

Abstract
Primary immunodeficiencies (PIDs) comprise a diverse group of over 400 genetic disorders that result in clinically apparent immune dysfunction. Although classically considered mendelian disorders with complete penetrance, we now understand that absent or partial clinical disease is often noted in individuals harboring disease-causing genotypes. Despite the frequency of incomplete penetrance in PID, no conceptual framework exists from which to categorize and explain these occurrences. Here, by reviewing decades of reports on incomplete penetrance in PID we identify four recurrent themes of incomplete penetrance, namely genotype quality, (epi)genetic modification, environmental influence, and mosaicism. For each of these principles, we review what is known, underscore what remains unknown, and propose future experimental approaches to fill the gaps in our understanding. Although the content herein relates specifically to inborn errors of immunity, the concepts are generalizable across genetic disease.
Keywords: penetrance, variable expressivity, primary immunodeficiency, human genetics, mosaicism
Introduction
Primary immunodeficiencies (PIDs) are a heterogeneous group of inborn genetic errors that result in broad or narrow susceptibility to infections, predisposition to malignancy, or disorders of immune overactivation. Since the first descriptions of inherited immunodeficiency in the mid-20th century (Wiskott, 1937; Lutz, 1946; Kostmann, 1950; Bruton, 1952; Aldrich, Steinberg and Campbell, 1954), PIDs were branded as mendelian disorders. In the subsequent decades, the number of recognized PIDs has exploded, now exceeding over 400 unique entities (Tangye et al., 2020). Immense progress has been achieved in our understanding of the basic immunology and clinical pathogenesis surrounding these diseases. However, these advances have cast a shadow: although originally ascribed mendelian inheritance, these disorders often display imperfect segregation of gene mutations with disease traits.
Known as incomplete penetrance, this term can be broadly defined as any absence of clinical disease in individuals harboring a known disease-causing genotype. Precise terminology is crucial. Incomplete penetrance is synonymous with reduced penetrance. While penetrance measures the binary presence or absence of the disease trait, a genetic defect may also present with a range of disease severity or varying clinical phenotypes—a concept known as variable expressivity. For the purposes of this review, variable expressivity will be discussed under the umbrella of incomplete penetrance, as the origins can be largely overlapping. Lastly, fully-penetrant and monogenic are nonsynonymous, as either may occur without the other. Yet, both are often considered requirements for a mendelian trait.
Although incomplete penetrance is often encountered, the exact incidence is difficult to quantify. Most often, penetrance is assessed in family members of affected patients when the segregation of disease is traced from the proband. In such reports, rates of penetrance in specific PIDs range from as low as 5–10% (Yel, 2010; Kong et al., 2013) to nearly 100% (Vihinen et al., 1997; Candotti, 2017). Composite estimates across all PIDs indicate that ~9% families display some degree of incomplete penetrance (Stray-Pedersen et al., 2017). It should be noted several inherent biases likely exist that lead to underreporting of incomplete penetrance. These include a failure to pursue or publish variants with highly reduced penetrance (reporting bias), and an inability to detect asymptomatic individuals carrying mutations in the general population (ascertainment bias).
Because this topic is often overlooked or even avoided, more is unknown than known regarding incomplete penetrance. However, the study of PID is now at a crossroads where enough cases of incomplete penetrance exist that patterns are beginning to emerge. From this vantage point, the article herein will review incomplete penetrance across PIDs and put forth 4 existing principles of incomplete penetrance that create a conceptual framework from which to categorize incomplete penetrance in PID. Within each of these, what is known will be reviewed and followed by what is unknown with future testable hypotheses that attempt to advance our current understanding. These existing principles and new ideas will lay the groundwork for future studies specifically aimed at incomplete penetrance—a focus which currently does not exist. Although the content is specific to inborn errors of immunity, the concepts can easily be generalized to genetic disease broadly.
Principle I. The quality of genetic defect affects penetrance
Known
An important distinction first needs to be defined between cellular penetrance and clinical penetrance. Cellular penetrance refers to the extent of perturbation in biological pathways or processes relevant to the mutated gene product, often measured when cells are studied in isolation with a specific assay (e.g. the signal transduction pathway of a mutated receptor). The cellular phenotype is naturally a direct function of the severity of the genetic defect. Complete protein absence, for example, results in more serious biological aberrances than hypomorphic mutations. Further, penetrance at a clinical level often, but not always, tracks with cellular penetrance. Thus, a simple model can be put forth: severity of genetic defect determines molecular dysfunction, which governs the degree of perturbation in immune cells, and therefore the tendency for clinical manifestations.
There are many cases in which this simplification holds true. Most famously, this occurs with IFNGR1-deficiency, the first described specific genetic etiology of Mendelian Susceptibility to Mycobacterial Disease, either from environmental mycobacteria (EM) or bacille Calmette-Guerin (BCG) immunization (Jouanguy et al., 1996; Newport et al., 1996). Autosomal recessive (AR) IFNGR1 defects with complete signaling deficiencies invariably develop BCG or EM infections by age 5 years and the clinical penetrance is complete (Bustamante et al., 2014; Rosain et al., 2019). On the contrary, subjects with partial IFNGR1 deficiency, which are typically autosomal dominant (AD), often remain asymptomatic for longer periods of time, have milder disease, or in some cases never develop disease (Jouanguy et al., 1999; Dorman et al., 2004; Bustamante et al., 2014). It logically follows, and was proven experimentally, that these AD forms retain some activity when IFN- γ signaling is assayed in patient cells in vitro. (Jouanguy et al., 1996; Dorman et al., 2004).
A similar hierarchy is noted with STAT1 defects: AR complete deficiency with absent type I-III interferon signaling leads to complete penetrance of lethal intracellular bacterial and viral infections (Dupuis et al., 2003; Chapgier et al., 2006; Vairo et al., 2011); AR partial deficiency with reduced signaling leads to penetrant but milder intracellular bacterial disease (Chapgier et al., 2009; Kong et al., 2010; Kristensen et al., 2011); whereas AD forms with LoF point mutations retaining some function cause predominantly mycobacterial disease but with incomplete penetrance (Dupuis et al., 2001; Chapgier et al., 2006; Sampaio et al., 2012; Tsumura et al., 2012). While this demonstrates that partial deficiencies of essential genes can potentiate incomplete penetrance, so can complete deficiencies of non-essential genes of type I-III IFN signaling, which leave some signaling still intact. This can readily be observed in the deficiencies of STAT2, TYK2 and IFNAR2, with severe viral disease affecting 3–5/7, 8/10, and 1/2 individuals receptively (Minegishi et al., 2007; Hambleton et al., 2013; Duncan et al., 2015; Kreins et al., 2015; Moens et al., 2017; Taft and Bogunovic, 2018; Sarrafzadeh et al., 2019).
Beyond infectious susceptibility, allele-penetrance associations also exist in other PIDs, including mutations of STAT3 (Jägle et al., 2019) PRF1 (Feldmann et al., 2002; Molleran Lee et al., 2004), and AIRE (Oftedal et al., 2015). While the functional impact of each variation must be studied in isolation, at times, genetics alone can lead the way. For example, a recent cohort of congenital asplenia owing to RPSA mutations revealed marked incomplete penetrance, but no differences in functional prediction were apparent between incompletely penetrant and fully penetrant mutations. Yet, all missense mutations with incomplete penetrance were structurally located together, as were those with complete penetrance. Likewise, an RPSA mRNA noncoding structural defect resulted in incomplete penetrance, whereas a noncoding variant that lead to complete transcript decay conferred complete penetrance (Bolze et al., 2018). These findings suggest that milder hypomorphic variants likely retain some residual function, which in some individuals is sufficient for normal spleen development. These cases illustrate that, even when incompletely understood, the severity of the defect associates with penetrance.
Upon first approximation, autoimmune lymphoproliferative syndrome (ALPS) also fits this mold in that penetrance appears to be a function of the location of the FAS mutation, the most common cause of ALPS. Homozygous or compound heterozygous forms of ALPS-FAS are fully penetrant and especially severe, early in onset, and often lethal (Rieux-Laucat et al., 1995; Le Deist et al., 1996; Kasahara et al., 1998; van der Burg et al., 2000). Heterozygous mutations are less penetrant. Among these AD variants, an additional hierarchy exists: missense variants of the intracellular domain are more highly penetrant (63%−90%) as compared to those in the extracellular domain (30% – 52%). It follows that the dominant-negative mechanism of ICD mutations leads to more severe apoptosis defects that those of the ECD, which act by haploinsufficiency (Kuehn et al., 2011). Clearly, a relationship exists between the nature of the variant and disease probability. However, some defective Fas-mediated apoptosis can be identified in nearly 100% of individuals across all variants. Furthermore, some ‘asymptomatic’ individuals even exhibit lymphocyte expansions or autoantibody presence without clinical autoimmunity or true lymphoproliferation (Infante et al., 1998; Jackson et al., 1999; Bleesing et al., 2001). Defining the threshold after which these subclinical cellular defects cross into clinically-apparent disease will be key for better understanding this model.
Unknown
However, there are many instances in which there is no apparent association between the magnitude of the pathogenic variant and penetrance. In the most extreme case, this model fails to explain that complete genetic deficiencies (deletions, frameshifts, etc) can exhibit variable penetrance. This discrepancy is well captured in CTLA4 haploinsufficiency; despite large, intensively-studied cohorts, no associations between genotype and penetrance have been observed (Kuehn et al., 2014; Schubert et al., 2014; Schwab et al., 2018). For example, in a recent analysis of 133 subjects from 54 unrelated families carrying 45 different heterozygous CTLA4 mutations, only 90 exhibited disease features. Whether the pathogenic variant was missense, nonsense or frameshift had no apparent bearing on penetrance. Further, both unaffected and affected mutation carriers exhibit similar immunologic phenotyping and in vitro CTLA4 dysfunction, suggesting complete cellular penetrance (Schwab et al., 2018). However, cellular penetrance is fully dependent on the phenotype in question and the sensitivity of the assay implemented. Thus, if examined more closely, the loss of surface CTLA4 expression was, in fact, less severe in unaffected carriers (Schwab et al., 2018). This indicates that although the CTLA4 genotype cannot explain disease segregation, the degree of CTLA4 cellular perturbation still correlates with disease presentation. Along these lines, related deficiencies in T-cell regulation that represent more severe phenocopies of CTLA4-haploinsufficiency (LRBA deficiency, IPEX) demonstrate nearly complete penetrance (Lopez-Herrera et al., 2012; Gámez-Díaz et al., 2016). Therefore, additional disease modifiers must exist that influence the degree of T-reg dysfunction. Yet, their exact identity remains unknown, as neither secondary germline hits, somatic variation or reversion, viral exposure history nor HLA restriction can solely explain disease segregation (Schwab et al., 2018).
These cases and others (Fieschi et al., 2003; de Beaucoudrey et al., 2011; Prando et al., 2013), illustrate that the most severe genetic defects don’t always equate with propensity for disease, and therefore demand alternate hypotheses. Of note, it has recently become evident that, in fact, severe genetic mutations may lead to more robust compensatory responses (El-Brolosy et al., 2019; Ma et al., 2019). Transcriptional adaptation, the process by which frameshifts/nonsense mutations activate transcription of homologous genes, may rescue these complete deficiencies. In these instances, either by experimental knockout or disease mutations, nonsense-mediated decay triggers upregulation of sequence-similar genes that are predicted to have partially overlapping function (El-Brolosy et al., 2019; Ma et al., 2019). This phenomenon poses an enticing and testable hypothesis to explain incomplete penetrance in asymptomatic disease carriers of nonsense mutations. The ability for this “genomic compensation” to rescue disease phenotypes represents an important focus of future studies.
Principle II: Genetic and epigenetic modifiers can impact penetrance of a mutation
Known
Despite sparse evidence historically, one of the often-cited mechanisms for incomplete penetrance is the presence of potential modifier genes and epigenetic regulation. With next generation sequencing (NGS) becoming commonplace, and epigenetic techniques introduced into the study of PID, we are now beginning to substantiate these occurrences.
Common variable immunodeficiency (CVID), represents a great platform from which to examine incomplete penetrance. Monogenic mutations causing CVID have been identified but explain only a fraction of cases—in fact, when identified, these single mutations are reclassified as specific diagnoses and are instead deemed “CVID-like’ disorders. In some instances, epigenetic phenomena seem to predominate, as evidenced in a recent report of CVID-discordant monozygotic twins that show heightened DNA methylation in critical B-cell genes (PIK3CD, BCL2L1, RPS6KB2, TCF3 and KCNN4) (Rodríguez-Cortez et al., 2015). Likewise, a follow-up analysis on 23 CVID patients revealed defective demethylation of selected CpG sites during the transition from naïve to switched memory B cells (Del Pino-Molina et al., 2019). Although further concrete evidence is largely nonexistent, such epigenetic changes are particularly enticing given the close association of CVID and aging.
In other cases, especially when CVID-like disease is inherited (~10–20%), the etiology is often hypothesized to be polygenic. Gene variants, like those of TNFRSF13B, MSH5, BAFFR, are enriched in cohorts of CVID patients but also exist in healthy populations, so are insufficient to solely drive disease (Bogaert et al., 2016; de Valles-Ibáñez et al., 2018). Instead, this predisposition from any one gene is thought to be driven by epistatic interactions with another (i.e. the manifestation of a genotype depends on the genotype of another gene), only some of which we’ve begun to define. Massaad et al. first demonstrated this effect in homozygous NEIL3 mutations manifesting in one family as a uniformly fatal immune disease (recurrent infections and severe autoimmunity) but as a biologically-similar but clinically-silent immune dysfunction in an unrelated healthy individual. Genetic reanalysis uncovered a cryptic duplicated homozygous mutation in LRBA, defects of which are known to cause systemic autoimmunity, recurrent infections, and hypogammaglobulinemia. To further this “double-hit hypothesis,” the authors generated Neil3-deficient mice, which like humans, showed no overt signs of autoimmunity unless challenged with a second insult. However, genetic epistasis was not directly documented (Massaad et al., 2016).
Subsequently, epistasis was concretely evidenced in CVID by the discovery of a de novo Transcription Factor 3 (TCF3) mutation in a family already carrying a mutation of the CVID-associated TNFRSF13B gene. The proband with both mutations exhibited a severe CVID-like disorder and systemic lupus erythematosus. Family members with just the TNFRSF13B mutation displayed mild or absent disease and the proband’s son with just TCF3 mutation exhibited a partial clinical phenotype (Ameratunga et al., 2017). The true synergy of epistasis was evidenced for disease severity (by clinical scoring) and biological phenotype (by in vitro studies). It thus logically follows that the function of proteins encoded by these two genes converge on immunoglobulin class switching pathways. A similar convergence of alike pathways and putative epistasis has also been noted in many other conditions, including: ALPS with concurrent mutations in FAS and PRF1 (Clementi et al., 2004) or FAS and CASP10 (Cerutti et al., 2007); Hyperimmunoglobulinaemia D and periodic fever syndrome from MVK and TNFRSF1A mutation (Hoffmann et al., 2005); broad infectious susceptibility associated with IFNAR1 and IFNGR2 mutations (Hoyos-Bachiloglu et al., 2017); X-linked immunodeficiency caused by XIAP mutation and a CD40LG polymorphism (Rigaud et al., 2011); and in pediatric IBD in which known NOD2 mutation likely interacting with variants in GSDMB, ERAP2 or SEC16A (Christodoulou et al., 2013).
Interestingly in most of these cases, one of the two hits were previously reported as a causative variant in isolation. This poses an apparent paradox, because true epistasis requires a synergistic interaction of >2 genetic loci resulting in unmasking or increased severity of disease. Thus, one might suppose that these isolated cases (e.g. TCF3 (Boisson et al., 2013) and LRBA alone (Lopez-Herrera et al., 2012; Revel-Vilk et al., 2015; Gámez-Díaz et al., 2016)) were either milder disease forms or that these “isolated” cases actually contained an unknown modifier gene. For example, in a family of 4 with homozygous LoF LRBA mutations, the one unaffected sibling showed an intermediate apoptotic defect, suggesting that other genetic differences likely exist that regulate penetrance (Revel-Vilk et al., 2015).
Alternatively, rather than epistatic interactions triggering a single disease etiology, these combinatorial genetic defects may instead produce blended phenotypes, so called because of the overlapping clinical disease that results from the co-occurrence of two independent monogenic defects. Blended phenotypes are remarkably frequent across clinical genetics (~5% of rare disease diagnoses) (Yang et al., 2014; Driggers et al., 2016; Posey et al., 2016), and have recently been identified in unique immunodeficiencies caused by separate and distinct genetic defects (Chinn et al., 2017; Rae et al., 2017). Whether by blended phenotypes or epistasis, digenic inheritance is becoming a well-recognized determinant of expressivity and penetrance.
Unknown
The examples above point to combinatorial genetic hits, each of which are seemingly rare. However, what remains unknown is the role of common variation in the incomplete penetrance of rare disease. In regard to epistasis, it is plausible that the modifier gene could be a common variant that only plays a pathogenic role in combination with a rare mutation. This is likely to be the case in monogenic forms of autoimmunity (e.g. APS1, IPEX, CTLA4), in which relatively common autoimmunity-associated HLA alleles likely modify risk for specific autoantigens (Oftedal et al., 2015). More substantively, it has been suggested that X-linked variable immunodeficiency segregates with relatively common variations in CD40LG (Rigaud et al., 2011) and susceptibility to familial Mediterranean fever is modified by the interactions of MEFV mutations with polymorphisms in SAA1 (Migita et al., 2013). Although not a PID, non-syndromic midline craniosynostosis caused by rare SMAD6 mutations with common BMP2 variations remains the most well-substantiated example of this inheritance pattern and demonstrates that future studies in PID will require careful study of exceptionally large cohorts (Timberlake et al., 2016). With such large-scale studies, investigation of other nuanced phenomena becomes possible as well. In particular, mutational burden, in which the aggregate effect of many minor deleterious variants regulates disease risk, may prove relevant for PID, as it has for other types of rare disease (Cady et al., 2015; Girard et al., 2015; Guo et al., 2018).
These examples also suggest that the exact division between rare and common or monogenic and polygenic are unknown. For example, rare mutations leading to complete TYK2 deficiency result in monogenic susceptibility to TB and MSMD with relatively high penetrance (~80%) (Kreins et al., 2015; Boisson-Dupuis et al., 2018). Conversely, it was recently demonstrated that a common TYK2 variant (4.2% allele frequency in Europeans) predisposes to tuberculosis (OR 89.3) and MSMD (OR 23.5) at the homozygous state in endemic regions. The estimated penetrance was ~80% and 0.05% for TB and MSMD respectively (Boisson-Dupuis 2019, (Kerner et al., 2019). Although not considered a PID, these studies suggest that susceptibility to common infections can be caused by relatively frequent AR disorders in a proportion of patients. As more patients and healthy individuals are sequenced, with computational advances in tandem, many more such disorders on the borders of rare / common and monogenic / polygenic will likely be identified.
As we expand these NGS approaches to cover the whole genome, it is likely that we also uncover significant frequencies of noncoding variants with strong effects on characterized pathogenic mutations in the coding space. To date, less than 29 PIDs are associated with pathogenic variants in the noncoding genome—further, the bulk of these variants are located immediately proximal to exons (Telenti, 2019). In addition, the existence of compound heterozygosity in which a coding mutation and a noncoding cis regulatory variant cooperate to cause PIDs was recently documented (Thaventhiran et al., 2018). Whether these cases represent rare oddities or the tip of an iceberg remains unknown. With further exploration by whole genome sequencing, we are likely to not only identify causative variants in the regulatory regions, but also noncoding modifier alleles in cis and trans that change the expression of already pathologic mutations and regulate penetrance.
Akin to noncoding variation, copy number variations (CNVs) affecting critical immune genes could potentiate or curb the effects of deleterious alleles by gene dosage effects. Several global and site-specific CNVs have already been linked to PIDs (Green et al., 2011; Orange et al., 2011; Keller et al., 2014; Al-Mousa et al., 2016; Bradshaw et al., 2018). However, the impact of CNV on penetrance remains unexplored and will require tailored studies technically equipped to capture large structural variation.
Lastly, we postulate that protective variants—coding and noncoding—also exist that are capable of rescuing aberrant biology in cis and in trans. It is easy to conceive that common variants in proteins that interact with the mutated proteins may have significantly more or less functional capability and can therefore rescue the deficient function of the defective gene product. However, given the complexity of interactions, these discoveries will likely prove especially evasive.
Principle III. Defined environmental exposures can shape manifestations of immune defects.
Known
Differences in environment are frequently offered as explanations for phenotypic discrepancies. Their net effect, now labeled the “exposome,” encompasses many factors relevant to the immune system including infections, resident microbes, diet/metabolism, and radiation. In fact, many of these insults are sufficient to trigger a secondary immunodeficiency in previously healthy individuals (Chinen and Shearer, 2010). Despite these robust effects, which are burgeoning disciplines in the broader field of immunology, only few well-substantiated examples for environmental modifiers in PIDs exist to date.
These factors are most readily appreciable in the case of susceptibility to infection, as exposure to infectious agents can vary greatly. In the simplest case, individuals harboring mutations that confer susceptibility to specific pathogens do not present if never exposed to that microbe. This is most readily appreciated in individuals with mutations linked to BCG-disease that did not receive BCG vaccine (Zhang et al., 2015).
Yet, variable microbial exposures not only influence primary infections but also subsequently shape adaptive responses. At least for invasive pneumococcal disease from IRAK4- and MyD88-deficiencies, age and the immunity built with it are known to be major determinants of disease. Penetrance is highest by age 10, but IPD recurrence and mortality subsequently falls with age—presumably from acquired anti-pneumococcal immunity (Picard et al., 2010). This example raises the possibility that environmental exposures, which are thought to incite clinical presentations, can also protect. The best demonstration of this phenomenon exists in the incomplete penetrance seen in deficiency of TIRAP, a critical adapter in TLR-based sensing. Despite this complete innate immune defect, only 1/8 TIRAP-deficient homozygotes exhibited staphylococcal disease. In the other 7, acquired anti-LTA (staphylococcal lipoteichoic acid (LTA) Abs) rescued TLR-dependent susceptibility to staphylococcus (Israel et al., 2017). This remains one of the most clearly substantiated demonstrations of environmental factors that explain disease segregation.
Infection may also trigger immune dysregulation beyond acute infection, as we now understand that pathogens are often the inciting event to autoimmune and autoinflammatory disorders. This notion is well captured in familial haemophagocytic lymphohistiocytosis (HLH), a previously fatal disease characterized by excessive macrophage and lymphocyte activity. When identified early in life, individuals with disease-causing mutations exhibit the hallmark cellular dysfunction prior to the development of clinical disease (Feldmann et al., 2002). Further, upper respiratory or gastrointestinal infections tend to be present around the onset of HLH (Sung et al., 2001). This sequence suggests that a known, and likely nonspecific infectious trigger is required for presentation. Such factors may account for variable disease presentation when mutation-carrying individuals are compared at a single point in time.
Other forms of environmental influence can also modulate penetrance in PID. LIG4-mutated individuals, who suffer lymphocyte deficiencies and nonimmune features due to DNA repair defects, are often clinically unremarkable until treated with chemotherapy and radiotherapy. Therefore, asymptomatic carriers of LIG4 mutations, may have yet to receive sufficient double-strand break insults to cross the threshold of disease (Felgentreff et al., 2016). As these events are difficult to pinpoint in human patients, animal models have begun to interrogate these matters experimentally. In addition to the above example in Neil3-deficient mice (Massaad et al., 2016), families with Schimke immune-osseus dysplasia (SIOD) display reduced penetrance that is insufficiently explained by their biallelic mutations in SMARCAL1, a conserved chromatin regulator (Bökenkamp et al., 2005; Dekel et al., 2008; Elizonod et al., 2009). Drosophila and murine models for SMARCAL1 deficiency, which recapitulate the chromatin and transcriptional aberrances of SIOD, suggest that an additional environmental (heat-shock or pharmacologic) or genetic insult to transcription is required for disease manifestation (Baradaran-Heravi et al., 2012).
Unknown
Immunization both provides answers and poses mysteries to the role of environmental exposures in variable penetrance. BCG vaccination for instance, represent an extremely well-controlled “experiment” of penetrance in which all individuals receive an identical pathogen at a similar age. Yet, we still observe incomplete penetrance for MSMD, as in IL12RB1 deficiency (~70% penetrance), suggesting that simple environmental differences cannot fully explain penetrance (Bustamante et al., 2014). Likewise, some PID-specific pathogens are nearly ubiquitous, as in herpes simplex encephalitis (HSE), a sporadic disease with known monogenic etiologies (Abel et al., 2010; Bradley et al., 2014). Despite nearly complete cellular defects in TLR3-dependent IFN immunity, 4/6 TRIF-deficient, 2/3 UNC-93B-deficient, 3/8 TLR3-deficient, 3/4 IRF3-mutant, and 2/3 TBK1-hypomorphic reported individuals have developed HSE (Casrouge et al., 2006; Zhang et al., 2007; Guo et al., 2011; Sancho-Shimizu et al., 2011; Herman et al., 2012; Lim et al., 2014; Andersen et al., 2015; Mørk et al., 2015).
In these cases, incomplete penetrance may instead be a function of other factors, including age at exposure. In support of this, HSE patients are most often young and recurrence is rare (Abel et al., 2010). In cases with repeated HSE in childhood, patients have survived to adulthood and no longer develop HSE episodes (Whitley and Kimberlin, 2005; Zhang et al., 2007; Lim et al., 2014). This suggests that prior exposures may effectively immunize and regulate disease penetrance. We speculate that asymptomatic mutation carriers may have previously received non-infectious or exceedingly low-quantity exposures to HSV1 that are insufficient for productive infection but capable of inducing adaptive immune responses that potently neutralize future challenges that are truly infectious. A similar effect may occur with IL12RB1 deficiency, as patients that acquire BCG disease tend to be mutually exclusive with patients with environmental mycobacteriosis, suggesting that one may immunize against the other (Fieschi et al., 2003; de Beaucoudrey et al., 2011). The same mechanisms could apply to other infectious susceptibilities, both broad and narrow, but of course will require experimental evidence.
Yet, perhaps the most important interactions with microbial species occur with those bacteria, fungi and viruses that naturally colonize our tissues. The microbiome represents our first and most abundant exposure to microbial organisms. It is therefore unsurprising that in the last decade the microbiome has proven to be a major determinant of immune function and disease (Belkaid, 2015; Gilbert et al., 2018). Despite these strong associations, the relevance of the microbiome in PIDs, the most extreme immune pathologies, is yet unknown. Recent studies demonstrating altered bacterial microbiota in CVID, which correlate with immune activation (Jørgensen et al., 2016; Fiedorová et al., 2019) have begun to scratch the surface, but the direction of causality remains unclear. We hypothesize that specific microbiota regulates the penetrance of PIDs by shaping the relative tolerance and reactivity of the innate or adaptive immune system. The divergence of the microbiome with respect to geography and diet may underlie PID phenotypes that vary across populations with similar monogenic lesions. However future studies that include microbial sequencing in PID cohorts will be required moving forward.
Lastly, it should also be noted that the environment of modern times differs markedly from that which our ancestral immune systems evolved. Just a century ago, when mortality from infectious causes was 200-fold more frequent, up to one-third of children died before the age of five (Roser, Ritchie and Dadonaite, 2019). It is likely that a study on the genetics of infectious disease a century ago, with the tools of today, would have identified far more common alleles as causative. Yet in modern times, these genetic susceptibilities are likely masked by sanitation, vaccination and antibiotic usage. Close examination for these potential alleles in isolated systems may be informative and contribute to our understanding of incomplete penetrance. On the other side of the coin though, our recent use of immunosuppressants in the clinic may draw out new and old genetic susceptibilities with surprising frequency and pathogen specificity.
Principle IV. Mosaicism of disease-causing alleles reduces clinical penetrance.
Known
The above discussions assume that all cells in affected individuals carry the same mutation, but we now understand that cells differ genetically with surprising frequency within a single individual. Genetic mosaicism originates from post-zygotic (de novo) mutations that arise during the embryonic or postnatal period. At first believed to be a rare occurrence in PIDs, somatic mutation is now understood to be rather common. In fact, a recent systematic analysis across PIDs using targeted deep sequencing in 128 families estimated mosaicism to be 23.4% (Mensa-Vilaró et al., 2019).
Disease occurrence, onset or severity are often less intense in cases of mosaicism, as a direct consequence of gene dosage. Reduced penetrance in mosaic PID was first documented in an extraordinary case followed through the 1980s and 1990s of delayed-onset ADA-deficiency, which is typically a severe form of SCID. ADA mosaicism was directly observed in peripheral blood cells, and overtime, ADA-normal populations overtook as clinical disease resolved (Uberti et al., 1983; Arredondo-Vega et al., 1990; Hirschhorn et al., 1994). After this discovery, several other documentations of mosaicism followed (Puck et al., 1995; O’Marcaigh et al., 1997), with many presenting as mild or atypical disease phenotypes, including mutations in NLRP3 (de Koning et al., 2015; Rowczenio et al., 2017), STAT3 (Hsu et al., 2013; Walker et al., 2016), FAS (Holzelova, Vonarbourg, M.-C. Stolzenberg, et al., 2004; Dowdell et al., 2010), CYBB (Wolach et al., 2005), and TNFAIP3 (Kadowaki et al., 2018). Curiously, these appear to be predominantly disorders of immune hyperactivation rather than deficiency.
As these cases accumulate, the evidence for mosaicism as a mechanism for reduced penetrance has strengthened. In a recent systematic analysis of 10 families where one member carried a post-zygotic mutation for a PID gene, 80% of mosaic individuals were asymptomatic. The remaining mosaic individuals exhibited only partial clinical disease, whereas their progeny with germline mutation status demonstrated complete disease (Mensa-Vilaró et al., 2019). Likewise, examination of variant read frequencies in a family with PIK3CD mutations revealed that affected siblings harbor more mutant cells than their mildly-affected father, with allele fractions of 37%−54% and 15%, respectively (Stray-Pedersen et al., 2017). However, when mutant cells predominate in the relevant cell compartment, differences between germline and somatic cases fade. For example, ALPS patients harboring FAS mutations in ~100% of their double-negative T-cells (the pathologic cell-type of ALPS) demonstrate complete disease despite undetectable levels of mutation in whole blood. (Holzelova, Vonarbourg, M. C. Stolzenberg, et al., 2004; Dowdell et al., 2010).
Somatic mutations may also create “second hits” that allow an otherwise clinically-silent disease to manifest. For example, ALPS patients have been documented to carry both an inherited heterozygous FAS mutation and a somatic event in the second FAS allele, including missense mutations, nonsense mutations or loss of heterozygosity (Magerus-Chatinet et al., 2011; Neven et al., 2011; Hauck et al., 2013). Alternatively, the second hit may be at a different locus, as in a recent report of a somatic FAS mutation complexed with an existing CASP10 mutation (Martínez-Feito et al., 2016). In all cases, relatives who never developed a second mutation post-zygotically remained asymptomatic or partially affected, substantiating the importance of ‘second hit’ mosaicism on incomplete penetrance.
Yet, somatic mutation can also rescue disease rather than cause it. Several cases have been documented in which somatic reversions underlie mild or absent clinical disease that appears as variable expressivity or incomplete penetrance. For instance, reversion in DOCK-8 deficiency, which occurs in roughly half of affected patients, associates with longer survival and less severe allergic disease, albeit similar infectious susceptibility (Jing et al., 2014). Several other examples exist of reversions underlying incompletely penetrant clinical disease, including in ADA (Ariga, Oda, et al., 2001), XLA (Stephan et al., 1996; Speckmann et al., 2008), WASP (Ariga, Kondoh, et al., 2001; Boztug et al., 2007), leukocyte adhesion deficiency (Tone et al., 2007), X-linked immunodeficiency with ectodermal dysplasia due to mutations in NEMO Nishikomori 2004, Omenn syndrome with CARD11 deficiency (Fuchs 2015), and IKBKG-associated immunodeficiency (Stray-Pedersen et al., 2017). Interestingly, these reversions may even be second-site mutations of the mutated gene that create altered non-WT, but still functional, gene products (Boztug et al., 2008).
Although most mosaic PIDs appear stable with time (Rowczenio et al., 2017; Mensa-Vilaró et al., 2019), somatic reversions that bestow a fitness advantage allow reverted cells to selectively expand and reestablish healthy immune cell populations. For instance, reversions of mutations in JAK3, an essential mediator of lymphocyte development, can repair immune cell proliferation and differentiation. In one family with JAK3 hypomorphic mutations, the asymptomatic sibling showed CD4+ T-Cell reversion, whereas a brother without this reversion suffered recurrent respiratory tract infections (Ban et al., 2014). In an extreme case, McDermott and colleagues reported a WHIM patient cured by a process known as chromothripsis, or “chromosome shattering,” in which chromosomes undergo massive deletion and rearrangement. Fortuitously, this event deleted the mutated CXCR4 allele in a single hematopoietic stem cell, which then took over the bone marrow and reconstituted immune function (McDermott et al., 2015). However, if the selective pressure is removed, as in the case of enzyme replacement therapy in ADA-deficiency with reversions or allogeneic stem cell therapy, the WT cells seemingly lose their selective advantage and proportionally decline (Ariga, Kondoh, et al., 2001).
Unknown
Despite strong associations between mosaicism and mild disease, there are cases in which mosaic mutations lead to severe disease (Niemela et al., 2011; Takagi et al., 2011; Shiota et al., 2015; Walker et al., 2016; Ma et al., 2017; Gruber et al., 2019). Of note, some of these cases are the first and only reports of patients with such mutations, suggesting that these defects may be embryonically/perinatally lethal at germline status. Although these instances may be the exception that proves the rule of mosaicism in incomplete penetrance, it fails to explain many others (Del Bel et al., 2017; Gruber et al., 2019; Mensa-Vilaró et al., 2019). Evolving technologies and expanded cohorts will be central to answering the remaining questions. As Sanger sequencing previously failed to detect many low-frequency somatic variants, standard WES and WGS also suffer inherent limits of detection. This is not simply a function of total mosaic fractions, but also of the tissue being assayed. Many somatic mutations have been documented to be only detectable in specific immune cell types (Dowdell et al., 2010; de Koning et al., 2015; Walker et al., 2016), and it is likely that many more such cell-type specific mutations exist, including extra-haematopoietically.
Yet beyond the genotype, it has recently become clear that mosaicism can also exist at the transcript level across genetically identical cells (Reinius and Sandberg, 2015). Similar to X-inactivated genes, autosomal genes can be expressed from a single allele in a random fashion across cells from one individual. This restriction to one allele can occur dynamically, as in transcriptional bursting (Deng et al., 2014; Reinius et al., 2016), or remain fixed over time in what is designated as monoallelic expression (MAE) (Gimelbrant et al., 2007; Jeffries et al., 2012; Borel et al., 2015; Reinius and Sandberg, 2015). Remarkably, up to 10% of the autosomal genome exhibits this phenomenon (Gimelbrant et al., 2007). For these genes, allelic bias is established in lineage differentiation by a unique chromatin signature and persists with subsequent cell division (Nag et al., 2013; Gendrel et al., 2014).
Although we increasingly understand the nature of this epigenetic phenomenon, we have yet to grasp the functional consequences, especially in light of genetic disease. Heterozygous mutations occurring in MAE genes will create a mixture of both WT- and mutant-expressing cells with divergent phenotypes in affected individuals. We hypothesize that, by creating this mosaic “transcriptotype”, MAE can modulate the functional impact of disease-causing mutations. Because the proportions of cells expressing one allele or another vary at random (Gimelbrant et al., 2007; Jeffries et al., 2012; Borel et al., 2015), MAE may help explain phenotypic variation in genetic disease. For AR disease, this phenomenon will present in affected carriers, whereas in AD disease, mosaicism will reduce penetrance of disease phenotypes in patients.
While this hypothesis remains unsubstantiated, supporting evidence has begun to accumulate. Computational predictions suggest an enrichment of MAE in genes for which gain-of-function variants with AD inheritance are linked to neuropsychiatric disease (Savova et al., 2017). Experimentally, disease-related genes have been demonstrated to undergo MAE (Adegbola et al., 2015) and, recently, the first gene mutation with allelic bias was documented (Gruber et al., 2019). Clearly, future studies aimed at testing MAE in PID genes are warranted, which will require careful and intensive experimentation to compare expression across single cells. As most single cell technologies currently lack sufficient depth and breadth of transcript coverage for meaningful study, and clonal systems are biased, significant technological advance will be required. Nevertheless, if certain PID genes undergo MAE, our classical notion of heterozygosity would need to be redefined.
Conclusions
Failing to understand penetrance in PID has hindered our advance in human genetics. By amassing the cases of variable penetrance in PID, this review aimed to illuminate where connections lie, and gaps persist. It is clear that four major influences continually reduce penetrance (partial genetic defects, (epi)genetic modifiers, environmental influences and mosaicism), while others remain unexplored (genomic compensation, protective variants, sub-infectious inoculations, monoallelic expression) (Figure 1). Although discussed separately, these driving principles likely work in tandem and interact. It should be noted that the biggest breakthroughs in these domains have come not only from reports of large cohorts, but also intense study of single patients. Thus, furthering our understanding of penetrance will require studies of comprehensive depth and breadth.
Figure 1. Four principles of incomplete penetrance in primary immunodeficiency.
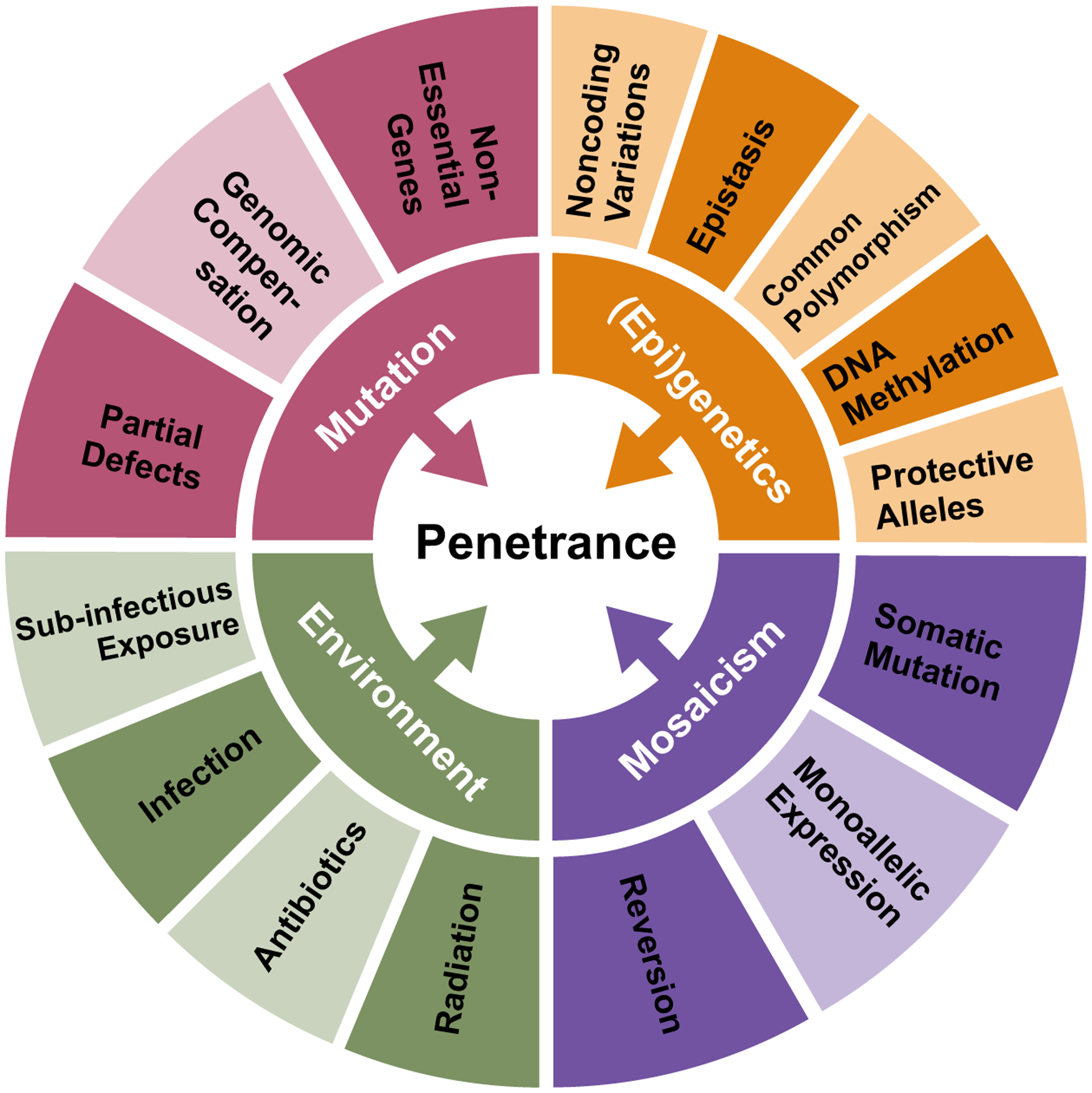
Inner circle represents the four broad principles of incomplete penetrance, with the outer circle denoting specific processes that are established (dark) or hypothesized (light) to contribute.
Acknowledgements:
This research was supported by National Institute of Allergy and Infectious Diseases Grants R01AI127372, R21 AI134366 and R21AI129827, and funding from the March of Dimes, awarded to DB. CG was supported by T32 training grant 5T32HD075735-07.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest Statement
On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- Abel L et al. (2010) ‘Age-dependent mendelian predisposition to herpes simplex virus type 1 encephalitis in childhood’, Journal of Pediatrics, 157(4). doi: 10.1016/j.jpeds.2010.04.020. [DOI] [PubMed] [Google Scholar]
- Adegbola AA et al. (2015) ‘Monoallelic expression of the human FOXP2 speech gene’, Proceedings of the National Academy of Sciences, 112(22), pp. 6848–6854. doi: 10.1073/pnas.1411270111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mousa H et al. (2016) ‘Unbiased targeted next-generation sequencing molecular approach for primary immunodeficiency diseases’, Journal of Allergy and Clinical Immunology. Elsevier Ltd, 137(6), pp. 1780–1787. doi: 10.1016/j.jaci.2015.12.1310. [DOI] [PubMed] [Google Scholar]
- Aldrich RA, Steinberg AG and Campbell DC (1954) ‘Pedigree demonstrating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis and bloody diarrhea.’, Pediatrics, 13(2), pp. 133–9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/13133561. [PubMed] [Google Scholar]
- Ameratunga R et al. (2017) ‘Epistatic interactions between mutations of TACI (TNFRSF13B) and TCF3 result in a severe primary immunodeficiency disorder and systemic lupus erythematosus’, Clinical & Translational Immunology. Nature Publishing Group, 6(10), p. e159. doi: 10.1038/cti.2017.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen LL et al. (2015) ‘Functional IRF3 deficiency in a patient with herpes simplex encephalitis’, Journal of Experimental Medicine, 212(9), pp. 1371–1379. doi: 10.1084/jem.20142274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariga T, Kondoh T, et al. (2001) ‘Spontaneous In Vivo Reversion of an Inherited Mutation in the Wiskott-Aldrich Syndrome’, The Journal of Immunology, 166(8), pp. 5245–5249. doi: 10.4049/jimmunol.166.8.5245. [DOI] [PubMed] [Google Scholar]
- Ariga T, Oda N, et al. (2001) ‘T-cell lines from 2 patients with adenosine deaminase (ADA) deficiency showed the restoration of ADA activity resulted from the reversion of an inherited mutation’, Blood, 97(9), pp. 2896–2899. doi: 10.1182/blood.V97.9.2896. [DOI] [PubMed] [Google Scholar]
- Arredondo-Vega FX et al. (1990) ‘Paradoxical expression of adenosine deaminase in T cells cultured from a patient with adenosine deaminase deficiency and combined immunodeficiency’, Journal of Clinical Investigation, 86(2), pp. 444–452. doi: 10.1172/JCI114730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban SA et al. (2014) ‘Combined Immunodeficiency Evolving into Predominant CD4+ Lymphopenia Caused by Somatic Chimerism in JAK3’, Journal of Clinical Immunology, 34(8), pp. 941–953. doi: 10.1007/s10875-014-0088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baradaran-Heravi A et al. (2012) ‘Penetrance of biallelic SMARCAL1 mutations is associated with environmental and genetic disturbances of gene expression’, Human Molecular Genetics, 21(11), pp. 2572–2587. doi: 10.1093/hmg/dds083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Beaucoudrey L et al. (2011) ‘Revisiting Human IL-12Rbeta1 Deficiency: A Survey of 141 Patients From 30 Countries’, Medicine, 89(5), pp. 381–402. doi: 10.1097/MD.0b013e3181fdd832.Revisiting. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bel KL et al. (2017) ‘JAK1 gain-of-function causes an autosomal dominant immune dysregulatory and hypereosinophilic syndrome’, Journal of Allergy and Clinical Immunology, 1(June). doi: 10.1016/j.jaci.2016.12.957. [DOI] [PubMed] [Google Scholar]
- Belkaid Y and H. T (2015) ‘Role of the Microbiota in Immunity and inflammation Yasmine’, Cell, 157(1), pp. 121–141. doi: 10.1016/j.cell.2014.03.011.Role. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleesing JJH et al. (2001) ‘Immunophenotypic profiles in families with autoimmune lymphoproliferative syndrome’, Blood, 98(8), pp. 2466–2473. doi: 10.1182/blood.V98.8.2466. [DOI] [PubMed] [Google Scholar]
- Bogaert DJA et al. (2016) ‘Genes associated with common variable immunodeficiency: One diagnosis to rule them all?’, Journal of Medical Genetics, 53(9), pp. 575–590. doi: 10.1136/jmedgenet-2015-103690. [DOI] [PubMed] [Google Scholar]
- Boisson-Dupuis S et al. (2018) ‘Tuberculosis and impaired IL-23–dependent IFN-γ immunity in humans homozygous for a common TYK2 missense variant’, Science Immunology, 3(30), p. eaau8714. doi: 10.1126/sciimmunol.aau8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson B et al. (2013) ‘A recurrent dominant negative E47 mutation causes agammaglobulinemia and BCR- B cells’, Journal of Clinical Investigation, 123(11), pp. 4781–4785. doi: 10.1172/JCI71927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bökenkamp A et al. (2005) ‘R561C missense mutation in the SMARCAL1 gene associated with mild Schimke immuno-osseous dysplasia’, Pediatric Nephrology, 20(12), pp. 1724–1728. doi: 10.1007/s00467-005-2047-x. [DOI] [PubMed] [Google Scholar]
- Bolze A et al. (2018) ‘Incomplete penetrance for isolated congenital asplenia in humans with mutations in translated and untranslated RPSA exons’, Proceedings of the National Academy of Sciences of the United States of America, 115(34), pp. E8007–E8016. doi: 10.1073/pnas.1805437115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borel C et al. (2015) ‘Biased allelic expression in human primary fibroblast single cells’, American Journal of Human Genetics. The American Society of Human Genetics, 96(1), pp. 70–80. doi: 10.1016/j.ajhg.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boztug K et al. (2007) ‘Large granular lymphocyte proliferation and revertant mosaicism: two rare events in a Wiskott-Aldrich syndrome patient.’, Haematologica, 92(3), pp. 43–45. doi: 10.3324/haematol.11222. [DOI] [PubMed] [Google Scholar]
- Boztug K et al. (2008) ‘Multiple independent second-site mutations in two siblings with somatic mosaicism for Wiskott-Aldrich syndrome’, Clinical Genetics, 74(1), pp. 68–74. doi: 10.1111/j.1399-0004.2008.01019.x. [DOI] [PubMed] [Google Scholar]
- Bradley H et al. (2014) ‘Seroprevalence of herpes simplex virus types 1 and 2-United States, 1999–2010’, Journal of Infectious Diseases, 209(3), pp. 325–333. doi: 10.1093/infdis/jit458. [DOI] [PubMed] [Google Scholar]
- Bradshaw G et al. (2018) ‘Exome sequencing diagnoses X-linked moesin-associated immunodeficiency in a primary immunodeficiency case’, Frontiers in Immunology, 9(March). doi: 10.3389/fimmu.2018.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruton O (1952) ‘Agammaglobulinemia’, Pediatrics, 9(6). [PubMed] [Google Scholar]
- van der Burg M et al. (2000) ‘Autoimmune Lymphoproliferative Syndrome (ALPS) in a Child from Consanguineous Parents: A Dominant or Recessive Disease?’, Pediatric Research, 47(3), pp. 336–343. doi: 10.1203/00006450-200003000-00009. [DOI] [PubMed] [Google Scholar]
- Bustamante J et al. (2014) ‘Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity’, Semin Immunol, 26(6), pp. 454–470. doi: 10.1016/j.smim.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady J et al. (2015) ‘Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes’, Annals of Neurology, 77(1), pp. 100–113. doi: 10.1002/ana.24306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candotti F (2017) ‘Clinical Manifestations and Pathophysiological Mechanisms of the Wiskott-Aldrich Syndrom’, Journal of Clinical Immunology, 38(1). [DOI] [PubMed] [Google Scholar]
- Casrouge A et al. (2006) ‘Herpes simplex virus encephalitis in human UNC-93B deficiency’, Science, 314(5797), pp. 308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- Cerutti E et al. (2007) ‘Co-inherited mutations of Fas and caspase-10 in development of the autoimmune lymphoproliferative syndrome’, BMC Immunology, 8, pp. 1–9. doi: 10.1186/1471-2172-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapgier A et al. (2006) ‘Human Complete Stat-1 Deficiency Is Associated with Defective Type I and II IFN Responses In Vitro but Immunity to Some Low Virulence Viruses In Vivo’, The Journal of Immunology, 176(8), pp. 5078–5083. doi: 10.4049/jimmunol.176.8.5078. [DOI] [PubMed] [Google Scholar]
- Chapgier A et al. (2009) ‘A partial form of recessive STAT1 deficiency in humans’, Journal of Clinical Investigation, 119(6), pp. 1502–1514. doi: 10.1172/JCI37083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinen J and Shearer WT (2010) ‘Secondary immunodeficiencies, including HIV infection’, Journal of Allergy and Clinical Immunology. Elsevier Ltd, 125(2 SUPPL. 2), pp. S195–S203. doi: 10.1016/j.jaci.2009.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinn IK et al. (2017) ‘Novel combined immune deficiency and radiation sensitivity blended phenotype in an adult with biallelic variations in ZAP70 and RNF168’, Frontiers in Immunology, 8(MAY). doi: 10.3389/fimmu.2017.00576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christodoulou K et al. (2013) ‘Next generation exome sequencing of paediatric inflammatory bowel disease patients identifies rare and novel variants in candidate genes’, Gut, 62(7), pp. 977–984. doi: 10.1136/gutjnl-2011-301833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementi R et al. (2004) ‘Inherited Perforin and Fas Mutations in a Patient with Autoimmune Lymphoproliferative Syndrome and Lymphoma’, New England Journal of Medicine, 351(14), pp. 1419–1424. doi: 10.1056/NEJMoa041432. [DOI] [PubMed] [Google Scholar]
- Le Deist F et al. (1996) ‘Clinical, immunological, and pathological consequences of Fas-deficient conditions’, The Lancet, 348(9029), pp. 719–723. doi: 10.1016/S0140-6736(96)02293-3. [DOI] [PubMed] [Google Scholar]
- Dekel B et al. (2008) ‘Schimke immuno-osseous dysplasia: Expression of SMARCAL1 in blood and kidney provides novel insight into disease phenotype’, Pediatric Research, 63(4), pp. 398–403. doi: 10.1203/PDR.0b013e31816721cc. [DOI] [PubMed] [Google Scholar]
- Deng Q et al. (2014) ‘Single-Cell RNA-Seq Reveals Dynamic, Random Monoallelic Gene Expression in Mammalian Cells’, 343(January). doi: 10.1126/science.1245316. [DOI] [PubMed] [Google Scholar]
- Dorman SE et al. (2004) ‘Clinical features of dominant and recessive interferon γ receptor 1 deficiencies’, Lancet, 364(9451), pp. 2113–2121. doi: 10.1016/S0140-6736(04)17552-1. [DOI] [PubMed] [Google Scholar]
- Dowdell KC et al. (2010) ‘Somatic FAS mutations are common in patients with genetically undefined autoimmune lymphoproliferative syndrome’, Blood, 115(25), pp. 5164–5169. doi: 10.1182/blood-2010-01-263145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driggers RW et al. (2016) ‘Zika Virus Infection with Prolonged Maternal Viremia and Fetal Brain Abnormalities.’, The New England journal of medicine, p. NEJMoa1601824. doi: 10.1056/NEJMoa1601824. [DOI] [PubMed] [Google Scholar]
- Duncan CJA et al. (2015) ‘Human IFNAR2 deficiency: lessons for antiviral immunity Europe PMC Funders Group’, Sci Transl Med, 7(307), pp. 307–154. doi: 10.1126/scitranslmed.aac4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis S et al. (2001) ‘Impairment of Mycobacterial But Not Viral Immunity by a Germline Human STAT1 Mutation’, Science, 293(5528), pp. 300–303. doi: 10.1126/science.1061154. [DOI] [PubMed] [Google Scholar]
- Dupuis S et al. (2003) ‘Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency’, Nature Genetics, 33(3), pp. 388–391. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- El-Brolosy M et al. (2019) ‘Genetic compensation is triggered by mutant mRNA degradation’, Nature, p. 328153. doi: 10.1101/328153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elizonod LI et al. (2009) ‘Schimke immuno-osseous dysplasia: SMARCAL1 loss-of-function and phenotypic correlation’, Journal of Medical Genetics, 46(1), pp. 49–59. doi: 10.1136/jmg.2008.060095. [DOI] [PubMed] [Google Scholar]
- Feldmann J et al. (2002) ‘Functional consequences of perforin gene mutations in 22 patients with familial haemophagocytic lymphohistiocytosis’, British Journal of Haematology, 117(4), pp. 965–972. doi: 10.1046/j.1365-2141.2002.03534.x. [DOI] [PubMed] [Google Scholar]
- Felgentreff K et al. (2016) ‘Ligase-4 Deficiency Causes Distinctive Immune Abnormalities in Asymptomatic Individuals’, Journal of Clinical Immunology. Journal of Clinical Immunology, 36(4), pp. 341–353. doi: 10.1007/s10875-016-0266-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedorová K et al. (2019) ‘Bacterial but not fungal gut microbiota alterations are associated with common variable immunodeficiency (CVID) phenotype’, Frontiers in Immunology, 10(August). doi: 10.3389/fimmu.2019.01914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieschi C et al. (2003) ‘Low penetrance, broad resistance, and favorable outcome of interleukin 12 receptor β1 deficiency: Medical and immunological implications’, Journal of Experimental Medicine, 197(4), pp. 527–535. doi: 10.1084/jem.20021769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gámez-Díaz L et al. (2016) ‘The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency’, Journal of Allergy and Clinical Immunology, 137(1), pp. 223–230. doi: 10.1016/j.jaci.2015.09.025. [DOI] [PubMed] [Google Scholar]
- Gendrel AV et al. (2014) ‘Developmental dynamics and disease potential of random monoallelic gene expression’, Developmental Cell. Elsevier Inc., 28(4), pp. 366–380. doi: 10.1016/j.devcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- Gilbert JA et al. (2018) ‘Current understanding of the human microbiome’, Nature Medicine, 24(4), pp. 392–400. doi: 10.1038/nm.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimelbrant A et al. (2007) ‘Widespread monoallelic expression on human autosomes’, Science, 318(5853), pp. 1136–1140. doi: 10.1126/science.1148910. [DOI] [PubMed] [Google Scholar]
- Girard SL et al. (2015) ‘Mutation burden of rare variants in schizophrenia candidate genes’, PLoS ONE, 10(6), pp. 1–11. doi: 10.1371/journal.pone.0128988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MR et al. (2011) ‘A novel immunodeficiency disorder characterized by genetic amplification of interleukin 25’, Genes and Immunity. Nature Publishing Group, 12(8), pp. 663–666. doi: 10.1038/gene.2011.50. [DOI] [PubMed] [Google Scholar]
- Gruber C et al. (2019) ‘Complex Autoinflammatory Syndrome Unveils Fundamental Principles of JAK1 Transcriptional and Biochemical Function’, bioRxiv, p. 807669. doi: 10.1101/807669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo MH et al. (2018) ‘Burden Testing of Rare Variants Identified through Exome Sequencing via Publicly Available Control Data’, American Journal of Human Genetics. ElsevierCompany, 103(4), pp. 522–534. doi: 10.1016/j.ajhg.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y et al. (2011) ‘Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity’, Journal of Experimental Medicine, 208(10), pp. 2083–2098. doi: 10.1084/jem.20101568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambleton S et al. (2013) ‘STAT2 deficiency and susceptibility to viral illness in humans’, Proceedings of the National Academy of Sciences, 110(8), pp. 3053–3058. doi: 10.1073/pnas.1220098110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauck F et al. (2013) ‘Somatic loss of heterozygosity, but not haploinsufficiency alone, leads to full-blown autoimmune lymphoproliferative syndrome in 1 of 12 family members with FAS start codon mutation’, Clinical Immunology. Elsevier Inc., 147(1), pp. 61–68. doi: 10.1016/j.clim.2013.02.019. [DOI] [PubMed] [Google Scholar]
- Herman M et al. (2012) ‘Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood’, Journal of Experimental Medicine, 209(9), pp. 1567–1582. doi: 10.1084/jem.20111316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn R et al. (1994) ‘Somatic mosaicism for a newly identified splice-site mutation in a patient with adenosine deaminase-deficient immunodeficiency and spontaneous clinical recovery’, American Journal of Human Genetics, 55(1), pp. 59–68. [PMC free article] [PubMed] [Google Scholar]
- Hoffmann F et al. (2005) ‘Identification of a novel mevalonate kinase gene mutation in combination with the common MVK V3771 substitution and the low-penetrance TNFRSF1A R92Q mutation’, European Journal of Human Genetics, 13(4), pp. 510–512. doi: 10.1038/sj.ejhg.5201352. [DOI] [PubMed] [Google Scholar]
- Holzelova E, Vonarbourg C, Stolzenberg MC, et al. (2004) ‘Autoimmune lymphoproliferative syndrome with somatic Fas mutations’, New England Journal of Medicine, 351(14), pp. 1409–1418. doi: 10.1056/NEJMoa040036. [DOI] [PubMed] [Google Scholar]
- Holzelova E, Vonarbourg C, Stolzenberg M-C, et al. (2004) ‘Autoimmune Lymphoproliferative Syndrome with Somatic Fas Mutations’, New England Journal of Medicine, 351(14), pp. 1409–1418. doi: 10.1056/NEJMoa040036. [DOI] [PubMed] [Google Scholar]
- Hoyos-Bachiloglu R et al. (2017) ‘A digenic human immunodeficiency characterized by IFNAR1 and IFNGR2 mutations’, Journal of Clinical Investigation, 127(12), pp. 4415–4420. doi: 10.1172/JCI93486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu AP et al. (2013) ‘Intermediate phenotypes in patients with autosomal dominant hyper-IgE syndrome caused by somatic mosaicism’, Journal of Allergy and Clinical Immunology, 131(6), pp. 1586–1593. doi: 10.1016/j.jaci.2013.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante AJ et al. (1998) ‘The clinical spectrum in a large kindred with autoimmune lymphoproliferative syndrome caused by a Fas mutation that impairs lymphocyte apoptosis’, Journal of Pediatrics, 133(5), pp. 629–633. doi: 10.1016/S0022-3476(98)70102-7. [DOI] [PubMed] [Google Scholar]
- Israel L et al. (2017) ‘Human Adaptive Immunity Rescues an Inborn Error of Innate Immunity’, Cell, 168(5), pp. 789–800.e10. doi: 10.1016/j.cell.2017.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CE et al. (1999) ‘Autoimmune lymphoproliferative syndrome with defective Fas: Genotype influences penetrance’, American Journal of Human Genetics, 64(4), pp. 1002–1014. doi: 10.1086/302333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jägle S et al. (2019) ‘Distinct molecular response patterns of activating STAT3 mutations associate with penetrance of lymphoproliferation and autoimmunity’, Clinical Immunology. Elsevier, 210(November 2019), p. 108316. doi: 10.1016/j.clim.2019.108316. [DOI] [PubMed] [Google Scholar]
- Jeffries AR et al. (2012) ‘Stochastic choice of allelic expression in human neural stem cells’, Stem Cells, 30(9), pp. 1938–1947. doi: 10.1002/stem.1155. [DOI] [PubMed] [Google Scholar]
- Jing H et al. (2014) ‘Somatic reversion in dedicator of cytokinesis 8 immunodeficiency modulates disease phenotype’, Journal of Allergy and Clinical Immunology. Elsevier Ltd, 133(6), pp. 1667–1675. doi: 10.1016/j.jaci.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen SF et al. (2016) ‘Altered gut microbiota profile in common variable immunodeficiency associates with levels of lipopolysaccharide and markers of systemic immune activation’, Mucosal Immunology, 9(6), pp. 1455–1465. doi: 10.1038/mi.2016.18. [DOI] [PubMed] [Google Scholar]
- Jouanguy E et al. (1996) ‘Interferon-γ –Receptor Deficiency in an Infant with Fatal Bacille Calmette–Guérin Infection’, New England Journal of Medicine, 335(26), pp. 1956–1962. doi: 10.1056/NEJM199612263352604. [DOI] [PubMed] [Google Scholar]
- Jouanguy E et al. (1999) ‘A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection’, Nature Genetics, 21(4), pp. 370–378. doi: 10.1038/7701. [DOI] [PubMed] [Google Scholar]
- Kadowaki T et al. (2018) ‘Haploinsufficiency of A20 causes autoinflammatory and autoimmune disorders’, Journal of Allergy and Clinical Immunology, 141(4), pp. 1485–1488.e11. doi: 10.1016/j.jaci.2017.10.039. [DOI] [PubMed] [Google Scholar]
- Kasahara Y et al. (1998) ‘Novel Fas (CD95/APO-1) mutations in infants with a lymphoproliferative disorder’, International Immunology, 10(2), pp. 195–202. doi: 10.1093/intimm/10.2.195. [DOI] [PubMed] [Google Scholar]
- Keller M et al. (2014) ‘Burden of copy number variation in common variable immunodeficiency’, Clinical and Experimental Immunology, 177(1), pp. 269–271. doi: 10.1111/cei.12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerner G et al. (2019) ‘Homozygosity for TYK2 P1104A underlies tuberculosis in about 1% of patients in a cohort of European ancestry’, Proceedings of the National Academy of Sciences of the United States of America, 116(21), pp. 10430–10434. doi: 10.1073/pnas.1903561116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong XF et al. (2010) ‘A novel form of human STAT1 deficiency impairing early but not late responses to interferons’, Blood, 116(26), pp. 5896–5906. doi: 10.1182/blood-2010-04-280586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong XF et al. (2013) ‘Haploinsufficiency at the human IFNGR2 locus contributes to mycobacterial disease’, Human Molecular Genetics, 22(4), pp. 769–781. doi: 10.1093/hmg/dds484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning HD et al. (2015) ‘Myeloid lineage–restricted somatic mosaicism of NLRP3 mutations in patients with variant Schnitzler syndrome’, Journal of Allergy and Clinical Immunology, 135(2), pp. 561–564.e4. doi: 10.1016/j.jaci.2014.07.050. [DOI] [PubMed] [Google Scholar]
- Kostmann R (1950) ‘Hereditär reticulos-en ny systemsjukdom’, Svenska Läkartideningen. [Google Scholar]
- Kreins AY et al. (2015) ‘Human TYK2 deficiency: Mycobacterial and viral infections without hyper-IgE syndrome’, The Journal of Experimental Medicine, 212(10), pp. 1641–1662. doi: 10.1084/jem.20140280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen IA et al. (2011) ‘Novel STAT1 alleles in a patient with impaired resistance to mycobacteria’, Journal of Clinical Immunology, 31(2), pp. 265–271. doi: 10.1007/s10875-010-9480-8. [DOI] [PubMed] [Google Scholar]
- Kuehn HS et al. (2011) ‘FAS Haploinsufficiency Is a Common Disease Mechanism in the Human Autoimmune Lymphoproliferative Syndrome’, The Journal of Immunology, 186(10), pp. 6035–6043. doi: 10.4049/jimmunol.1100021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn HS et al. (2014) ‘Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4’, Science, 345(6204), pp. 1623–1627. doi: 10.1126/science.1255904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HK et al. (2014) ‘TLR3 deficiency in herpes simplex encephalitis’, Neurology, 83, pp. 1888–1897. doi: 10.1212/WNL.0000000000000999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Herrera G et al. (2012) ‘Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity’, American Journal of Human Genetics, 90(6), pp. 986–1001. doi: 10.1016/j.ajhg.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz W (1946) ‘A propos de l’Epidermodysplasie verruciforme’, Dermatology, 92(1), pp. 30–43. doi: 10.1159/000255805. [DOI] [PubMed] [Google Scholar]
- Ma CA et al. (2017) ‘Somatic STAT5b gain-of-function mutations in early onset nonclonal eosinophilia, urticaria, dermatitis, and diarrhea.’, Blood, 129(5), pp. 650–653. doi: 10.1182/blood-2016-09-737817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z et al. (2019) ‘PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components’, Nature. Springer US, 12. doi: 10.1038/s41586-019-1057-y. [DOI] [PubMed] [Google Scholar]
- Magerus-Chatinet A et al. (2011) ‘Onset of autoimmune lymphoproliferative syndrome (ALPS) in humans as a consequence of genetic defect accumulation’, Journal of Clinical Investigation, 121(1), pp. 106–112. doi: 10.1172/JCI43752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Feito A et al. (2016) ‘Autoimmune lymphoproliferative syndrome due to somatic FAS mutation (ALPS-sFAS) combined with a germline caspase-10 (CASP10) variation’, Immunobiology, 221(1), pp. 40–47. doi: 10.1016/j.imbio.2015.08.004. [DOI] [PubMed] [Google Scholar]
- Massaad MJ et al. (2016) ‘Deficiency of base excision repair enzyme NEIL3 drives increased predisposition to autoimmunity’, Journal of Clinical Investigation, 126(11), pp. 4219–4236. doi: 10.1172/JCI85647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott DH et al. (2015) ‘Chromothriptic Cure of WHIM Syndrome’, Cell, 160(4), pp. 686–699. doi: 10.1016/j.cell.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensa-Vilaró A et al. (2019) ‘Unexpected relevant role of gene mosaicism in patients with primary immunodeficiency diseases’, Journal of Allergy and Clinical Immunology, 143(1), pp. 359–368. doi: 10.1016/j.jaci.2018.09.009. [DOI] [PubMed] [Google Scholar]
- Migita K et al. (2013) ‘The Contribution of SAA1 Polymorphisms to Familial Mediterranean Fever Susceptibility in the Japanese Population’, PLoS ONE, 8(2), pp. 1–7. doi: 10.1371/journal.pone.0055227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minegishi Y et al. (2007) ‘Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome’, Nature, 448(August). doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- Moens L et al. (2017) ‘A novel kindred with inherited STAT2 deficiency and severe viral illness’, Journal of Allergy and Clinical Immunology, 139(6), pp. 1995–1997.e9. doi: 10.1016/j.jaci.2016.10.033. [DOI] [PubMed] [Google Scholar]
- Molleran Lee S et al. (2004) ‘Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis’, Journal of Medical Genetics, 41(2), pp. 137–144. doi: 10.1136/jmg.2003.011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mørk N et al. (2015) ‘Mutations in the TLR3 signaling pathway and beyond in adult patients with herpes simplex encephalitis’, Genes and Immunity, 16(8), pp. 552–566. doi: 10.1038/gene.2015.46. [DOI] [PubMed] [Google Scholar]
- Nag A et al. (2013) ‘Chromatin signature of widespread monoallelic expression’, eLife, 2013(2), pp. 1–19. doi: 10.7554/eLife.01256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neven B et al. (2011) ‘Asurvey of 90 patients with autoimmune lymphoproliferative syndrome related to TNFRSF6 mutation’, Blood, 118(18), pp. 4798–4807. doi: 10.1182/blood-2011-04-347641. [DOI] [PubMed] [Google Scholar]
- Newport MJ et al. (1996) ‘A Mutation in the Interferon-γ –Receptor Gene and Susceptibility to Mycobacterial Infection’, New England Journal of Medicine, 335(26), pp. 1941–1949. doi: 10.1056/NEJM199612263352602. [DOI] [PubMed] [Google Scholar]
- Niemela JE et al. (2011) ‘Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis’, Blood, 117(10), pp. 2883–2886. doi: 10.1182/blood-2010-07-295501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Marcaigh AS et al. (1997) ‘Maternal mosaicism for a novel interleukin-2 receptor gamma-chain mutation causing X-linked severe combined immunodeficiency in a Navajo kindred.’, Journal of clinical immunology, 17(1), pp. 29–33. doi: 10.1023/a:1027332327827. [DOI] [PubMed] [Google Scholar]
- Oftedal BE et al. (2015) ‘Dominant Mutations in the Autoimmune Regulator AIRE Are Associated with Common Organ-Specific Autoimmune Diseases’, Immunity, 42(6), pp. 1185–1196. doi: 10.1016/j.immuni.2015.04.021. [DOI] [PubMed] [Google Scholar]
- Orange JS et al. (2011) ‘Genome-wide association identifies diverse causes of common variable immunodeficiency’, Journal of Allergy and Clinical Immunology, 127(6), pp. 1360–1367.e6. doi: 10.1016/j.jaci.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard C et al. (2010) ‘Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency’, Medicine, 89(6), pp. 403–425. doi: 10.1097/MD.0b013e3181fd8ec3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Pino-Molina L et al. (2019) ‘Impaired CpG demethylation in common variable immunodeficiency associates with B cell phenotype and proliferation rate’, Frontiers in Immunology, 10(April), pp. 1–11. doi: 10.3389/fimmu.2019.00878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey JE et al. (2016) ‘Molecular diagnostic experience of whole-exome sequencing in adult patients’, Genetics in Medicine, 18(7), pp. 678–685. doi: 10.1038/gim.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prando C et al. (2013) ‘Inherited IL-12p40 deficiency: Genetic, immunologic, and clinical features of 49 patients from 30 kindreds’, Medicine, 92(2), pp. 109–122. doi: 10.1097/MD.0b013e31828a01f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puck JM et al. (1995) ‘Female germ line mosaicism as the origin of a unique IL-2 receptor G- chain mutation causing X-linked severe combined immunodeficiency’, Journal of Clinical Investigation, 95(2), pp. 895–899. doi: 10.1172/jci117740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae W et al. (2017) ‘Clinical efficacy of a next-generation sequencing gene panel for primary immunodeficiency diagnostics’, Clinical Genetics, 93(3), pp. 647–655. doi: 10.1111/cge.13163. [DOI] [PubMed] [Google Scholar]
- Reinius B et al. (2016) ‘Analysis of allelic expression patterns in clonal somatic cells by single-cell RNA–seq’, Nature Genetics, 48(11), pp. 1430–1435. doi: 10.1038/ng.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius B and Sandberg R (2015) ‘Random monoallelic expression of autosomal genes: Stochastic transcription and allele-level regulation’, Nature Reviews Genetics. Nature Publishing Group, 16(11), pp. 653–664. doi: 10.1038/nrg3888. [DOI] [PubMed] [Google Scholar]
- Revel-Vilk S et al. (2015) ‘Autoimmune lymphoproliferative syndrome-like disease in patients with LRBA mutation’, Clinical Immunology. Elsevier Inc., 159(1), pp. 84–92. doi: 10.1016/j.clim.2015.04.007. [DOI] [PubMed] [Google Scholar]
- Rieux-Laucat F et al. (1995) ‘Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity’, Science, 268(5215), pp. 1347–1349. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- Rigaud S et al. (2011) ‘Human X-linked variable immunodeficiency caused by a hypomorphic mutation in XIAP in association with a rare polymorphism in CD40LG’, Blood, 118(2), pp. 252–261. doi: 10.1182/blood-2011-01-328849. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Cortez VC et al. (2015) ‘Monozygotic twins discordant for common variable immunodeficiency reveal impaired DNA demethylation during naïve-to-memory B-cell transition’, Nature Communications, 6. doi: 10.1038/ncomms8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosain J et al. (2019) ‘Mendelian susceptibility to mycobacterial disease: 2014–2018 update’, Immunology and Cell Biology, 97(4), pp. 360–367. doi: 10.1111/imcb.12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roser M, Ritchie H and Dadonaite B (2019) ‘Child & Infant Mortality’, Our World In Data. [Google Scholar]
- Rowczenio DM et al. (2017) ‘Late-onset cryopyrin-associated periodic syndromes caused by somatic NLRP3 mosaicism-UK single center experience’, Frontiers in Immunology, 8(October). doi: 10.3389/fimmu.2017.01410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio EP et al. (2012) ‘A novel STAT1 mutation associated with disseminated mycobacterial disease’, Journal of Clinical Immunology, 32(4), pp. 681–689. doi: 10.1007/s10875-012-9659-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho-Shimizu V et al. (2011) ‘Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency’, Journal of Clinical Investigation, 121(12), pp. 4889–4902. doi: 10.1172/JCI59259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrafzadeh SA et al. (2019) ‘A New Patient with Inherited TYK2 Deficiency’, pp. 10–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savova V et al. (2017) ‘Risk alleles of genes with monoallelic expression are enriched in gain-of-function variants and depleted in loss-of-function variants for neurodevelopmental disorders’, Molecular Psychiatry, 22(12), pp. 1785–1794. doi: 10.1038/mp.2017.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D et al. (2014) ‘Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations’, Nature Medicine, 20(12), pp. 1410–1416. doi: 10.1038/nm.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab C et al. (2018) ‘Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4–insufficient subjects’, Journal of Allergy and Clinical Immunology, 142(6), pp. 1932–1946. doi: 10.1016/j.jaci.2018.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota M et al. (2015) ‘Somatic Mosaicism for a NRAS Mutation Associates with Disparate Clinical Features in RAS-associated Leukoproliferative Disease: a Report of Two Cases’, Journal of Clinical Immunology, 35(5), pp. 454–458. doi: 10.1007/s10875-015-0163-3. [DOI] [PubMed] [Google Scholar]
- Speckmann C et al. (2008) ‘Clinical and immunologic consequences of a somatic reversion in a patient with X-linked severe combined immunodeficiency’, Blood, 112(10), pp. 4090–4097. doi: 10.1182/blood-2008-04-153361. [DOI] [PubMed] [Google Scholar]
- Stephan V et al. (1996) ‘Atypical X-Linked Severe Combined Immunodeficiency Due to Possible Spontaneous Reversion of the Genetic Defect in T Cells’, New England Journal of Medicine, 335(21), pp. 1563–1567. doi: 10.1056/NEJM199611213352104. [DOI] [PubMed] [Google Scholar]
- Stray-Pedersen A et al. (2017) ‘Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders’, Journal of Allergy and Clinical Immunology, 139(1), pp. 232–245. doi: 10.1016/j.jaci.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung L et al. (2001) ‘The Role of Infections in Primary Hemophagocytic Lymphohistiocytosis: A Case Series and Review of the Literature’, Clinical Infectious Diseases, 33(10), pp. 1644–1648. doi: 10.1086/323675. [DOI] [PubMed] [Google Scholar]
- Taft J and Bogunovic D (2018) ‘The Goldilocks Zone of Type I IFNs: Lessons from Human Genetics’, The Journal of Immunology, 201(12), pp. 3479–3485. doi: 10.4049/jimmunol.1800764. [DOI] [PubMed] [Google Scholar]
- Takagi Masatoshi et al. (2011) ‘Autoimmune lymphoproliferative syndrome–like disease with somatic KRAS mutation’, Blood, 117(10), pp. 2887–2890. doi: 10.1182/blood-2010-08-301515. [DOI] [PubMed] [Google Scholar]
- Tangye SG et al. (2020) ‘Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee’, Journal of Clinical Immunology. Journal of Clinical Immunology. doi: 10.1007/s10875-019-00737-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telenti A (2019) ‘Regulatory genome variants in human susceptibility to infection’, Human Genetics. Springer Berlin Heidelberg, (0123456789). doi: 10.1007/s00439-019-02091-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaventhiran JED et al. (2018) ‘Whole Genome Sequencing of Primary Immunodeficiency reveals a role for common and rare variants in coding and non-coding sequences’, bioRxiv, p. 499988. doi: 10.1101/499988. [DOI] [Google Scholar]
- Timberlake AT et al. (2016) ‘Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles’, eLife, 5(September2016), pp. 1–19. doi: 10.7554/eLife.20125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tone Y et al. (2007) ‘Somatic revertant mosaicism in a patient with leukocyte adhesion deficiency type 1’, Blood, 109(3), pp. 1182–1184. doi: 10.1182/blood-2007-08-039057. [DOI] [PubMed] [Google Scholar]
- Tsumura M et al. (2012) ‘Dominant-negative STAT1 SH2 domain mutations in unrelated patients with mendelian susceptibility to mycobacterial disease’, Human Mutation, 33(9), pp. 1377–1387. doi: 10.1002/humu.22113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uberti J et al. (1983) ‘A phenotypically normal revertant of an adenosine deaminase-deficient lymphoblast cell line.’, Journal of Immunology, 130(6), pp. 2866–2870. [PubMed] [Google Scholar]
- Vairo D et al. (2011) ‘Severe impairment of IFN-γ and IFN-α responses in cells of a patient with a novel STAT1 splicing mutation’, Blood, 118(7), pp. 1806–1817. doi: 10.1182/blood-2011-01-330571. [DOI] [PubMed] [Google Scholar]
- de Valles-Ibáñez G et al. (2018) ‘Evaluating the genetics of common variable immunodeficiency: Monogenetic model and beyond’, Frontiers in Immunology, 9(May), pp. 1–15. doi: 10.3389/fimmu.2018.00636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihinen M et al. (1997) ‘BTKbase, mutation database for X-linked agammaglobulinemia (XLA)’, Nucleic Acids Research, 25(1), pp. 166–171. doi: 10.1093/nar/25.1.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker S et al. (2016) ‘Identification of a gain-of-function STAT3 mutation (p.Y640F) in lymphocytic variant hypereosinophilic syndrome’, Blood, 127(7), pp. 360–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley RJ and Kimberlin DW (2005) ‘Herpes simplex: Encephalitis children and adolescents’, Seminars in Pediatric Infectious Diseases, 16(1), pp. 17–23. doi: 10.1053/j.spid.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Wiskott A (1937) ‘Familiärer, angeborener Morbus Werlhofii?’, Monatsschr Kinderheilkd, 68(212). [Google Scholar]
- Wolach B et al. (2005) ‘Unusual late presentation of X-linked chronic granulomatous disease in an adult female with a somatic mosaic for a novel mutation in CYBB’, Blood, 105(1), pp. 61–66. doi: 10.1182/blood-2004-02-0675. [DOI] [PubMed] [Google Scholar]
- Yang Y et al. (2014) ‘Molecular findings among patients referred for clinical whole-exome sequencing’, JAMA - Journal of the American Medical Association, 312(18), pp. 1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yel L (2010) ‘Selective IgA Deficiency’, Journal of Clinical Immunology, 30(1), pp. 10–16. doi: 10.1007/s10875-009-9357-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SY et al. (2007) ‘TLR3 deficiency in patients with herpes simplex encephalitis’, Science, 317(5844), pp. 1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- Zhang X et al. (2015) ‘Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation.’, Nature, 517(7532), pp. 89–93. doi: 10.1038/nature13801. [DOI] [PMC free article] [PubMed] [Google Scholar]