

Abstract
Supramolecular and dynamic biomaterials hold promise to recapitulate the time-dependent properties and stimuli-responsiveness of the native extracellular matrix (ECM). Host–guest chemistry is one of the most widely studied supramolecular bonds, yet the binding characteristics of host–guest complexes (β-CD/adamantane) in relevant biomaterials have mostly focused on singular host–guest interactions or nondiscrete multivalent pendent polymers. The stepwise synergistic effect of multivalent host–guest interactions for the formation of dynamic biomaterials remains relatively unreported. In this work, we study how a series of multivalent adamantane (guest) cross-linkers affect the overall binding affinity and ability to form supramolecular networks with alginate-CD (Alg-CD). These binding constants of the multivalent cross-linkers were determined via NMR titrations and showed increases in binding constants occurring with multivalent constructs. The higher multivalent cross-linkers enabled hydrogel formation; furthermore, an increase in binding and gelation was observed with the inclusion of a phenyl spacer to the cross-linker. A preliminary screen shows that only cross-linking Alg-CD with an 8-arm-multivalent guest results in robust gel formation. These cytocompatible hydrogels highlight the importance of multivalent design for dynamically cross-linked hydrogels. These materials hold promise for development toward cell- and small molecule-delivery platforms and allow discrete and fine-tuning of network properties.
Introduction
Hydrogels are the most widely used class of materials for three-dimensional cell culture, stimuli-responsive biomaterials, and drug delivery. A current major aim within this area is to create synthetically tailorable hydrogels capable of mimicking a cell’s native extra cellular matrix (ECM), a complex biopolymer hydrogel network. Since the 1960s, significant work has been put forth providing researchers the possibility to design and tailor the chemical and physical properties of hydrogels.1 However, to date most of the development focused on the biocompatibility and mechanical properties of covalently cross-linked hydrogels, which possess networks that are inherently static. This hardly allows the recapitulation of native ECM properties; the native ECM relies on covalent, dynamic covalent and supramolecular interactions for its complex properties and responsiveness. With increasing progress in supramolecular and dynamic covalent biomaterials,2 more emphasis can be seen in designing hydrogels that are not only biocompatible but also capable to dynamically reconfigure and respond to cell behavior.3−5
Cyclodextrins (CD) are cyclic oligosaccharides constituting a “host” supramolecular cavity capable to bind “guest” molecules. Comprised of glucopyranose units linked by α-1,4-glucosidic bonds, CD is typically depicted as a ring, with the inner side hydrophobic (C3 and C5 hydrogens) while the hydrophilic secondary hydroxyl groups (on C2 and C3) and primary (C6 hydroxyl groups) are positioned on the outside.6 The availability of the hydrophobic cavity allows cyclodextrins to serve as “hosts” that provide an environment suitable for inclusion of “guest” hydrophobic compounds in aqueous environments.7 This complexation offers the ability to create supramolecular polymers for drug delivery,8 hydrogels,9,10 polyrotaxanes,11 and surface functionalization12 among other applications.13 The reversibility of the complexation can be an advantage in material design providing shear-thinning10 and self-healing properties.14 When used in the presence of cells, this dynamic environment provides a closer mimic to the natural cellular matrix where cells have the possibility to remodel and interact with their surroundings. However, some of the drawbacks are the lack of stability and mechanical integrity of these materials.
To increase network stability, one possible approach is to create a multivalent synergistic system with binding energies contributed by multiple individual complexes.15−18 This multivalent approach to increase the strength of interactions is seen throughout Nature and conserved across many structural binding interactions. The complexation of a single CD/guest moiety would ease the inclusion of other surrounding guest moieties, via a decrease in the entropic binding penalty for subsequent interactions. The decrease in conformational space of the polymer chains increases local concentration of the remaining free CD and guest moieties, thus favoring the inclusion of the guests into the CD cavities. Multivalency is well characterized in model supramolecular systems; yet, the effect of multivalency can be significant and should be considered as we design dynamic biomaterials. This use of multivalency can be seen throughout the construction of supramolecular hydrogels19 and dynamic matrices for cell culture.20 For example, hyaluronic acid functionalized with pendent CD and adamantane (ADA) moieties have been shown to complex and at higher concentrations of polymer mixtures, form self-standing hydrogels, enable new 3D bioprinting modalities for tissue engineering, and create tough double network hydrogels for cell encapsulation.10,21,22 Multiarm PEG hydrogels formed between cucurbiturils and various guests have been shown to facilitate fine control over dynamic network properties and allow or prevent tissue regrowth within a mouse model.23 In addition, multivalent systems can comprise more than a single host–guest chemistry. Highly elastic and self-healing polyacrylamide-based networks cross-linked via pendent CD/ADA complexes were developed by the Harada group.24,25 Further development produced a multivalent system, composed of multiple host–guest CD/ADA and CD/ferrocene cross-links that demonstrated shape memory behavior due to the redox-responsive complexation of CD/ferrocene.26 Despite the promise of dynamic and multivalent host–guest hydrogels in biomedical and performance materials, there exist few systematic studies on the discrete effect of multivalency on materials properties.
In this study, we have chosen to work with alginate, a naturally derived FDA approved component (from food to medical devices) and biobased polymer that is extensively used in the medical and bioengineering fields.27 Because of alginate’s lack of cell-interaction motifs and antifouling nature, it has also been a material of choice in the rational design of synthetic extracellular matrices (ECM). The alginate copolymer is comprised of β-d-mannuronic acid (M units) and α-l-guluronic acid (G units), and cross-linking can easily be introduced through the addition of multivalent ions such as Ca2+ ions (cross-linking the G-blocks of the polymer). Chemical modification is also possible via the free hydroxyl and carboxyl groups present on the alginate backbone to introduce different functionality, or possibly to present bioactivity to the material.28 Alginate-based cyclodextrin systems have already shown promise for drug-delivery devices29 and for dynamic display of cell adhesion motifs30 in hydrogels.
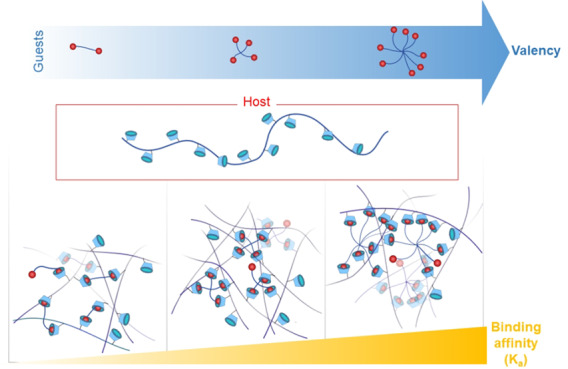
We set out to quantitatively study the effect of multivalency within a PEG-adamantane/β-cyclodextrin-alginate system toward the creation of supramolecular hydrogels as cell matrices (Figure 1). Host β-cyclodextrin (CD) moieties were introduced onto the backbone of alginate, while the end groups of multiarm poly(ethylene glycol) (PEG) are functionalized with guest adamantane (ADA) moieties. CD and ADA were chosen as the host/guest pair as ADA is known to have a strong affinity to CD and is one of the most widely used guests in the design of biomaterials.30−32 The design of our network relies on the complexation of pendent host moieties (on the alginate) to end group guest moieties (on the PEG), as opposed to complexation of host–guest moieties both attached as pendent groups, allowing us to discretely change the valence of the system in a stepwise fashion. We investigated the effect of multivalency by varying the number of PEG arms (2, 4, and 8) to increase the binding affinity of CD/ADA. The molecular weight of the multiarm PEGs used in this work was selected in a way that each arm was approximately 2.5 kDa to maintain a consistent distance between complexed units. The overall strategy employed for the current work is depicted in Figure 1.
Figure 1.

A general overview of the synthetic strategy in this work. Alginate functionalized with CD was mixed with different PEG-ADA cross-linkers of different valencies to create a library of dynamically cross-linked materials.
Materials
All materials were acquired from suppliers indicated and used without further purification unless stated otherwise: toluene (>99.8%, Acros Organics), 2-arm PEG–OH (4.6 kDa, Sigma-Aldrich, PEG2-OH), 4-arm PEG–OH (10 kDa, 97.5%, Creative PEGWorks, PEG4-OH), 8-arm PEG–OH (hexaglycerol core, 20 kDa, 99.1%, Creative PEGWorks, PEG8-OH), 1-adamantane methylamine (>98.0%, TCI Europe, ADA), triethylamine (>99%, Merck, Et3N), 1,4-dioxane (99.8%, Sigma-Aldrich), 1,6-hexadiamine (98%, Sigma-Aldrich), 4-toluenesulfyl chloride (>98%, Sigma-Aldrich, OTs) anhydrous chloroform (>99%, Sigma-Aldrich), anhydrous dimethylformamide (99.8%, Sigma-Aldrich, DMF), 1-(3-(dimethylamino)propyl)-3-ethylcarbodiimide hydrochloride (98+%, VWR, EDC-HCl), N-hydroxysulfosuccinimide sodium salt (≥98%, Sigma-Aldrich, sulfo-NHS), β-cyclodextrin (99%, Sigma-Aldrich, CD), carbonyl diimidazole (>90%, Sigma-Aldrich, CDI), deuterated chloroform (>99.8%, Sigma-Aldrich, CDCl3), deuterium oxide (99.9%, Sigma-Aldrich, D2O), Snakeskin MWCO 10,000 dialysis tubes (Thermo Scientific U.S.A.), diethyl ether (>99%, VWR). Sodium hydride (NaH, 60 wt % in mineral oil) was washed with hexane and tetrahydrofuran (THF) under an argon atmosphere. All used THF was freshly distilled over fresh NaH. 4-(1-Adamantyl) phenol was prepared as described by Jensen et al.33 6-(6-Aminohexyl)amino-6-deoxy-β-cyclodextrin (CD-HA) was prepared via a two-step synthetic route.34,35Supporting Information contains synthesis and NMR spectra. Alginate (Manugel GMB, FMC, Lot No. G9402001) was purified with activated charcoal Norit (Sigma-Aldrich) and characterized before use via 1H NMR and GPC as described in previous work.36 The Mn of alginate was 258 kDa (Đ = 2.0) as measured by GPC (100 mM sodium nitrate) and with an estimated ratio of G- and M-blocks of 74% and 26% determined by 1H NMR. 2-(N-Morpholino)ethanesulfonic acid (MES) buffer (0.100 M MES, 0.300 M NaCl) was prepared by dissolution of 4.88 g of MES hydrate (≥99.5%, Sigma) and 4.38 g of NaCl (Bioxtra, Sigma-Aldrich) in 250 mL of deionized water. The pH of the buffer was adjusted to 6.5 with 50% (w/v) NaOH before use.
Methods
General Synthesis of Multiarm PEG-ADA
Synthesis of PEG-ADA was performed in a two-step reaction. As a typical example, PEG2-OH (4.6 kDa, 1.0 g, 0.435 mmol OH) was dissolved in toluene and dried using a rotary evaporator. This step was repeated two times. Subsequently, the dried PEG was dissolved in 4 mL of anhydrous 1,4-dioxane. CDI (0.33 g, 2.04 mmol) was dissolved in 4 mL of dry 1,4-dioxane and added to the PEG solution. Next, the reaction mixture was stirred at 37 °C for 2 h. The sample was precipitated four times in 30 mL of cold diethyl ether (−20 °C) and subsequently centrifuged for 15 min at 7200×g. The supernatant was decanted to yield a white solid. Residual diethyl ether was removed using a rotary evaporator. The polymer was characterized by NMR and GPC. Yield (PEG2-CDI = 0.93 g, 93%). Synthesis of PEG4-CDI and PEG8-CDI were carried out using similar conditions.
In the second step of the reaction, PEG2-CDI (1.0 g, 0.417 mmol active end groups) was dissolved in 12.5 mL of anhydrous DMF followed by the addition of 1-adamantane methylamine (0.30 g, 1.82 mmol) dissolved in 12.5 mL of anhydrous DMF. The reaction was stirred at 70 °C for 24 h under N2. The product was concentrated to 10 mL using a rotary evaporator and subsequently precipitated four times in 30 mL of cold diethyl ether (−20 °C). The polymer was characterized using NMR and GPC. Yield (PEG2-ADA = 0.56 g, 53%). Samples were stored at −20 °C until further use. Synthesis of PEG4-ADA and PEG8-ADA were carried out using similar conditions.
Synthesis of PEG-OTs
PEG8-OH (6.87 g, 2.75 mmol OH) and toluene-4-sulfonyl chloride (5.24 g, 27.5 mmol) were dissolved in CH2Cl2 (80 mL) under an argon atmosphere. Triethylamine (2.0 mL, 27.5 mmol) was dissolved in 30 mL of CH2Cl2 and added dropwise to the above solution at 0 °C. The mixture was stirred for 48 h at room temperature and subsequently precipitated into diethyl ether. After filtering and drying in vacuo at 25 °C, the product PEG8-OTs was obtained as a white powder in a yield of 100% (7.27 g) with a degree of substitution of 85%.
Synthesis of PEG-pheADA
Phenyladamantyl functionalized 8-arm PEG was prepared by the reaction of PEG8-OTs with adamantylphenolate. In a round bottomed flask, NaH (50 mg, 2.08 mmol) was suspended in 20 mL of THF under an argon atmosphere. To the suspension, 4-(1-adamantyl)-phenol (560 mg, 2.5 mmol) dissolved in 10 mL of THF was added. After 1 h, a solution of 8-arm PEG-OTs (1.0 g (0.37 mmol OTs groups) in 10 mL of THF was added dropwise to the adamantylphenolate and the resulting solution was heated to 40 °C and stirred for 48 h. The reaction mixture was left to cool to room temperature and precipitated in diethyl ether. The crude product was dialyzed against 30% ethanol toward pure water and obtained as a fluffy white solid after lyophilization. All tosylated groups were replaced by phenyladamantyl, resulting in a degree of substitution of 85%.
Synthesis of β-cyclodextrin Conjugated Alginate (Alg-CD)
Purified alginate (0.4010 g, 2.30 mmol COOH groups) was weighed into a 100 mL Schott flask and dissolved in 80 mL of MES buffer. Sulfo-NHS (0.5271 g, 2.43 mmol) and EDC-HCl (0.3990 g, 2.08 mmol) were added and the reaction was left to stir for 30 min. 6-(6-Aminohexyl)amino-6-deoxy-β-cyclodextrin (CD-HA, 0.9953 g, 0.807 mmol) was added to the reaction mixture and the pH of the solution was adjusted to 8 with 50% (w/v) NaOH. The reaction was left to stir for 18 h at room temperature. The solution was then transferred to a 10 kDa MWCO dialysis tube and dialyzed against NaCl solutions, starting from 100, 50, 25 mM, and finally deionized water, with change of dialysate every 10 to 18 h. A white fluffy solid was yielded after lyophilization.
Preparation of Hydrogel Samples
Molar ratios (1:1) of CD and ADA were calculated based on the degree of functionalization of the Alg-CD and PEG-ADA. Hydrogels were prepared by weighing PEG-ADA into a 2 mL Eppendorf tube, followed by the addition of a 4% (w/v) alginate stock solution in Ca2+/Mg2+ free phosphate buffered saline (PBS). Samples were supplemented with PBS to adjust the total volume to 50 μL. Gel formation was tested by a tube inversion test.
Characterization
Nuclear Magnetic Resonance (NMR)
NMR analysis was performed with a Bruker Ascend 700 MHz NMR Spectrometer (Bruker, Germany) and data were analyzed with the TopSpin 3.5 Software (Bruker, Germany). NMR samples were prepared by dissolving 2–4 mg of polymer in either 500 μL of CDCl3 (PEG derivates) or D2O (Alginate-CD). Spectra of the PEG derivatives were acquired at 299.7 K. For alginate samples, water suppression pulse sequence was applied, and measurements were carried out at 325 K. Spectra were calibrated with respect to nondeuterated solvent (CHCl3, 7.26 ppm or H2O, 4.29 ppm).
Gel Permeation Chromatography (GPC)
GPC was performed using N,N-dimethylformamide (DMF) containing 0.1 wt % LiBr as eluent and sample concentration of 2 mg/mL The Shimadzu system comprised of an autosampler, a Shodex KD-G 4A guard column (4.6 × 10 mm) with 8 μm beads, followed by two Shodex KD-802 (5 μm, 8 × 300 mm) and KD-804 (7 μm, 8 × 300 mm) columns, a refractive index detector and a photodiode array detector at 50 °C. A flow rate of 1 mL min–1 was applied. The GPC system was calibrated against linear poly(methyl methacrylate) (PMMA) standards with molecular weights ranging from 600 to 265 300 g mol–1. Samples were filtered through polytetrafluoroethylene (PTFE) membranes with a pore size of 0.2 μm prior to injection.
Dynamic Light Scattering (DLS)
Alg-CD, PEG8-ADA, and PEG8phe-ADA were dissolved in deionized water to prepare 0.06 mM stock solutions. The solutions were filtered through 0.2 μm cellulose filters prior to measurement. Measurements were performed on a Malvern Zetasizer Nano ZSP (He–Ne laser 633 nm).
Scanning Electron Microscopy (SEM)
Hydrogels of Alg-CD/PEG8-ADA and Alg-CD/PEG8-pheADA were lyophilized to remove the water content. Following this, samples were carefully mounted on stubs and gold-sputtered (Cressington Sputter Coater 108 auto) for 60 s at 30 mA. Images were acquired with a SEM (Philips XL-30 ESEM, Philips) at 10 kV.
Rheology
Rheological analysis was performed on a DHR-2 rheometer at 20 °C using a 20 mm cone–plate geometry with a 2.002° angle. Samples were loaded and a time sweep was measured at 1% strain and 10 rad s–1 to allow samples to equilibrate. Subsequently a frequency sweep from 100 to 0.1 rad s–1 with a strain of 1% followed by a strain sweep from 1 to 100% strain at 10 rad s–1 were taken.
Cell Viability Assay
In general, cell-laden hydrogels were prepared by adding a L929 fibroblast (cell line from mouse, passage 4) cell suspension in cell culture medium (Dulbecco’s Modified Eagle’s Medium (DMEM, ThermoFisher) with glutamax, 1% penicillin–streptomycin, and 10% fetal bovine serum) to 100 μL of hydrogel in a tissue culture treated clear-bottom 96-well black plate. The final cell concentration in the hydrogels was 106 cells mL–1. The samples were incubated for 24 h at 37 °C with 5% CO2. Cell viability was evaluated using a LIVE/DEAD viability/cytotoxicity kit (Thermofisher). A stock solution of ethidium homodimer (2.5 μM) and calcein-AM (1 μM) was prepared in Ca2+/Mg2+ free phosphate buffered saline (PBS). Then, 100 μL of the ethidium/calcein stock solution was added to each hydrogel and incubated for 30 min at 37 °C. Prior to imaging, the dye solutions were aspirated carefully from the wells before adding 100 μL of culture medium without phenol red to wells. Imaging was carried out on a Nikon TI-E with environmental control using a 10× objective (WD = 15, NA = 0.3). Live cells were stained by Calcein AM with green fluorescence (ex/em = 495/515 nm) and dead cells were stained by ethidium homodimer with red fluorescence (ex/em = 495/635 nm).
Results and Discussion
Synthesis of PEG-ADA and Alg-CD Precursors
To synthesize PEG-ADA, the hydroxyl groups on PEG were first converted to better leaving groups to allow subsequent substitution reaction with adamantane methylamine. Our initial attempt was to introduce tosylate groups through the reaction of PEG with 4-toluenesulfonyl chloride. Even though the reaction did proceed, the degree of functionalization was low (less than 40%) and there were inconsistencies in reproducibility. We then decided to use a carbonyl diimidazole (CDI) coupling strategy. The reaction of PEG with CDI forms a imidazolyl carbamate intermediate (Figure 2A), and subsequent reaction with 1-adamantane methylamine results in a carbamate bond (Figure 2B). The same reaction strategy was employed to prepare PEG4-ADA and PEG8-ADA.
Figure 2.
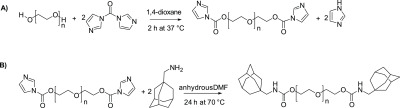
Synthesis of PEG2-ADA, starting from (A) activation of PEG2-OH with CDI (B) reaction of the PEG2-CDI with 1-adamantane methylamine to give PEG2-ADA.
The success of the two-step reaction was followed by NMR through the integration of the protons from the polymer end groups relative to the protons of the PEG repeating units (Figure 3). In Figure 3B, the aromatic protons of CDI were observed at 8.34, 7.51, and 7.15 ppm, along with upfield shifted α- and β-methylene protons of PEG (3.85 and 4.59 ppm, respectively) after reaction. The degree of functionalization (DoF) of PEG2-CDI was estimated by comparing these peaks to that of the PEG backbone and determined to be 95%. In our hands, this reaction has proven reproducible (DoF = 86–97%). Similar reaction sequences and analysis was carried out to create the PEG4-CDI (DoF = 85%) and PEG8-CDI (DoF = 81%) (Supporting Information, Figures S3 and S4, NMR spectra). PEG-CDI samples were also analyzed via GPC (Supporting Information, Figure S6) and as expected no significant differences in molecular weight were observed.
Figure 3.

1H NMR spectrum (CDCl3) of (A) PEG2, (B) PEG2-CDI, and (C) PEG2-ADA. The appearance of CDI specific peaks (h, i, and j) and α and β methylene protons (c, b) were observed after reaction of PEG2 with CDI. Following reaction of PEG2-CDI with adamantane methylamine, appearance of adamantane specific peaks (d, e, f, and g) and disappearance of CDI peaks were observed. X denotes presence of residual diethyl ether.
After activating the OH end groups of the multivalent PEGs with CDI, the products were reacted with 1-adamantane methylamine under dry conditions to give the adamantane end functionalized PEGs. For all end group modified PEGs prepared, 1H NMR was used to estimate the DoF and if any unreacted starting material remained. As an example, the 1H NMR spectrum of PEG2-ADA is presented in Figure 3C. After the reaction, adamantane peaks were observed at 1.97, 1.72–1.61, and 1.46 ppm and the CDI aromatic peaks were absent. Comparing the integral values for the for the PEG protons to the adamantane aliphatic protons, the DoF was estimated to be 84% for PEG2-ADA. This reaction was also successfully carried out to produce PEG4-ADA with a DoF of 85% and PEG8-ADA with a DoF of 81% (Supporting Information, Figure S3 and Figure S4, 1H NMR spectra, Figure S6 GPC).
The β-CD conjugated alginate was prepared via activation of the carboxylic acid groups of alginate by an EDC/NHS reaction and subsequent reaction with the amine group of a mono functionalized cyclodextrin-hexyldiamine. The choice of buffer and reaction conditions were adapted from previous work.36 We incorporated a six-carbon spacer between the alginate and cyclodextrin in order to increase the conformational freedom of the cyclodextrin when accompanying guests. Conveniently, this hexamethylene spacer also facilitated characterization of the conjugated alginate by NMR. The peaks attributed to the methylene protons of the hexylamine spacer (1.2–2.2 ppm) are well separated from the proton signals of the alginate backbone (3.4–5.2 ppm) (Figure 4). By adding a known concentration of dimethylformamide as an internal standard in the NMR measurements, we estimated the amount of CD functionalization on alginate, or degree of functionalization, to be approximately 5% (1 CD for every 20 alginate repeat units or 0.19 mmol of CD per gram of functionalized alginate; Supporting Information, calculation included).
Figure 4.

1H NMR spectrum (D2O) of (A) alginate and (B) alginate modified β-CD (Alg-CD). Peaks at 1.2 to 2.2 ppm corresponding to methylene protons of the spacer between β-CD and the alginate backbone were observed. In (B) dimethylformamide (6.5 mM) was used as internal standard for quantification.
Host–Guest Complexation of Components
In model experiments, the complexation of PEG-ADA precursors with β-CD was first evaluated. Using a 1:1 molar ratio of adamantyl to cyclodextrin groups, nuclear Overhauser effect spectroscopy (NOESY) was performed on a solution (D2O) of PEG2-ADA and β-CD. The cross-correlation peaks between the β-CD inner protons on C3 and C5 (3.8 to 3.9 ppm)37 and the cycloalkane protons of ADA (2.18, 1.75, and 1.57 ppm) were clearly present as shown in Figure 5. On the contrary, no significant correlation peaks were observed between the β-CD outer C2 and C5 protons (3.6 to 3.7 ppm) and the ADA protons. This indicates a close proximity of the relevant protons (within 4 Å)38 of the adamantyl groups in the cavity of the cyclodextrins as a result of the complexation. Furthermore, the differences in chemical shifts observed when comparing the 1H NMR spectra of β-CD, PEG2-ADA, and the mixture of PEG2-ADA/β-CD also support the formation of host guest complexes. Peaks corresponding to adamantanyl groups of PEG2-ADA (1.5 to 2.0 ppm) and β-CD (3.5 to 4.0 ppm) showed distinctive shifts after complexation (Supporting Information, Figure S5, NMR spectra).
Figure 5.

NOESY spectrum of a 1.25 mM solution of PEG2-ADA/β-CD in D2O. The red areas are signals specific to adamantane and the blue area is specific to CD.
Next, the binding affinity of the multivalent PEG-ADA cross-linkers to Alg-CD was investigated. Both components were dissolved at low concentration in deuterated water and the Alg-CD (host) was titrated (0 to 4 mM of CD) against fixed concentrations (2 mM of ADA) of the multivalent PEG-ADA cross-linkers (guest). Since the overlap concentration of the alginate polymer is expected to be around 0.5 wt %,39 these binding measurements are considered to be performed in the semidilute polymer regime. Because of complexities in discerning inter- versus intramolecular binding and effective molarities in these polymeric solutions, the values reported here should only be treated as apparent binding constants. An example of a series of 1H NMR spectra obtained, when PEG8-ADA was titrated with Alg-CD at various CD to ADA (host–guest) molar ratios is presented in Figure 6. The NMR chemical shifts of the CH2 and CH adamantyl protons (δ0 = 1.21 and 1.70) were monitored to calculate the binding affinity (Ka) of the ADA-CD complexes. The changes in the chemical shifts (Δδ) of the adamantyl protons as a function of the CD concentration according the Benesi–Hildebrand equation10,40,41 (eq 1) afforded the affinity constants. The maximum change in chemical shift (Δδmax) was extrapolated from the plot.
| 1 |
An increase in binding affinity was observed with increasing numbers of ADA moieties on the PEG (Table 1). The observed increase of binding affinity upon increasing multi-valency may be attributed to a high local concentration of guest moieties. After initial host–guest complexes are formed, a cooperative effect eases inclusion of guests into CD cavities. However, compared to binding affinities reported in literature for CD/ADA complexes (∼104 M–1),42−45 the obtained values were lower. This observed weaker binding affinity may be caused by several factors. Alginate chains carry significant charge (negative carboxylate) and could experience electrostatic repulsion when bundled by the multivalent cross-linkers. Considering the amphiphilic nature of the PEG-ADA conjugates, these will be present as micellar-like aggregates in solution (vide infra). The host–guest association may be lowered due to this aggregation, resulting in an overall lower observed binding affinity.46 Steric hindrance47 as shown for systems in which the CD moieties were conjugated without a spacer is likely minimized in our system, which is based on spacer-conjugated Alg-CD and ADA conjugated PEGs. Interestingly, PEG8-pheADA showed an order of magnitude higher binding affinity compared to PEG8-ADA. This result can be attributed to the hydrophobic phenyl attached adjacent to the guest moiety promoting inclusion in the host’s cavity.
Figure 6.

1H NMR spectra of PEG8-ADA (guest) titrated with Alg-CD (host). Chemical shifts of ADA peaks (1.21 and 1.70 ppm) were monitored for determination of binding constant.
Table 1. Binding Constant (Ka) of Multivalent PEG-ADA and Alg-CD Complexes.
| complex | Ka (M–1) | R2 |
|---|---|---|
| PEG-2ADA + Alg-CD | 10 | 0.99 |
| PEG-4ADA + Alg-CD | 130 | 0.97 |
| PEG-8ADA + Alg-CD | 390 | 0.95 |
| PEG-8pheADA + Alg-CD | 3900 | 0.94 |
Formation of Dynamic Hydrogels
The marked effect of multivalency on the binding constants of the PEG-ADAs/Alg-CD host–guest complexes was expected to be reflected in the formation and stability of hydrogels. The efficiency of gelation was qualitatively characterized via the tube inversion method. If gels remained intact with no flow when the tube was inverted, the sample was considered to be in the gel state. The shape integrity of the complexes was also reassessed after five minutes by tapping the inverted tube, followed by dislocating the gel using a pipet tip to evaluate a self-healing capacity. We first tested the ability of the PEG8phe-ADA/Alg-CD to form hydrogels at higher concentrations. Initial attempts with a 1:1 ADA/CD molar ratio at a 4 wt/v% alginate concentration showed the formation of a relatively strong hydrogel, which was resistant to mechanical disruption, and was capable of self-healing. This hydrogel was formed quickly and spontaneously upon mixing solutions of the individual macromolecules.
Realizing the broad formulation space possible with these molecular architectures (multivalency, cross-linking equivalents, concentration), we next set out to qualitatively characterize the gelation in a qualitative manner using the tube inversion method. A library comprising 48 combinations of mixtures was assessed by varying the concentration of alginate (1, 2, 3, 4 wt/v%), the ratio of ADA,CD (2:1, 1:1, 1:2), and the cross-linker (PEG2-ADA, PEG4-ADA, PEG8-ADA, and PEG8-pheADA). Mixtures that appeared gel-like but were unable to retain their shape integrity when their tubes were inverted and manually disrupted were classified as “weak”. On the contrary, mixtures that maintained a solid gel form even upon tube inversion and manual disruption were considered as “strong” hydrogels (Figure S11, Table S1).
We observed that formulations containing the 8-arm PEG cross-linkers formed hydrogels at higher concentrations of Alg-CD. Mixtures comprising the lower valence structures, PEG2-ADA and PEG4-ADA, remained mostly liquidlike after mixing with Alg-CD at all concentrations and ADA-CD molar ratios. Interestingly, we observed that the PEG8-pheADA formulations formed hydrogels above 2 wt/v%, while the PEG8-ADA formulations only formed hydrogels at 4 wt/v%, reflecting the higher binding constants in pheADA. All gels formed were observed to be self-healing. The 8-arm hydrogels interestingly appeared stronger at a 2:1 AD/CD ratio. We initially attribute this to a larger amount of solids in the hydrogel (higher wt% polymer due to increased PEG-ADA); however, with these low apparent binding constants the addition of guest can also significantly increase the amount of bound cross-linkers.
Since the PEG8-ADA and the PEG8phe-ADA formed the most stable hydrogels in the series, these were chosen for further studies on the hydrogel microstructure and mechanical properties.
Characterization of 8-Arm Hydrogels
SEM images of the PEG8-ADA and PEG8phe-ADA hydrogels after lyophilization (Figure 7) revealed highly porous structures. The PEG8-ADA was comprised of interconnected beadlike fibrous structure whereas the Alg-CD/PEG8phe-ADA formed a somewhat denser, more bundled fibrous structure. In addition, DLS experiments (Supporting Information, Figures S9−11) showed larger aggregate formation for the PEG8phe-ADA system, even in dilute solutions. These observations correspond to the results obtained from the NMR titration experiments, whereby stronger complexation was observed between Alg-CD and PEG8phe-ADA.
Figure 7.

SEM images of hydrogels formed via complexation of PEG8-ADA/Alg-CD (a) and complexation of PEG8phe-ADA/Alg-CD (b). Scale bar is 10 μm.
To investigate the 8-arm hydrogels’ mechanical properties, oscillatory rheology experiments were performed (Figure 8). Immediately apparent, the parent Alg-CD (control) is a liquid (loss moduli above storage moduli), while the addition of the supramolecular cross-linker triggers the formation of a hydrogel via an increase of storage modulus over 2 orders of magnitude. Both systems formed hydrogels with a storage modulus of approximately 1000 Pa with a low tan δ. A frequency sweep of these materials showed no significant trends across the investigated frequency range with only modest drops in the apparent stiffness in the low-frequency regime. Furthermore, strain sweeps revealed these hydrogels lost their mechanical properties around 10% strain with a gradual and continuous inversion of storage and loss moduli. Interestingly, the higher binding constant cross-linker (PEG8phe-ADA) created a hydrogel with a higher strain at break.
Figure 8.

Oscillatory rheology of 8-arm PEG adamantanes and Alg-CD. Top is PEG8-ADA gel, and bottom is PEG8phe-ADA. Controls are 4 wt % Alg-CD solutions with no cross-linker. Gels are at 4 wt %.
Both the PEG8-ADA and PEG8phe-ADA hydrogels were observed to be self-healing (vide supra) and able to dynamically adapt to stress. A simple stress relaxation test on the rheometer showed both gels were able to rapidly dissipate an applied stress with half-lives (t1/2) on the order of 101 seconds (Supporting Information Figures S14 and S15). Interestingly the weaker binding PEG8-ADA (∼26 s) exhibited a slightly slower relaxation rate as compared to the strong binding PEG8phe-ADA (∼9 s). During attempts at hydrogel swelling measurements, no equilibrium was reached, further reflecting the dynamic nature of these systems.
Cell Viability
The biocompatibility of the 3 wt % Alg-CD/PEG8phe-ADA gel was studied with L929 fibroblast cells. After 24 h of incubation of fibroblasts embedded in the supramolecular hydrogels, a calcein/ethidium live/dead assay was performed. The images from the live/dead assay showed high viability of cells with few dead cells observed (Figure 9). These materials show promise for use in cell-delivery and cell-culture applications; future studies will elaborate on the cytocompatibility and use of these materials as biomaterials.
Figure 9.
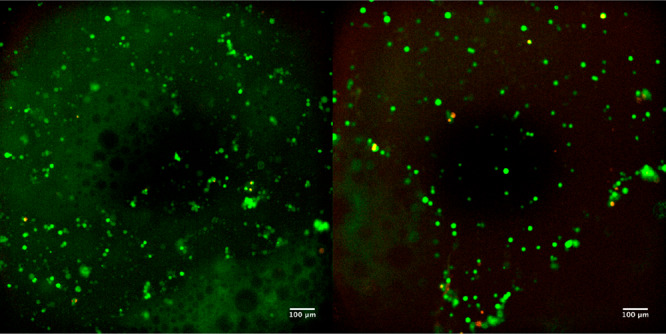
Live/dead assay of L929 cells cultured on TCP in the presence of 3% Alg-CD and PEG8phe-ADA. Images were after 24 h incubation (green = live cells and red = dead cells; scale bars = 100 μm).
Conclusions
Herein we have shown the drastic effect that multivalency of cross-linking architecture can have on the formation of a model series of host–guest hydrogels. Supramolecular alginate hydrogels based on multivalent host–guest chemistry were developed and studied. Multiarm PEG cross-linkers with ADA (guest moieties) were successfully synthesized in high yields and consistency. Multivalency was found to play a significant part in influencing the strength of binding affinities. Only with the 8-arm cross-linkers were strong and stable gels formed. These multivalent cross-linkers enabled a 100-fold increase in the storage moduli (compared to an uncross-linked sample), while creating fibrous and cytocompatible hydrogel architectures. The supramolecular hydrogel system developed shows potential for possible use as injectable biomaterial platform and emphasizes the key role of multivalency in the design of supramolecular and dynamically cross-linked hydrogels.
Acknowledgments
The authors thank Prof. G. Julius Vansco for guidance in development of the cross-linkers utilized in this study. The authors also thank SyMO-Chem, Joost van Dongen (Eindhoven University of Technology), and Birgit Huber (Karlsruhe Institute of Technology) for assistance with aqueous GPC of these systems.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.0c00148.
Synthetic procedures, NMRs, GPCs, NMR binding titrations, DLS, gelation table, and stress-relaxation rheometry (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Wichterle D. L. Hydrophilic Gels for Biological Use. Nature 1960, 185 (4706), 117–118. 10.1038/185117a0. [DOI] [Google Scholar]
- Webber M. J.; Appel E. A.; Meijer E. W.; Langer R. Supramolecular Biomaterials. Nat. Mater. 2016, 15 (1), 13–26. 10.1038/nmat4474. [DOI] [PubMed] [Google Scholar]
- Ooi H. W.; Hafeez S.; Van Blitterswijk C. A.; Moroni L.; Baker M. B. Hydrogels That Listen to Cells: A Review of Cell-Responsive Strategies in Biomaterial Design for Tissue Regeneration. Mater. Horiz. 2017, 4 (6), 1020–1040. 10.1039/C7MH00373K. [DOI] [Google Scholar]
- Wang H.; Heilshorn S. C. Adaptable Hydrogel Networks with Reversible Linkages for Tissue Engineering. Adv. Mater. 2015, 27 (25), 3717–3736. 10.1002/adma.201501558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick J. A.; Murphy W. L. Moving from Static to Dynamic Complexity in Hydrogel Design. Nat. Commun. 2012, 3, 1269. 10.1038/ncomms2271. [DOI] [PubMed] [Google Scholar]
- Del Valle E. M. M. M. Cyclodextrins and Their Uses: A Review. Process Biochem. 2004, 39 (9), 1033–1046. 10.1016/S0032-9592(03)00258-9. [DOI] [Google Scholar]
- Saenger W. Cyclodextrin Inclusion Compounds in Research and Industry. Angew. Chem., Int. Ed. Engl. 1980, 19 (5), 344–362. 10.1002/anie.198003441. [DOI] [Google Scholar]
- Wenz G. An Overview of Host-Guest Chemistry and Its Application to Nonsteroidal Anti-Inflammatory Drugs. Clin. Drug Invest. 2000, 19 (2), 21–25. 10.2165/00044011-200019002-00003. [DOI] [Google Scholar]
- van de Manakker F.; Kroon-Batenburg L. M. J.; Vermonden T.; van Nostrum C. F.; Hennink W. E. Supramolecular Hydrogels Formed by β-Cyclodextrin Self-Association and Host–Guest Inclusion Complexes. Soft Matter 2010, 6 (1), 187–194. 10.1039/B916378F. [DOI] [Google Scholar]
- Rodell C. B.; Kaminski A. L.; Burdick J. A. Rational Design of Network Properties in Guest-Host Assembled and Shear-Thinning Hyaluronic Acid Hydrogels. Biomacromolecules 2013, 14 (11), 4125–4134. 10.1021/bm401280z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X.; Yang F. Sliding Hydrogels with Mobile Molecular Ligands and Crosslinks as 3D Stem Cell Niche. Adv. Mater. 2016, 28, 7257–7263. 10.1002/adma.201601484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolino M.; Ennen F.; Lamponi S.; Cernescu M.; Voit B.; Cappelli A.; Appelhans D.; Komber H. Cyclodextrin-Adamantane Host–Guest Interactions on the Surface of Biocompatible Adamantyl-Modified Glycodendrimers. Macromolecules 2013, 46 (9), 3215–3227. 10.1021/ma400352m. [DOI] [Google Scholar]
- Tan S.; Ladewig K.; Fu Q.; Blencowe A.; Qiao G. G. Cyclodextrin-Based Supramolecular Assemblies and Hydrogels: Recent Advances and Future Perspectives. Macromol. Rapid Commun. 2014, 35, 1166–1184. 10.1002/marc.201400080. [DOI] [PubMed] [Google Scholar]
- Harada A.; Takashima Y.; Nakahata M. Supramolecular Polymeric Materials via Cyclodextrin-Guest Interactions. Acc. Chem. Res. 2014, 47 (7), 2128–2140. 10.1021/ar500109h. [DOI] [PubMed] [Google Scholar]
- Hashidzume A.; Harada A. Multivalency in Cyclodextrin/Polymer Systems. Multivalency 2017, 121–142. 10.1002/9781119143505.ch5. [DOI] [Google Scholar]
- Badjić J. D.; Nelson A.; Cantrill S. J.; Turnbull W. B.; Stoddart J. F. Multivalency and Cooperativity in Supramolecular Chemistry. Acc. Chem. Res. 2005, 38 (9), 723–732. 10.1021/ar040223k. [DOI] [PubMed] [Google Scholar]
- Kitov P. I.; Bundle D. R. On the Nature of the Multivalency Effect: A Thermodynamic Model. J. Am. Chem. Soc. 2003, 125 (52), 16271–16284. 10.1021/ja038223n. [DOI] [PubMed] [Google Scholar]
- Mulder A.; Huskens J.; Reinhoudt D. N. Multivalency in Supramolecular Chemistry and Nanofabrication. Org. Biomol. Chem. 2004, 2 (23), 3409–3424. 10.1039/b413971b. [DOI] [PubMed] [Google Scholar]
- Appel E. A.; Del Barrio J.; Loh X. J.; Scherman O. A. Supramolecular Polymeric Hydrogels. Chem. Soc. Rev. 2012, 41 (18), 6195–6214. 10.1039/c2cs35264h. [DOI] [PubMed] [Google Scholar]
- Rosales A. M.; Anseth K. S. The Design of Reversible Hydrogels to Capture Extracellular Matrix Dynamics. Nat. Rev. Mater. 2016, 1 (2), 1–15. 10.1038/natrevmats.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highley C. B.; Rodell C. B.; Burdick J. A. Direct 3D Printing of Shear-Thinning Hydrogels into Self-Healing Hydrogels. Adv. Mater. 2015, 27 (34), 5075–5079. 10.1002/adma.201501234. [DOI] [PubMed] [Google Scholar]
- Rodell C. B.; Dusaj N. N.; Highley C. B.; Burdick J. A. Injectable and Cytocompatible Tough Double-Network Hydrogels through Tandem Supramolecular and Covalent Crosslinking. Adv. Mater. 2016, 28, 8419–8424. 10.1002/adma.201602268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L.; Braegelman A. S.; Webber M. J. Dynamic Supramolecular Hydrogels Spanning an Unprecedented Range of Host-Guest Affinity. ACS Appl. Mater. Interfaces 2019, 11, 5695–5700. 10.1021/acsami.8b22151. [DOI] [PubMed] [Google Scholar]
- Kakuta T.; Takashima Y.; Harada A. Highly Elastic Supramolecular Hydrogels Using Host-Guest Inclusion Complexes with Cyclodextrins. Macromolecules 2013, 46 (11), 4575–4579. 10.1021/ma400695p. [DOI] [Google Scholar]
- Kakuta T.; Takashima Y.; Nakahata M.; Otsubo M.; Yamaguchi H.; Harada A. Preorganized Hydrogel: Self-Healing Properties of Supramolecular Hydrogels Formed by Polymerization of Host-Guest-Monomers That Contain Cyclodextrins and Hydrophobic Guest Groups. Adv. Mater. 2013, 25 (20), 2849–2853. 10.1002/adma.201205321. [DOI] [PubMed] [Google Scholar]
- Miyamae K.; Nakahata M.; Takashima Y.; Harada A. Self-Healing, Expansion-Contraction, and Shape-Memory Properties of a Preorganized Supramolecular Hydrogel through Host-Guest Interactions. Angew. Chem., Int. Ed. 2015, 54 (31), 8984–8987. 10.1002/anie.201502957. [DOI] [PubMed] [Google Scholar]
- Lee K. Y.; Mooney D. J. Alginate: Properties and Biomedical Applications. Prog. Polym. Sci. 2012, 37 (1), 106–126. 10.1016/j.progpolymsci.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar S. N.; Edgar K. J. Alginate Derivatization: A Review of Chemistry, Properties and Applications. Biomaterials 2012, 33 (11), 3279–3305. 10.1016/j.biomaterials.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Izawa H.; Kawakami K.; Sumita M.; Tateyama Y.; Hill J. P.; Ariga K. β-Cyclodextrin-Crosslinked Alginate Gel for Patient-Controlled Drug Delivery Systems: Regulation of Host-Guest Interactions with Mechanical Stimuli. J. Mater. Chem. B 2013, 1 (16), 2155–2161. 10.1039/c3tb00503h. [DOI] [PubMed] [Google Scholar]
- Boekhoven J.; Rubertpérez C. M.; Sur S.; Worthy A.; Stupp S. I. Dynamic Display of Bioactivity through Host-Guest Chemistry. Angew. Chem., Int. Ed. 2013, 52 (46), 12077–12080. 10.1002/anie.201306278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. L.; Highley C. B.; Yeh Y. C.; Galarraga J. H.; Uman S.; Burdick J. A. Three-Dimensional Extrusion Bioprinting of Single- and Double-Network Hydrogels Containing Dynamic Covalent Crosslinks. J. Biomed. Mater. Res., Part A 2018, 106 (4), 865–875. 10.1002/jbm.a.36323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao R.; Lv P.; Wang Q.; Zheng J.; Feng B.; Yang B. Cyclodextrin-Based Biological Stimuli-Responsive Carriers for Smart and Precision Medicine. Biomater. Sci. 2017, 5 (9), 1736–1745. 10.1039/C7BM00443E. [DOI] [PubMed] [Google Scholar]
- Jensen J. J.; Grimsley M.; Mathias L. J. No Title. J. Polym. Sci., Part A: Polym. Chem. 1996, 34 (3), 397–402. . [DOI] [Google Scholar]
- Amajjahe S.; Choi S.; Munteanu M.; Ritter H. Pseudopolyanions Based on Poly(NIPAAM-Co-Beta-Cyclodextrin Methacrylate) and Ionic Liquids. Angew. Chem., Int. Ed. 2008, 47, 3435–3437. 10.1002/anie.200704995. [DOI] [PubMed] [Google Scholar]
- Miao T.; Fenn S. L.; Charron P. N.; Oldinski R. A. Self-Healing and Thermoresponsive Dual-Cross-Linked Alginate Hydrogels Based on Supramolecular Inclusion Complexes. Biomacromolecules 2015, 16 (12), 3740–3750. 10.1021/acs.biomac.5b00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi H. W.; Mota C.; ten Cate A. T.; Calore A.; Moroni L.; Baker M. B. Thiol–Ene Alginate Hydrogels as Versatile Bioinks for Bioprinting. Biomacromolecules 2018, 19 (8), 3390–3400. 10.1021/acs.biomac.8b00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S.; Roy A.; Roy K.; Roy M. N. Study to Explore the Mechanism to Form Inclusion Complexes of β -Cyclodextrin with Vitamin Molecules. Sci. Rep. 2016, 6 (1), 1–12. 10.1038/srep35764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt B. V. K. J.; Barner-Kowollik C. Supramolecular X- and H-Shaped Star Block Copolymers via Cyclodextrin-Driven Supramolecular Self-Assembly. Polym. Chem. 2014, 5 (7), 2461–2472. 10.1039/c3py01580g. [DOI] [Google Scholar]
- Wedlock D. J.; Fasihuddin B. A.; Phillips G. O. Comparison of Molecular Weight Determination of Sodium Alginate by Sedimentation-Diffusion and Light Scattering. Int. J. Biol. Macromol. 1986, 8 (1), 57–61. 10.1016/0141-8130(86)90072-3. [DOI] [Google Scholar]
- Benesi H. A.; Hildebrand J. H. A Spectrophotometric Investigation of the Interaction of Iodine with Aromatic Hydrocarbons. J. Am. Chem. Soc. 1949, 71 (8), 2703–2707. 10.1021/ja01176a030. [DOI] [Google Scholar]
- Atkins P.; Paula J. De.. Physical Chemistry, 9th ed.; W.H. Freeman and Company, 2010. [Google Scholar]
- Granadero D.; Bordello J.; Pérez-Alvite M. J.; Novo M.; Al-Soufi W. Host-Guest Complexation Studied by Fluorescence Correlation Spectroscopy: Adamantane-Cyclodextrin Inclusion. Int. J. Mol. Sci. 2010, 11 (1), 173–188. 10.3390/ijms11010173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schibilla F.; Voskuhl J.; Fokina N. A.; Dahl J. E. P.; Schreiner P. R.; Ravoo B. J. Host–Guest Complexes of Cyclodextrins and Nanodiamonds as a Strong Non-Covalent Binding Motif for Self-Assembled Nanomaterials. Chem. - Eur. J. 2017, 23 (63), 16059–16065. 10.1002/chem.201703392. [DOI] [PubMed] [Google Scholar]
- Carrazana J.; Jover A.; Meijide F.; Soto V. H.; Tato J. V. Complexation of Adamantyl Compounds by β-Cyclodextrin and Monoaminoderivatives. J. Phys. Chem. B 2005, 109 (19), 9719–9726. 10.1021/jp0505781. [DOI] [PubMed] [Google Scholar]
- Sadrerafi K.; Moore E. E.; Lee M. W. Association Constant of β-Cyclodextrin with Carboranes, Adamantane, and Their Derivatives Using Displacement Binding Technique. J. Inclusion Phenom. Macrocyclic Chem. 2015, 83 (1–2), 159–166. 10.1007/s10847-015-0552-5. [DOI] [Google Scholar]
- Winiewska M.; Bugajska E.; Poznañski J. ITC-Derived Binding Affinity May Be Biased Due to Titrant (Nano)-Aggregation. Binding of Halogenated Benzotriazoles to the Catalytic Domain of Human Protein Kinase CK2. PLoS One 2017, 12 (3), e0173260. 10.1371/journal.pone.0173260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X.; Wang J.; Li L.; Pham D. T.; Clements P.; Lincoln S. F.; May B. L.; Chen O.; Zheng L.; Prud’Homme R. K. Tailoring Polymeric Hydrogels through Cyclodextrin Host-Guest Complexation. Macromol. Rapid Commun. 2010, 31 (3), 300–304. 10.1002/marc.200900560. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.