

Abstract
‘T cell exhaustion’ is a broad term that has been used to describe the response of T cells to chronic antigen stimulation, first in the setting of chronic viral infection but more recently in response to tumours. Understanding the features of and pathways to exhaustion has crucial implications for the success of checkpoint blockade and adoptive T cell transfer therapies. In this Viewpoint article, 18 experts in the field tell us what exhaustion means to them, ranging from complete lack of effector function to altered functionality to prevent immunopathology, with potential differences between cancer and chronic infection. Their responses highlight the dichotomy between terminally differentiated exhausted T cells that are TCF1– and the self-renewing TCF1+ population from which they derive. These TCF1+ cells are considered by some to have stem cell-like properties akin to memory T cell populations, but the developmental relationships are unclear at present. Recent studies have also highlighted an important role for the transcriptional regulator TOX in driving the epigenetic enforcement of exhaustion, but key questions remain about the potential to reverse the epigenetic programme of exhaustion and how this might affect the persistence of T cell populations.
What do we mean by T cell exhaustion and/or dysfunction and how would you define this state?
Nicholas P. Restifo and Rachel C. Lynn.
It is important to start off by stating that the term ‘T cell exhaustion’ is a basket term that describes various distinct epigenetic and metabolic states of post-thymic T cells. This term was popularized by viral immunologists and anthropomorphizes chromatin states that are characteristic of mice experiencing chronic viral infection, mainly with lymphocytic choriomeningitis virus (LCMV), where T cells are thought to be unable to clear a chronic infection.
Axel Kallies and Dietmar Zehn.
The term ‘exhaustion’ is used mainly to refer to effector T cells with a reduced capacity to secrete cytokines and increased expression of inhibitory receptors. These cells were thought to be hypofunctional effector T cells that differentiate from normal effector T cells in response to a chronically high antigen load. However, several observations have challenged this view and suggest that exhausted T cells are heterogeneous, have crucial roles in limiting viral infection or tumour growth1 and may develop independently from normal effector T cells, as outlined in the responses below (FIG. 1).
Fig. 1 |. The knowns and unknowns of T cell exhaustion.
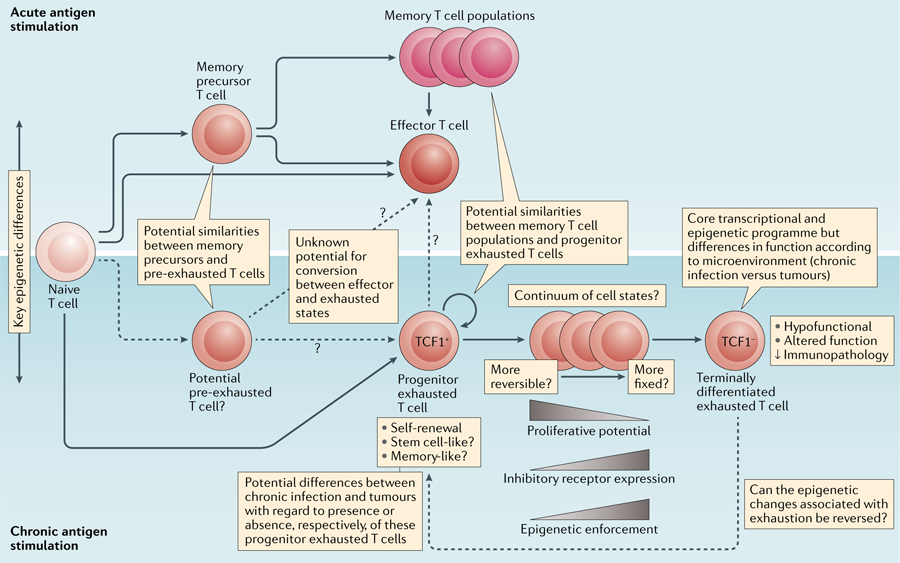
Potential developmental relationships between and features of exhausted T cell subsets compared with effector and memory T cell suswbsets during chronic versus acute antigen stimulation.
W. Nicholas Haining and Arlene H. Sharpe.
When an infection cannot be cleared by the host, a détente can occur whereby pathogen-specific T cells curtail their antipathogen function to avoid causing damage to normal tissues. Importantly, T cell exhaustion does not involve the complete absence of function: exhausted T cells can proliferate in vivo2, produce effector molecules, including inflammatory cytokines and granzymes, and exert some control over pathogens or tumours3.
E. John Wherry.
I agree that T cell exhaustion is an evolutionarily conserved adaptation to chronic antigen stimulation that is probably important to limit immunopathology or autoreactivity; thus, exhausted T cells are not inherently good or bad.
Pamela L. Schwartzberg.
Yes, although exhaustion is often seen as a dysfunctional state, it also allows T cells to persist and partially contain chronic infections without causing immunopathology.
Mary Philip and Andrea Schietinger.
Like our colleagues, we define T cell exhaustion as a differentiation state that is observed during chronic infections in the presence of persistent antigen and chronic T cell receptor (TCR) stimulation. Exhausted T cells express inhibitory receptors but can retain some antipathogen effector function, resulting in a pathogen–host ‘stalemate’4.
E.J.W.
There is general consensus that some features of exhausted T cells, compared with effector or memory T cells, include altered, sometimes reduced, effector functions, such as decreased (but not absent) cytokine production; increased chemokine expression; persistently high levels of expression of multiple inhibitory receptors, such as PD1, TIM3, LAG3, CTLA4 and TIGIT; reduced proliferative capacity when stimulated; an altered transcriptional programme involving the transcription factor TOX; and a unique epigenetic landscape4.
N.P.R. and R.C.L.
Chronic TCR signalling as a core mechanistic driver of exhaustion is highlighted by the well-established role of the calcineurin-dependent transcription factor nuclear factor of activated T cells (NFAT)5 and other NFAT-driven, TCR-responsive transcription factors (such as IRF4, BATF, nuclear receptor subfamily 4 group A (NR4A) and TOX)6–12 in both upregulating the expression of inhibitory receptors and maintaining the long-term survival of exhausted T cells.
Patrick G. Hogan and Anjana Rao.
We believe that for both tumour-infiltrating and virus-specific T cells, the most useful definition of exhaustion is an operational one: when exhausted T cells are present in the same environment or are stimulated under the same conditions as fully functional effector T cells, they have reduced responses (are hyporesponsive) to antigen. In addition to the features described in the responses above, it might also be worth expanding the definition of exhausted T cells to include criteria such as reduced levels of signalling proteins associated with activation and/or increased levels of negative regulatory proteins such as diacylglycerol kinases, phosphatases and E3 ubiquitin ligases13,14.
Enrico Lugli and Benjamin A. Youngblood.
We think it is important to note that although many investigators assign exhaustion status to T cells simply on the basis of their expression of inhibitory receptors, this is not a definitive feature of exhausted T cells as many highly functional effector T cells also express inhibitory receptors.
M.P. and A.S.
Another caveat to consider is that although antigen-specific T cells in tumours have many immunophenotypic and molecular features of exhausted T cells in chronic infection, they often completely lack effector function and fail to control tumour growth; therefore, we refer to these T cells as being dysfunctional rather than exhausted15.
Christian U. Blank and Ton N. Schumacher.
Instead, we would rather argue that the analysis of T cells in human and mouse tumours by single-cell sequencing and functional assays has provided strong evidence that T cells with high levels of expression of inhibitory receptors should not be considered exhausted (meaning ‘used up’ or inert) but rather should be considered dysfunctional or divergent (by which we mean that they have assumed an unconventional functional state). For example, this is shown by the ongoing proliferation in part of this T cell pool and the capacity of these cells to produce the chemokine CXCL13 (REFS16–18). Human tumours can contain large numbers of bystander T cells, and intrinsic tumour reactivity (the presence of a tumour-reactive TCR) is enriched among cells with a dysfunctional phenotype19,20. Although the in vivo development of a dysfunctional state was initially described under conditions of chronic antigen exposure21,22, the fact that dysfunctional T cell states are observed early after tumorigenesis23 and viral infection7,24, and can be induced by removing CD4+ T cell help25, suggests that the quality of costimulatory and/or inhibitory signals during TCR triggering may be an equally important factor.
N.P.R. and R.C.L.
We agree that the epigenetic state of T cells in chronic viral infection is similar but not identical to the epigenetic state of tumour-infiltrating lymphocytes (TILs), which are exposed to additional stimuli that may contribute to their dysfunction, including metabolites or lack thereof, immunosuppressive cytokines and chemokines and ionic disturbances. Thus, exhausted T cells are dysfunctional, but not all dysfunctional T cells are exhausted. Alternative mechanisms are at work, particularly in the case of TILs. We think it would be more constructive to use tumour systems to describe the epigenetic states of exhausted TILs rather than extrapolating from LCMV infection to describe these states.
Werner Held and Daniel E. Speiser.
As can be seen from the answers to the following question, recent studies have suggested that there are two distinct subsets of exhausted T cells — a progenitor (or precursor) subset, which has variably been referred to as being stem cell-like and/or memory-like, and a terminally differentiated subset. In contrast to some of our colleagues, we use the term ‘exhausted’ to refer only to the subset of chronically stimulated PD1+ cells that has limited recall expansion capacity and reduced effector functions (despite the expression of effector genes) and is irreversibly differentiated.
What do we know about the developmental relationships between exhausted T cells, their precursors and effector and memory T cell subsets?
E.L. and B.A.Y.
The initial hypothesis was that exhausted T cells share features with terminally differentiated T cells, which suggested a developmental relationship between these cell types (reviewed in REF.26).However, as outlined in the responses from my colleagues, subsequent studies showed that exhausted T cells seem to be derived from effector cells that retain the capacity to be long-lived (in other words, that survive the contraction stage of the immune response)27.
M.P. and A.S.
During both acute and chronic infections, naive antigen-specific T cells clonally expand and differentiate into effector cells. During chronic infection, effector T cells are persistently stimulated and become exhausted. The exhausted T cell population is heterogeneous, with progenitor T cells that self-renew and give rise to and maintain terminally differentiated exhausted T cells28–30.
W.H. and D.E.S.
The terminally differentiated exhausted T cell subset coexpresses multiple inhibitory receptors with effector genes and transcription factors (such as those encoding granzyme B (GZMB) and PRDM1) and has limited expansion capacity. This terminally differentiated subset is continuously derived from PD1+ cells that express and depend on the transcription factor TCF1 (REFS28,30,31). These progenitor cells, which also express SLAMF6 (REFS17,30,31) and CXCR5 (REFS28,32), lack expression of effector genes but express genes that are characteristic of central memory T cells (such as those encoding TCF1 (REF.33) and BCL-6 (REF.34)) in combination with a limited set of inhibitory receptors (for example, they lack expression of TIM3). The PD1+TCF1+ progenitor cells have stem cell-like properties as they have the capacity to proliferate, self-renew and produce terminally differentiated PD1+TCF1– GZMB+ cells.
P.G.H. and A.R.
We concur with the existence of subsets of progenitor cells (typically PD1hiTIM3lowTCF1+ cells) and terminally differentiated cells (typically PD1hiTIM3hiTCF1– cells, with partial effector function). The progenitor subset, which has characteristics of both stem cells and memory cells (such as self-renewal and rapid recall responses), can expand and give rise to effector cells after vaccination or PD1 checkpoint blockade. When transplanted into recipient infected or tumour-bearing mice, progenitor cells can proliferate and yield terminally differentiated exhausted cells, but transplanted terminally differentiated cells expand only marginally and remain PD1hiTIM3hi (REFS28,30,31,34–37). Single-cell RNA sequencing and mass cytometry have shown that a functionally exhausted T cell population still expresses a wide spectrum of genes and proteins associated with effector function16,38,39, which confirms earlier biological observations that exhausted T cells retain some effector function.
N.P.R. and R.C.L.
We agree that a TCF1+ stem cell-like progenitor population exists within the exhausted T cell subset and is likely responsible for the proliferative and functional responses that occur following checkpoint blockade. Having a more stem cell-like population of tumour-responsive T cells also allows better antitumour immune responses following adoptive cell transfer40,41. However, the ontogeny of endogenous stem cell-like exhausted T cells remains incompletely elucidated. We suggest that exhaustion is a parallel programme that can be overlaid onto ‘normal’ T cell differentiation, in which the exhausted state can be induced in T cells of any differentiation state and these cells continue to produce progeny with characteristics of exhaustion.
E.J.W.
In terms of the differences from other T cell functional states, exhausted T cells arise from cells that have undergone productive initial activation, which distinguishes exhaustion from tolerance or anergy. Exhausted T cells develop from the overstimulation of precursors, which results in early divergence between memory T cell precursors and precursors of the exhausted T cell lineage6. Terminally differentiated effector T cells (KLRG1hi cells in mice) cannot form exhausted T cells7,27 and exhausted T cells are distinct from senescent T cells in humans. Both progenitor and terminally differentiated subsets of exhausted T cells are epigenetically distinct from effector T cells or memory T cells.
W.N.H. and A.H.S.
Similarly to E.J.W., we note that unlike anergy, which is a state of dysfunction imprinted on naive T cells, exhaustion arises from effector T cells responding to chronic infection, which undergo progressive and hierarchical loss of function. Features of T cell exhaustion can be recognized within days after the onset of chronic infection, but exhaustion does not become irreversibly distinct from the effector T cell differentiation elicited by acute infection for approximately 2 weeks. T cell exhaustion, like many cellular differentiation processes, develops with a continuum of phenotypic and functional intermediate states.
A.K. and D.Z.
A further point of discussion is the relationship between the subsets of T cells found among antigen-specific cells responding to acute or chronic infection. T cells activated during an acute infection segregate into subsets of effector and memory precursor T cells. As we have learned recently, a related branching occurs also in chronic infections and in tumours28,30,34,42 with a segregation into cells that retain proliferative capacity (which we refer to as precursors rather than progenitors) and cells that are terminally differentiated. In both acute and chronic infections, the development of T cells with re-expansion potential depends on a similar set of regulators, such as TCF1, BCL-6 and FOXO1, and in both conditions these T cells express the IL-7 receptor and have a similar function in the generation of new T cells (secondary effector T cells or terminally differentiated exhausted T cells)28,30,34,42. Thus, despite some phenotypic differences, we think that memory precursor T cells in acute infection and the precursor population of exhausted T cells in chronic infection share important features and are related.
P.L.S.
I agree that the TCF1+ progenitor exhausted T cells resemble memory T cells transcriptionally to some extent, but both progenitor and terminally exhausted T cell populations also differ from memory and effector T cells, respectively, both transcriptionally and epigenetically6,43. Recent studies from multiple groups indicate that one key difference is the expression of the transcription factor TOX by exhausted T cells, which is crucial for epigenetic remodelling6–10.
M.P. and A.S.
We believe that there are important differences between chronic infection and the tumour microenvironment that should be taken into account. During tumour development, antigen-specific T cells are inadequately primed and fail to proliferate or become functional effector cells; instead they become dysfunctional (lacking effector function). Progressive dysfunctional ‘states’ are determined by tumour antigen chronicity: initially, T cells are in a plastic dysfunctional state and can be functionally rescued, but with continued tumour antigen exposure T cells undergo chromatin remodelling to a fixed dysfunctional state and cannot be functionally rescued15,44.
C.U.B. and T.N.S.
We agree with M.P. and A.S. that there is compelling evidence in mouse and human tumours that dysfunctional T cells are derived from a subset of what may be called ‘predysfunctional’ cells16,17,31,36. We note that it has not been established whether (pre)dysfunctional T cells in human tumours evolve from a functional effector cell or take a distinct differentiation pathway shortly after activation25.
P.G.H. and A.R.
It is widely accepted that exhausted CD8+ T cells arise in a matter of days or weeks from effector T cells exposed to chronic antigen stimulation4. However, we agree with M.P. and A.S. that an alternative but not mutually exclusive hypothesis, which has been proposed in the case of a spontaneously arising tumour model, is that exhausted T cells can arise directly from naive T cells subjected to continuing TCR stimulation without adequate costimulation23. A mechanism that can explain both of these routes to exhaustion involves NFAT. Continued activation of NFAT downstream of the TCR in the absence of AP-1 — owing to inadequate costimulation or the transitory expression of AP-1 proteins — turns on a negative regulatory programme that dampens the immune response. Indeed, we and others6–11,45–48 have shown that the secondary transcription factors induced by NFAT without AP-1 include NR4A and TOX, which impose and/or maintain the exhaustion programme. Non-transcriptional mechanisms — including modulation of the levels or activities of numerous signalling proteins — are also likely to have a role. Many studies have shown that exhausted T cells can alter their characteristics over time, as shown, for example, by progressive changes in chromatin accessibility of TILs44.
What have been the most important recent developments in the field?
P.L.S.
Recent discoveries include the description of a progenitor cell population that is characterized by high-level expression of TCF1 and low-level expression of PD1, which is responsible for responses to PD1 blockade28,30,34; the use of epigenetic analyses and single-cell transcriptomics to evaluate T cell exhaustion and its stability, as well as to delineate distinct cell populations in mice and humans4,6,43; and the description of TOX as a key transcriptional regulator of T cell exhaustion that also has a role in T cell persistence during chronic infection and cancer6–10.
E.J.W.
I have a similar list of major advances, including the demonstration of a distinct epigenetic landscape consistent with exhausted T cells, representing a unique immune cell fate43,44,49; the identification of progenitor and terminally differentiated subsets of exhausted T cells29,50 and the role of TCF1 (REFS28,30,34); the importance of exhausted T cells in the response to PD1 pathway blockade in humans; emerging single-cell RNA sequencing data that allow transcriptional signatures of exhausted T cells to be parsed in humans; the identification of TOX as a major driver of the epigenetic changes associated with exhaustion6–10; and the lack of epigenetic reprogramming of exhausted T cells by PD1 pathway blockade43. These general themes of single-cell analysis, epigenetics and checkpoint blockade are reiterated in the responses of my colleagues.
E.L. and B.A.Y.
Yes, we now know that many of the functional properties that are used to define T cell exhaustion become ‘imprinted’, being reinforced by epigenetic programmes51. These data have collectively advanced the concept that T cell exhaustion is a distinct stage of T cell differentiation.
M.P. and A.S.
Recent technological advances, including single-cell genomic analyses, epigenetic analyses and TCR sequencing, have enabled us to appreciate the heterogeneity and lineage relationships in antigen-specific T cell populations in tumours and infections6,16,44. The demonstration that T cell functional states are largely defined by the underlying epigenetic states, which ultimately regulate T cell plasticity and reprogrammability, has important implications for understanding and predicting responses to immunotherapeutic interventions44.
N.P.R. and R.C.L.
Indeed, the most significant advance in the field is the demonstration that the effectiveness of exhausted T cells after adoptive transfer or checkpoint blockade is related to their underlying epigenetic stem cell-like state.
W.N.H. and A.H.S.
PD1 blockade does not alter the underlying epigenetic state of exhausted T cells to durably restore effector functions17,43. Rather, checkpoint blockade induces the proliferation and differentiation of the progenitor exhausted T cells into terminally differentiated exhausted T cells17,43.
W.H. and D.E.S.
Thus, the identification of stem cell-like PD1+TCF1+CD8+ T cells in chronic infection28,30 and cancer17,31 explains how the immune response to persisting antigens can be sustained long-term and which cells mediate cellular expansion in response to immunotherapy. Their discovery reveals that checkpoint blockade does not reverse a terminal T cell exhaustion programme but rather expands and differentiates a stem cell-like progenitor subset. Finally, the presence of such progenitor cells among TILs provides a likely explanation for the efficacy of adoptive T cell therapy.
A.K. and D.Z.
In addition to the development of checkpoint blockade, which has revolutionized cancer therapy, the field has made substantial progress in understanding how exhausted T cells are generated and maintained, in particular by regulators directly downstream of TCR signalling, such as NFAT, TOX, NR4A1 and IRF4 (REFS5–12,45). Similarly, we have learned that exhausted and non-exhausted T cells have distinct metabolic and epigenetic features. All of these observations provide powerful opportunities to develop new therapeutic strategies. Arguably the most important recent discovery is that the maintenance and targeting of long-term T cell responses require the memory-like TCF1+ exhausted T cells28,30,34.
What are the key controversies and outstanding research questions?
P.G.H. and A.R.
The two most consequential unanswered questions are what are the cellular signalling and transcriptional pathways that drive the conversion to an exhausted T cell phenotype, and how can the chromatin and transcriptional changes of exhaustion be reversed in individual exhausted cells? Checkpoint blockade therapies, vaccination and treatment with costimulatory agonist antibodies have had real successes in restoring T cell function at the population level, but a detailed understanding of the cellular processes that initiate and sustain exhaustion will provide the foundation for a new generation of effective therapeutic strategies.
P.L.S.
Similarly, from my perspective, a key issue concerns the signalling pathways that contribute to the development of exhaustion, as well as the related but different issue of maintaining progenitor cells. In particular, whether and how we can manipulate signalling pathways to both activate and maintain T cell responses remain open questions, as does the question of whether pharmacological manipulations can reverse the epigenetic changes associated with exhaustion versus expand less-exhausted populations. The similarities and differences between exhausted T cells and other types of dysfunctional T cells (such as anergic or senescent T cells) is also an important issue to investigate further, given that overlapping signals contribute to these states (for example, NFAT activation is involved in both anergy and exhaustion, although these states differ in arising from naive and activated cells, respectively)1.
N.P.R. and R.C.L.
We need to define better the effects of the microenvironment on the induction of T cell exhaustion, the developmental trajectories of exhaustion and the point at which and extent to which exhaustion can be reversed. Understanding the consequences of unleashing T cells from exhaustion will also be crucial to designing the most effective therapeutic interventions. The inhibitory programmes driven by exhaustion prevent overactivation that leads to terminal differentiation and apoptotic death; thus, deletion of exhaustion-associated genes may ultimately accelerate T cell differentiation and death. Indeed, significant evidence shows that disruption of the genes encoding PD1, TOX and IRF4 ultimately reduces survival of the exhausted T cell pool7,8,10,12,52.
E.L. and B.A.Y.
Can exhaustion be reversed? TCR signalling contributes to exhaustion, but what else? Are signal 3 cytokines, such as type I interferon53 or transforming growth factor-β54, involved? Further resolution of these questions, as well as others that provide general insight into the mechanisms that reinforce exhausted T cells, will advance our efforts to generate long-lived therapeutic responses from adoptively transferred T cells or endogenous T cells55.
M.P. and A.S.
The outstanding question in tumour immunology is which T cells mediate antitumour activity in patients who respond to checkpoint blockade therapy: dysfunctional TILs that are reinvigorated or (non-dysfunctional) tumour-specific T cells that are newly recruited from the periphery? A related, highly debated question is whether there is a stable tumour-reactive stem cell-like progenitor T cell population that responds to checkpoint blockade. Whereas the concept of progenitor exhausted T cells is well established in chronic infection, we do not think there is any definitive evidence for their existence in TIL populations. TCF1+ T cells are observed in human tumours, but their tumour reactivity is largely unknown; on the basis of immunophenotype, they probably include non-tumour-reactive, bystander naive or memory T cells16,20 or recently infiltrated tumour-specific, functional T cells (TCF1hi), which will become dysfunctional (TCF1low) with continued exposure to tumour antigen15.
C.U.B. and T.N.S.
Similarly, we note that although there is strong evidence that predysfunctional T cells are required for long-term responses to PD1 blockade in mouse models17,31, it is unclear whether the predysfunctional T cells themselves, or their more differentiated (and phenotypically dysfunctional) progeny, form the ultimate effector pool for control of human tumours. On a related note, it is unclear what constitutes the better predictive biomarker for response to checkpoint blockade — the presence of a dysfunctional T cell pool that is likely to contain intrinsic tumour reactivity or the presence of a pool of less differentiated TCF1+ cells that may have the capacity for long-term reinvigoration but can be ‘contaminated’ by bystander T cells16,18,20,35.
W.N.H. and A.H.S.
We agree that T cell exhaustion needs to be defined more clearly in human tumours. In chronic infections, T cells can be isolated using MHC tetramers and compared with fully functional T cells elicited by an analogous acute infection. However, in human tumours, it is difficult to isolate unperturbed antigen-specific T cells that are unequivocally tumour specific or to identify an ideal comparator population. How do the functions and states (subpopulations) of exhausted T cells change over time? Can the epigenetic state of exhaustion be reversed to form true effector or memory T cells, and is this required for improved cancer immunotherapy? What is the relationship between T cell tolerance and exhaustion56, and how can the beneficial effects of checkpoint blockade on antitumour and antipathogen immunity be separated from immunopathology and autoimmunity?
E.J.W.
The presence of exhausted T cells in cancer probably indicates that there is a partially useful ongoing immune response and that tumour antigens that are recognized by the immune system exist. However, identifying exhausted T cells in humans remains challenging, as does evaluating their functionality. There is no definitive marker for exhausted T cells, although TOX may prove to be useful. Transcriptional profiles are informative, but epigenetic changes are more specific and robust43,44,49. A major clinical question is whether exhausted T cells can be, or indeed need to be, reprogrammed to achieve therapeutic benefit.
W.H. and D.E.S.
Does the presence, the abundance or other properties of PD1+TCF1+ progenitor cells predict the response to immunotherapy in human patients? How are the generation, maintenance and differentiation of PD1+TCF1+ cells into terminally differentiated PD1+TCF1– cells controlled? Do PD1+TCF1+ cells exist in additional pathological disorders with persisting antigen, such as graft rejection or autoimmunity, and what are the relevant similarities and differences between PD1+TCF1+CD8+ T cells present in distinct pathological situations.
A.K. and D.Z.
A key question that is still controversially discussed is when and how exhausted T cell populations are formed. The original view that they are terminally differentiated descendants of formerly ‘normal’ effector T cells has been challenged in different ways. First, T cells with features of exhausted T cells can be found in the early phase of infection, which suggests that their formation is independent of that of normal effector T cells. Second, we have recently learned that exhausted or terminally differentiated cells are derived from proliferation-competent TCF1+ cells28,30,34 that stably propagate the exhausted phenotype30,57. This indicates that exhausted T cells are maintained actively and independently from normal effector cells. Another central question is which signals establish and maintain the particular functional features of exhausted T cells.
E.L. and B.A.Y.
What is the relationship between the memory-like progenitor exhausted T cells and conventional, long-lived memory T cells?
E.J.W.
Indeed, there may well be similarities between resident memory T cells and exhausted T cells, but these similarities remain poorly understood.
What are the drivers of the similarities and differences in exhausted T cells in tumours compared with infection?
P.G.H. and A.R.
Of course there are differences, but it makes more sense to focus on the similarities — the common core programme of exhaustion that has been identified by several studies9,23,39 — as these are the shared characteristics that can be manipulated for therapeutic purposes.
E.J.W.
In global comparisons of RNA sequencing or epigenetic data, exhausted T cells share a core molecular programme in both tumours and chronic infections, in both mice and humans39.
W.N.H. and A.H.S.
In both cases, chronic antigen stimulation and inflammation result in the shared expression of transcription factors (such as TOX, TCF1 and NFAT) and coinhibitory receptors (such as PD1, TIM3 and TIGIT)10. Similarly, we think that progenitor and terminally differentiated exhausted T cell subsets are found in both contexts, although this is disputed by M.P. and A.S. However, there are also likely to be important differences driven by the tissue microenvironment. For example, exhausted T cells from chronic viral infection have a stronger type I interferon signal, whereas the tumour microenvironment probably puts more metabolic constraints on exhausted T cells17.
C.U.B. and T.N.S.
Similarly, we think that the development of the dysfunctional T cell state is driven by (continued) TCR triggering under conditions in which costimulation may be suboptimal and other inhibitory signals may be present. Whereas TCR affinity and antigen availability may have a role in certain cases, the difference between chronic infection and tumours is likely to lie primarily in the nature of the auxiliary signals, and such signals may potentially even differ between tumour microenvironments. This is an area that is only starting to be addressed17,23 and where our understanding is still limited.
E.J.W.
I agree that comparisons across different disease settings reveal disease-specific features of exhausted T cells that may relate to the inflammatory environment, antigen levels or tissue-specific effects. Thus, a core epigenetic and transcriptional programme of T cell exhaustion exists across diseases and species, but specific environments shape some features of exhaustion.
P.L.S.
Yes, as in real estate, location cannot be ignored. Although transcriptionally similar, the development of exhaustion may be very different in the tumour environment and in different tumours compared with during a chronic infection10.
M.P. and A.S.
During chronic infections, naive T cells encounter antigen acutely in an inflammatory context and differentiate into functional effector cells, which then become exhausted. By contrast, during tumour development, there is usually a long latency period after an initiating clonal oncogenic event during which tumour antigens are presented to the immune system in a non-inflammatory, non-stimulatory context58. Thus, an important difference is that exhausted T cells in chronic infection go through an initial effector phase and may retain some function, as evidenced by the rapid progression of infection when exhausted T cells are depleted, whereas dysfunctional TILs have little or no function as evidenced by continued tumour outgrowth. We do not think there is solid evidence of a stable, progenitor population of exhausted T cells within tumours.
A.K. and D.Z.
With further reference to potential differences in the TCF1+ population between chronic infection and tumours, TCF1+ T cells in tumours lack expression of CXCR5 (REF.31), which is expressed on these cells when localized to lymphoid tissues28,42. Nonetheless, in our opinion, TCF1+ T cells in tumours seem to be the precursors (progenitors) of exhausted effector T cells, which is similar to the precursor–progeny relationship that is observed in chronic infection.
W.H. and D.E.S.
Common features of the transcriptomes of memory-like progenitor cells from chronic infection and tumours include the expression of gene signatures of haematopoietic progenitor cells and follicular helper T cells28,31,34. However, the formation of memory-like cells in chronic infection depends on TCF1 (REFS28,30), whereas memory-like cells in tumours arise in the absence of TCF1 but their stem cell-like properties require TCF1 (REF.31).
N.P.R. and R.C.L.
Although we agree that there is a common core signature of exhaustion present in both tumours and chronic infection (imposed by chronic TCR signalling), given the differences that have been pointed out here between the tumour microenvironment and chronic infection, there is no reason in the current era to study tumour immunology using data obtained from LCMV as a model system. Untangling the diverse and overlapping biological phenomena that ultimately result in dysfunctional T cells in the tumour microenvironment will be key to developing efficacious immunotherapies for solid tumours. It seems unnecessary to put so much research effort in continuing to characterize T cell exhaustion in mouse models of chronic viral infection in the hope that these findings will translate to human tumour biology.
Can we manipulate the exhausted T cell states pre-emptively or therapeutically, and what is the durable effector potential of these exhausted cells?
P.G.H. and A.R.
Of course — all biological processes can be manipulated once we know what the players and signals are. However, what we do not know is whether it is possible to directly convert exhausted T cells into durable effector cells or to increase their effector potential, or whether it is better to focus on increasing the representation of the TCF1+ stem cell-like progenitor population.
A.K. and D.Z.
Although exhausted T cells are limited in their effector capacity, they undoubtedly contribute to tumour and virus control. Mounting evidence shows that exhaustion enables an organism to re-establish an immune equilibrium that ensures some level of virus control while limiting T cell-mediated immunopathology8,59,60.
E.L. and B.A.Y.
Yes, exhaustion seems to be a compromise between the host’s need to resolve the infection or tumour and the amount of damage that can be tolerated61. Because exhaustion limits the T cell response to chronic infection with pathogens or to tumours, it is thought to be a major barrier limiting the long-term efficacy of adoptive T cell therapies or therapies that rely on the host’s established endogenous T cell response (such as immune checkpoint blockade).
C.U.B. and T.N.S.
It is evident that the formation of the dysfunctional state can be influenced pre-emptively by modifying the conditions of T cell priming25. From a therapeutic perspective, there is evidence that at least a transient restoration of effector functions of late dysfunctional T cells derived from human tumours can be achieved18. However, durable restoration of T cell activity upon PD1 blockade relies on the progeny of early dysfunctional or predysfunctional T cells16,17. The development of technologies to determine the intrinsic tumour recognition potential of individual intratumoural CD8+ T cells as well as their capacity for durable reinvigoration upon immune checkpoint blockade would be a major step forward.
N.P.R. and R.C.L.
Disinhibition of T cells can clearly be accomplished with checkpoint blockade immunotherapy. The efficacy of using TILs therapeutically suggests that their optimized expansion outside the hostile tumour microenvironment can also reinvigorate dysfunctional T cells62. However, the extent to which the efficacy of these interventions relies on the prevalence of the more stem cell-like exhausted T cells is yet to be fully understood. The most promising opportunity for pre-emptive application of these strategies is adoptive T cell therapy, in which transferred T cells can be either generated under conditions that maintain the stem cell-like properties40,63 or genetically engineered with intrinsic exhaustion resistance64.
P.L.S.
There are two issues here. The first is how to invigorate exhausted T cells, or perhaps more accurately the TCF1+ progenitor-like cells, as has been achieved using checkpoint inhibitors. The second is how to have those effects persist. Recent work on TOX highlights this dichotomy. TOX is required for the exhausted phenotype, and TOX-deficient CD8+ T cells have increased effector function6–10. However, loss of TOX also prevents the persistence of antigen-specific CD8+ T cells in chronic infection, probably as a result of both loss of the TCF1+ progenitor population and promotion of a terminal effector phenotype that is more susceptible to cell death. Stepwise manipulations or the transfer of combinations of distinctly treated T cells may therefore be beneficial.
A.K. and D.Z.
A particular challenge is the epigenetic enforcement of the exhausted T cell phenotype51. Intuitively, this calls for strategies to prevent this epigenetic fixation. However, this may be difficult to achieve and could also lead to impaired persistence of T cells. All of this underlines that better immune therapies will require a much deeper understanding of the molecular and cellular ‘set points’ that establish T cell exhaustion in chronic infection and in cancer.
E.J.W.
I also note that the reinvigoration of exhausted T cells after PD1 blockade is temporary because the cells are not epigenetically reprogrammed43. Indeed, reinvigorated exhausted T cells are still exhausted, although the balance of subsets and aspects of transcriptional and functional activities may temporarily change. Targeting molecular programmers such as TOX might prevent the induction of exhaustion but two potential challenges with such an approach are immunopathology and/or loss of durability owing to differentiation to a terminal cell state7,8,10.
W.N.H. and A.H.S.
Because exhaustion is a state of differentiation that is enforced transcriptionally and epigenetically, its reversal is unlikely to be straightforward. Indeed, there are few examples of successful therapies that involve the dedifferentiation of a cell state. Knowledge of how exhaustion develops and the inherent states of exhausted subpopulations could guide therapeutic development. For example, epigenetic drugs can modify the development of T cell exhaustion51, but their selectivity for exhaustion (and not for other differentiation processes) remains to be determined. Alternatively, genome-editing technology offers the opportunity to manipulate exhaustion-specific regulators or enhancer elements49 to generate ‘exhaustion-proof ‘ T cells for adoptive cell therapy, albeit at the risk of enhanced immunopathology.
M.P. and A.S.
Similarly, we believe that in tumours, different epigenetic states of T cell dysfunction define the reprogrammability of a T cell: an earlier more plastic chromatin state can respond to immunotherapeutic interventions, whereas a fixed state is resistant to therapeutic reprogramming44. Because most T cells in tumours are largely in a fixed, non-reprogrammable cell state, we think that immunotherapy mainly functions through the recruitment of functional T cells or dysfunctional but reprogrammable T cells from the periphery and through preventing the dysfunction of newly recruited T cells. It remains to be seen whether ‘fixed’ dysfunctional T cells can in fact be reprogrammed through epigenetic targeting or other means.
W.H. and D.E.S.
There is already evidence from preclinical mouse models that memory-like progenitor T cells can be targeted therapeutically31 and used for adoptive cell therapy17. These cells can undergo intratumoural proliferation and differentiation into exhausted T cells in response to vaccination or checkpoint blockade, which is associated with improved tumour control31. Available evidence from chronic infections suggests that the functional improvements after checkpoint blockade are transient43, although the impact on memory-like progenitor cells needs to be addressed directly.
Do we need a new nomenclature for exhausted T cell subsets?
W.N.H. and A.H.S.
Yes, given the multiple names that are used in the literature and confusion about their respective functionalities. However, until we understand these subsets better and the spectrum of heterogeneity in different disease states, it will be difficult to assign a nomenclature to these subsets.
C.U.B. and T.N.S.
Non-experts in particular will view the term ‘exhausted’ as referring to a state in which a T cell is fully inert, and this is simply not in line with recent experimental data. We think that the term ‘dysfunctional’ better captures biology, by indicating that this involves a T cell population with properties that are distinct from those of the classic CD8+ T cell subsets.
P.G.H. and A.R.
The exhaustion programme seems to be an exaggerated and abnormally sustained manifestation of a normal negative-feedback programme that is turned on in all activated T cells. Unlike C.U.B. and T.N.S., we do not think that ‘dysfunctional’ effectively describes this adaptation to chronic antigen stimulation. Thus, we believe that broad definitions of exhausted T cells are best, particularly when functionally validated as part of a meaningful physiological response. The simple terms ‘hyporesponsive’ and ‘exhausted’ are sufficient to indicate the deviation of a T cell population from full effector capability, with the understanding that these classifications embrace a wide spectrum of functional impairments in the individual cells. However, as expressed by our colleagues below, we recognise that it has been useful to divide the exhausted T cell population into a stem cell-like progenitor subset and a more terminally differentiated subset, which can be distinguished by expression of a functional marker, TCF1.
P.L.S.
These two subsets were first delineated in terms of their responses to PD1 blockade and differential expression of eomesodermin and T-bet29, and, more recently, by the description of a TCF1+ progenitor population that can both renew and repopulate terminally differentiated exhausted T cells28,30,34. However, single-cell analyses suggest that this may be an oversimplification6, an idea that is supported by the lack of correlation between expression of TCF1 and expression of eomesodermin. Thus, there may be multiple stages of differentiation of exhausted T cells, leading to an epigenetically distinct terminally differentiated state. These stages may differ between tumours and chronic infections.
E.L. and B.A.Y.
We agree that there are at least two distinct subsets of exhausted T cells that are organized in a hierarchy similar to that of conventional long-lived memory T cells and terminally differentiated effector T cells, albeit with unique features. Therefore, these subsets warrant unique descriptors. Notably, recent data suggest that epigenetic mechanisms are crucially involved in the developmental changes that delineate these two subsets51. As such, a molecular definition of the differentiation status of a T cell in the context of exhaustion should include epigenetic programmes.
N.P.R. and R.C.L.
We agree with P.L.S. that while any proposed nomenclature should include an understanding of the stem cell-like versus terminally differentiated exhausted T cells, this should not imply only two differential states but rather should imply what is likely to be a continuous trajectory. In addition, states derived from chronic antigen exposure (‘exhaustion’) should be parsed from states describing T cell dysfunction derived from ‘suppression’ or ‘ignorance’65.
C.U.B. and T.N.S.
On the basis of the observation of a continuum of cell states with, for example, increasing levels of inhibitory receptor expression and variable levels of proliferation16, it may not be appropriate to consider the dysfunctional T cell pool as a single well-defined subset, and it may be more productive to think of the process as involving transit through a dysfunctional gradient.
W.N.H. and A.H.S.
Yes, the primary issue is less about assigning phenotypic ‘buckets’ for what is in effect a continuum of substates but more about identifying the fundamental biological regulators of T cell functional impairment. The latter is the brass ring that will determine whether and how reversal of exhaustion could be exploited to treat human disease.
W.H. and D.E.S.
A new nomenclature would be warranted, but will likely be hard to agree. We refer to the PD1+TCF1+ progenitor cells as being memory-like, because they have a unique phenotype, gene expression profile28,30 and epigenetic signature17 and because these cells persist in the presence of antigen. However, memory is defined by the absence of antigen. Irrespective of the presence of antigen, memory-like PD1+TCF1+ T cells and central memory T cells share stem cell-like properties. By contrast, we use the term ‘exhausted’ to refer selectively to the terminally differentiated progeny of the memory-like PD1+TCF1+ T cells.
E.J.W.
Subsets of exhausted T cells were originally defined as progenitor cells or terminally differentiated cells on the basis of their ability to undergo proliferation (and differentiation) in response to PD1 blockade50 or persisting antigen29. Other terms have also been used to describe the progenitor cells, including ‘stem cell-like’ and ‘memory-like’. However, until we have a more complete understanding of the number of truly distinct subsets of exhausted T cells, their diversity and underlying epigenetics, it may be challenging to assign nomenclature that accurately matches the biology.
M.P. and A.S.
We find it useful to use distinct terms for hyporesponsive T cells in chronic infection and cancer (exhausted T cells and dysfunctional T cells, respectively) because, despite some similarities, there are important differences in their ontogeny and function. It remains to be seen whether the conceptual framework of progenitor and terminally differentiated subsets applies to dysfunctional TILs. An important point is that memory T cells by definition are antigen-experienced cells that persist for a long time after antigen clearance in acute infection; thus, in contrast to W.H. and D.E.S., we do not think that antigen-specific T cells present during chronic infection or in tumours can be defined as memory T cells.
A.K. and D.Z.
We agree with P.G.H. and A.R. that the terms ‘exhausted’ and ‘dysfunctional’ somewhat contrast with the view that these T cells seem to be tightly regulated and functionally adapted to the conditions of chronic antigen stimulation. However, as ‘exhausted’ is widely used and can easily accommodate the particular aspects of functionally impaired T cells in chronic infection and in tumours, we would prefer to continue using this term. What we definitively need is a nomenclature for the specialized TCF1+ cells that maintain long-term T cell responses by constantly replacing the pool of short-lived cells. Currently, these cells are being referred to as progenitor cells, memory-like cells, stem cell-like cells or follicular cytotoxic T cells1,8,12,28,30,34,42,57. Yet, these closely related cells constitute a unique entity, for which we lack an appropriate term. Despite their self-renewing capacity, these cells are committed to the production of terminally exhausted T cells. Therefore, our suggestion is to use the term ‘precursors’ of exhausted T cells.
Acknowledgements
P.G.H. and A.R. are funded by National Institutes of Health grants AI109842, AI040127 and AI084167, and U01 DE028227. A.K. is supported by a fellowship by the National Health and Medical Research Council of Australia. E.L. is supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC IG20607).
Footnotes
Competing interests
W.N.H. is an employee of Merck & Co. and a founder of Arsenal Biosciences. The La Jolla Institute for Immunology (LJI) has received a research grant from Lyell lmmunopharma to support aspects of the work of P.G.H. on transcriptional mechanisms in CD8+ T cells. E.L. receives preclinical research funding from Bristol-Myers Squibb. LJI is the recipient of a research grant from Lyell lmmunopharma, which supports studies in the laboratory of A.R. using mouse models to elucidate the transcriptional and epigenetic programmes operating in CD8+ tumour-infiltrating T cells. LJI has a pending patent, PCT/US201B/062354, covering the use and production of engineered immune cells to disrupt NFAT–AP-1 pathway transcription factors, including NR4A1/2/3, TOX and TOX2, with A.R. listed as one of the inventors. LJI is the recipient of a research grant from the Takeda-Sanford Innovation Alliance for research in the laboratory of A.R. related to NR4A in human CD8+ T cells. N.P.R. and R.C.L. are employees of Lyell Immunopharma. A.H.S. has patents on the PD1 pathway licenced by Roche/Genentech and Novartis, consults for Novartis, is on the scientific advisory boards for Surface Oncology, Sqz Biotech, Elstar Therapeutics, Elpiscience, Selecta and Monopteros, and has research funding from Novartis, Roche, Ipsen, UCB, Quark Ventures and Merck. E.J.W. is a member of the Parker Institute for Cancer lmmunotherapy, which supported the present study. E.J.W. has consulting agreements with and/or is on the scientific advisory board for Merck, Roche, Pieris, Elstar and Surface Oncology. E.J.W. has a patent licensing agreement on the PD1 pathway with Roche/Genentech. E.J.W. is a founder of Arsenal Biosciences. B.A.Y. has patents associated with epigenetic programming of T cells. He has received honoraria for speaking at companies (less than US$5,000). The other authors declare no competing interests.
References
- 1.Speiser DE et al. T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nat. Rev. Immunol 14, 768–774 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Jin X et al. Dramatic rise in plasma viremia after CD8+ T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med 189, 991–998 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wherry EJ T cell exhaustion. Nat. Immunol 12, 492–499 (2011). [DOI] [PubMed] [Google Scholar]
- 4.McLane LM, Abdel-Hakeem MS & Wherry EJ CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol 37, 457–495 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Martinez GJ et al. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 42, 265–278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao C et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat. Immunol 20, 890–901 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan O et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alfei F et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Seo H et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl Acad. Sci. USA 116, 12410–12415 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott AC et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J et al. Nr4a transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Man K et al. Transcription factor IRF4 promotes CD8+ T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity 47, 1129–1141 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Macian F et al. Transcriptional mechanisms underlying lymphocyte tolerance. Cell 109, 719–731 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Heissmeyer V et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat. Immunol 5, 255–265 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Philip M & Schietinger A Heterogeneity and fate choice: T cell exhaustion in cancer and chronic infections. Curr. Opin. Immunol 58, 98–103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 176, 775–789 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller BC et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thommen DS et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med 24, 994–1004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheper W et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med 25, 89–94 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Simoni Y et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Wherry EJ et al. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol 77, 4911–4927 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zajac AJ et al. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med 188, 2205–2213 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schietinger A et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 45, 389–401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Youngblood B et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8+ T cells. Immunity 35, 400–412 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahrends T et al. CD4+ T cell help confers a cytotoxic T cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness. Immunity 47, 848–861 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Akbar AN & Henson SM Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol 11, 289–295 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Angelosanto JM, Blackburn SD, Crawford A & Wherry EJ Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J. Virol 86, 8161–8170 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Im SJ et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paley MA et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338, 1220–1225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Utzschneider DT et al. T cell factor 1-expressing memory-like CD8+ T cells sustain the immune response to chronic viral infections. Immunity 45, 415–427 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Siddiqui I et al. Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211.e10 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Brummelman J et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8+ T cells infiltrating human tumors. J. Exp. Med 215, 2520–2535 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jefvannet G et al. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc. Natl Acad. Sci. USA 107, 9777–9782 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu T et al. The TCF1–Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol 1, eaai8593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sade-Feldman M et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurtulus S et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1– CD8+ tumor-infiltrating T cells. Immunity 50, 181–194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jadhav RR et al. Epigenetic signature of PD-1+TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade. Proc. Natl Acad. Sci. USA 116, 14113–14118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bengsch B et al. Epigenomic-guided mass cytometry profiling reveals disease-specific features of exhausted CD8 T cells. Immunity 48, 1029–1045 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gattinoni L et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med 15, 808–813 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gattinoni L et al. A human memory T cell subset with stem cell-like properties. Nat. Med 17, 1290–1297 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leong YA et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nat. Immunol 17, 1187–1196 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Pauken KE et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Philip M et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 567, 525–529 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X et al. TOX promotes the exhaustion of antitumor CD8+ T cells by preventing PD1 degradation in hepatocellular carcinoma. J. Hepatol 10.1016/j.jhep.2019.05.015 (2019). [DOI] [PubMed]
- 47.Scott-Browne JP et al. Dynamic changes in chromatin accessibility occur in CD8+ T cells responding to viral infection. Immunity 45, 1327–1340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mognol G et al. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc. Natl Acad. Sci. USA 114, E2776–E2785 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sen DR et al. The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blackburn SD, Shin H, Freeman GJ & Wherry EJ Selective expansion of a subset of exhausted CD8 T cells by αPD-L1 blockade. Proc. Natl Acad. Sci. USA 105, 15016–15021 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghoneim HE et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell 170, 142–157 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Odorizzi PM et al. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med 212, 1125–1137 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Teijaro JR et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340, 207–211 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stephen TL et al. SATB1 expression governs epigenetic repression of PD-1 in tumor-reactive T cells. Immunity 46, 51–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pilipow K et al. Antioxidant metabolism regulates CD8+ T memory stem cell formation and antitumor immunity. JCI Insight 3, 122299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKinney EF, Lee JC, Jayne DR, Lyons PA & Smith KG T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 523, 612–616 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Utzschneider DT et al. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat. Immunol 14, 603–610 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Willimsky G & Blankenstein T Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature 437, 141–146 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Doedens AL et al. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat. Immunol 14, 1173–1182 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frebel H et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J. Exp. Med 209, 2485–2499 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moskophidis D, Lechner F, Pircher H & Zinkernagel RM Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 362, 758–761 (1993). [DOI] [PubMed] [Google Scholar]
- 62.Rosenberg SA & Restifo NP Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vodnala SK et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 363, eaau0135 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lynn RC et al. c-Jun overexpressing CAR-T cells are exhaustion-resistant and functionally superior. Nature (in the press).
- 65.Overwijk WW et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp. Med 198, 569–580 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]