

Abstract
Prostate cancer remains among the leading causes of cancer-related deaths in men. Patients with aggressive disease typically undergo hormone-deprivation therapy. While treatment is initially very successful, these men commonly progress to lethal, castration resistant prostate cancer in 2–3 years. Standard therapies for castration resistant prostate cancer include second-generation anti-androgens, which prolong patient lifespan by only several months. It is imperative to advance our understanding of the mechanisms leading to resistance to identify new therapies for aggressive prostate cancer. This study identifies Notch1 as a therapeutic target in prostate cancer. Loss of Notch1 in aggressive prostate cancer cells decreases proliferation, invasion and tumorsphere formation. Therapeutic inhibition of Notch1 activity with gamma secretase inhibitors RO4929097 or DAPT in prostate cancer cells further results in decreased proliferative abilities. Loss of Notch1 and treatment of immunocompromised mice bearing prostate cancer xenografts with RO4929097 display significantly impaired tumor growth. Loss of Notch1 additionally decreased metastatic potential of prostate cancer cells in invasion assays in vitro as well as in vivo experiments. Moreover, treatment with gamma secretase inhibitors, or Notch1 gene deletion synergized with anti-androgen therapies, Enzalutamide or Abiraterone, to decrease the growth of prostate cancer cells. Combination of gamma secretase inhibitors with Abiraterone significantly inhibited cell migration and invasion, while combination with Enzalutamide reversed Enzalutamide induced migration and invasion. These collective findings suggest loss of Notch1 delays growth of CRPC, inhibits metastasis, and inhibition of Notch1 activation in conjunction with second-generation anti-androgen therapies could delay growth and progression of prostate cancer.
Keywords: Prostate cancer, Notch, molecular biology, drug therapy, combination therapy
Introduction
Prostate cancer is the most common cancer affecting men, and the second leading cause of cancer associated mortalities in men in the United States (1). Current treatments for aggressive prostate cancer, including androgen ablation therapy, have great short-term success yet frequently results in recurrence, at which point it is termed castration-resistant prostate cancer (CRPC) (2). Treatment options include second-generation anti-androgens Enzalutamide, Abiraterone, or the recently FDA approved Apalutamide; chemotherapeutic agents Docetaxel or Cabazitaxel; or Provenge prostate cancer immunotherapy (2–9). Ultimately due to the aggressive and highly metastatic nature of CRPC, patients often develop resistance to individual therapies, and to date, CRPC remains incurable. This highlights the urgent need to define new pathways that can drive the occurrence of advanced prostate cancer and evaluate novel therapies for advanced disease and metastatic CRPC.
Notch1 is a transmembrane glycoprotein belonging to the Notch family of receptors (Notch1/2/3/4). Involved in cell-cell signaling, Notch1 receptor is activated by ligands (Jagged 1/2 and Delta 1/3/4), initiating a multi-step cleavage by members of the Disintegrin Metalloprotease (ADAMs) families as well as the gamma secretase complex (10). This results in the cleavage of the intracellular domain of Notch1 (NICD1), which translocates to the nucleus (activated Notch1) as a transcriptional co-activator, regulating self-renewal, cell maintenance and tumorigenesis (11–14). Alterations of the Notch1 receptor are found in many malignancies, acting in a context-dependent manner as either oncogenes or tumor suppressors (15).
In prostate cancer, while no genetic alterations of Notch1 have been described, down-regulation of Notch1 and its ligand Jagged1 decrease cell invasion, growth, and migration (16–18). Notch1 expression promotes epithelial-to-mesenchymal transition (EMT), seminal vesicle transformation, and metastatic occurrence (18–20). Further, we previously demonstrated that Notch1 plays an important oncogenic role in the development of CRPC (21). We identified increased nuclear activated NICD1 levels in high-risk prostate cancer and CRPC patient specimens over benign and low-risk disease, and determined NICD1 acts a driver for aggressive prostate adenocarcinoma and metastatic CRPC in conjunction with other common alterations observed in prostate cancer (Myc over-expression, loss of PTEN, or activation of the Ras signaling pathway) (21). These findings indicate Notch1 is involved in the progression of prostate cancer and targeting Notch1 signaling may represent an effective therapy for aggressive disease (21).
Multiple strategies to inhibit the Notch pathway have been generated including targeting the activation of Notch receptors via gamma secretase inhibition (GSI) (22–26). In preclinical studies of epithelial cancers, GSIs as single agents have demonstrated promising anti-tumorigenic abilities, as was the case for one of the GSI’s described in this study, RO4929097 (27). Using two GSIs, RO4929097 and DAPT as well as loss of Notch1 via CRISPR-Cas9 deletion, we aimed to determine the potential for loss of Notch1 as a strategy to inhibit the growth of aggressive prostate cancer, metastasis, as well as Notch1 inhibition synergy with anti-androgen therapies.
Materials and Methods
Cell Lines and Culture.
22RV1 cells were obtained from ATCC (Manassas, VA) and C4–2 cells were a gift from Dr. Owen Witte’s laboratory at UCLA. 22RV1 Delta-Notch1 cells were generated using CRISPR-Cas9 knockout of Notch1 as previously described (21). 22RV1-Luc and 22RV1 Delta-Notch1-Luc cells were generated with lentiviral transduction of pHIV-Luc-ZsGreen, a kind gift from Bryan Welm (Addgene plasmid # 39196). All cell lines are annually authenticated. Most recently, through the Stanford Functional Genomics Facility. Cells are tested for presence of Mycoplasma biannually using the Lonza Mycoalert Detection Kit (Lonza). Cells were cultured in RPMI supplemented with 10% FBS, 1% Penicillin/Streptomycin, and 1% L-Glutamine, with warmed Trypsin/EDTA (0.25%) used for dissociation. Cells were incubated at 37C with 5% CO2.
Colony Formation Assay.
5×102 22RV1, C4–2, or 22RV1 Delta-Notch1 cells were plated per well of a six-well plate in triplicate. Cells were treated with DMSO vehicle control, Enzalutamide (5μM), Abiraterone (5μM), DAPT (50μM), or RO4929097 (20μM) (All compounds were purchased from Selleckchem, Houston, TX, catalog numbers S1250, S2246, S2215 and S1575 respectively) (28–30, 27). Cells were cultured 9 days, with media and compounds changed every third day. Colonies were then fixed with methanol and stained with 0.1% crystal violet one hour at room temperature. Plates were washed by submerging in a water bath for one hour. Colonies were counted, and colony formation rate determined (percentage), quantified as number of colonies per 5×102 cells X 100, as previously described (21).
Tumorsphere Formation Assay.
1×104 22RV1 or Delta-Notch1 cells were plated in 50% Matrigel (Corning Inc, Fremont, CA), 50% supplemented RPMI in individual wells of a 24 well plate in triplicate. Cells were grown 15 days with media changed every third day. Wells were imaged on Leica stereomicroscope. 20x images were acquired per well in four quadrants and tumorsphere number per field was quantified, including any growth larger than 100 micron largest diameter, as measured in FIJI (31).
Cell viability assay.
1×104 22RV1 or C4–2 cells were plated per well in a 96 well plate overnight. The following day media was changed to 100μl of media containing the indicated concentrations of drugs. 72 hours later, 100μl fresh media was added + 20μl Cell-Titer Blue viability reagent (Promega, Madison, WI), and plate with reagent was incubated 2 hours at 37C prior to reading fluorescence on Tecan plate reader. Relative fluorescence units (RFU’s) were graphed as fold change over vehicle control.
Cell invasion assay.
22RV1, Delta-Notch1 or C4–2 cells were serum starved overnight in RPMI. Cells were then trypsinized, counted, and 5×104 were plated on top of Matrigel coated Costar Transwell (Sigma-Aldrich, St. Louis, MO) invasion chambers that were pre-incubated with serum free RPMI. The bottom chamber with filled with RPMI supplemented with 10% FBS as a chemoattractant. In the case of drug treated conditions, cells were pre-treated 72 hours prior to plating chambers, and both supplemented and unsupplemented media were treated with appropriate inhibitors to maintain treatment conditions. Chambers were incubated 24 hours for C4–2 cells or 36 hours for 22RV1 and Delta-Notch1, then fixed with ice cold methanol. Non-invading cells were removed from top of chamber with cotton swab, then chambers were stained with 0.1% crystal violet for one hour. Chambers were washed in water, air dried over-night, then imaged under Leica stereomicroscope at 161X. Five representative images were taken per chamber (three chambers per condition), then cells were counted in each image and averaged. Experiments were performed in triplicate and analyzed using a Student’s t-test.
Matrigel Dot Assay.
2×105 C4–2 cells were resuspended in 20μl Matrigel and gently pipetted into empty well of a 12 well plate, forming 3-D dots. The dots were placed in incubator at 37°C for 20 minutes to solidify Matrigel. Media supplemented with inhibitors at aforementioned concentrations was then added, and subsequently changed every other day. Matrigel dots were imaged on Day 0 at 20X for whole dot and 80X, measuring the leading edge of the dot in four quadrants. At 5 days after plating, dots were again imaged at the leading edge in the same locations. Images were overlaid, and distance migrated was measured using FIJI. Each treatment condition was performed in triplicate. For each dot, the average of all measurements was calculated. Triplicates for each condition were then averaged and standard deviation was quantified. Student’s t-test was used for analysis. Representative images are shown. Dots were then fixed with 2% Paraformaldehyde in PBS for 10 minutes at room temperature. Wells were washed 3 X 5 minutes in PBS, and permeabilized 5 minutes with 0.1% Triton-X100. Beta-Actin (Santa-Cruz, sc-47778) was used to stain the dots overnight, followed by Alexa Fluor 594 (Abcam, Cambridge, MA), and counterstained with DAPI. Dots were re-imaged at 100X.
Immunoblotting.
Cells were harvested by scraping in PBS and lysed with RIPA lysis buffer containing protease and phosphatase inhibitors (Thermo Fisher Scientific, Carlsbad, CA). Tumor tissues were snap frozen in liquid nitrogen at time of harvest. Tissue was later homogenized in RIPA, and sonicated. BCA assay was used to quantify protein, then samples diluted in 4X Lamelli Buffer and boiled. Samples were run on Novex Tris-Glycine gels (Invitrogen, Carlsbad, CA), blocked for one hour in milk, and incubated in primary antibodies overnight in Tris buffered saline containing 0.1% Tween-20. Secondary antibodies were applied for one hour after washing, then Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, Waltham, MA) was used to develop chemiluminescent signals on IVIS Lumina imager, with Living Image Software. Antibodies include: anti-GAPDH sc-47724 (Santa Cruz Biotechnology, Dallas, TX); anti-Notch1 D1E11 #3608, anti-Cleaved Notch1 (Val1744) D3B8 #4147, (Cell Signaling Technology, Danvers, MA); anti-AR N-20, anti-Notch3 (Abcam), and secondary HRP conjugated mouse and rabbit antibodies (Santa Cruz Biotechnology, Dallas, TX).
Animals
In conducting research using animals, the investigators adhere to the laws of the United States and regulations of the Department of Agriculture. Further, all animal studies and procedures have been approved and performed in accordance with Stanford Administrative Panel on Laboratory Animal Care (APLAC), IACUC, as well as the USAMRMC Animal Care and Use Review Office (ACURO). NSG (NOD-SCID-IL2Rγ–null) mice (Jackson Laboratory, Sacramento, CA), 6–8 weeks old, were used for all experiments, and housed at Stanford University animal facilities. The length (L), width (W), and height (H) of tumors as well as animal weights were measured every third day for subcutaneous tumors. Tumor volume was calculated by (L × W × H)/2.
GSI Tumor Treatment.
Mice were castrated one week prior to cell implantation. 22RV1 or C4–2 cells (5 × 105) were injected subcutaneously (s.c.) into the flank in Matrigel. When tumors reached palpable size (50 mm3 on average), animals were randomly assigned to treatment with vehicle (control) or with RO4929097 (10 mg/kg), purchased from Selleckchem. Vehicle and RO4929097 were dissolved in 2% DMSO + 30% PEG 300 + 5% Tween + ddH20 and administered by oral gavage daily. Tumors graphed as fold change over the first measurable tumor volume of each tumor.
Delta-Notch1 Tumors.
Mice were castrated one week prior to cell implantation. 5 × 105 22RV1 (n=10) or Delta-Notch1 (n=9 due to one tumor not grafting) cells were injected s.c. into the flank in Matrigel. Measurements began 3 days after implantation. Tumors were graphed as fold change.
Intracardiac Metastasis Model.
1×105 22RV1-Luc or Delta-Notch1-Luc cells were injected into the left ventricle of the heart under isoflurane anesthesia. Bioluminescence signal in animals was imaged at 1 hour following injection procedure to ensure proper flow of cells into circulation, then at 7 and 14 days post-procedure.
Bioluminescent Imaging.
Mice were injected s.c. with 150mg/kg D-Luciferin (Perkin Elmer) in 1xPBS, and placed into isoflurane anesthesia chamber. Five minutes post-injection, animals were transferred to Lago for bioluminescent imaging (Spectral Instruments Imaging, Tucson, AZ) at Stanford Preclinical Imaging Facility at Porter Drive. Aura software was used for image acquisition and analysis. Briefly, animals were imaged 4 minutes at high binning. At time of sacrifice, animals were injected with Luciferin, euthanized, then organs were excised and ex-vivo imaging was performed on tissues, placed in a petri dish, in a solution of 300μg/ml D-Luciferin and imaged on IVIS Lumina imager.
Histology.
At time of harvest, tumor or organ tissues were fixed in 10% formalin overnight at 4°C. Tissue was then transferred to 70% ethanol, processed into paraffin and embedded. Formalin-fixed paraffin embedded (FFPE) tissue was sliced to 4 microns and attached to charged glass slides. Slides were heated to 65°C for one hour prior to deparaffinization, followed by hydration. Antigen retrieval was performed using a steamer, with 10mM sodium citrate buffer pH 6.0, or 1mM EDTA pH 8.0 (for Cleaved Notch1 Val1744 antibody only). After washing, 0.3% Hydrogen peroxide solution was applied to slides to inactivate endogenous peroxidases, then slides were blocked in 2.5% horse (rabbit antibodies) or goat serum (mouse antibodies) (Vector Laboratories, Burlingame, CA) for 1 hour at room temperature. Antibodies were applied in corresponding serum overnight at 4C in a humidified chamber. The following day, after washing, secondary mouse or rabbit HRP (Vector Laboratories) was then applied for 1 hour. DAB (Dako Laboratories/Agilent, Santa Clara, CA) was used to stain the tissue, followed by counterstain in hematoxylin, and subsequent dehydration. Slides were cover-slipped in Faramount (Dako). Imaging was performed on Hamamatsu nanozoomer and photos were taken at 40X magnification. Anti-Cleaved Notch1, anti-E-Cadherin #3195, anti-Vimentin #5741 (Cell Signaling Technology); anti-Hes1, anti-Ki67, anti-Ku70 (Santa Cruz Biotechnologies) were used for immunohistochemistry.
Results
Loss of Notch1 impairs CRPC cellular proliferation and invasion.
Based on our previous studies and others implicating Notch1 in prostate cancer progression, invasion, metastasis and CRPC development, we posited that inhibition of Notch1 would impair prostate cancer cell growth. Two GSIs, RO4929097 and DAPT, were tested for their ability to decrease activated Notch1 levels in C4–2 and 22RV1 CRPC cells, as well as our previously described 22RV1 CRISPR-Cas9 Notch1 Knockout cells (Delta-Notch1) (21). C4–2 and 22RV1 express high endogenous Notch1 and abundant activated, or cleaved NICD1 (NICD1 Val1744) (Figure 1A and Supplementary Figure 1A). GSI treatment of 22RV1 and C4–2 cells abolished activated NICD1 levels by 24 hours treatment, and was observed through 72 hours, while the two tested GSIs had no effect on NICD3 previously implicated in prostate cancer (Figure 1B and Supplementary Figure 1B).
Figure 1. Gamma Secretase Inhibition as a single therapeutic agent in prostate cancer cells impairs colony and tumorsphere formation.
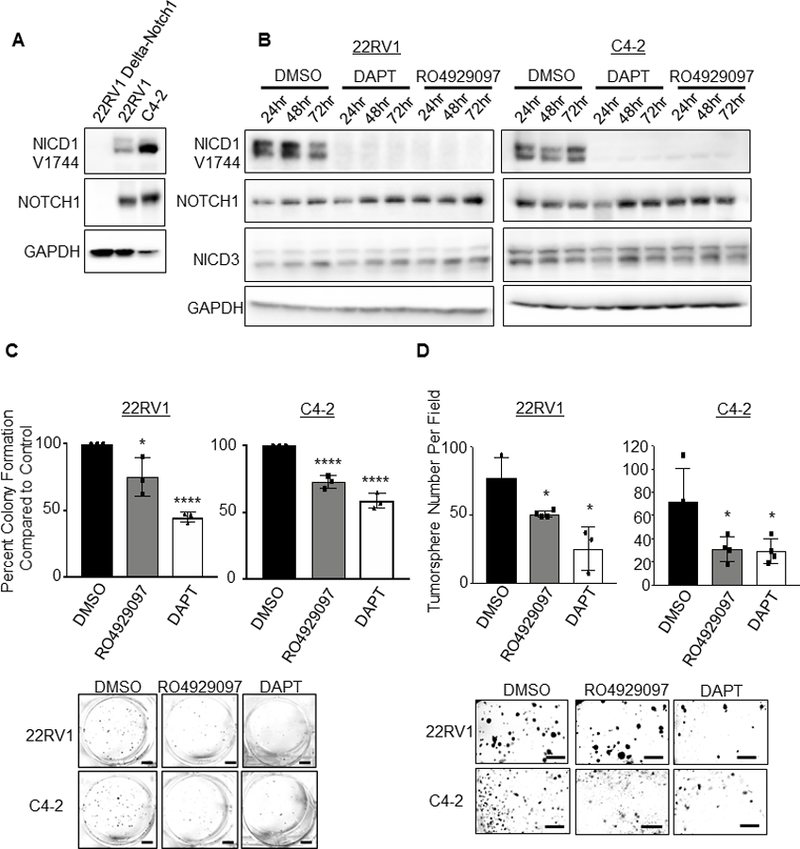
A. Immunoblot of human prostate cancer cells: 22RV1, C4–2 and 22RV1 Notch1 knock-out, Delta-Notch1. B. Immunoblot of time-course Gamma Secretase Inhibitors (GSIs) treatment in 22RV1 or C4–2 prostate cancer cells. Cells were treated 24, 48 or 72 hours with Inhibitors DAPT (50 μM) or RO4929097 (20 μM). Staining was performed for activated NICD1 (NICD1 Val1744), Notch1, NICD3, and GAPDH. C. Colony formation assay- 500 cells were plated per well in 6 well dish in triplicate. Cells were grown 9 days, with media and drugs changed every third day. Colonies were then fixed with methanol and stained with 0.1% crystal violet. Colonies were hand counted and graphed as percent colony formation over control treatment. Control treatment (DMSO) was normalized to 100%. Scale bar represents 100mm. Experiment is representative of three, performed in triplicate. D. Tumorsphere formation assay was performed with 1×104 22RV1 or C4–2 cells plated in 50% Matrigel in 24 well plate. Cells were treated with DAPT or RO4929097, then grown for 15 days with media and inhibitors changed every third day. Scale bar represents 250 mm. Error bars are ± SD. Using two-tailed Students t-test: *=P<0.05; ****=P<0.001; ns= no significance.
Extended treatment with either GSI in 22RV1 and C4–2 cells decreased the colony formation abilities of both CRPC lines (Figure 1C), as well as their ability to form tumorspheres (Figure 1D).
To eliminate concern of off-target effects of GSIs, experiments were performed with Delta-Notch1 cells compared against 22RV1 parental cells. Loss of Notch1 decreased colony formation over parental 22RV1 cells (Figure 2A). Treatment of Delta-Notch1 cells with either RO4929097 or DAPT had no effect on colony formation, suggesting the observed effects from GSI treatment appear to be dependent on Notch1 expression (Figure 2B). Loss of Notch1 was additionally responsible for decreased tumorsphere formation (Figure 2C) of 22RV1 cells. Delta-Notch1 tumorsphere numbers were not affected by the addition of either RO4929097 or DAPT (Figure 2D). These data indicate NICD1 expression to be important for CRPC cellular proliferation, and enforce the specificity of the Delta-Notch1 knock-out cells.
Figure 2. Loss of Notch1 inhibits prostate cancer cell proliferation and tumorsphere formation.
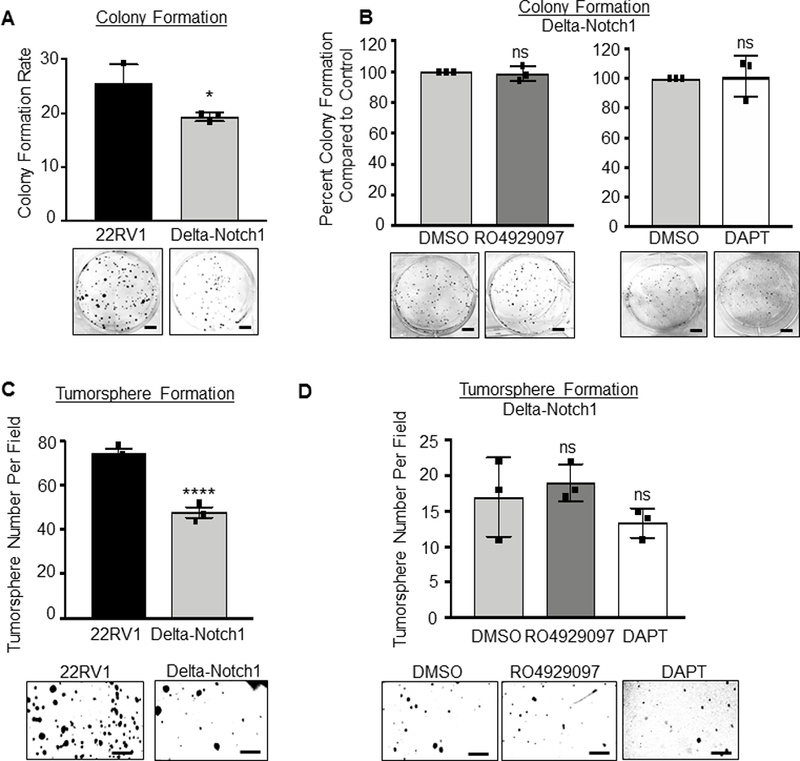
In vitro assays were performed on 22RV1 and Delta-Notch1 cells. A, B. Colony formation assay- 5×102 cells were plated per well in 6 well dish. Cells were grown 9 days, with media changed every third day. Colonies were then fixed with methanol and stained with 0.1% crystal violet. Colonies were hand counted and graphed as Colony Formation Rate (A) for 22RV1 and Delta-Notch1 cells, or percent colony formation compared to vehicle control (DMSO, normalized to 100%) for treated Delta-Notch1 cells (B). Delta-Notch1 cells were vehicle treated, RO4929097 treated (20 μM) or DAPT treated (50 μM). Scale bars represent 100mm. C, D. Tumorsphere formation assay- 1×104 22RV1 and Delta-Notch1 cells (C), or Delta-Notch1 cells treated with vehicle, RO4929097 (20 μM) or DAPT (50 μM) (D) were plated in 50% Matrigel with RPMI in a 24 well plate. Cells were grown 15 days with media changed every third day. Scale bar represents 250 mm. Experiments performed in triplicate with representative experiments shown. Using two-tailed Students t-test: *=P<0.05; ****=P<0.001; ns= no significance. Error bars are ± SD.
Loss of Notch1 by gene deletion or therapeutic inhibition impairs CRPC tumor growth.
To determine the effects of GSIs in vivo, subcutaneous 22RV1 or C4–2 xenografts were implanted in castrated NSG mice to mimic androgen ablation in vivo. RO4929097 (10mg/kg) or vehicle control was administered via daily oral gavage. Fold change in tumor volume growth was markedly reduced in RO4929097 animals (Figure 3A) with no observed toxicity (Supplementary Figure 2). Notch1, cleaved Notch1 (NICD1 Val1744) and androgen receptor (AR) expression were confirmed by immunoblotting (Figure 3B). Interestingly, there was a decrease in AR in C4–2 cells known to express full length AR, which was not observed in 22RV1 expressing AR and AR-V7 (Figure 3A, B Supplementary Figure 3). The mechanism through which inhibition of Notch activation regulates AR remains to be determined. Next, 22RV1 or Delta-Notch1 cells were implanted into castrated NSG mice. Delta-Notch1 tumors immediately displayed delayed growth, continued until time of sacrifice- 30 days post-implantation when compared to the control animal tumor volume (Figure 3C). As expected, Notch1 and NICD1 expression were undetectable in Delta-Notch1 tumors (Figure 3D). Immunoblots were quantified in Supplementary Figure 3. Histological analysis of all tissues demonstrated reduction or loss of NICD1 in all treatment groups (Figure 3E). This corresponded to a decrease of common Notch1 target gene, Hes1. Proliferation was reduced in RO4929097 treatment as evidenced by Ki67 staining. We further noted substantial reversal of EMT markers upon loss of Notch1 or treatment with R04929097 observed via increased E-Cadherin corresponding to decreased Vimentin in all treatment groups (Figure 3E). These experiments demonstrate that Notch1 may potentially represent a therapeutic target for CRPC.
Figure 3. Therapeutic inhibition of Notch1 impairs tumor formation of castration resistant prostate cancer.
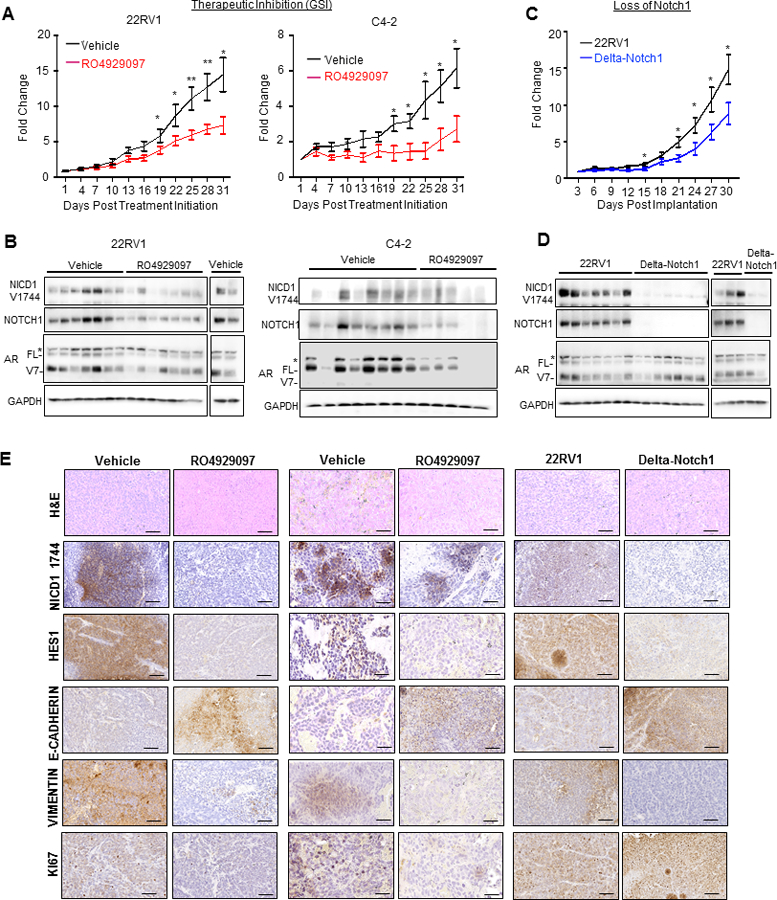
A. 5×105 22RV1 or C4–2 cells were implanted s.c. into castrated NSG mice (22RV1 N=9 Vehicle, 22RV1 N=7 RO4929097; C4–2 N=11 Vehicle; C4–2 N=10 RO4929097). Tumors were grown to palpable size (50 mm3) and randomly distributed. At 50 mm3 treatment was initiated (plotted as Day 1). Animals were gavaged daily at 10 mg/kg of RO4929097 or vehicle control. B. Immunoblot analysis of 22RV1, or C4–2 tumor tissue for NICD1 V1744, Notch1, AR and GAPDH. * represents a non-specific band, FL= Androgen Receptor full length, and V7 =Androgen Receptor splice variant 7. C. 5×105 22RV1 or Delta-Notch1 cells were implanted s.c. into castrated NSG Mice (n=10). Tumor height (h), width (w) and length (l) were measured every third day and tumor volumes calculated as: (h x w x l)/2. Tumors graphed as fold change ± SEM, and analyzed for each time point by Student’s t-test. *=P<0.05; **=P<0.01. D. Immunoblot analysis of 22RV1 or Delta-Notch1 tumor tissue for NICD1 V1744, Notch1, AR and GAPDH. * represents a non-specific band, FL= Androgen Receptor full length, and V7 =Androgen Receptor splice variant 7. E. Immunohistochemistry of FFPE tumor tissue from 22RV1 and C4–2 xenografts treated with RO4929097 as well as Delta-Notch1 for NICD1 1744, Hes1, E-Cadherin, Vimentin and Ki67, as well as histological analysis with H&E. Images were taken at 40X and are representative of treatment group. Scale bars represent 50μm.
Loss of Notch1 decreases metastatic potential of CRPC.
CRPC is highly metastatic in patients, with metastases most commonly occurring in bone, and secondarily in soft tissues such as liver (32). We first tested whether loss of Notch1 or therapeutic inhibition with GSI’s would decrease CRPC cells invasive behavior in vitro. Inhibition of Notch1 through treatment with DAPT (50μM) or RO4929097 (20μM) significantly impaired invasion in 22RV1 and C4–2 cells (Figure 4A). Similar effect was observed in Delta-Notch1 cells (Figure 4B).
Figure 4. Loss of Notch1 decreased metastatic potential of CRPC.
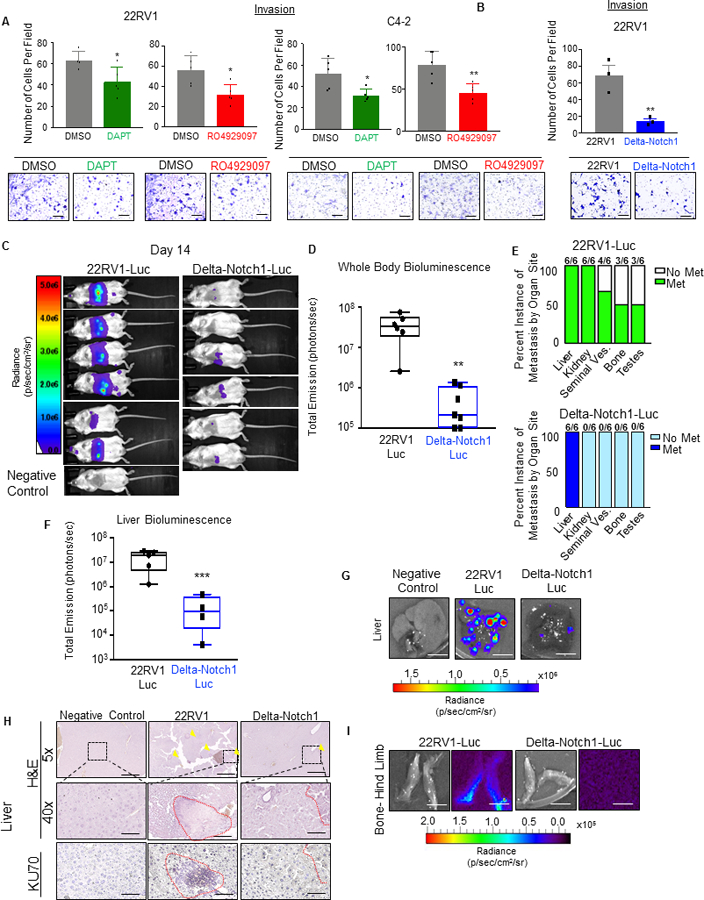
A. 22RV1 or C4–2 cells were pre-treated 72 hours with RO4929097 (20 μM) or DAPT (50 μM). Cells were serum starved overnight and 5×104 were plated in Matrigel invasion chambers and incubated 36 or 24 hours respectively. Scale bars = 250μm. B. 22RV1 or Delta-Notch1 cells were serum starved overnight, then 5×104 were plated in Matrigel transwell chambers. Chambers were incubated 36 hours, then fixed with methanol, and stained with crystal violet. For each condition, 5 fields were captured per chamber, number of cells per field were counted, then averaged for three chambers. Error bars represent ±SD. Scale bars = 250 μm C. 1×105 22RV1-Luc or Delta-Notch1-Luc cells were injected intracardiac into NSG mice. Animals were subjected to intraperitoneal (i.p.) injection with 150mg/kg D-Luciferin and imaged at 0 hours, 7 days and 14 days. Final imaging at 14 days is pictured with all animals set to the same radiance scale. D. Quantification of whole animal bioluminescence is plotted as total emission (photons/sec), representing each value in a box and whisker plot on Log10 scale. E. Ex vivo imaging was performed on all organs, with signal maintained in 300μg/ml D-Luciferin after harvest. Instance of organs with positive bioluminescence was graphed out of 6 animals. F. Liver bioluminescence was set to the same radiance scale for all animals, then quantified as radiance (p/sec/cm2/sr) and graphed on Log10 scale in box and whisker plot. G. Representative bioluminescent images of liver from 22RV1-Luc, Delta-Notch1-Luc or negative control (uninjected with cells) animals. Scale bar = 1cm. H. Excised livers were fixed in formalin and paraffin embedded. Tissues were used to perform H&E (5X and 40X) as well as IHC for human KU70. Yellow arrows indicate visible metastatic lesions. Scale bars are 500μm and 50 μm, respectively. I. Representative bioluminescent images of bone metastasis from 22RV1-Luc or Delta-Notch1 conditions. Scale bar = 1cm. *=P<0.05; **=P<0.01; ***=P<0.005
Preclinical models of prostate cancer often struggle to recapitulate the human metastatic phenomenon of bone metastasis. However, 22RV1 cells implanted through intracardiac injection do commonly colonize in the hind limb of NSG mice as well as soft tissue (33). Using this model, we implanted 22RV1-Luc or Delta-Notch1-Luc cells via intracardiac injection to determine whether loss of Notch1 impairs metastasis (Figure 4C–I). Two weeks after implantation, Delta-Notch1-Luc animals had markedly lower detectable bioluminescence than their 22RV1-Luc counterparts, indicative of significantly reduced metastasis (Figure 4C) and quantified as photons/second (Figure 4D). The following day, mice were sacrificed and ex vivo imaging was performed on the mice, 5 minutes post Luciferin injection (Figure 4E). Organs imaged included liver, kidneys, heart, lungs, prostate, seminal vesicles, bladder, and hind limbs, and the remainder of the animal was imaged to ensure no bioluminescent signal remained (Figure 4E). Strikingly, Delta-Notch1-Luc cells only colonized the mouse liver (6/6 animals), while the 22RV1-Luc group presented metastases in the liver (6/6), kidney (6/6), seminal vesicles (4/6), bone (3/6) and testes (3/6) (Figure 4E).
Average liver bioluminescence was quantified and compared between the two groups after subtracting the background signal of a negative control (uninjected) animal from each liver signal (Figure 4F). As apparent in whole body images and liver sections, liver colonization was drastically impaired by the loss of Notch1 (Figure 4F–H). Representative liver bioluminescence is depicted in Figure 4G. Bone samples were also collected (representative luminescent images in Figure 4I). Liver samples (FFPE) were used to perform histological comparisons between 22RV1-Luc, Delta-Notch1-Luc and a negative uninjected control. Human Ku70, a protein involved with non-homologous end-joining and the repair of DNA double-strand breaks, was used as proxy for human specific staining of 22RV-Luc and Delta-Notch1-Luc cells (Figure 4H). We observed highly infiltrated livers in the 22RV1-Luc group depicted in histological invasion of human epithelial cells into mouse livers, with significantly fewer observations in Delta-Notch1 tissue, consistent with bioluminescent findings (Figure 4H). Collectively, these results demonstrate that loss of Notch1 significantly impaired the metastatic potential of 22RV1 CRPC cells.
Notch1 inhibition synergizes with anti-androgens Enzalutamide and Abiraterone to inhibit prostate cancer cell proliferation in vitro.
Inhibition of Notch receptor activation has demonstrated a unique ability to sensitize tumors to chemotherapies (34–36). We previously demonstrated expression of nuclear NICD1 in CRPC patient samples, reflecting Notch1 activation is significantly elevated in CRPC (21). To determine whether Notch1 inhibition could sensitize CRPC to second-generation anti-androgens, we utilized RO4929097 and DAPT to treat 22RV1 cells in vitro alone, or in conjunction with Enzalutamide (ENZ) or Abiraterone (ABI). Effects of combination therapy on cellular viability were modest suggesting low toxicity, with the greatest combined impact from Abiraterone combined with either GSI (Supplementary Figure 4).
Cells were then subjected to a colony formation assay with either RO4929097 or DAPT in combination with ENZ or ABI. As expected, treatment with ENZ as an independent drug in 22RV1 cells did not impact cell growth due to the known presence of Androgen Receptor Splice Variant 7 (AR-V7) (Figure 5A, B and Supplementary Figure 5). In contrast to the short-term viability assay, GSI combination with ENZ synergized to decrease CRPC cell growth measured by colony formation (Figure 5A, B). These effects were even more pronounced when performed with ABI (Figure 5C, D and Supplementary Figure 5).
Figure 5. Notch1 inhibition synergizes with anti-androgens Enzalutamide and Abiraterone to inhibit prostate cancer cell proliferation in vitro.
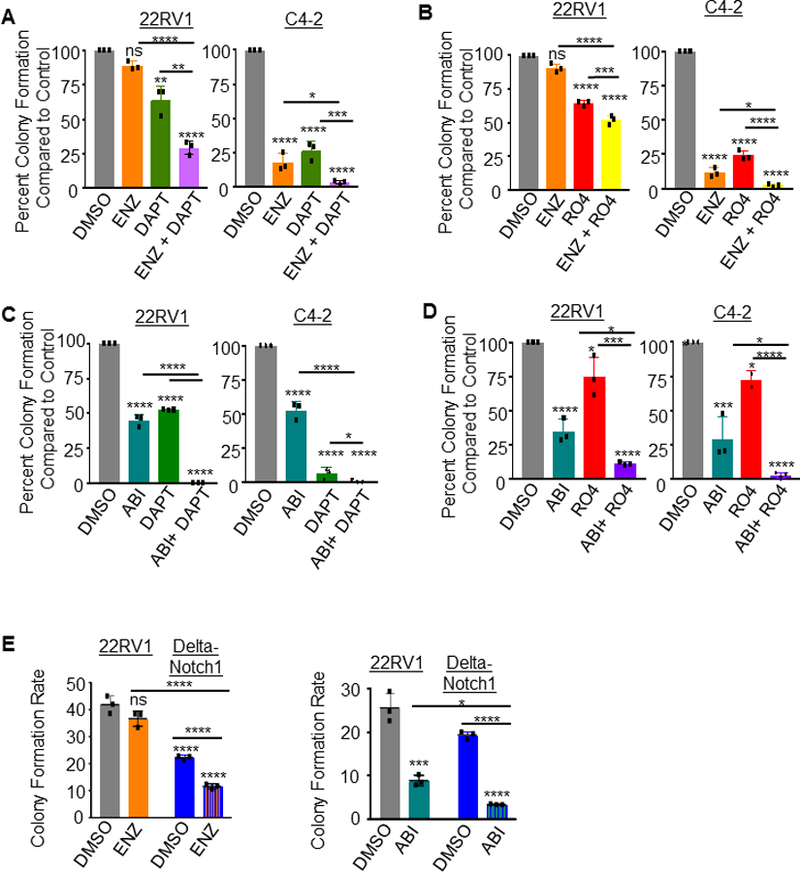
A. Colony formation assay- 5×102 22RV1 or C4–2 cells were plated per well in 6 well dish, treated with vehicle (DMSO) control, Enzalutamide (ENZ) 5 μM; DAPT 50 μM, ENZ + DAPT, or B. DMSO; ENZ; RO4929097 (RO4) 20 μM; or ENZ + RO4. Cells were grown 9 days, with media and drugs changed every third day. Colonies were then fixed with methanol and stained with 0.1% crystal violet. Control treatment (DMSO) was normalized to 100%. C. Colony formation assay was performed for DMSO control, Abiraterone (ABI) 5 μM, DAPT 50 μM, ABI + DAPT, or D. DMSO, ABI, RO4929097 (RO4) 20 μM, or ABI + RO4. Colonies were hand counted and graphed as Percent Colony Formation compared to control (vehicle). Control treatment (DMSO) was normalized to 100%. E. Colony formation of 22RV1 compared against Delta-Notch1 cells in the presence of ENZ, ABI or DMSO control. Graphed as colony formation rate to compare across cell lines. All experiments performed in triplicate with representative images shown. *=P<0.05; ***=P<0.005; ****=P<0.001; ns=no significance. Error bars +/− SD.
To confirm the specificity of this effect to inhibition of Notch1, we performed the colony formation combination therapy experiments in Delta-Notch1 cells. Delta-Notch1 cells exhibited decreased colony formation potential compared to 22RV1 parental cells as previously observed, and further synergized with the addition of ENZ or ABI to impair proliferative colony formation (Figure 5E and Supplementary Figure 5).
Notch1 inhibition synergizes with anti-androgens Enzalutamide and Abiraterone to inhibit prostate cancer cell invasion and migration.
As we have observed, loss of Notch1 leads to a significant decrease in the metastatic potential of CRPC in vitro and in vivo. We further assessed whether combinatorial treatment of GSIs with anti-androgen therapy would affect invasive potential of CRPC cells. We utilized two assays of cell motility. First, traditional Matrigel transwell assays were plated with 22RV1 or C4–2 cells pretreated for 72 hours with the same concentrations of DAPT, RO4929097, ENZ, ABI or combination thereof as described above. At the time of plating for invasion, cells were counted again to ensure plating of equal number of cells. After 24 hour incubation for C4–2 cells and 36 hour incubation for 22RV1 cells, chambers were harvested, fixed and stained with crystal violet. As we already established, DAPT and RO4929097 inhibited invasion in both 22RV1 and C4–2 cells. ENZ, while an effective short-term therapeutic for CRPC patients, it was recently reported that ENZ increases CRPC invasiveness (37). Enzalutamide displayed a consistent increase in invasion in our assays (Figure 6A, B). To this end, combination of ENZ with either GSI had invasion levels similar to that of DAPT or RO4929097 (Figure 6A, B). This suggested that co-treatment with GSI’s ablated the effects of ENZ induced cellular invasion. ABI had no effect on cellular invasive potential, and in combination, ABI with DAPT or RO4929097 synergized to significantly decrease invasion (Figure 6A, B).
Figure 6. Notch1 inhibition synergizes with anti-androgens Enzalutamide and Abiraterone to inhibit prostate cancer cell invasion and migration in vitro.
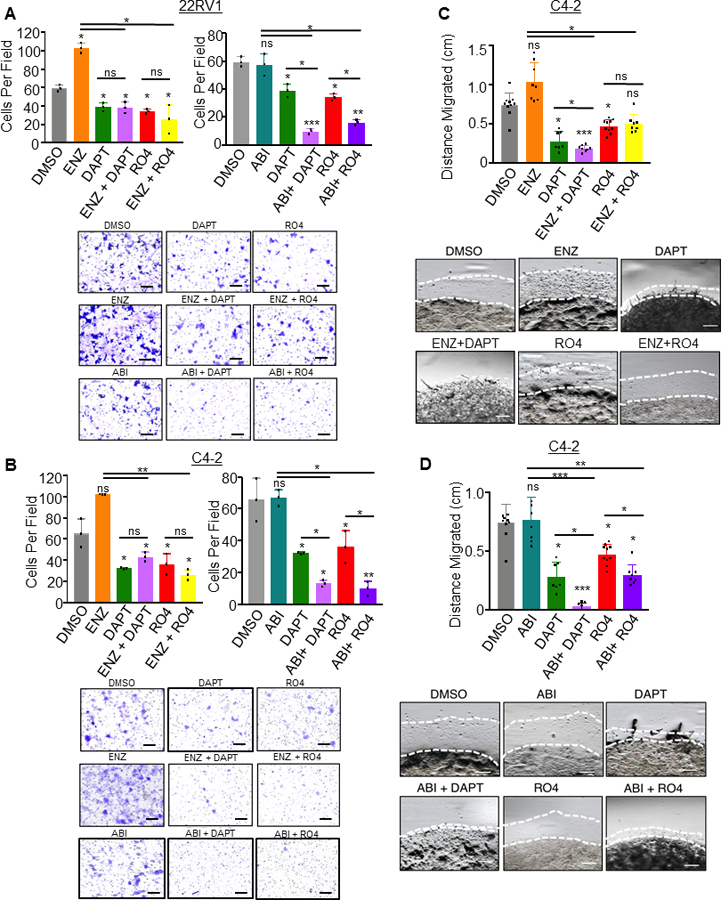
For invasion assays, 5×104 cells were plated in Matrigel coated transwell invasion chambers. Cells were drug treated 72 hours prior to serum starving overnight, and plating in chambers. A. 22RV1 cells were treated with DAPT (50 μM), ENZ (5 μM), ABI (5 μM), RO4929097 (RO4) (20 μM), or combination of ENZ with DAPT or RO4 and ABI with DAPT or RO4. B. C4–2 cells were treated with DAPT (50 μM), ENZ (5 μM), ABI (5 μM), RO4929097 (RO4) (20 μM), or combination of ENZ with DAPT or RO4 and ABI with DAPT or RO4. Chambers were incubated 36 hours for 22RV1 and 24 hours for C4–2 cells, then fixed in methanol and stained in 0.1% crystal violet. Five images were captured per chamber at 161X, performed in triplicate chambers and averaged. Experiments were performed concurrently and graphed separately for each cell line for ease of visualization, thus DMSO, DAPT and RO4 conditions are based on the same samples in these graphs. C,D. 2×105 C4–2 cells were resuspended in 20 μl Matrigel and plated as a 3D dot on a 12 well plate. After Matrigel solidified, media was added and wells were treated with ENZ, DAPT, RO4, combined ENZ with DAPT or RO4 (C) or ABI, DAPT, RO4 or combined ABI with DAPT or RO4. Media was changed every 48 hours. Dots were imaged at four leading edges at Day 0 and Day 5 and distance migrated (millimeters mm) was calculated. ENZ and ABI experiments were performed concurrently, then graphed separately for ease of visualization, thus in (C) and (D), DMSO, DAPT and RO4 conditions are based on the same samples. Scale bars = 250μm. All experiments performed in triplicate with representative images shown. *=P<0.05; **=P<0.01; ***=P<0.005; ns= no significance. Error bars +/− SD.
To further validate this phenotype, we performed a Matrigel Dot assay in which C4–2 cells were plated in 100% Matrigel as a 3D dot on a cell culture plate. These dots were treated with GSI and anti-androgen combination therapy for a total of 5 days. The dots were imaged at Day 0 and Day 5 allowing us to measure the distance cells migrated outside of the Matrigel Dot. Results for C4–2 cell migration confirmed the observed invasion phenotype in that either DAPT or RO4929097 decreased cell migration (Figure 6C,D, and Supplementary Figure 6). Further, ENZ again increased cell migration ability, and GSIs were able to reverse and significantly decrease migratory ability of ENZ treated cells (Figure 6C and Supplementary Figure 6A). While ABI had no effect on migration, ABI strongly synergized with GSIs to decrease migration (Figure 6D and Supplementary Figure 6B). This experiment was performed for 22RV1 cells as well but they did not escape the Matrigel capsule at the experimental time-point and thus migration measured by Matrigel Dot assay was not assessed in these cells (Supplementary Figure 7).
Our study suggests that loss of Notch1 through genetic deletion or therapeutic inhibition sensitized CRPC cells to anti-androgens to decrease cell growth and in the case of ABI, invasion and migration and inhibit ENZ driven invasion and migration. While GSI in combination with ENZ is also a potent inhibitor of cell growth, these combinations play an additional role in reversing an ENZ induced increase invasive phenotype in vitro.
Discussion
A recent study corroborates our findings in which GSI inhibits oncogenic activity associated with prostate cancer cells (38). This study suggests the effect of GSI treatment on prostate cancer cells is due to inhibition of Notch3, based on an observation of upregulated Notch3 mRNA in publicly available prostate cancer patient datasets. Our studies previously implicated Notch1 upregulation as a driver of prostate adenocarcinoma in conjunction with other alterations of prostate cancer, with increased Notch1 levels correlating with prostate cancer Gleason grade and CRPC (21). Herein, we inhibited Notch through GSI treatment as well as Notch1 knockout, and observe comparable phenotypes of decreased oncogenesis, supporting Notch1 as a therapeutic target, but not Notch3.
Work by Mohamed et al. found GSI can synergize with ENZ, ABI, as well as another anti-androgen, Bicalutamide to decrease cell proliferation, viability, and apoptosis of ERG positive prostate cancer cells (VCaP), as Notch1 and 2 were found to be targets of ERG (39). ERG is an ETS family transcription factor commonly found increased in prostate cancer in roughly 50% of patients due to gene fusions with TMPRSS2 serine protease (40). While this study suggests there is no impact of ERG negative cells when GSI therapy is attempted, CRPC cell lines were not tested. 22RV1 and C4–2 are both ERG-negative CRPC cell lines. These cells being affected by Notch1 loss or inhibition, paired with our previous work observing increased NICD1 in patient samples in progression to CRPC, reflect Notch1 signaling increases in CRPC, likely through an alternative ERG independent mechanism.
In this manuscript we determined that loss of Notch1 impaired in vitro invasion and in vivo metastatic colonization of 22RV1 cells. Our previous work determined that Notch1, when acting as a driver of prostate cancer progression to CRPC in combination with other known alterations in prostate cancer enriches for an epithelial to mesenchymal transition (EMT) (21). Further, overexpression of Notch1 increases invasion of prostate cancer cells (18). We verified this signature in Delta-Notch1 tumors as well as C4–2 or 22RV1 tumors treated with RO4929097. Not only were tumor volumes less with RO4929097 treatment or loss of Notch1, but upon analyzing the pathology of the tumors we observed increases in epithelial marker E-Cadherin, as well as a decrease of Vimentin, a mesenchymal cell marker, indicating an EMT reversal. This observation of Notch1 inhibition reversing EMT could attribute to the decreased invasive and metastatic properties we observed herein.
Loss of Notch1 in this study synergized with ABI or ENZ to decrease prostate cancer growth and invasion. These anti-androgen therapies offered as treatment in hormone refractory prostate cancer patients often result in resistance. Several studies have determined that androgen deprivation therapies such as ENZ can themselves cause EMT and lead to resistance to ENZ therapy (37). Interestingly, co-treatment with GSI prevented the ENZ induced increase of prostate cancer cell invasion, and in the case of ABI, synergize to decrease cellular invasion and migration. Notch1 and AR, both transcription factors, have no known direct interactions, or reported overlapping downstream targets in prostate cancer. One mechanism in which Notch1 and AR have cross-talk is hypoxia, involved in prostate cancer progression and is associated with hormone therapy resistance due to regulation of PSA and AR target genes (41). It has been previously reported that hypoxia requires Notch signaling to maintain undifferentiated cell states common to hypoxic environments (42). Therefore, it stands to reason that in hypoxic environments, Notch1 may be involved in hypoxia-associated androgen-resistance. While transcription factor cross-talk amongst Notch1 and AR target genes could be involved, Notch receptors have been implicated in chemoresistance independent of AR. In these studies, it was shown that another GSI, PF-03084014 targets a well-known facet of Notch1 signaling, stem cells. Inhibition of prostate cancer stem cells with GSI was responsible for sensitizing chemoresistant cells to Docetaxel (34, 36). These mechanisms should be explored to determine the full potential of Notch1 regulated hormone therapy resensitization. Another possibility is that Notch1 regulation of stem cell renewal is independent of AR. Zhang et al. demonstrated that introduction of NICD1 into AR negative PC3 cells increased tumorsphere formation as well as cancer stem cell surface markers, contributing to self-renewal (18). In the prostate, Notch1 signaling can occur downstream of TGFβ, a well-known signal transduction pathway which maintains prostate stem cell quiescence (43). Together, Notch1 and TGFβ aid in maintaining prostate basal cell lineage, such that inhibition on Notch1 signaling may delay prostate cancer by delaying basal cell to luminal cell differentiation (43).
Although GSI’s have been tested in clinical trials for epithelial cancers with promising results, they have not demonstrated durable anti-tumor effects as single agents (22). Despite common high toxicity issues surrounding GSIs, RO4929097 was safely administered to the animals in our preclinical studies, as well as in clinical trials co-administered with multiple cancer treatments (44–47). Combination therapy with GSI has shown promising partial responses in solid tumors, even sensitizing chemoresistant CRPC (34, 35). Several new strategies are being examined for clinical inhibition of Notch1. Small interfering RNA (siRNA) for Notch1 have shown promising in vivo results in models of prostate cancer, where siRNA was delivered to prostate specific membrane antigen (PSMA) positive prostate cancer cells, inducing apoptosis (48). Notch1 siRNA therapy has additionally demonstrated oncogenic inhibition of in vivo models of gastric cancer and melanoma (49, 50). Likewise, monoclonal antibodies specifically inhibiting Notch1 by targeting the negative regulatory region (NRR) of the Notch1 receptor have shown promising tumor growth inhibition in adenoid cystic carcinoma (ACC) and T-cell acute lymphoblastic leukemia (T-ALL) and some are undergoing clinical trials for treatment of solid tumors (25, 51–53).
Our study demonstrates the merit of further studying Notch1 inhibition as a treatment for CRPC as a single agent, as well as in combination with anti-androgen therapy. These treatments have the potential for delaying resistance to effective CRPC therapies, and resensitization of patients who are hormone refractory. It is likely that the benefit of Notch1 targeted therapies could extend to an even broader patient population, as PTEN loss, one of the most common genetic aberrations in prostate cancer has been linked to Notch1 expression, and Notch1 inhibition has been demonstrated to synergize with anti-androgen therapies to induce apoptosis (39, 54). These findings in conjunction with our results demonstrate that Notch1 inhibition may represent a promising targeting strategy in a wide array of prostate cancer patients.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. James Brooks and Dr. Donna Peehl for their thoughtful guidance and suggestions and the continued support of the Canary Foundation.
Financial support: T.S. is supported by the Canary Foundation, the National Institute of Health/National Cancer Institute (NCI) K99/R00 Pathway to Independence Award 4R00CA184397 and R03CA230819, the U.S. Army Medical Research Acquisition Activity through the Congressionally Directed Medical Research Program (CDMRP) under Award No. W81XWH1810323, the McCormick and Gabilan Faculty Award. M.A.R. is supported by the U.S. Army Medical Research Acquisition Activity, through the Congressionally Directed Medical Research Program (CDMRP) under Award No. W81XWH1810141. Opinions, interpretation, conclusions and recommendations are those of the authors and not necessarily endorsed by the U.S. Army.
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Siegel RL, Miller KD, & Jemal A. Cancer statistics, 2018 CA: A Cancer Journal for Clinicians; 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Feldman B & Feldman D. The development of androgen-independent prostate cancer. Nature Reviews 2001;1:34–45. [DOI] [PubMed] [Google Scholar]
- 3.Smith MR, Saad F, Chowdhury S, Oudard S, Hadaschik BA, Graff JN, et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N Engl J Med 2018;378:1408–1418. [DOI] [PubMed] [Google Scholar]
- 4.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CB, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012;367:1187–1197. [DOI] [PubMed] [Google Scholar]
- 5.Beer TM, Armstrong AJ, Ranthkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 2014;371(5):424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hussain M, Fizazi K, Saad F, Rathenbord P, Shore N, Ferreira U, et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N Engl J Med 2018;378:2465–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 2011;364:1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010;363:411–422. [DOI] [PubMed] [Google Scholar]
- 9.Oudard S, Fizazi K, Sengelov L, Daugaard G, Saad F, Hansen S, et al. Cabazitaxel Versus Docetaxel As First-Line Therapy for Patients With Metastatic Castration-Resistant Prostate Cancer: A Randomized Phase III Trial-FIRSTANA. J Clin Oncol 2017;35:3189–3197. [DOI] [PubMed] [Google Scholar]
- 10.Schroeter E, Kisslinger J, & Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 1998;393:382–386. [DOI] [PubMed] [Google Scholar]
- 11.Artavanis-Tsakonas S, Rand MD & Lake RJ. Notch Signaling: Cell Fate Control and Signal Integraton in Development. Science 1999;284:770–776. [DOI] [PubMed] [Google Scholar]
- 12.Struhl G & Adachi A. Nuclear access and action of Notch in vivo. Cell 1998;93:649–660. [DOI] [PubMed] [Google Scholar]
- 13.De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 1999;398:512–522. [DOI] [PubMed] [Google Scholar]
- 14.Jarriault S, Brou C, logeat F, Schroeter EH, Kopan R & Israel A. Signaling downstream of activated mammalian Notch. Nature 1995;37:355–358. [DOI] [PubMed] [Google Scholar]
- 15.Ranganathan P, Weaver KL, & Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer 2011;11:338–351. [DOI] [PubMed] [Google Scholar]
- 16.Santagata S, Demichelis F, Riva A, Varambally S, Hofer MD, Kutok JL, et al. JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res 2004;64:6854–6857. [DOI] [PubMed] [Google Scholar]
- 17.Zhu H, Zhou X, Redfieid S, Lewin J & Miele L. Elevated Jagged-1 and Notch-1 expression in high grade and metastatic prostate cancers. Am. J. Transl. Res 2013;5:368–378. [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, Sha J, Yang G, Huang X, Bo J & Huang Y. Activation of Notch pathway is linked with epithelial-mesenchymal transition in prostate cancer cells. Cell Cycle 2017;16:999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwon OJ, Zhang L, Wang J, Su Q, Feng Q, Zhang XH, et al. Notch promotes tumor metastasis in a prostate-specific Pten-null mouse model. J Clin Invest 2016;126:2626–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng G, Zheng X, Jiang P, Chen K, Wang X, Jiang K, et al. Notch1 suppresses prostate cancer cell invasion via the metastasis-associated 1-KiSS-1 metastasis-suppressor pathway. Oncol Lett 2017;14:4477–4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stoyanova T, Riedinger M, Lin S, Faltermeier CM, Smith BA, Zhang KX, et al. Activation of Notch1 synergizes with multiple pathways in promoting castration-resistant prostate cancer. Proc Natl Acad Sci U S A 2016;113:E6457–E6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purow B Notch inhibition as a promising new approach to cancer therapy. Adv. Exp. Med. Biol 2012;727:305–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takebe N, Nguyen D, & Yang S. Targeting notch signaling pathway in cancer: clinical development advances and challenges. Pharmacology & Therapeutics 2014;141:140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 2006;444:1083–1087. [DOI] [PubMed] [Google Scholar]
- 25.Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010;464:1052–1057. [DOI] [PubMed] [Google Scholar]
- 26.Moellering R, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC et all. Direct inhibition of the NOTCH transcription factor complex. Nature 2009;462:182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luistro L, He W, Smith M, Packman K, Vilenchik M, Carvajal D, et al. Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Res 2009;69:7672–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009;324:787–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pinto-Bazurco Mendieta MA, Megri M, Jagusch C, Muller-Vleira U, Lauterbach T & Hartmann RW. Synthesis, biological evaluation, and molecular modeling of abiraterone analogues: novel Cyp17 inhibitors for the treatment of prostate cancer. J Med Chem 2008;51:5009–18. [DOI] [PubMed] [Google Scholar]
- 30.Dovey HF, John V, Anderson JP, Chen LZ, de Saint Andrieu P, Fang LY, et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. Journal of Neurochemistry 2001;76:173–181. [DOI] [PubMed] [Google Scholar]
- 31.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halabi S, Kelly WK, Ma H, Zhou H, Solomon NC, Fizazi K, et al. Meta-Analysis Evaluating the Impact of Site of Metastasis on Overall Survival in Men With Castration-Resistant Prostate Cancer. J Clin Oncol 2016;34:1652–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drake JM, Gabriel CL, & Henry MD. Assessing tumor growth and distribution in a model of prostate cancer metastasis using bioluminescence imaging. Clin Exp Metastasis 2015;22:674–684. [DOI] [PubMed] [Google Scholar]
- 34.Cui D, Dai J, Keller JM, Mizokami A, Xia S & Keller ET. Notch pathway inhibition using PF03084014, a gamma-secretase inhibitor (GSI), enhances the anti-tumor effect of docetaxel in prostate cancer. Clin Cancer Res 2015;21:4619–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang W, Wang L, Mizokami A, Shi J, Zou C, Dai J, et al. Down-regulation of E-cadherin enhances prostate cancer chemoresistance via Notch signaling. Chin J Cancer 2017;36:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, et al. Suppression of Acquired Docetaxel Resistance in Prostate Cancer through Depletion of Notch- and Hedgehog-Dependent Tumor-Initiating Cells. Cancer Cell 2012;22:373–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J, Li L, Yang Z, Luo J, Yeh S & Chang C. Androgen-deprivation therapy with enzalutamide enhances prostate cancer metastasis via decreasing the EPHB6 suppressor expression. Cancer Lett 2017;408:155–163. [DOI] [PubMed] [Google Scholar]
- 38.Cui J, Wanh Y, Dong B, Qin L, Wang C, Zhou P, et al. Pharmacological inhibition of the Notch pathway enhances the efficacy of androgen deprivation therapy for prostate cancer. Int J Cancer 2018;143:645–656. [DOI] [PubMed] [Google Scholar]
- 39.Mohamed AA, Tan SH, Xavier CP, Huang W, Ravindranath L, Jamal M, et al. Synergistic Activity with NOTCH Inhibition and Androgen Ablation in ERG-Positive Prostate Cancer Cells. Mol Cancer Res 2017;15:1308–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshimoto M, Joshua AM, Chilton-Macneill S, Bayani J, Selvarajah S, Evans AJ, et al. Three-color FISH analysis of TMPRSS2/ERG fusions in prostate cancer indicates that genomic microdeletion of chromosome 21 is associated with rearrangement. Neoplasia 2006;8:465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horii K, Suzuki Y, Kondo Y, Akimoto M, Nishimura T, Yamabe Y, et al. Androgen-dependent gene expression of prostate-specific antigen is enhanced synergistically by hypoxia in human prostate cancer cells. Mol Cancer Res 2007;5:383–391. [DOI] [PubMed] [Google Scholar]
- 42.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell 2005;9:617–628. [DOI] [PubMed] [Google Scholar]
- 43.Valdez JM, Zhang L, Su Q, Dakhova O, Zhang Y, Shahi P, et al. Notch and TGFbeta form a reciprocal positive regulatory loop that suppresses murine prostate basal stem/progenitor cell activity. Cell Stem Cell 2012;11:676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richter S, Bedard PL, Chen EX, Clarke BA, Tran B, Hotte SJ, et al. (2014) A phase I study of the oral gamma secretase inhibitor R04929097 in combination with gemcitabine in patients with advanced solid tumors. Invest New Drugs 2014;32:243–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.LoConte NK, Razak AR, Ivy P, Tevaarwerk A, Leverence R, Kolesar J, et al. A multicenter phase 1 study of gamma -secretase inhibitor RO4929097 in combination with capecitabine in refractory solid tumors. Invest New Drugs 2015;33:169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sahebjam S, Bedard PL, Castonguay V, Chen Z, Reedjik M, Liu G, et al. A phase I study of the combination of ro4929097 and cediranib in patients with advanced solid tumours Br J Cancer 2013;109:943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diaz-Padilla I Hirte H, Oza AM, Clarke BA, Cohen B, Reedjik M, Zhang T, et al. A phase Ib combination study of RO4929097, a gamma-secretase inhibitor, and temsirolimus in patients with advanced solid tumors. Invest New Drugs 2013;31:1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su Y, Yu L, Liu N, Guo Z, Wang G, Zheng J, et al. PSMA specific single chain antibody-mediated targeted knockdown of Notch1 inhibits human prostate cancer cell proliferation and tumor growth. Cancer Lett 2013;338:282–291. [DOI] [PubMed] [Google Scholar]
- 49.Yang Z, Qi Y, Zhang J, Luo R, & Kang S. Small interfering RNA (siRNA)-mediated knockdown of Notch1 suppresses tumor growth and enhances the effect of IL-2 immunotherapy in malignant melanoma. J BUON 2015;20:1553–1564. [PubMed] [Google Scholar]
- 50.Zhao EH, Jin X, Shen ZY, Liu H, & Cao H (2012) Influence of silencing Notch1 on proliferation, migration and invasion of BGC-823 gastric cancer cells. Zhonghua Wei Chang Wai Ke Za Zhi 2012;15:1296–1300. [PubMed] [Google Scholar]
- 51.Ferrarotto R, Eckhardt G, Patnaik A, LoRusso P, Faoro L, Heymach JV, et al. A phase I dose-escalation and dose-expansion study of brontictuzumab in subjects with selected solid tumors. Ann Oncol 2018;29:1561–1568. [DOI] [PubMed] [Google Scholar]
- 52.Ferrarotto R, Mitani Y, Diao L, Guijarro I, Wang J, Zweidler-McKay P, et al. Activating NOTCH1 Mutations Define a Distinct Subgroup of Patients With Adenoid Cystic Carcinoma Who Have Poor Prognosis, Propensity to Bone and Liver Metastasis, and Potential Responsiveness to Notch1 Inhibitors. J Clin Oncol 2017;35:352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gordon WR & Aster JC. Application and evaluation of anti-Notch antibodies to modulate Notch signaling. Methods Mol Biol 2014;1187:323–333. [DOI] [PubMed] [Google Scholar]
- 54.Revandkar A, Perciato ML, Toso A, Alajati A, Chen J, Gerber H, et al. Inhibition of Notch pathway arrests PTEN-deficient advanced prostate cancer by triggering p27-driven cellular senescence. Nat Commun 2016; 7:13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.