

Abstract
Germline mutations in GATA2 are associated with an inherited predisposition to bone marrow failure (BMF), myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Hematopoietic stem cell transplantation (HSCT) remains the only curative therapy. However, patients may be at an increased risk for transplant-related toxicity (TRT) and mortality (TRM) due to their underlying disease biology. We performed a retrospective case-control study of pediatric BMF/MDS/AML patients with germline GATA2 mutations, comparing HSCT outcomes to randomly selected patients without germline GATA2 mutations and BMF/MDS (control A) and acute leukemia (control B). The 5-year overall and disease-free survival rates in the GATA2 cohort (65%, 51%) were similar to control A (58%, 49%) and B (45%, 43%) cohorts. In contrast, the 5-year event-free survival rate was significantly lower in the GATA2 cohort (7%±6%, 28%±10% and 33%±8% for GATA2, A, and B, respectively), due to an increased number of unique TRT. Specifically, neurologic toxicities occurred significantly more frequently in GATA2 patients than in the control groups and post-HSCT thrombotic events occurred only in the GATA2 cohort. There was no difference in TRM, infections or graft-versus-host disease (GVHD) across groups. The higher incidence of thrombotic and neurological events specific to GATA2 patients warrants further investigation and has potential treatment ramifications.
Keywords: GATA2, MonoMac syndrome, Emberger syndrome, bone marrow failure, myelodysplastic syndromes, stem cell transplantation, pediatric MDS
INTRODUCTION
Pediatric myelodysplastic syndromes (MDS) represent a spectrum of rare hematopoietic stem cell disorders characterized by cytopenias, ineffective hematopoiesis, marrow dysplasia and risk for leukemic transformation. While some cases are likely acquired due to de novo transformation, germline mutations in DNA repair pathways, ribosomal pathways, the telomere complex or in hematopoietic transcription factors (e.g., CEBPA, RUNX1, GATA2 and ETV6) are predisposition syndromes underlying many pediatric cases of MDS and acute myeloid leukemia (AML).
It is now well recognized that GATA2 deficiency is an inherited syndrome that can lead to marrow dysfunction, MDS/AML, as well as defects in the endothelial-vascular and immune systems. Patients with GATA2 deficiency often show multisystem organ involvement analogous to other inherited bone marrow failure syndromes (IBMFS) such as Fanconi anemia (FA), Shwachman-Diamond Syndrome (SDS), and dyskeratosis congenita (DC). While it is clearly established that patients with the above IBMFS experience increased transplant-related toxicities (TRT) and mortality (TRM) when exposed to standard myeloablative conditioning regimen, whether this is true for patients with GATA2 deficiency has not been completely assessed. We previously reported on a cohort of pediatric MDS patients with germline GATA2 mutations and described a high transplant-related mortality (TRM) rate, low relapse rate and frequent unique toxicities, particularly thrombosis and neurological and pulmonary toxicities.1 We hypothesized that this was the result of cellular defects analogous to those seen in other IBMF.
To rigorously assess the specific contribution of germline GATA2 mutations to TRT/TRM, we performed a single institution retrospective case-control study comparing GATA2 patients undergoing myeloablative allogeneic HSCT to 2 cohorts of patients with similar diagnoses but known wildtype GATA2: 1) bone marrow failure (BMF)/MDS and 2) acute leukemia. We intentionally selected two different control cohorts to increase the generalizability of the results. Control A is comprised of patients with BMF/MDS without a known underlying IBMFS based on comprehensive clinical, laboratory evaluation and molecular genetic studies. Control B was a heavily pretreated group of patients with acute leukemia where a higher rate of TRT and TRM would be expected compared to the untreated patients in control A.
MATERIALS AND METHODS
Patient Population and Study Design
A total of 80 patients met study eligibility for this retrospective case-control study. We report on 15 consecutive patients (ages 1-21 years) with BMF/MDS/leukemia associated with germline GATA2 mutations (GATA2 cohort) undergoing HSCT at our institution between 2000-2014. Eligible controls without GATA2 mutations were identified and randomly selected: control A are pediatric patients with BMF/MDS (n=25), control B are patients with de novo AML or acute lymphoblastic leukemia (ALL) (n=40). Controls were matched for age (0-10, 11-20 years), prior chemotherapy (yes/no, control A only), conditioning regimen, stem cell source and donor match (Supplemental Table 1). Patients with a pre-HSCT Lansky score <80%, uncontrolled infection or molecular or clinical diagnosis of other underlying IBMFS were excluded. GATA2 sequencing and molecular testing for IBMFS were performed in a Clinical Laboratory Improvement Amendments certified laboratory. All patients in the GATA2 and control A group underwent comprehensive clinical testing and molecular genetic testing for IMBMFS which included chromosomal breakage for Fanconi Anemia, telomere length analysis to rule out dyskeratosis congenita and single gene or next generation sequencing analysis. Patients in control B had de novo leukemia. Therefore, the majority of patients did not undergo molecular testing to rule out IBMFS. A few patients were tested for selected IBMFS or GATA2 based on their history, family history or clinical presentation and found to be negative. The study was approved by the local IRB and conducted according to the Declaration of Helsinki.
Due to previously observed HSCT-related specific complications in GATA2 patients1, in this study we defined TRT as experiencing at least one of the following: thrombosis, infection, organ toxicities ≥3 (Common Terminology Criteria for Adverse Events v4.0) and secondary malignancies. TRM was defined as death not due to relapse.
The primary objective was to determine if the proportion of patients who experienced a) TRT, b) acute grade II-IV or chronic graft-versus-host disease (GVHD), and c) TRM differed between the three cohorts. The secondary objectives were to perform a descriptive comparison of overall survival (OS), disease-free survival (DFS) and event-free survival (EFS) rates between cases and controls.
Statistical Analysis
Two-sided Fisher’s exact and two-sided Wilcoxon rank-sum tests were used to examine the differences in baseline characteristics. A one-sided Fisher’s exact test compared the proportion with high grade TRT for GATA2 vs control groups (p<0.1 considered statistically significant; otherwise p<0.05). Kaplan-Meier curves (point estimates at 5 years; standard errors per Greenwood) were compared with log-rank tests. We used SAS version 9.4 (SAS Institute Inc., Cary, NC, USA).
RESULTS
Pre-HSCT Patient Characteristics
For the entire cohort the overall median follow-up of patients last reported as alive was 5.9 years (range=1-12.1, n=42). Patients in the GATA2 cohort were significantly older than control A patients at time of HSCT (15.7 vs 12 years, p=0.03). Patients in the GATA2 and control A cohorts showed a similar spectrum of disease phenotypes (Table 1). One GATA2 patient had MDS/MonoMac syndrome with B-cell ALL, reported previously.2 Control B had equal representation of ALL and AML. As previously reported, monosomy 7 was the most common cytogenetic abnormality in the GATA2 cohort, more frequent than in control B (p=0.0001).3, 4 Patients with GATA2 had a higher platelet count prior to transplant compared to control A patients (p=0.015). Absolute neutrophil and monocyte counts were significantly lower in the GATA2 cohort compared to control B (p=0.0008 and 0.04, respectively). GATA2 patients had a significantly higher occurrence of lymphedema and warts, consistent with previous reports (Table 1).3, 4
Table 1.
Patient, disease and stem cell transplant characteristics of the cases with pediatric MDS/BMF due to germline GATA2 mutation, control A (pMDS/BMF without GATA2 mutations) and control B (ALL/AML without GATA2 mutations)
| GATA2 cohort n=15 | Control A n=25 | Control B n=40 | p-value for GATA2 vs Control A | p-value for GATA2 vs Control B | ||
|---|---|---|---|---|---|---|
| Follow-up, years | Median (range) | 5 (1-8.5) (n=9) | 5.8 (1.1-12.1) (n=14) | 6.5 (1.4-10.9) (n=19) | - | - |
| Patient age*, years | Median (range) | 15.7 (5.7-20) | 12 (0.9-22.2) | 13.2 (2.8-20.3) | 0.03 | 0.3 |
| Patient age at diagnosis, years | Median (range) | 15.2 (5.4-19.8) | 9 (0.3-22) | 12.3 (0.6-20.2) | 0.07 | 0.14 |
| Sex, n (%) | Male | 10 (67) | 15 (60) | 24 (60) | 0.7 | 0.8 |
| Female | 5 (33) | 10 (40) | 16 (40) | |||
| Diagnosis, n(%) | RCC | 8 (53) | 9 (36) | 0 (0) | – | – |
| RAEB/RAEB-T/AML-MRC | 4 (27) | 11 (44) | 0 (0) | |||
| SAA | 0 (0) | 4 (16) | 0 (0) | |||
| BMF, NOS# | 0 (0) | 1 (4) | 0 (0) | |||
| ALL | 1 (7) | 0 (0) | 20 (50) | |||
| AML | 2 (13) | 0 (0) | 20 (50) | |||
| Karyotype, n (%) | 5q- | 0 (0) | 0 (0) | 1 (3) | – | – |
| +8 (trisomy 8) | 5 (33) | 2 (8) | 2 (5) | 0.08 | 0.01 | |
| Other | 2 (13) | 10 (4) | 22 (55) | - | - | |
| Monosomy 7# | 9 (60) | 9 (36) | 9 (25) | 0.19 | 0.0001 | |
| -7 | 7 (47) | 9 (36) | 3 (8) | - | - | |
| 7q | 2 (13) | 0 (0) | 0 (0) | - | - | |
| CBC, median (ranges) | WBC | 2.2 (0.7-39) | 3.5 (1.4-12.8) | 3.2 (0.8-62.4) | 0.07 | 0.16 |
| Hgb | 0.8 | 0.7 | ||||
| Platelets | 10.2 (3.5-13.9) | 9.9 (2.7-17.4) | 10.5 (7.9-13.8) | 0.015 | 0.11 | |
| ANC | 0.2 | 0.0008 | ||||
| AMC | 98 (12-443) | 41.5 (9-269) | 151 (16-759) | 0.3 | 0.04 | |
| 0.6 (0.1-17) | 1 (0-7.2) | 2.2 (0.3-8.7) | ||||
| 0.1 (0-72) | 0.2 (0-1.6) | 0.4 (0-441) | ||||
| Associated pathologies/Pre-SCT morbidities, n (%) | Lymphedema | 4 (27%) | 0 (0%) | 0 (0%) | 0.015 | 0.004 |
| Warts | 4 (27%) | 1 (4%) | 0 (0%) | 0.056 | 0.004 | |
| PAP | 1 (7%) | 0 (0%) | 0 (0%) | 0.4 | 0.3 | |
| Polyneuropathy | 1 (7%) | 0 (0%) | 1 (3%) | 0.4 | 0.5 | |
| Hearing loss | 1 (7%) | 0 (0%) | 0 (0%) | 0.4 | 0.3 | |
| Infection | 3 (2%) | 5 (2%) | 5 (13%) | 1 | 0.7 | |
| Other | 5 (33%) | 9 (36%) | 23 (58%) | 1 | 0.14 | |
| Pre-SCT chemo, n (%) | 4 (27) | 9 (36) | 40 (100) | 0.7 | <0.0001 | |
| Conditioning, n (%) | CY/TBI +/− ATG | 11 (73) | 17 (68) | 31 (78) | 0.9 | 0.9 |
| BU/CY +/− ATG | 2 (13) | 5 (20) | 4 (10) | |||
| Flu/Cy/TBI | 2 (13) | 3 (12) | 5 (13) | |||
| Cell source, n (%) | BM-MRD (10/10) | 3 (20) 5 (33) |
4 (16) 11 (44) |
10 (25) 11 (28) |
0.7 | 1 |
| BM-MUD (10/10) | 3 (20) | 7 (28) | 8 (20) | |||
| BM-mmURD (8 or 9/10) | 4 (27) | 3 (12) | 11 (28) | |||
| UCB | ||||||
| GVHD prophylaxis§, n (%) | Steroids | 10 (67) | 20 (80) | 24 (60) | - | - |
| CSA | 15 (100) | 22 (88) | 38 (95) | |||
| Tacrolimus | 0 (0) | 3 (12) | 2 (5) | |||
| MTX | 9 (60) | 20 (80) | 28 (70) | |||
| MMF | 3 (20) | 2 (8) | 5 (13) | |||
p-values were generated using Wilcoxon rank sum or Fisher exact test (two-sided).
“–” indicates not performed
Patient age at the time of transplantation.
Monosomy 7 is defined as having -7 or -7q.
GVHD prophylaxis agents are not mutually exclusive.
ALL – acute lymphoblastic leukemia; AML – acute myeloid leukemia excluding inherited bone marrow failure syndromes; AM-MRC – acute myeloid leukemia with myelodysplastic related changes; AMC – absolute monocyte count; ANC – absolute neutrophil count; ATG – antithymoglobulin; BM – bone marrow; BMF – bone marrow failure; BMF, NOS – bone marrow failure, not otherwise specified (excluding inherited bone marrow failure syndromes); BU – busulfan; CBC – complete blood count (baseline, prior to SCT); CSA – cyclosporine A; CY – cyclophosphamide; Hgb – hemoglobin; Flu – fludarabine; MMF – mycophenolate mofetil; mmURD – mismatched unrelated donor; MRD – matched related donor; MTX – methotrexate; MUD – matched unrelated donor; PAP – pulmonary alveolar proteinosis; RAEB – refractory anemia with excess blasts; RAEB-T – refractory anemia with excess blasts in transformation; RCC – refractory cytopenia of childhood; SAA – severe aplastic anemia; SCT – stem cell transplant; TBI – total body irradiation; UCB – umbilical cord blood.
Other GATA2 associated pathologies prior to transplant (n=5) included hypoplastic kidney (n=1), ventricular septal defect and benign mass at elbow (n=1), elliptocytosis (n=1), undescended testis and developmental delay (n=1), Klinefelter syndrome (n=1).
HSCT Outcomes and Toxicities
The OS and DFS curves of patients with GATA2 mutations appeared similar to those of the control groups (Figures 1A and 1B). TRT was not different between groups for commonly observed HSCT organ toxicities (grade ≥3) such as renal or liver complications. However, we observed a number of unique, less commonly observed TRT events in GATA2 patients. This resulted in a lower EFS (7%±6%) compared to control A (28%±10%, p=0.003) and B (33%±8%, p=0.001) (Figure 1C).
Figure 1.
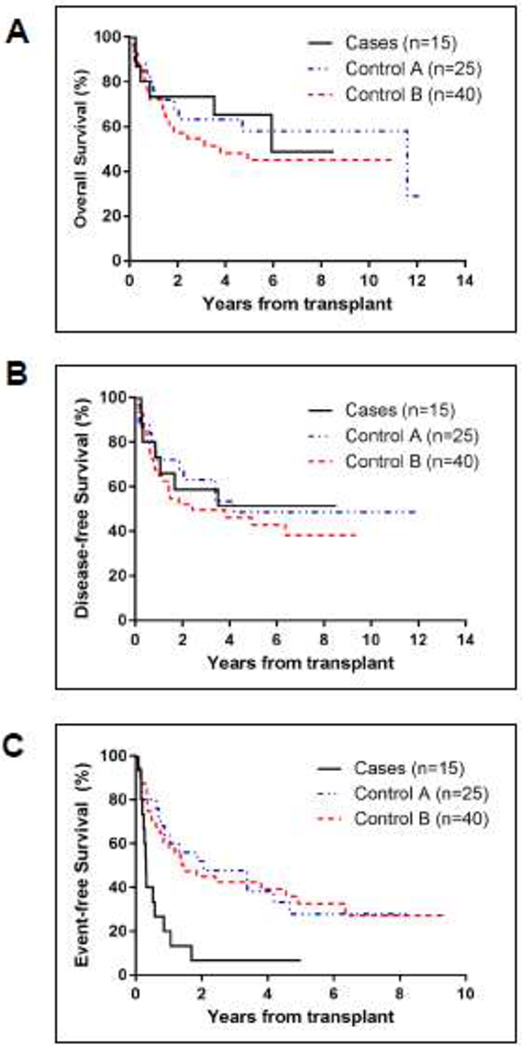
Outcomes of HSCT for patients with germline GATA2 mutations compared to patients without GATA2 mutations (control A and control B).
(A-C) Outcomes after HSCT in patients with GATA2 mutations compared to control A and B. Kaplan-Meier curves of overall survival (OS) (A), disease-free survival (DFS) (B), and event-free survival (EFS) (C) starting from day 0 of HSCT. For OS, death from any cause was the only event considered. DFS time was the time from transplant to the first occurrence of transplant related mortality (TRM) or relapse. EFS time was the time from transplant to the first occurrence of TRM, relapse, acute graft versus host disease (GVHD) grade II-IV, extensive chronic GVHD, secondary malignancy, thromboembolic event or graft failure. If no event occurred, the time was censored at last patient contact.
Those unique TRT included a higher rate of infectious/immunologic complications in GATA2 patients compared to controls. One patient experienced necrotizing fasciitis and deep myositis with bacillus cereus day +188 post-HSCT requiring leg amputation 3 days later. He ultimately succumbed from complications of this infection on day +311 post-HSCT. Another patient experienced prolonged daily persistent fevers for about 10 weeks starting 2 weeks after his engraftment (engrafted day +15) until day +75 following his HSCT. Despite a comprehensive infectious disease workup including serial blood cultures, viral serologies and PCR titers, lumbar puncture and CT scans to rule out occult infection an etiology was not identified. Fevers ultimately improved after removal of the central venous catheter and resolved a few weeks after that without an identified cause. Another patient had severe ischemia-related gastrointestinal bleeding requiring a partial bowel resection 154 days following HSCT. Interestingly the histology showed evidence of arterial and venous thrombosis within the resected segment. Ultimately the patient recovered from his surgery with no further GI bleeding and restoration of bowel function.
Interestingly, the occurrence of neurologic toxicities was significantly higher in the GATA2 cohort [6 (40%) in GATA2 vs 2 (8%) and 0 (0%) in controls A and B, respectively; p=0.02 and 0.0002], which has not been previously described. Events included severe peripheral polyneuropathy (n=3), non-infectious encephalopathy (n=1) and unexplained mental status changes/delirium (n=2) in a total of 4 patients. One patient experienced multiple complications including delirium of unclear etiology and severe polyneuropathy with neuropathic pain and demyelination approximately 75 days following HSCT as demonstrated by magnetic resonance imaging (MRI) and electromyography (EMG). The EMG showed severe symmetric sensorimotor polyneuropathy and axonal demyelination while the MRI showed a degenerative process. This patient required lidocaine infusions followed by oxcarbazepine for his pain control. He ultimately fully recovered after one year of intensive rehabilitation with physical therapy. The second patient developed mental status changes day +58 following HSCT and was found to be encephalopathic possible due to adenovirus and/or aspergillus infection – however, that could not be confirmed diagnostically. An MRI of the brain was non-diagnostic. This patient ultimately passed away from respiratory failure/acute respiratory distress syndrome and secondary multiorgan failure day +88 post HSCT. He ultimately passed away due to complications of extensive chronic GVHD 3.5 years post HSCT. The third patient had polyneuropathy and developed subsequent altered mental status 86 days following HSCT leading to severe non-infectious encephalitis. A brain biopsy was performed to determine the cause of the encephalitis after an infectious work-up, including a lumbar puncture, was not revealing. Histology of the brain tissue demonstrated a nonspecific infiltration by macrophages and reactive gliosis. A specific etiology of the encephalitis could not be identified. High dose steroid therapy was attempted with no improvement. The patient died from encephalitis and pulmonary failure 94 days following HSCT. This patient is the single patient reported with ALL described in detailed elsewhere. 2 The fourth patient had polyneuropathy of unclear etiology, which did not contribute to his TRM.
The rates of acute and chronic GVHD were similar across groups. Secondary malignancies were rare in all groups. None of the patients experienced primary graft failure. Three patients in control A and one in control B had secondary graft failure. One of the GATA2 patients experienced both a desmoid tumor of the chest wall and a squamous cell carcinoma about 3 years following his matched unrelated bone marrow transplant. Both were successfully resected and required no further therapy. TRM was the most common cause of death in all groups at 1 year and there was no difference in TRM between the GATA2 cohort and controls (Table 2).
Table 2.
Transplant-related toxicities and mortalities in patients with pMDS/BMF due to germline GATA2 mutations compared to control A (pMDS/BMF without GATA2 mutations) and control B (ALL/AML without GATA2 mutations)
| GATA2 cohort n=15 | Control A n=25 | Control B n=40 | p-value for GATA2 vs Control A** | p-value for GATA2 vs Control B** | ||
|---|---|---|---|---|---|---|
| Overall TRT*, n (%) | Overall | 14 (93) | 18 (72) | 28 (70) | 0.17 | 0.07 |
| Organ TRT‡, n (%) | Renal | 6 (40) | 6 (24) | 12 (30) | 0.24 | 0.3 |
| Liver | 6 (40) | 5 (20) | 15 (38) | 0.16 | 0.6 | |
| Lung | 6 (40) | 6 (24) | 14 (35) | 0.24 | 0.5 | |
| Lung Transplant | 2 (13) | 0 (0) | 1 (3) | 0.13 | 0.18 | |
| Neurologic | 6 (40) | 2 (8) | 0 (0) | 0.02 | 0.0002 | |
| Cardiac | 0 (0) | 1 (4) | 1 (3) | 1 | 1 | |
| GI | 1 (7) | 0 (0) | 1 (3) | 0.4 | 0.5 | |
| ID/Immunology | 4 (27) | 1 (4) | 1 (3) | 0.06 | 0.02 | |
| Hematologic | 0 (0) | 2 (8) | 2 (5) | 1 | 1 | |
| Lymphedema | 0 (0) | 0 (0) | 0 (0) | – | – | |
| Other | 4 (27) | 0 (0) | 1 (3) | 0.02 | 0.02 | |
| Infection TRT, n (%) | Overall | 13 (87)# | 18 (72) | 28 (70) | 0.25 | 0.18 |
| Viral | 7 (46) | 5 (20) | 10 (25) | 0.08 | 0.1 | |
| Bacterial | 5 (33) | 13 (52) | 18 (45) | 0.9 | 0.9 | |
| Secondary malignancies, n (%) | 1 (7) | 1 (4) | 2 (5) | 0.6 | 0.6 | |
| GVHD, n (%) | Overall± | 9 (60) | 9 (36) | 14 (35) | 0.13 | 0.09 |
| Acute (II-IV) | 5 (33) | 5 (20) | 10 (25) | 0.28 | 0.4 | |
| Chronic§ | 5 (33) | 8 (32) | 9 (23) | 0.6 | 0.3 | |
| Chronic, extensive | 4 (26) | 5 (20) | 6 (15) | – | – | |
| Relapse, n (%) | Occurrence | 3 (20) | 6 (24) | 9 (23) | 0.7 | 0.7 |
| TRM, n (%) | Death within 365 days | 3 (20) | 4 (16) | 7 (18) | 0.5 | 0.5 |
| Death within 100 days | 2 (13) | 2 (8) | 2 (5) | 0.5 | 0.3 | |
Overall TRT: a patient is counted if the patient has experienced at least one organ TRT, Infection TRT, or Other TRT.
p-values are one-sided, testing for higher proportion of toxicities, and complications in the case group compared to each control group.
“–” indicates not performed.
Organ toxicity is defined as grade 3-5 based on NCI CTCAE v4.03 criteria.
infection type for one infection was unknown.
Overall GVHD includes Acute (ll-IV) and Chronic GVHD.
Chronic GVHD includes limited and extensive.
GI – Gastrointestinal; GVHD – graft versus host disease; ID – infectious disease; NA – Not applicable; TRM – Treatment-related mortality. TRT – Transplant related toxicity
Secondary malignancies included squamous cell carcinoma and desmoid tumor (GATA2, n=1), high grade glioma (n=1, control A), post-transplant lymphoproliferative disorder (n=1, control B) and osteosarcoma (n=1, control B).
Other TRT in the GATA2 cohort included skin rash (non-GVHD) (n=1), hyperlipidemia and cataract (n=1), ICU transfer with multiorgan toxicity (n=1), hemorrhagic cystitis (non-BK virus, n=1).
Rate of thromboembolic events in GATA2 cohort compared to controls
The overall rate of thrombotic events (pre- and post-HSCT combined) occurred at a significantly higher frequency in GATA2 patients (8/15) compared to control A (0/25) and B (4/40) (p<0.0001 and p=0.0015), which has not previously been described. Two patients with GATA2 deficiency experienced a clot prior to HSCT while the remaining 6 occurred post-HSCT. A total of 9 thromboembolic events occurred in 8 of the 15 GATA2 patients. Interestingly, 7 out of the 9 thrombotic events in the GATA2 group occurred post-HSCT while only two occurred prior to HSCT. The two thromboembolic events pre-HSCT were comprised of a right upper extremity clot associated with a central venous line (CVL) and an unprovoked dural venous thrombosis that resolved prior to HSCT. The four thromboembolic events in control B patients all occurred prior to HSCT (Table 3).
Table 3.
Thromboembolic events pre- and post-hematopoietic stem cell transplant in patients with pMDS/BMF due to germline GATA2 mutations compared to control A (pMDS/BMF without GATA2 mutations) and control B (ALL/AML without GATA2 mutations)
| n (%) GATA2 cohort n=15 | n (%) Control A n=25 | n (%) Control B n=40 | p-value for GATA2 vs Control A* | p-value for GATA2 vs Control B* | ||
|---|---|---|---|---|---|---|
| Total number of patients with thrombosis n (%) | 8 (53) | 0 (0) | 4 (10) | <0.0001 | 0.0015 | |
| Number of patients with thromboembolic events in relation to HSCT, n (%) |
Pre-HSCT | 2 (13) | 0 (0) | 4 (10) | 0.13 | 0.5 |
| Post-HSCT | 6 (40) | 0 (0) | 0 (0) | 0.0013 | 0.0002 | |
| Number of patients with thromboembolic events post-HSCT, by type**, n (%) | DVT | 4 (27) | 0 (0) | 0 (0) | 0.015 | 0.004 |
| PE | 2 (13) | 0 (0) | 0 (0) | 0.13 | 0.071 | |
| Stroke | 1 (7) | 0 (0) | 0 (0) | 0.38 | 0.27 | |
| Number of patients with Post-HSCT thrombosis CVL associated, n (%) | Yes, n (%) | 1/6 (17) | NA | NA | NA | NA |
p-values are one-sided, testing for higher proportion of events in GATA2 group compared with control groups. HSCT – hematopoietic stem cell transplant; NA – Not applicable; CVL – central venous line
one patient had two types of thromboembolic events post-SCT: one DVT and one PE.
The seven thrombotic events following HSCT in the GATA2 cohort were characterized by deep venous thrombosis (DVT) in 4 patients, pulmonary emboli in 2 patients, and one patient experienced an embolic stroke. Thrombosis occurred between 58-608 days post HSCT (mean=194.7 days, median=111 days). Only one of the GATA2 patients had a thrombotic event associated with a CVL post-HSCT. In the other 5 patients (33%) thrombosis occurred spontaneously. In contrast none of the patients in controls A or B experienced thrombotic events following HSCT despite all of those having a CVL (Table 3). Six out of the 8 patients in the GATA2 cohort that experienced a clot had a thrombophilia workup performed. The thrombosis workup was normal in all, leaving the underlying etiology for the thromboembolic event uncertain. Seven patients received anticoagulation with enoxaparin and/or unfractionated heparin. All patients had complete resolution of their thrombosis and symptoms and none of those events contributed to an increased TRM.
DISCUSSION
Our analysis revealed that the OS, DFS, TRM, rates of GVHD and overall organ toxicity for patients with germline GATA2 mutations following a fully myeloablative HSCT for BMF/MDS/AML is similar to that of BMF/MDS or leukemia patients with wildtype GATA2. This is different from other IBMFS (FA, DC) where myeloablative conditioning often leads to severe organ toxicity and increased TRM and modification of the conditioning regimen has become standard. Based on our experience it appears that patients with GATA2 deficiency appear to tolerate standard myeloablative conditioning regimens. However, we did observe a small number of serious and relatively unusual infectious complications that may be unique to this population and will require further analysis as more patients with GATA2 mutations are identified.
Overall our transplant outcomes are comparable to those reported by others.3, 4 A few studies have reported superior outcomes of patients with GATA2 deficiency following HSCT.5–9 It is important to note that these studies represent a heterogenous group of patients with varying phenotypes, ages, HSCT-related risk factors, conditioning regimens and duration of follow up. Parta and colleagues reported on a group of 22 patients transplanted with a busulfan-based conditioning regimen with a considerably shorter median follow-up time of 2 years, compared to nearly 6 years in our studies. While the overall DFS was excellent at 86%, the incidence of grade III and IV acute GVHD and chronic GVHD was 26% and 46% respectively. This study included fewer patients with high risk cytogenetics, in particular monosomy 7, compared to our cohort. As monosomy 7 is well known to confer an inferior prognosis in patients with MDS irrespective of GATA2 mutation status this may account for the difference in results.4
In our retrospective case-control study all patients were treated with a myeloablative conditioning regimen. This is the standard approach in pediatric patients with myelodysplasia and leukemia particularly with advanced disease and/or high-risk features such as monosomy 7. While reduced intensity conditioning approaches have been utilized successfully in a few patients with GATA2 mutations it is important to note that these patients were transplanted primarily for the associated immunodeficiency syndrome and not, as in our group, for high-risk MDS or acute leukemia.9 In such cases, a reduced intensity approach might be beneficial, in particular if there are associated infectious complications or pulmonary dysfunction that would increase the risk of myeloablative conditioning.
The role of GATA2 is incompletely understood and its described functions are protean.3 This may explain the unique post-HSCT toxicities we describe in patients with GATA2 insufficiency undergoing HSCT. In terms of thrombotic events, especially following HSCT, our findings are consistent with those from a larger cohort of GATA2 patients reported by Spinner and colleagues in a series of 57 patients, 14 (25%) were found to develop venous thrombosis and two patients had thrombotic events following HSCT.3 However, of the 57 patients with GATA2 mutations only 21 underwent HSCT, estimating that approximately 10% of patients developed thrombosis after HSCT, which is lower than the rate observed in our cohort. Similarly, Donadieu and colleagues reported on a cohort of 79 patients with GATA2 mutation, of which 7 experienced a thromboembolic or vascular event (8.8%) as part of their disease phenotype.10 GATA2 is expressed in endothelial cells and is important in the regulation of vascular integrity and cell adhesion.3, 11 In addition it appears involved in the regulation of genes modulating the inflammatory response.12 These observations perhaps provide an explanation for the increased thrombotic events observed in GATA2 patients following the inflammatory environment associated with HSCT. Further studies will be needed to elucidate any mechanistic link between the increased thrombosis and an inflammatory state following HSCT.
Neurologic dysfunction has not previously been described in patients with GATA2 mutations. We previously reported on patient 4 in this cohort who died 3 months post-HSCT with severe encephalopathy. Brain biopsy pre-mortem revealed an aseptic, leukocytic brain infiltrate.2 Our findings uniquely suggest that neurologic complications occur more commonly in these patients post-HSCT than in other patients with similar diagnoses. Interestingly, GATA2 is expressed in neural tissues.11, 13–16 One could speculate that patients with GATA2 mutations may have changes in GATA2 expression in neural tissues that could lead to neurological deficits. However, to our knowledge this has not been investigated and we are not aware of any mechanistic studies elucidating the reasons for the neurotoxicity following HSCT observed in our cohort or the neuro-behavioral changes observed in a small subset of patients by Wlodarski and colleagues.4 Of note, behavioral challenges (e.g., autism and aggressive behavior) were reported in 19% of the GATA2 patients by Wlodarski and colleagues.4 This certainly needs to be investigated further and confirmed in larger studies of patients with germline GATA2 mutations.
Our study suggests that patients with germline GATA2 mutations undergoing allogeneic HSCT do not require a different conditioning regimen and likely have comparable rates of graft failure, GVHD and TRM to those of pediatric patients with BMF/MDS or leukemia and wildtype GATA2. In addition, the relapse rates do not appear to be higher than those in GATA2 wildtype patients, perhaps because full dose conditioning can be tolerated. While our study is not powered to demonstrate that MDS subtype (advanced MDS) and monosomy 7 rather than GATA2 mutation status are relevant prognostic factors for patients with GATA2 deficiency undergoing HSCT, we concur with others that timing, conditioning regimen and decision to transplant should be based on the patient’s MDS phenotype and cytogenetics. Timing to HSCT is ideal when patients experience BMF/hypocellular MDS prior to disease progression to advanced MDS/AML. Given the reports of donor-derived MDS/AML in families with germline GATA2 mutations we want to stress the importance of early patient and potential related donor testing for GATA2 mutation.17
We acknowledge that our study has some obvious limitations such as a small sample size often observed in rare conditions and the retrospective nature of the case control study. The institutional practice to performed genetic testing for GATA2 and other IBMFS for marrow failure, MDS and leukemia patients was based on an increased clinical suspicion based on clinical presentation (such as features of GATA2 deficiency or other bone marrow failure syndromes), laboratory findings (such as monocytopenia or immunological defects), presence of monosomy 7 on karyotype or FISH given the association of GATA2 and monosomy 7, and a family history concerning for a cancer, leukemia or marrow failure predisposition syndromes. Despite a relatively aggressive approach to testing for an underlying genetic predisposition syndrome and GATA2 deficiency in control cohort B we cannot exclude that there might have been a small subset of patients within control B where an underlying GATA2 deficiency or IBMFS was missed especially prior to the disease being recognized in 2010 with subsequent gene discovery in 2011.18–21 Nevertheless, our findings of increased thromboembolic events and neurological toxicities in patients with GATA2 deficiency are intriguing and contribute novel observations to the disease phenotype.
Given the increased frequency of unique TRT observed in our GATA2 patient cohort, providers should perform a tailored pre-HSCT baseline evaluation and have a high index of suspicion for thrombotic, neurologic and perhaps infectious complications during the HSCT period. Following HSCT, given that only the hematopoietic compartment has been repaired, patients remain at risk for non-hematopoietic GATA related complications and will need lifelong follow-up by practitioners experienced in inherited cancer predisposition syndromes. Based on the limited available data we cannot recommend changes in the management or preventative strategies such as anticoagulation in this patient population. However, careful long-term observation and systematic analysis of these unique transplant related complications in the GATA2 patient population would help us to further elucidate unique phenotypes and vulnerabilities during and following HSCT in these patients.
Supplementary Material
Supplemental Table 1: Phenotype, genotype, treatment and clinical outcomes characterization of pediatric MDS/BMF patients with GATA2 mutations (GATA2), control A and control B
Highlights.
Transplant outcomes were similar in patients with GATA2 mutations compared to wild type.
Patients with GATA2 mutation experience unique transplant toxicities.
Neurologic toxicities occurred more frequently in GATA2 patients.
Post-transplant thrombotic events occurred only in the GATA2 cohort.
Acknowledgements
The study team would like to thank the clinical research coordinator Nolan Neu for data management and entry and James Zacny, Ph.D. for assistance with line editing of the manuscript.
This study was supported (in part) by research funding from the Natalie Fund [W.L., D.G., H.A.] to support statistical analysis. The Pediatric MDS Registry and I.H. were supported by NIH grant R24 DK 099808-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial disclosure statement: The authors declare no competing financial interests
References
- 1.Hofmann I, Stetson A, Lee M, Williams DA, M.D. F, Lehmann L. Outcomes of allogeneic stem cell transplantation in pediatric patients with myelodysplastic syndrome and bone marrow failure due to GATA2 mutation. Bone Marrow Transplant. 2015;50:S72–S73.26039213 [Google Scholar]
- 2.Koegel AK, Hofmann I, Moffitt K, Degar B, Duncan C, Tubman VN. Acute lymphoblastic leukemia in a patient with MonoMAC syndrome/GATA2 haploinsufficiency. Pediatr Blood Cancer. 2016;63:1844–1847. [DOI] [PubMed] [Google Scholar]
- 3.Spinner MA, Sanchez LA, Hsu AP, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123:809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wlodarski MW, Hirabayashi S, Pastor V, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016;127:1387–1397; quiz 1518. [DOI] [PubMed] [Google Scholar]
- 5.Grossman J, Cuellar-Rodriguez J, Gea-Banacloche J, et al. Nonmyeloablative allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Biol Blood Marrow Transplant. 2014;20:1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parta M, Shah NN, Baird K, et al. Allogeneic Hematopoietic Stem Cell Transplantation for GATA2 Deficiency Using a Busulfan-Based Regimen. Biol Blood Marrow Transplant. 2018;24:1250–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rastogi N, Abraham RS, Chadha R, et al. Successful Nonmyeloablative Allogeneic Stem Cell Transplant in a Child With Emberger Syndrome and GATA2 Mutation. J Pediatr Hematol Oncol. 2018;40:e383–e388. [DOI] [PubMed] [Google Scholar]
- 8.Simonis A, Fux M, Nair G, et al. Allogeneic hematopoietic cell transplantation in patients with GATA2 deficiency-a case report and comprehensive review of the literature. Ann Hematol. 2018;97:1961–1973. [DOI] [PubMed] [Google Scholar]
- 9.Tholouli E, Sturgess K, Dickinson RE, et al. In vivo T-depleted reduced-intensity transplantation for GATA2-related immune dysfunction. Blood. 2018;131:1383–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donadieu J, Lamant M, Fieschi C, et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica. 2018;103:1278–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson KD, Hsu AP, Ryu MJ, et al. Cis-element mutated in GATA2-dependent immunodeficiency governs hematopoiesis and vascular integrity. J Clin Invest. 2012;122:3692–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linnemann AK, O’Geen H, Keles S, Farnham PJ, Bresnick EH. Genetic framework for GATA factor function in vascular biology. Proc Natl Acad Sci U S A. 2011;108:13641–13646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Craven SE, Lim KC, Ye W, Engel JD, de Sauvage F, Rosenthal A. Gata2 specifies serotonergic neurons downstream of sonic hedgehog. Development. 2004;131:1165–1173. [DOI] [PubMed] [Google Scholar]
- 14.Nardelli J, Thiesson D, Fujiwara Y, Tsai FY, Orkin SH. Expression and genetic interaction of transcription factors GATA-2 and GATA-3 during development of the mouse central nervous system. Dev Biol. 1999;210:305–321. [DOI] [PubMed] [Google Scholar]
- 15.Scherzer CR, Grass JA, Liao Z, et al. GATA transcription factors directly regulate the Parkinson’s disease-linked gene alpha-synuclein. Proc Natl Acad Sci U S A. 2008;105:10907–10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Y, Yamamoto M, Engel JD. GATA2 is required for the generation of V2 interneurons. Development. 2000;127:3829–3838. [DOI] [PubMed] [Google Scholar]
- 17.Galera P, Hsu AP, Wang W, et al. Donor-derived MDS/AML in families with germline GATA2 mutation. Blood. 2018;132:1994–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hahn CN, Chong CE, Carmichael CL, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43:1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu AP, Sampaio EP, Khan J, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118:2653–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ostergaard P, Simpson MA, Connell FC, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet. 2011;43:929–931. [DOI] [PubMed] [Google Scholar]
- 21.Vinh DC, Patel SY, Uzel G, et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood. 2010;115:1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: Phenotype, genotype, treatment and clinical outcomes characterization of pediatric MDS/BMF patients with GATA2 mutations (GATA2), control A and control B