
Fig. 1. Pedigree structures and clinical findings in NAFLD families with ABHD5 mutations.
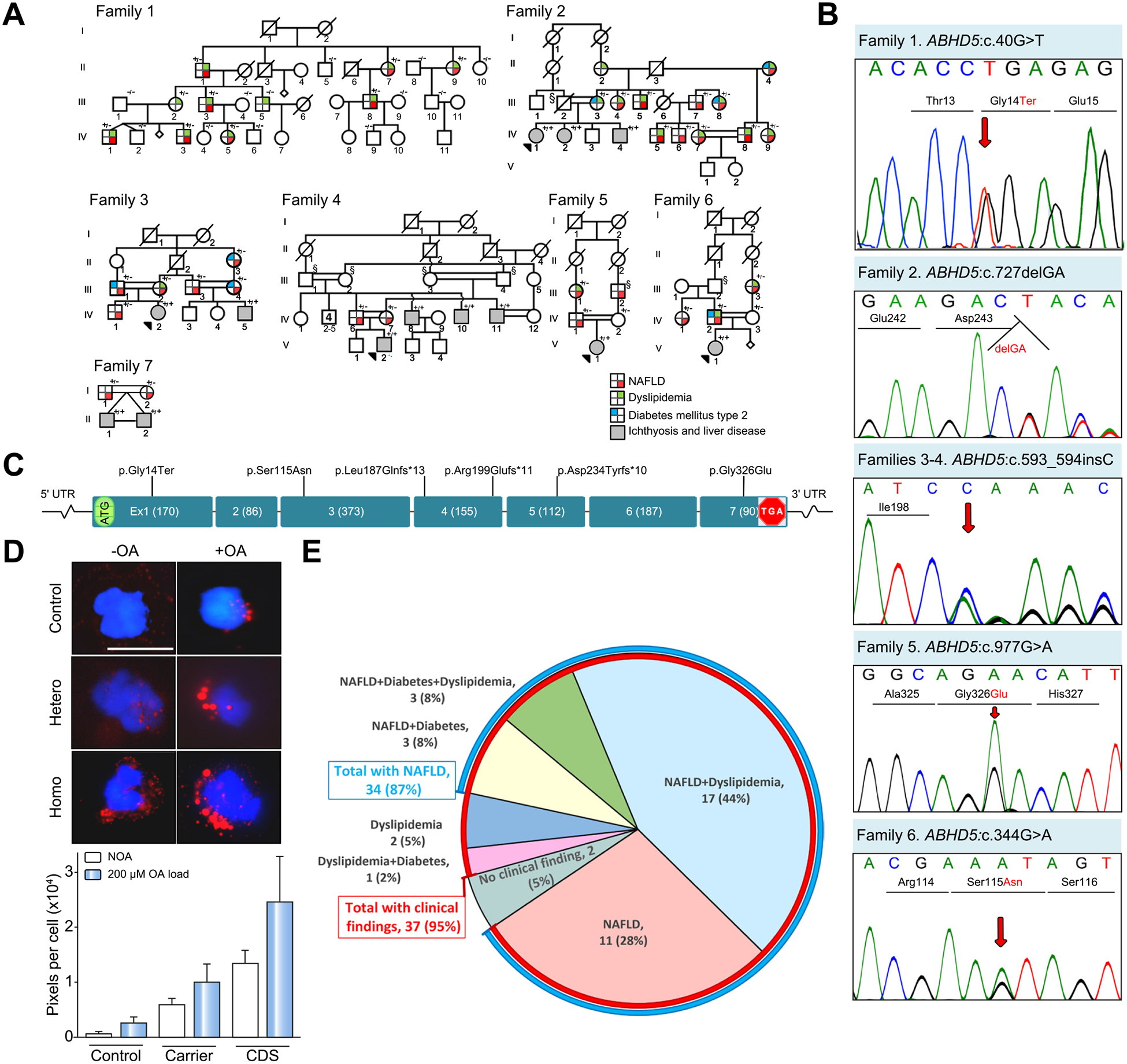
(A) Family 1 is of Italian ancestry with a monoallelic mutation in ABHD5. Note the lack of consanguinity in this family. Families 2–7 of Iranian ancestry show extensive consanguinity. Heterozygous carriers (+/−)show evidence of NAFLD and/or dyslipidemia and type 2 diabetes mellitus, and patients with bi-allelic mutations (+/+) manifest with CDS with neonatal ichthyosis and NAFLD. Individuals marked with § are presumed obligatory carriers of the mutation. For the presence of clinical manifestations in individuals tested, see the color code. (B) Sanger sequencing of mutations in Families 1–6. The mutation of Family 7, ABHD5:c.560_578 del19 was published previously.17 (C) Positions of the distinct mutations along the ABHD5 consisting of 7 exons drawn to scale; the introns are not in scale. (D) Presence of lipid droplets (red) in leukocytes from a control (upper panels), a heterozygous carrier (middle panel), and a homozygous individual (lower panel) after incubation without (-OA, left) or with (+OA, right) 200 lM OA. The lipid content was quantitated by assay of pixel density of Oil red O and DAPI stained cells (bar graph). The values represent the mean ± SD of 105–125 cells for each sample. (E) Relative distribution of clinical findings and their combinations in 39 ABHD5 carriers. Note that 35 individuals had NAFLD (87%, blue line) while 37 had clinical findings (95%, red line). CDS, Chanarin-Dorfman syndrome; NAFLD, non-alcoholic fatty liver disease; OA, oleic acid; UTR, untranslated region.