT |
| TBX3 | AAACTCTGCGCGGAGAAAGA | CCCCCAGTAGCTCAATGCAA |
| HNF6 | ATGTCCAGCGTCGAACTCTAC | TGCTTTGGTACAAGTGCTTGAT |
| LDHA | GGAGATCCATCATCTCTCCC | GGCCTGTGCCATCAGTATCT |
| SLC16A1 | CACTTAAAATGCCACCAGCA | AGAGAAGCCGATGGAAATGA |
| MAFA | GAGAGCGAGAAGTGCCAACT | TTCTCCTTGTACAGGTCCCG |
| UCN3 | GGAGGGAAGTCCACTCTCG | TGTAGAACTTGTGGGGGAGG |
Acknowledgments
This work was supported by the JDRF Career Development Award (5-CDA-2017–391-A-N), NIH (5R01DK114233), and startup funds from Washington University School of Medicine Department of Medicine. N.J.H. was supported by the NIH (T32DK007120). K.G.M. was supported by the NIH (T32DK108742). L.V.C. was supported by the NIH (R25GM103757). Microscopy was performed through the Washington University Center for Cellular Imaging (WUCCI), which is supported by Washington University School of Medicine, CDI (CDI-CORE-2015–505) and the Foundation for Barnes-Jewish Hospital (3770). Microscopy was supported by the Washington University Diabetes Research Center (P30DK020579). Sequencing work was performed by the Washington University Genome Technology Access Center in the Department of Genetics (NIH P30CA91842 and UL1TR000448) and supported by the Washington University Institute of Clinical and Translational Sciences (NIH UL1TR002345). We thank Michelle Kim for technical assistance.
Footnotes
Competing interests
N.J.H., L.V.C., and J.R.M. are inventors on patents and patent applications related to SC-β cell differentiation approaches described in this manuscript.
References
- 1.Millman JR & Pagliuca FW Autologous pluripotent stem cell-derived β-like cells for diabetes cellular therapy. Diabetes 66, 1111–1120 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Tomei AA, Villa C & Ricordi C. Development of an encapsulated stem cell-based therapy for diabetes. Expert Opin. Biol. Ther 15, 1321–1336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jennings RE, Berry AA, Strutt JP, Gerrard DT & Hanley NA Human pancreas development. Development 142, 3126–3137 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Pagliuca FW et al. Generation of functional human pancreatic β cells in vitro. Cell 159, 428–439 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Velazco-Cruz L et al. Acquisition of Dynamic Function in Human Stem Cell-Derived β Cells. Stem Cell Reports 12, 351–365 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rezania A et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 32, 1121–1133 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Russ HA et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 34, 1759–1772 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nair GG et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat. Cell Biol. 21, 263–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sui L et al. β-Cell replacement in mice using human type 1 diabetes nuclear transfer embryonic stem cells. Diabetes 67, 26–35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghazizadeh Z et al. ROCKII inhibition promotes the maturation of human pancreatic beta-like cells. Nat. Commun. 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nostro MC et al. Efficient generation of NKX6–1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Reports 4, 591–604 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rezania A et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes 61, 2016–2029 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Amour KA et al. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 23, 1534–1541 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Amour KAD et al. Production of pancreatic hormone – expressing endocrine cells from human embryonic stem cells. 24, 1392–1401 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Kroon E et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 26, 443–452 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Spence JR et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470, 105–110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng X et al. Self-renewing endodermal progenitor lines generated from human pluripotent stem cells. Cell Stem Cell 10, 371–384 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu G, Dubauskaite J & Melton DA Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 129, 2447–57 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Johansson KA et al. Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev. Cell 12, 457–465 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Nelson SB, Schaffer AE & Sander M. The transcription factors Nkx6.1 and Nkx6.2 possess equivalent activities in promoting beta-cell fate specification in Pdx1+ pancreatic progenitor cells. Development 134, 2491–2500 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Schulz TC et al. A scalable system for production of functional pancreatic progenitors from human embryonic stem cells. PLoS One 7, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engler AJ, Sen S, Sweeney HL & Discher DE Matrix elasticity directs stem cell lineage specification. Cell 126, 677–89 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Hogrebe NJ & Gooch KJ Direct influence of culture dimensionality on hMSC differentiation at various matrix stiffnesses using a fibrous self-assembling peptide hydrogel. J. Biomed. Mater. Res. Part A 104, 2356–68 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Kilian KA, Bugarija B, Lahn BT & Mrksich M. Geometric cues for directing the differentiation of mesenchymal stem cells. Proc. Natl. Acad. Sci. U. S. A. 107, 4872–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McBeath R, Pirone DM, Nelson CM, Bhadriraju K & Chen CS Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 6, 483–95 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Ahn EH et al. Spatial control of adult stem cell fate using nanotopographic cues. Biomaterials 35, 2401–10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frith JE, Mills RJ & Cooper-White JJ Lateral spacing of adhesion peptides influences human mesenchymal stem cell behaviour. J. Cell Sci. 125, 317–27 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Hogrebe NJ et al. Independent control of matrix adhesiveness and stiffness within a 3D self-assembling peptide hydrogel. Acta Biomater. 70, 110–119 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Janmey PA & Miller RT Mechanisms of mechanical signaling in development and disease. J. Cell Sci. 124, 9–18 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swift J et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341, 1240104 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dupont S et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–83 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Olson EN & Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 11, 353–65 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharon N et al. Wnt Signaling Separates the Progenitor and Endocrine Compartments during Pancreas Development. Cell Rep. 27, 2281–2291 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Veres A et al. Charting cellular identity during human in vitro β -cell differentiation. Nature 569, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leong WS et al. Thickness sensing of hMSCs on collagen gel directs stem cell fate. Biochem. Biophys. Res. Commun. 401, 287–92 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Peng GE, Wilson SR & Weiner OD A pharmacological cocktail for arresting actin dynamics in living cells. Mol. Biol. Cell 22, 3986–3994 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brenner SL & Korn D. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 2, 316–318 (1987). [DOI] [PubMed] [Google Scholar]
- 38.Chang YC et al. GEF-H1 couples nocodazole-induced microtubule disassembly to cell contractility via RhoA. Mol. Biol. Cell 19, 2147–2153 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adkar SS et al. Step-wise chondrogenesis of human induced pluripotent stem cells and purification via a reporter allele generated by CRISPR-Cas9 genome. Stem Cells 37, 65–76 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hohwieler M et al. Human pluripotent stem cell-derived acinar/ductal organoids generate human pancreas upon orthotopic transplantation and allow disease modelling. Gut 66, 473–486 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCracken KW, Howell JC, Wells JM & Spence JR Generating human intestinal tissue from pluripotent stem cells in vitro. Nat. Protoc. 6, 1920–1928 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ang LT et al. A roadmap for human liver differentiation from pluripotent stem cells. Cell Rep. 22, 2190–2205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin X et al. Niche-independent high-purity cultures of Lgr5 + intestinal stem cells and their progeny. Nat. Methods 11, 106–112 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X et al. Point mutations in the PDX1 transactivation domain impair human β-cell development and function. Mol. Metab. 24, 80–97 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balboa D et al. Insulin mutations impair beta-cell development in a patient-derived iPSC model of neonatal diabetes. Elife 7, 1–35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma S et al. β cell replacement after gene editing of a neonatal diabetes-causing mutation at the insulin locus. Stem Cell Reports 11, 1407–1415 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen KG, Mallon BS, McKay RDG & Robey PG Human pluripotent stem cell culture: Considerations for maintenance, expansion, and therapeutics. Cell Stem Cell 14, 13–26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kesavan G et al. Cdc42/N-WASP signaling links actin dynamics to pancreatic cell delamination and differentiation. J. Cell Sci. 127, 685–694 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seymour PA Sox9: A master regulator of the pancreatic program. Rev. Diabet. Stud. 11, 51–83 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mamidi A et al. Mechanosignalling via Integrins Directs Fate Decisions of Pancreatic Progenitors. Nature 564, 114–118 (2018). [DOI] [PubMed] [Google Scholar]
Additional References
- 51.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCarthy DJ, Chen Y & Smyth GK Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson MD, McCarthy DJ & Smyth GK edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–50 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mootha VK et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–73 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Liberzon A et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liberzon A et al. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uhlén M et al. Tissue-based map of the human proteome. Science. 347, 1–9 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Takebe T et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 499, 481–484 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Finkbeiner SR et al. Transcriptome-wide analysis reveals hallmarks of human intestine development and maturation in vitro and in vivo. Stem Cell Reports 4, 1140–1155 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCracken KW et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 516, 400–404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rosekrans SL, Baan B, Muncan V & van den Brink GR Esophageal development and epithelial homeostasis. Am. J. Physiol. Liver Physiol. 309, G216–G228 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Trisno SL et al. Esophageal organoids from human pluripotent stem cells delineate Sox2 functions during esophageal specification. Cell Stem Cell 23, 501–515 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Willet SG & Mills JC Stomach organ and cell lineage differentiation: from embryogenesis to adult homeostasis. Cell Mol Gastroenterol Hepatol 2, 546–559 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All single-cell and bulk RNA sequencing data have been deposited to the Gene Expression Omnibus under accession number GSE137659. Any other data and protocol information used in the present the manuscript are available from the corresponding author upon reasonable request.