

Abstract
Recurrence and drug resistance are major challenges in the treatment of acute myeloid leukemia (AML) that spur efforts to identify new clinical targets and active agents. STAT3 has emerged as a potential target in resistant AML, but inhibiting STAT3 function has proven challenging. This paper describes synthetic studies and biological assays for a naphthalene sulfonamide inhibitor class of molecules that inhibit G-CSF-induced STAT3 phosphorylation in cellulo and induce apoptosis in AML cells. We describe two different approaches to inhibitor design: First, variation of substituents on the naphthalene sulfonamide core allows improvements in anti-STAT activity and creates a more thorough understanding of anti-STAT SAR. Second, a novel approach involving hybrid sulfonamide–rhodium(II) conjugates tests our ability to use cooperative organic–inorganic binding for drug development, and to use SAR studies to inform metal conjugate design. Both approaches have produced compounds with improved binding potency. In vivo and in cellulo experiments further demonstrate that these approaches can also lead to improved activity in living cells, and that compound 3aa slows disease progression in a xenograft model of AML.
Keywords: STAT3, Leukemia, naphthylsulfonamide, iminonaphthoquinone
Graphical Abstract
Introduction
Signal transducer and activator of transcription (STAT) proteins are intracellular signaling proteins that mediate response to extracellular stimuli.1–3 Extracellular signaling proteins, such as cytokines and growth factors, activate membrane-bound receptors that then recruit and tyrosine phosphorylate the STAT3 loop domain immediately C-terminal to the SH2 domain.4 Phosphorylated STAT3 (pSTAT3) dimerizes and translocates to the nucleus where it activates transcription of tumor survival pathways.5,6 The aberrant activation of STAT3 is a common occurrence in many cancers.6,7 Inhibition of STAT3 activity has been shown to lead to apoptosis in cancers such as breast,8,9 head and neck,10,11 colon,12 liver,13 acute myeloid leukemia (AML),14,15 and prostate.16 Thus, STAT3 has been implicated as a promising protein target for the development of broad-spectrum chemotherapeutics.17
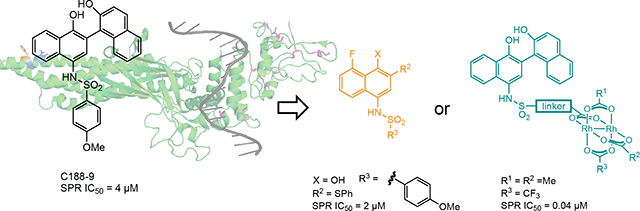
In spite of significant efforts, inhibiting STAT3 pathways has proven difficult.18 Blocking intracellular protein–protein interactions like those that mediate STAT3 activation remains a daunting pharmacological goal.19,20 We became interested in naphthalene sulfonamides as anti-STAT3 compounds,4,11,14,21 and sought to understand the molecular basis of STAT3 binding, to identify their molecular target on STAT3, and to assess the anti-AML activity of these compounds in molecular, cellular, and animal models of AML. We began this exploration assuming that the lead compound, C188–9 (a.k.a. TTI-101) (Figure 1b), competitively binds to the phosphotyrosine peptide binding site in SH2 domain, consistent with previous assumptions (Figure 1a). During the course of our investigations, rhodium-catalyzed proximity-driven covalent modification determined that modified naphthalene sulfonamides also target a new binding site in the coiled-coil domain (CCD), and that, importantly, binding to this site is not competed by the natural phosphotyrosine peptide (Figure 1a, orange residue).15 In part, current research seeks to investigate the therapeutic potential of targeting CCD.15
Figure 1.

(a) Crystal structure of a single STAT3 protein when bound as a dimer to duplex DNA.22 Nucleophilic ligands, cysteine and methionine residues of the SH2 domain (purple) and aspartate ligands of the coiled-coil domain (blue) are highlighted. Affinity labeling experiments with a rhodium inhibitor conjugate indicates that binding occurs at/near Phe174 (orange) in the coiled-coil domain. (b) A lead compound, C188–9 (a.k.a. TTI-101). (c) Rh-STAT3 conjugates use inorganic-organic cooperativity to bind STAT3.
At the same time, the challenges of STAT3 inhibition provided an opportunity to explore an unconventional approach to inhibitor development: employing rhodium–small molecule conjugates as anti-STAT agents. Conceptually, the approach is based on cooperative organic–inorganic binding that includes rhodium coordination to a Lewis-basic side chain near protein binding interface (Figure 1c).22,23 We have demonstrated this concept with rhodium-peptide conjugates, and this work provided an opportunity to explore the concept with non-peptide inhibitors (Figure 1c). Cellular studies provided an opportunity to assess function in living cells, as opposed to purely biophysical measurements, and these measurements shed light on the benefits and limitations of rhodium-based approaches, compared to traditional small-molecule STAT3 inhibitors.
Inhibitor Synthesis
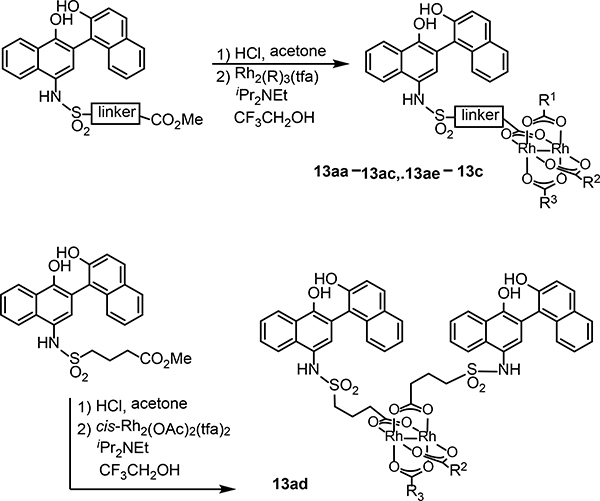
Naphthalene sulfonamides were identified as lead fragments through a combination of screening and limited structural variation.21 Compounds with STAT3-binding activity permit significant variation of the sulfonamide substituent (R3 in ), thus R3 was seen as the most straightforward site for incorporation of rhodium centers, via target structures such as 13aa-13c. Furthermore, the C5–C8 positions of the naphthalene ring (R4) were a largely unexplored site of variation. In the course of our studies, we became further interested in variation of the naphthol, and in assessing the function of redox variants of this core, such as the iminoquinone.
In the synthetic direction, the requisite 2-substituted naphthalene sulfonamides (e.g. 2a–4d, Scheme 1) are readily available via elaboration of simple naphthalene sulfonamides (1a–1f, Scheme 1) after sulfonamide formation from 4-amino-1-naphthol. Formation of the sulfonamide early in the synthesis is important to limit synthetic manipulations with oxidatively sensitive free 4-amino-1-naphthol derivatives. Elaboration of the 2-position of the naphthalene ring with halogens is directly achieved by electrophilic aromatic substitution to afford arylbromides (2a-2c). Alternatively, oxidative coupling with pro-nucleophiles (Nu–H = Ar–H, RS–H, RO–H) succeeds via the intermediacy of an iminobenzoquinone 1aox-1fox.25 This allowed preparation of a variety of additional S-linked (3aa-3cd) and C-linked (4a-4d) 2-substitution products.
Scheme 1.

Modular synthesis of STAT3 inhibitors (a) R1SO2Cl, pyridine, MeCN, MgSO4 (32–94%) (b) Br2, CH2Cl2 (25–60%) (c) PhI(OAc)2, acetone (d) PhSH, HCOOH (26–85%) (e) BF3·Et2O, 2-naphthol (30–70%). Yields for oxidative coupling were taken over 2 steps. (*) Inhibitor isolated as oxidized iminonaphthoquinone.
In tandem, we set our sights on 8-substituted derivatives that could provide electronic and steric perturbation of the naphthalene core, both to investigate SAR in this region and to perturb the redox stability of inhibitor molecules. In a first approach, (Scheme 2), we synthesized 1,8-difluoronaphthalene from 1,8-diaminonaphthalene,26 via a bis-diazonium intermediate. The 1,8-difluoronaphthalene was nitrated at the 4-position (5), and nucleophilic aromatic substitution of the fluorine para to nitro with an alkoxide delivered naphthyl ether, and subsequent hydrogenolysis yield 7. Efforts in this direction were limited by safety and scalability concerns: The bis-diazonium salt was prone to spontaneous and at times violent decomposition. As an alternative, we exploited more recent advances27 in nucleophilic aromatic fluorination to prepare 6 from 1,5-dinitronaphthalene (Scheme 2). This route gave better access to fluorinated naphthalene intermediates. The electrophilic naphthalene ring 6 could be oxidized with tert-butylhydroperoxide (TBHP) in liquid ammonia to install a hydroxyl group, affording naphthol 7. Etherification of the OH group was next performed, providing a benzyl (8) or methyl (10) ether. At this point, the nitro group could be reduced selectively in the presence of a platinum catalyst, and the resulting aniline then coupled with a sulfonyl chloride to provide sulfonamide 8. Deprotection of the benzyl group and oxidative coupling with thiophenol gave fluoride 9. Alternatively, the methyl ethers 12a and 12b were synthesized as analogues of compounds 2a-2c.
Scheme 2.

Synthesis of 5-fluoronaphthylsulfonamides (a) i) HBF4 (aq) NaNO2 ii) molten KHF2 (2–20%) (b) HNO3, NaNO2 (60%) (c) BnOH, NaH, CH2Cl2 (61%) (d) Pt/C, NaBH4 (99%) (e) CsF, DMSO, 100 °C (23%) (f) NH3, TBHP, NaOH, THF (70%) (g) BnBr, Cs2CO3, CH2Cl2 (75%) (h) i) Pt/C, NaBH4 ii) 4-methoxybenzenesulfonyl chloride, pyridine, MgSO4 (62%) (i) Pd/C, H2, (j) PhI(OAc)2, PhSH, (CF3)2CHOH (37%) (k) MeI, K2CO3, DMF (84%) (l) i) Pd/C, NaBH4 ii) R2SO2Cl, pyridine, MgSO4 (45–89%) (m) Br2, CH2Cl2 (56–75%)
Several structures with different ester functional groups serve as anchor points for rhodium conjugation (4a, 4b, 4c in Table 1). General methods that have been previously reported allow for facile cooperative rhodium(II) binding to neighboring residues that strongly coordinate to metal centers, such as methionine or histidine, allowed 50–500 fold increases in potency in other systems.23,24,28 The presumptive target of naphthalene sulfonamide inhibitors, the SH2 domain, contained several such residues (Figure 1a, purple). However, the coiled coil domain—later identified as another target of naphthalene sulfonamide inhibitors—is devoid of histidine, cysteine, and methionine residues. This discovery allowed us to assess the suitability of cooperative inhibition with rhodium conjugates toward binding sites that lack such strong metal-binding sites. (Figure 1a, blue). We chose to append esters onto the C4 substituent of the naphthalene ring (), a region of the inhibitor which was previously found to be tolerant to steric and electronic variations.21 We prepared several rhodium conjugates from ester-containing intermediates by acidic hydrolysis followed by metalation with a heteroleptic rhodium reagent containing labile trifluoroacetate groups (Table 2). Unfortunately, Rh-naphthylsulfonamide conjugates could not be made by traditional procedures that involve refluxing the carboxylate inhibitors in benzene. These conditions proved to be too harsh for the organic starting materials. Instead methodologies previously developed by our group were used to successfully couple these small molecules to rhodium.15
Table 1.
Preparation of rhodium conjugates by carboxylate exchange with heteroleptic rhodium trifluoroacetates.
 | ||||||
|---|---|---|---|---|---|---|
| entry |  |
product | R1 | R2 | R3 | yield (%) |
| 1 | 13aa | Me | Me | Me | 63 | |
| 2 | 13ab | nBu | nBu | nBu | 15 | |
| 3 | 13ac | fluorescein | Me | Me | 21 | |
| 4 | 13ad | n.a. | Me | Me | 15 | |
| 5 | 13ae | CF3 | Me | Me | 42 | |
| 6 | 13af | CF3 | CF3 | CF3 | 15 | |
| 7 |  |
13b | Me | Me | Me | 37 |
| 8 |  |
13c | Me | Me | Me | 8 |
Table 2.
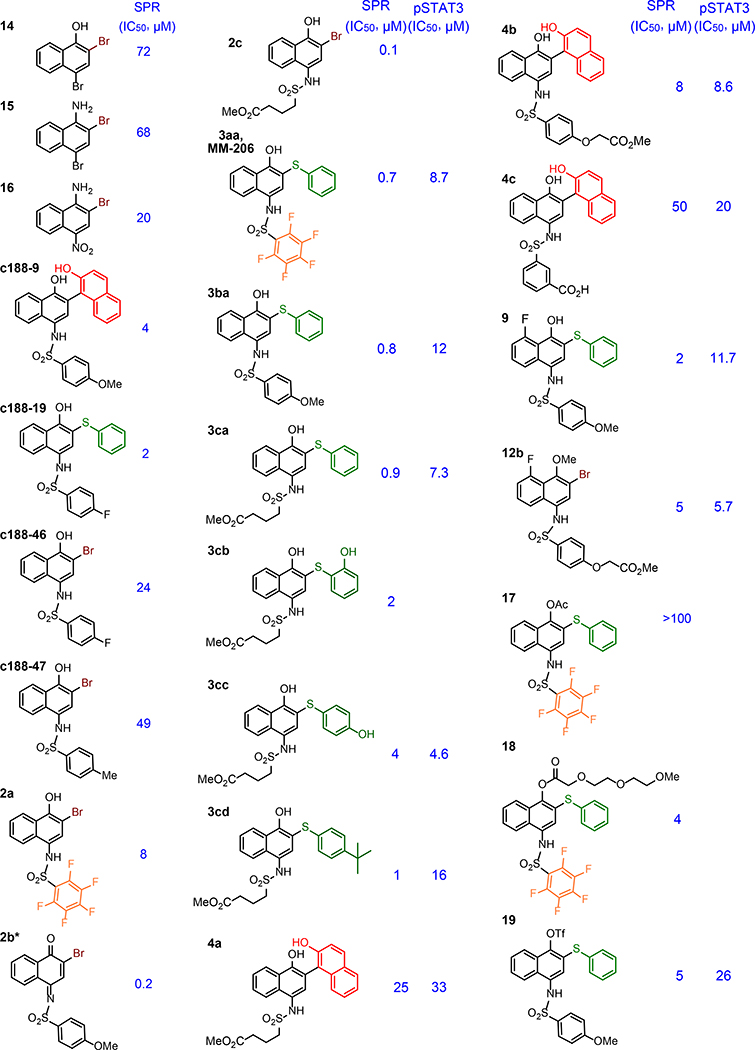
Inhibition of STAT3 phosphopeptide binding. Inhibitive potency (IC50) was determined by established SPR and/or a cellular STAT3 phosphorylation assay.
 |
SPR analysis
Potency was initially assessed by surface plasmon resonance (SPR), allowing quantification of binding and straightforward comparison to other studies.21 Variation of the R1 and R2 groups provided additional insight into the structural landscape of affinity. A perfluorinated ring within compound 2a—suggested by the appearance of this group in another STAT-binding compound,8,29 results in a 3-fold increase in potency from C188–46 (Table 2). When this change was combined with variation at the C2 position, significant gains in affinity were observed. Compound 3aa was found to be 10-fold more potent than 2a. Being inspired by this increase in potency, we made different analogues of the inhibitors. In general inhibitors with thiophenyl in the C2 position displayed greater potency. Even when the perfluorinate ring in the R1 position was replaced with another alkyl or aryl group, the potency remained similar. In addition, it seems the thiophenyl ring is functionally tolerant to hydroxylation and alkylation. Inhibitors (3cb-3cd) were only slightly less potent than their parent compound (3ca). In summary, SPR analysis demonstrates that molecular recognition of the inhibitor to STAT3 is dependent on the 1,2,4 substitution on the naphthalene ring. This is highlighted by the fact that smaller molecules without the sulfonamide-like moiety (14–16) still inhibit STAT3-phosphopeptide binding.
Naphthalene sulfonamide oxidation
The 1,4-substitution pattern of hydroxyl-naphthalenesulfonamides renders the core structure susceptible to oxidation. Some synthetic intermediates (i.e. 1b, 1c, and 1f) indeed proved quite prone to air oxidation and subsequent decomposition. In vivo oxidation could be a decomposition pathway, but it is also possible that oxidized iminoquinone species (e.g. Table 2, 2b*) may represented an active species, with naphthalenesulfonamides serving as a pro-drug. Iminoquinone structures might serve as reactive electrophiles for covalent inhibition pathways, and in this sense they might putatively share features in common with other known STAT3 inhibitors, such as LY530 and BA-TPQ,31 with structures consistent with electrophilic reactivity. Significant decomposition of C188–9 was observed within 4 hours in aqueous buffer under air (Figure S1), and crude NMR investigations showed new peaks consistent with a iminoquinone. In one case, an iminoquinone intermediate 2b* (Table 2) was stable enough to be isolated, characterized, and tested. This iminoquinone also demonstrated significant binding affinity in our SPR-based assay, suggesting that iminoquinone intermediates may contribute to at least some function in vivo.
To further probe these issues, we prepared a brief series of C188–9 analogues with an explicit goal of improving lifetime in media. Although some structural changes abrogated binding, a few did show promising binding by SPR, including fluoro-ether 12b and triflate 19 (Table 2). Interestingly, O-acylation of the naphthol hydroxyl group has a significant negative effect on binding in the case of 17, but substitution on oxygen is tolerated in other cases (e.g. 12b, 18, and 19). Unlike the parent C-188–9, fluoro-ether 12b and triflate 19 are indefinitely stable in aqueous buffer in air. The binding potency of 12b and 19 suggests that oxidation and/or accessing an iminoquinone structure is not required for binding or activity.
Inhibition of intracellular phosphorylation
The more promising molecules (3aa-3ca) were tested in intracellular STAT3 phosphorylation assays. To extrapolate intracellular potency, Kasumi-1 human AML cells were treated with differing concentrations of inhibitor (Figure 3a). Cells were incubated with granulocyte-colony stimulating factor (G-CSF) to induce STAT3 phosphorylation.32 After incubation, the cells were lysed and levels of STAT3 phosphorylated on tyrosine 705 (pSTAT3) were quantified with a fluorescent pSTAT3 antibody. Although 3aa, 3ba, and 3ca performed similarly by SPR (Table 2), their ability to inhibit STAT3 phosphorylation in cells was markedly different. This suggests that their molecular recognition of STAT3 in an isolated system like SPR is similar, however, in a protein-rich redox-buffered environment these inhibitors no longer behave the same. It is important to note that differences in small molecule specificity and cellular uptake can also affect the intracellular potency of the inhibitors. Nevertheless, 3aa was clearly the most potent small-molecule pSTAT3 inhibitor.
Figure 3.

Portions of this figure were adapted from a preliminary report.15 Compound 3aa inhibits G-CSF-induced STAT3 phosphorylation, induces apoptosis in human AML cells, and slows disease progression in a xenograft model of AML. (a) Histogram of decrease in pSTAT3 in Kasumi-1 cells. (b) 10 inhibits G-CSF-induced pSTAT3 in multiple AML cell lines. Legend values indicate IC50 values. Mean ±SD for n = 3. (c) Apoptosis quantified in Annexin V-FITC-labeled cells treated with 10 for 24 h. Spontaneous apoptosis in untreated cells was subtracted to yield the % apoptosis attributable to drug. Data shown for an AML cell line (MV4–11, HL-60) and primary patient-derived AML cells (p198) compared to ALL cell lines (KOPN-8, RS4–11) as a negative control. (d-e) NSG mice were injected iv with 106 MV4–11. ffluc AML cells at day 0. After two weeks, mice received compound 10, 30 mg/kg (n=8), or vehicle (n=6), ip daily 5 d/week for 4 weeks (weeks 2–6). (d) Luminescence images one week after treatment. Colorized signal intensity indicates amount of active disease, from low (blue) to high (red). (e) The percent of MV4–11 cells in bone marrow and spleen was evaluated by flow cytometry at the time of euthanasia. *P<0.05 (f) Disease burden was measured non-invasively by bioluminescence weekly. Each line represents the trajectory of an individual mouse. Mice were euthanized when moribund or at the end of 8 weeks.
We wanted to ensure that the molecule 3aa also inhibits inducible STAT3 phosphorylation in other AML cell lines (Figure 3b). Following a 30-minute treatment with 3aa prior to G-CSF stimulation, all three of the AML cell lines tested (HL-60, MOLM-13, and Kasumi-1) exhibited dose-dependent decreases in STAT3 phosphorylation with IC50 0.8–1.9 μM. This demonstrates that the anti-pSTAT3 activity of 3aa can be seen across different AML cell lines.
To further evaluate the therapeutic potential of 3aa, we confirmed its ability to induce apoptosis in different AML cell lines and primary tumor cells from pediatric AML patients. Cells were incubated with different doses of each inhibitor and subsequently incubated with fluorescent Annexin V. Annexin V binds to apoptotic cells, and FITC-Annexin V conjugates allow quantification of apoptosis by fluorescence assisted cell sorting (FACS). A 24-hour treatment with 3aa increased apoptosis in all AML cell lines and primary samples tested (Figure 3c). In contrast, acute lymphoblastic leukemia (ALL) cell lines—which do not have upregulated STAT3 activity33—were much less sensitive to apoptosis induction (Figure 3c, KOPN-8 and RS4–11). These findings are is consistent with the idea that 3aa inhibits STAT3 phosphorylation in AML cell lines and induces apoptosis through that mechanism of action.
After thoroughly investigating our 3aa in the in vitro models, we moved to investigate its potency in in vivo mouse models. STAT3-dependent cyctotoxicity in apoptosis assays suggested at least a half-log dosing window. Mice were injected intravenously with MV4–11 human AML cells. After two weeks, mice were treated for another 2–4 weeks with 3aa. Results from in vivo imaging, bone marrow and spleen harvesting indicate that 3aa increases mouse survival by decreasing the expansion of MV4–11 leukemia cells (Figure 3 d–f).
Rhodium conjugates as STAT3 inhibitors
After garnishing some promising results in the traditional small molecule studies with 3aa, we shifted our focus to the biological efficacy of small molecule-rhodium conjugates. Again, we used SPR to screen inhibitor potency (Table 3). Rhodium-inhibitor conjugate 13aa, 13ac, and 13b, showed potency increases over their analogous esters. Compared to previous studies with other binding sites, this affinity gain is rather modest, and is presumably consistent with at most very weak binding to side chains such as carboxylates (i.e. residue Asp 170 near the coiled-coil binding site) rather than stronger binding to histidine or methionine. A previous study found that sterically demanding carboxylates improved the decomposition half-life of rhodium complexes,34 but the sterically crowded rhodium complex 13ac had potency similar to other rhodium conjugates. The divalent structure 13ad did demonstrate moderately improved STAT3 affinity (0.1 μM).
Table 3.
SPR analysis of binding for carboxylate-containing and rhodium-conjugate compounds.
| SPR IC50 (uM) | ||||||
|---|---|---|---|---|---|---|
| entry | cmpd | R1 | R2 | R3 | ester | Rh(II) |
| 1 | 13aa | Me | Me | Me | 25 | 1 |
| 2 | 13ab | nBu | nBu | nBu | 25 | 0.6 |
| 3 | 13ac | fluorescein | Me | Me | 25 | 1 |
| 4 | 13ad | n.a. | Me | Me | 25 | 0.1 |
| 5 | 13ae | CF3 | Me | Me | 25 | 0.04 |
| 6 | 13af | CF3 | CF3 | CF3 | 25 | 0.2 |
| 7 | 13b | Me | Me | Me | 8 | 1 |
| 8 | 13c | Me | Me | Me | 50* | 0.3 |
SPR IC50 of the carboxylic acid derivative 4c was measured.
It seemed possible that a more Lewis-acidic rhodium center could benefit from more potent binding to peripheral residues.35 The coiled-coil domain does have several carboxylate side chains near the ligand-binding site (Figure 1a, blue), including Asp170, and we reasoned that carboxylates, while weaker ligands than histidine or methionine, might be sufficient ligands for a metal center with increased Lewis acidity. Alteration of the ancillary ligands on rhodium allows tuning the Lewis acidity of the metal center. An electron-poor variant, 13ae did show improved binding to STAT3 (IC50 = 0.04 μM, Table 3): compound 13ae is 100× more potent than C188–9, and 25× more potent than the nearly isosteric rhodium complex 13aa. This effect may be quite subtle: The analogous tris-trifluoroacetate complex (13af) exhibited a more modest affinity enhancement (IC50 = 0.2 μM, Table 3), perhaps reflecting a subtle balance of competitive water binding.34
Next, we examined the cellular activity of the lead rhodium conjugate on STAT3 phosphorylation and the induction of apoptosis in Kasumi cells (Figure 4). To provide a useful comparison, rhodium conjugates were directly compared against C188–9 and compound 4a, an isoelectric organic analogue without the capacity for coorperative organic–inorganic binding. In STAT3 intracellular phosphorylation assays (Figure 4b), the electronically-perturbed rhodium complex 13ae again exhibited significantly improved activity. The Kasumi cells were exceptionally resistant to both C188–9 and 4a, while 13ae inhibited STAT3 phosphorylation at lower micromolar potencies. In all, this is the first example of rhodium complexation enhancing the potency of any intracellular inhibitor in cellular assays andthese results indicate that rhodium conjugation is a viable strategy for increasing the potency of a biological probe for intracellular assays.
Figure 4.

(a) Small molecules inhibit STAT3/phosphopeptide (p1068) binding. STAT3 phosphopeptide binding was measured with established SPR protocols.21 (b) Kasumi cells were treated with G-CSF and then with inhibitors. Cells were analyzed for phosphorylated STAT3 (pSTAT3). (c) Apoptosis was measured 24 h after treatment of AML cells with STAT3 inhibitors.
In order to see if this increase phosphorylation inhibition translated to induction of apoptosis, we compared 13ae, 4a, and C188–9 in an Annexin assay (Figure 4c). The rhodium complex 13ae was more potent than both C188–9 and 4a, which is in agreement with the phosphorylation inhibition results. Therefore the fact that 13ae consistently outperformed C188–9 in these in vitro assays suggests further development of rhodium conjugates is warranted.
Naphthalene sulfonamides are a useful class of probes to alter STAT3 function. Optimization of the sulfonamide core allowed development of 3aa, an inhibitor with improved binding potency, and anti-STAT3 activity in cells. Some of the naphthalene sulfonamides also display in vivo activity in relevant cancer models.11,36 For example, pentafluoro compound 3aa, significantly decreases tumor progression in a mouse model of AML. Anti-phosphorylation activity correlates well with apoptosis induction in tumor models for the naphthalene sulfonamide compound class, again consistent with a specific STAT3-driven mode of action. Overall, optimization of the naphthalene sulfonamide core has resulted in modest gains in potency and activity, perhaps due to innate features of the coiled-coil binding site, which lacks a deep ligand-binding pocket. This limited optimization success led us to pursue tandem efforts to explore rhodium conjugates as potential solution to vexing inhibitor-development problems. The substantial increase in potency observed with rhodium complex 13ae indicates that significant gains in binding affinity are possible, even without proximal strong metal-binding residues (histidine, methionine, cysteine), by perturbing the electronic structure of the rhodium center in favor of increased Lewis acidity. Perhaps most importantly, the improvement in STAT3 binding carries through to cell-based assays, supporting our previous conclusions that rhodium complexes can enter and act within living cells. Complexes such as 13ae may serve as probe compounds with unique specificity; although many important questions remain, especially regarding in vivo application of these compounds.
Supplementary Material
Figure 2.

Previously established STAT3 inhibitor motif,21 and expanded motifs discovered in this work.
Acknowledgements
M.B.M. was supported by a Ruth L. Kirchstein National Service Award (NIH F31CA180696). We acknowledge support from the NIH (5R21CA170625), from the Robert A. Welch Foundation Research Grant C-1680 (Z.T.B.), the National Science Foundation (CHE-1904865, Z.T.B.), the Gillson Longenbaugh Foundation (M.S.R.), the Virginia and L.E. Simmons Family Foundation, and the MD Anderson Foundation (D.J.T).
References
- 1.Bromberg J, J. Clin. Invest, 2002, 109, 1139–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Darnell JE, Kerr IM and Stark GR, Science, 1994, 264, 1415–1421. [DOI] [PubMed] [Google Scholar]
- 3.Kisseleva T, Bhattacharya S, Braunstein J and Schindler CW, Gene, 2002, 285, 1–24. [DOI] [PubMed] [Google Scholar]
- 4.Chakraborty A, Dyer KF, Cascio M, Mietzner TA and Tweardy DJ, Blood, 1999, 93, 15–24. [PubMed] [Google Scholar]
- 5.Wu P, Wu D, Zhao L, Huang L, Shen G, Huang J and Chai Y, Oncotarget, 2016, 7, 19863–19883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamran MZ, Patil P and Gude RP, BioMed Res. Int,, DOI:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson DE, O’Keefe RA and Grandis JR, Nat. Rev. Clin. Oncol, 2018, 15, 234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X, Yue P, Page BDG, Li T, Zhao W, Namanja AT, Paladino D, Zhao J, Chen Y, Gunning PT and Turkson J, Proc. Natl. Acad. Sci, 2012, 109, 9623–9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, Ji M, Zhang S, Xue N, Xu H, Lin S and Chen X, J. Drug Target, 2018, 26, 920–930. [DOI] [PubMed] [Google Scholar]
- 10.Geiger JL, Grandis JR and Bauman JE, Oral Oncol, 2016, 56, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bharadwaj U, Eckols TK, Xu X, Kasembeli MM, Chen Y, Adachi M, Song Y, Mo Q, Lai SY and Tweardy DJ, Oncotarget, 2016, 7, 26307–26330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park JH, van Wyk H, McMillan DC, Quinn J, Clark J, Roxburgh CSD, Horgan PG and Edwards J, Clin. Cancer Res, 2017, 23, 1698–1709. [DOI] [PubMed] [Google Scholar]
- 13.He G and Karin M, Cell Res, 2011, 21, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Redell MS, Ruiz MJ, Alonzo TA, Gerbing RB and Tweardy DJ, Blood, 2011, 117, 5701–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minus MB, Liu W, Vohidov F, Kasembeli MM, Long X, Krueger M, Stevens A, Kolosov MI, Tweardy DJ, Redell MS and Ball ZT, Angew. Chem. Int. Ed, 2015, 54, 13085–13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abdulghani J, Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, Lisanti MP, Zellweger T, Alanen K, Mirtti T, Visakorpi T, Bubendorf L and Nevalainen MT, Am. J. Pathol, 2008, 172, 1717–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu H, Lee H, Herrmann A, Buettner R and Jove R, Nat. Rev. Cancer, 2014, 14, 736–746. [DOI] [PubMed] [Google Scholar]
- 18.Johnston PA and Grandis JR, Mol. Interv, 2011, 11, 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dang CV, Reddy EP, Shokat KM and Soucek L, Nat. Rev. Cancer, 2017, 17, 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verdine GL and Walensky LD, Clin. Cancer Res, 2007, 13, 7264–7270. [DOI] [PubMed] [Google Scholar]
- 21.Xu X, Kasembeli MM, Jiang X, Tweardy BJ and Tweardy DJ, PLoS ONE, 2009, 4, e4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren Z, Mao X, Mertens C, Krishnaraj R, Qin J, Mandal PK, Romanowski MJ, McMurray JS and Chen X, Biochem. Biophys. Res. Commun, 2008, 374, 1–5. [DOI] [PubMed] [Google Scholar]
- 23.Kundu R, Cushing PR, Popp BV, Zhao Y, Madden DR and Ball ZT, Angew. Chem. Int. Ed, 2012, 51, 7217–7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vohidov F, Knudsen SE, Leonard PG, Ohata J, Wheadon MJ, Popp BV, Ladbury JE and Ball ZT, Chem. Sci, 2015, 6, 4778–4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma H, Wu S, Sun Q, Li H, Chen Y, Zhao W, Ma B, Guo Q, Lei Z and Yan J, Synthesis, 2010, 2010, 3295–3300. [Google Scholar]
- 26.Mallory FB, Mallory CW and Fedarko MC, J. Am. Chem. Soc, 1974, 96, 3536–3542. [Google Scholar]
- 27.Schimler SD, Ryan SJ, Bland DC, Anderson JE and Sanford MS, J. Org. Chem, 2015, 80, 12137–12145. [DOI] [PubMed] [Google Scholar]
- 28.Popp BV and Ball ZT, J. Am. Chem. Soc, 2010, 132, 6660–6662. [DOI] [PubMed] [Google Scholar]
- 29.Page BDG, Fletcher S, Yue P, Li Z, Zhang X, Sharmeen S, Datti A, Wrana JL, Trudel S, Schimmer AD, Turkson J and Gunning PT, Bioorg. Med. Chem. Lett, 2011, 21, 5605–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao C, Wang W, Yu W, Jou D, Wang Y, Ma H, Xiao H, Qin H, Zhang C, Lü J, Li S, Li C, Lin J and Lin L, Oncotarget, 2016, 7, 12917–12926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang W, Rayburn ER, Velu SE, Chen D, Nadkarni DH, Murugesan S, Chen D and Zhang R, Breast Cancer Res. Treat, 2010, 123, 321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimozaki K, Nakajima K, Hirano T and Nagata S, J. Biol. Chem, 1997, 272, 25184–25189. [DOI] [PubMed] [Google Scholar]
- 33.Furuichi Y, Goi K, Inukai T, Sato H, Nemoto A, Takahashi K, Akahane K, Hirose K, Honna H, Kuroda I, Zhang X, Kagami K, Hayashi Y, Harigaya K, Nakazawa S and Sugita K, Cancer Res, 2007, 67, 9852–9861. [DOI] [PubMed] [Google Scholar]
- 34.Minus MB, Kang MK, Knudsen SE, Liu W, Krueger MJ, Smith ML, Redell MS and Ball ZT, Chem. Commun, 2016, 52, 11685–11688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coughlin JM, Kundu R, Cooper JC and Ball ZT, Bioorg. Med. Chem. Lett, 2014, 24, 5203–5206. [DOI] [PubMed] [Google Scholar]
- 36.Jung KH, Yoo W, Stevenson HL, Deshpande D, Shen H, Gagea M, Yoo S-Y, Wang J, Eckols TK, Bharadwaj U, Tweardy DJ and Beretta L, Clin. Cancer Res, 2017, clincanres.2253.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.