

Abstract
Objective
To investigate whether aquaporin-4–immunoglobulin G (AQP4-IgG) titers and measures of complement-mediated cell killing are clinically useful to predict the occurrence of relapse, relapse severity, and/or disability in neuromyelitis optica spectrum disorder (NMOSD).
Methods
We studied 336 serial serum specimens from 82 AQP4-lgG–seropositive patients. NMOSD activity at blood draw was defined as preattack (24 [7.1%], drawn within 30 days preceding an attack), attack (108 [32.1%], drawn on attack onset or within 30 days after), or remission (199 [59.2%], drawn >90 days after attack onset and >30 days preceding a relapse). For each specimen, we documented the attack type and severity and immunotherapy status. Complement-mediated cell killing was quantitated by flow cytometry using an M23-AQP4 cell-based assay.
Results
The estimated logarithmic means of AQP4-IgG titers in preattack, attack, and remission samples were 3.302, 3.657, and 3.458, respectively, p = 0.21. Analyses of 81 attack/remission pairs in 42 patients showed no significant titer differences (3.736 vs 3.472, p = 0.15). Analyses of 13 preattack/attack pairs in 9 patients showed no significant titer differences (3.994 vs 3.889, p = 0.67). Of 5 patients who converted to seronegative status, 2 continued to have attacks. Titers for major and minor attacks (n = 70) were not significantly different (3.905 vs 3.676, p = 0.47). Similarly, measures (titers) of complement-mediated cell killing were not significantly associated with disease course, attack severity, or disability at 5 years.
Conclusions and relevance
AQP4-IgG titer and complement-mediated cell killing lack significant prognostic or predictive utility in NMOSD. Although titers may drop in the setting of immunotherapy, seroconversion to negative status does not preclude ongoing clinical attacks.
Classification of evidence
This study provides Class II evidence that in patients with NMOSD, AQP4-IgG titers and measures of complement-mediated cell killing activity do not predict relapses, relapse severity, or disability.
Aquaporin-4–immunoglobulin G (AQP4-IgG)-positive neuromyelitis optica spectrum disorder (NMOSD) is a relapsing inflammatory demyelinating disease of the CNS.1,2 NMOSD relapses (also called attacks) tend to be more severe with less recovery than in MS.3 Neurologic disability in NMOSD is attack related and incremental, with no or little progressive worsening of disability between attacks.2,4 A potential biomarker of NMOSD activity that could be measured serially and would predict relapse would assist clinicians to add or increase immunotherapies at periods of greater risk. Small observational studies have reported that AQP4-IgG titer rises at the time of NMOSD attacks, suggesting that changes in titer may be a potential biomarker of NMOSD activity.5–10
It is well established that AQP4-IgG activates complement and induces cell killing of AQP4-expressing cells. Hinson et al.11 demonstrated complement-mediated cell killing of AQP4-expressing non-neural cells. Complement-mediated cell killing of rodent astrocytes was subsequently demonstrated in primary cell culture and animal models.12,13 More recently, Nishiyama et al.14 demonstrated injury of human astrocytes after in vitro application of AQP4-IgG–positive patient sera with human complement. Although complement activation is a major contributor to AQP4-IgG–positive NMOSD pathology, it remains to be determined whether complement activating potential predicts the occurrence of relapses or their severity. In a small study, Hinson et al.15 measured complement-mediated cell killing induced by sera from 12 patients with NMOSD during attacks and found increased cell killing of AQP4-transfected cells when exposed to sera from patients during a major attack compared with sera from patients during a minor attack.
Over the past 8 years, the Mayo Clinic NMOSD Biorepository has collected samples from AQP4-IgG–positive patients with NMOSD every 6 months and at the time of each attack. Some samples were collected in the 30-day period before an attack. We investigated whether AQP4-IgG titer or complement-mediated cell killing, measured by flow cytometry methodology, had any predictive or prognostic value.
Methods
Standard protocol approvals, registrations, and patient consents
This study was approved by the institutional review board. Written informed consent was obtained from all patients (or guardians of patients) enrolled in the Mayo Clinic and Martinique NMOSD biorepositories.
The primary research question was whether AQP4-IgG titers and measures of complement-mediated cell killing are clinically useful to predict the occurrence of relapse, relapse severity, and/or disability in NMOSD. This study provides Class II evidence that in patients with NMOSD, AQP4-IgG titers and measures of complement-mediated cell killing activity do not predict relapses, relapse severity, or disability.
Patients and sera
Inclusion criteria
We interrogated the Mayo Clinic and Martinique NMOSD biorepository database for patients fulfilling the following inclusion criteria: (1) seropositive for AQP4-IgG by any assay (tissue-based indirect immunofluorescence, cell-binding assay, or M1-AQP4–transfected cell-based flow cytometry assay), (2) at least 1 preattack/attack and 1 remission archived serum samples (stored at −80°C) available for testing, and (3) adequate clinical information available at the time of blood draw to allow the sample to be grouped into one of the following categories: preattack (drawn within 30 days preceding an attack), attack (drawn on attack onset or within 30 days after), or remission (drawn >90 days after attack onset).
All laboratory-based analyses were performed blinded to clinical data (J.P.F. and J.S. for AQP4-IgG titer and J.J. for AQP4-IgG complement activation assay). This study provides Class II evidence.
Description of patients
Inclusion criteria were fulfilled by 69 Mayo (60 female and 9 male) patients and 13 Martinique patients (7 female and 6 unknown). Baseline demographics (ethnicity, sex, and age at onset) of these 69 Mayo patients were collected. Clinical attack phenotypes included transverse myelitis (TM), optic neuritis (ON), area postrema syndrome, cerebral and brainstem syndromes, and multifocal attacks (any combination of symptoms). Long-term immunotherapy treatment included prednisolone, azathioprine, mycophenolate mofetil, cyclophosphamide, methotrexate, IV immunoglobulin, rituximab (no patients received ocrelizumab), eculizumab, and stem cell transplantation. No patients received ocrelizumab, inebilizumab, or satralizumab. Attack type and severity were documented. Classification of attack severity as major or minor was based on prior published criteria16 (supplement data, links.lww.com/NXI/A254).
Detection of AQP4-IgG and titer measurement
Samples were tested by an in-house–developed M1-AQP4–transfected cell-based flow cytometry assay validated and certified by the College of American Pathology.17,18 Such cell-based assays have been shown to have an optimized sensitivity and specificity.17–19 Samples were screened at 1:5 dilution and, if positive, retested at a dilution of 1:5, 1:10 and titrated further in 10-fold dilution steps. The farthest dilution yielding a positive result (IgG binding index ≥2.0) was recorded as the end point of positivity.
AQP4-IgG complement activation assay
All sera were tested for AQP4-IgG–linked complement activation using an in-house–developed flow cytometry–based assay using a stable HEK293 cell line expressing AQP4-M23.15,16,19 Heat-inactivated patient serum (56°C, 35 minutes) was serially diluted (doubling) from 1:5 to 1:100,000 in live cell-binding buffer. For analysis, 50 μL of each dilution was added to live AQP4-M23–transfected cells. After 10 minutes, 18 μL of rabbit Lo-Tox rabbit complement (CedarLane, Burlington, NC) was added, and the plate was kept at room temperature for 30 minutes. Buffer B supplemented with ethylenediaminetetraacetic acid (EDTA) and propidium iodide (PI; BD Biosciences, San Jose, CA) (75 mM EDTA and 0.5 μg PI) was added and held for 15 minutes in the dark. An additional 100 μL Buffer B–EDTA without PI was added before analysis by flow cytometry. Negative controls were included on every plate and were the basis for determining positive populations. The percent of positive events was used to evaluate the level of complement activation. Method reproducibility is shown in supplementary figure 1 (links.lww.com/NXI/A255).
Statistical analysis
All analyses of AQP4-IgG titer were performed on a logarithmic scale with base of 10 because of the 10-fold dilution steps for the titration assay of AQP4-IgG, and the data were normally distributed on the log scale (titer 1:5 transformed to 0.7, 1:10 to 1, 1:100 to 2, 1:1,000 to 3, 1:10,000 to 4, and 1:100,000 to 5, zero was left as zero). Analyses for M23-AQP4-IgG complement-mediated cell killing titers values were transformed as follows, based on a doubling distribution: 1:5 transformed to 1, 1:10 to 2, 1:20 to 3, 1:40 to 4, 1:80 to 5, 1:160 to 6, 1:320 to 7, 1:640 to 8, 1:1,280 to 9, 1:2,560 to 10, 1:5,120 to 11, and 1:10,240 to 12 (zero was left as zero). Unless otherwise indicated, all results with regard to the titers are reported with the transformed scale. Using the transformed scale, the titer levels were compared with patient characteristics using linear regression models with generalized estimating equations (GEEs) to account for repeated data within patient as well as within attack number. The sample type according to NMOSD activity (preattack, attack, and remission) was compared with the titer level in 2 ways. First, we compared the titer levels between sample types among all available samples (regardless of the timing of the samples). For this analysis, samples that could be classified as preattack and attack were included with the attack samples. Next, we focused on paired sample types (i.e., preattack vs subsequent attack or attack vs subsequent remission) among the subset of samples for which this was known. In this analysis, samples that could be classified as preattack and attack were considered for pairs as both types. For each pair, the difference in titer was calculated. Paired differences were then assessed using the GEE methods described above, with the paired difference as the outcome. The risk of developing an Expanded Disability Status Scale (EDSS) score of 6 or 8 was compared with the baseline AQP4-IgG complement-mediated cell killing titer level (<160 vs ≥160) with a likelihood ratio test from Cox proportional hazards regression.
All analyses were performed using SAS version 9.4 software (SAS Institute Inc., Cary, NC). p Values less than 0.05 were considered statistically significant.
Data availability
All authors have full access to all data sets and take full responsibility for the integrity of the data and accuracy of the data analysis. All data pertaining to this article are contained within or published as online supplement.
Results
We included 336 serial serum specimens in the analysis from 82 AQP4-IgG–positive patients with known sample type (preattack, attack, or remission). Median age at onset was 41 years (range 7–72 years), and females accounted for 88.2%. Ethnicities were Caucasian 59.8%, African American 28.0%, Hispanic 7.3%, Asian 3.7%, and unknown 1.2%. The median annualized relapse rate was 0.7 (range 0.1–4.5), and the median duration of follow-up was 8.0 years (range 0.4–40.8 years). Of the 336 serum samples for which the sample type was known, 24 (7.1%) were preattack samples, 108 (32.1%) were attack samples, 199 (59.2%) were remission samples, and the remaining 5 (1.5%) were considered both preattack and attack (drawn within 30 days after attack but also with subsequent attack within 30 days). For 97 attack samples from the Mayo Clinic cohort, attack types were TM in 58.8%, ON in 18.6%, multifocal attacks in 18.6%, and unknown in 4.1%. Among 97 attack samples, 87 samples could be identified to have one of the following treatments, IV methylprednisolone (IVMP) alone in 26.7%, oral prednisolone alone in 10.5%, plasma exchange (PLEX) alone in 2.3%, combination therapy of IVMP and PLEX in 57.0%, and no treatment in 3.5%. Among the samples collected for which maintenance immunotherapy status was known (N = 282), 74.1% were from patients receiving maintenance immunotherapy before blood collection and 62.8% were from patients receiving maintenance immunotherapy at blood collection.
AQP4-IgG titer
The median AQP4-IgG titer was 10,000 (range 0–100,000).
Attack and preattack AQP4-IgG titers were not significantly different from remission titers
AQP4-IgG titers did not differ significantly between preattack (estimated mean, 3.537), and attack (estimated mean, 3.698) compared with remission (estimated mean, 3.660), p = 0.79, figure 1A.a, table 1. Nine samples were drawn during NMOSD attacks, with at least 2 attacks occurring in 2 consecutive months. These samples had a median titer of 1:10,000.
Figure 1. AQP4-IgG (binding and complement-mediated cell killing) titers of specimens drawn at different time points (remission, preattack, and attack) in individual patients (original titer values shown in transformed scale).

(A.a) AQP4-IgG titers grouped into preattack, attack, and remission categories. (B.a) AQP4-IgG titers categorized according to baseline immunotherapy*. (C.a) AQP4-IgG titers categorized by attack severity type**. (A.b) AQP4 complement-mediated cell killing titers grouped into preattack, attack, and remission categories. (B.b) AQP4 complement-mediated cell killing titers categorized according to baseline immunotherapy*. (C.b) AQP4 complement-mediated cell killing titers categorized grouped into attack severity type**. *Immunotherapy included azathioprine, mycophenolate mofetil, cyclophosphamide, and rituximab. **Definition in the supplement. AQP4 = aquaporin-4; IgG = immunoglobulin G; IST = immunosuppressive therapy.
Table 1.
Aquaporin-4–immunoglobulin G titer (log base 10 scalea) of specimens from all available samples using estimated means


For individual patients, AQP4-IgG titer is not significantly increased during NMOSD attacks
Comparing the AQP4-IgG titers among 81 pairs of attack and subsequent remission samples, the mean titer level decreased from 3.736 to 3.472, but this was not statistically significant (p = 0.15; supplementary table, links.lww.com/NXI/A257). Of 5 patients who converted to seronegative status, 2 continued to have attacks.
For individual patients, neuromyelitis optica attacks are not preceded by an increase in AQP4-IgG titer
Analysis of 13 pairs of preattack to subsequent attack samples indicated that AQP4-IgG titers were not significantly different between samples drawn before an attack compared with samples drawn during an attack (mean, 3.994 and 3.889, respectively; p = 0.67; supplementary table, links.lww.com/NXI/A257).
Maintenance immunotherapy had no significant effect on AQP4-IgG titers
Estimated mean titers in sera from patients receiving immunosuppressive medications did not differ when compared with patients on no therapy (3.620 vs 3.546, p = 0.64, figure 1B.a, table 2).
Table 2.
Aquaporin-4–immunoglobulin G complement-mediated cell killing titers (doubling scalea) of specimens from all available samples using estimated means

Attack AQP4-IgG titers are similar across different attack types and severities
AQP4-IgG titers were similar across different attack types. The estimated mean AQP4-IgG titer was 3.868 in isolated TM, 3.589 in isolated ON, and 3.936 in multifocal attacks, p = 0.62, table 1. Furthermore, no significant differences in titers were observed between major vs minor attack severities (3.905 vs 3.676, respectively, p = 0.47; figure 1C.a, table 1).
AQP4-IgG complement-mediated cell killing titers
The median titer of AQP4-IgG complement-mediated cell killing ability was 1:160 (range 0–1:10,240).
Attack and preattack AQP4-IgG complement-mediated cell killing titers are not significantly different from remission titers
AQP4-IgG complement-mediated cell killing titers were not significantly different at preattack time (estimated mean, 4.654) and at attack time (estimated mean, 4.615) compared with titers at remission (estimated mean, 4.500), p = 0.92, figure 1A.b, table 2. Fifteen samples were drawn during a neuromyelitis optica attack, with at least 2 attacks in the same or consecutive months, and the median titer was 1:80.
For individual patients, AQP4-IgG complement-mediated cell killing titers are not significantly increased during NMOSD attacks
Considering attack and subsequent remission pairs (N = 80 sample pairs), the estimated mean AQP4-IgG complement-mediated cell killing titers were not significantly different in attack specimens (5.401) compared with subsequent remission specimens (5.140) as (p = 0.62; supplementary table, links.lww.com/NXI/A257).
For individual patients, NMOSD attacks are not preceded by an increase in AQP4-IgG complement-mediated cell killing titer
Considering 9 pairs of preattack and subsequent attack specimens, the estimated mean AQP4-IgG complement-mediated cell killing titers decreased slightly from 6.143 (preattack) to 5.429 (attack), but this was not statistically significant (p = 0.06; supplementary table, links.lww.com/NXI/A257). No significant difference was observed between preattack and remission samples. In supplementary figure 1 (links.lww.com/NXI/A255), binding and complement killing titers, respectively, for individual patients at disease course time points are highly variable. For some patients, titers are higher at times of remission compared with attack and vice versa in the same individual patient.
Considering 18 pairs of preattack and subsequent remission specimens, the estimated mean AQP4-IgG complement-mediated cell killing titers did not differ between 3.611 (preattack) and 3.834 (subsequent remission), p = 0.72 (supplementary table, links.lww.com/NXI/A257).
Maintenance immunotherapy had no significant effect on AQP4-IgG–induced complement-activated cell killing titers
Estimated mean titers in sera in samples from patients receiving immunosuppressant medication did not differ when compared with samples from patients on no therapy (4.665 vs 4.700, p = 0.95, figure 1B.b, table 2). AQP4-IgG complement-mediated cell killing titers of individual patients at different time points during disease course are shown in supplementary figure 1 (links.lww.com/NXI/A255).
Attack AQP4-IgG complement-mediated cell killing titers are similar across different attack types and severities
Cell killing induced by AQP4-IgG–mediated complement activation was similar across different attack types. The estimated means of AQP4-IgG complement-mediated cell killing titers among 75 attack samples analyzed were 5.260 in isolated TM, 4.316 in isolated ON, and 5.724 in multifocal attacks (p = 0.28, table 1). Furthermore, among 58 attack samples analyzed, no significant differences in titers were observed between major vs minor attack severities (5.421 and 4.293, respectively, p = 0.21; figure 1C.b, table 2).
No correlation of AQP4-IgG complement-mediated cell killing titers with disability
Testing of 139 samples showed no differences in AQP4-IgG complement-mediated cell killing titers stratified according to 3 EDSS groups (<3, 3–5.5, and ≥6), assessed at the time of sample collection. Calculated means were 4.657, 4.997, and 5.442, respectively, p = 0.55 (table 2). In addition, the baseline complement activation titer level was not significantly associated with the risk of developing an EDSS score of 6 or 8 within 5 years (p = 0.92 and p = 0.13, respectively, figure 2 and supplementary figure 2, links.lww.com/NXI/A256).
Figure 2. Kaplan-Meier estimates of time to develop EDSS score ≥ 6.
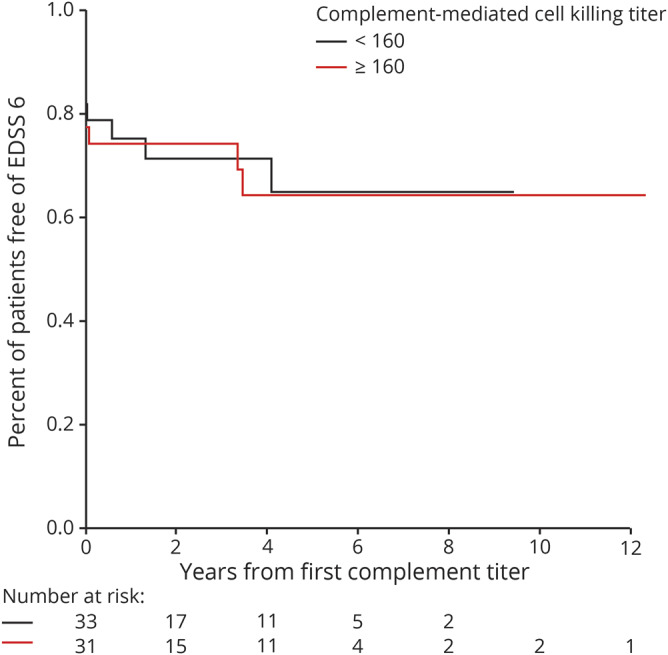
Years from the first aquaporin-4–immunoglobulin G complement-mediated cell killing titers to develop EDSS score ≥6. All Martinique patients, any patients with unknown status for each outcome, and any patients with missing complement values were excluded from the corresponding analysis. EDSS = Expanded Disability Status Scale.
Strong correlation between AQP4-IgG and complement killing titers
AQP4-IgG and complement-mediated cell killing titers, measured using AQP4-M23–transfected HEK293 cells, correlated strongly with each other (figure 3).
Figure 3. Correlation between M23-AQP4 end titer and M23-AQP4 complement-mediated cell killing titers.
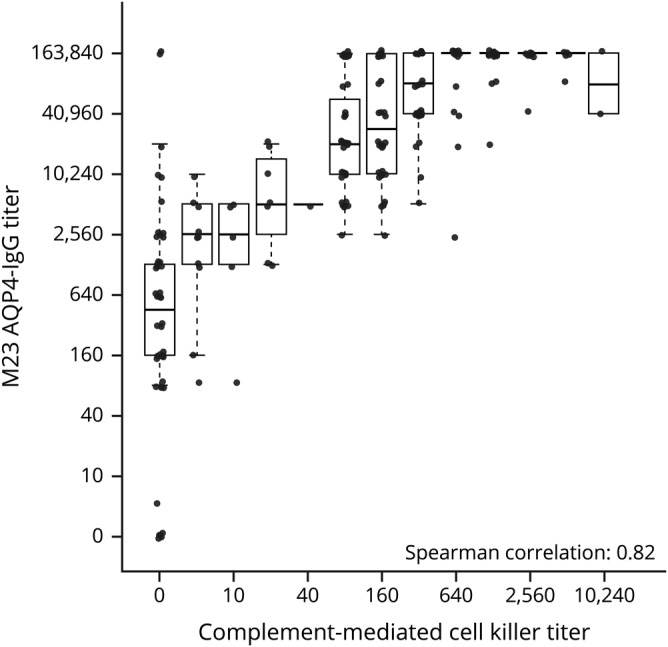
AQP4 = aquaporin-4; IgG = immunoglobulin G.
Discussion
This study failed to show any significant change in AQP4-IgG titers before or during an attack compared with the remission phase. Similarly, in vitro quantitation of complement activation by measuring cell killing of AQP4-expressing cells after exposure to patient serum samples and active complement varied from patient to patient. Longitudinal testing of samples in individual patients did not reveal changes preceding or during an attack compared with the remission phase. Thus, measurement of AQP4-IgG binding titer or complement-mediated cell killing by live cell flow cytometry shows no clinical utility as predictors of relapse or disease severity.
Previous studies have demonstrated conflicting results. Jarius et al.9 used an immunoprecipitation assay to measure AQP4-IgG titer of 96 serum samples taken from 8 patients. They reported that serum AQP4-IgG titers were significantly higher in attacks compared with remission phases. Nonetheless, some patients still had high AQP4-IgG titers after receiving long-term immunotherapy without clinical relapses.9 Similar to our findings, Kitley et al.20 reported that there M1-AQP4-IgG titers were generally higher but not significantly different at relapse compared with remission. As we found, Kitley et al.20 reported that some patients who seroconverted to negative status continued to relapse. AQP4-IgG titer did not correlate with disease severity. Dujmovic et al.7 demonstrated no correlation between serum AQP4-IgG and disability score in 12 patients, whereas CSF AQP4-IgG titer significantly decreased in the remission period. Paired attack-remission samples were analyzed in 11 and 12 patients in the 2 studies.7,11,20 Sato et al. suggested that during NMOSD relapses, the amount of astrocyte damage correlates with CSF AQP4-IgG titer. They studied CSF samples of 11 patients with NMOSD and showed that CSF AQP4-IgG titer correlates with amount of interleukin-6 and glial fibrillary acidic protein, a marker of astrocyte damage.21 However, no significant difference of serum AQP4-IgG titers was observed between the attacks and remissions.21 Our study analyzed 336 serum samples from 82 patients (137 of which were attack or preattack samples), providing significantly more power than prior published studies. We studied a broadly representative sampling of patients with both clinically mild and severe attacks, but did not find any correlation between serum AQP4-IgG titer and relapses.
We tested a much larger number of patient samples than previous studies to evaluate complement-mediated cell killing antibody titers.11–15 We found no correlation between a patient's serum ability to kill AQP4-transfected cells and disease course or severity. In contrast to previous studies, we evaluated not only the percentage of cell killing (data not shown) but also titrated the samples to 1:10,240, which probably provides more reliable data than determining the percentage of cells killed at a single serum dilution.
Studies investigating correlation between serum titers of pathogenic autoantibodies and clinical phenotype differ depending on the antibody-mediated neurologic disorder. In myasthenia gravis, titers of acetylcholine receptor (AChR) antibodies vary widely between patients and do not predict disease severity, treatment response, or likelihood of relapse.22
This may be partially explained in part by the recognized heterogeneous populations of AChR antibodies, limited sharing of epitope specificities, variability in light chain and subclass composition and in functional activities.22,23
In contrast to our findings in AQP4-IgG–positive NMOSD and prior published reports on myasthenia gravis, Gresa-Arribas et al.24 have reported that CSF and serum NMDA receptor (NMDAR) antibody titers were higher in patients with poor outcome or teratoma compared with good outcome or lacking tumor. Earlier and greater reductions in CSF titer were most closely associated with good outcome, and rising titers were associated with relapse. Why titers of immunopathogenic autoantibodies in CNS inflammatory disease should correlate with clinical course and outcome in anti-NMDAR encephalitis but not AQP4-IgG NMOSD remains unclear.
Significant differences in the immunopathogenicity of these conditions such as neuronal vs glial target, epitope specificity (main epitope region at GluN1 amino acid 369 in NMDAR encephalitis vs heterogeneous epitopes on extracellular loops of AQP4), differences in extent of intrathecal synthesis of antibody, functional effects of antibody (predominant modulation in anti-NMDAR encephalitis and complement activation in AQP4-IgG + NMOSD), and predominant monophasic vs relapsing nature of the disease may provide some clues. In our study, we did not interrogate CSFs as multiple CSFs are not preformed in patients with NMOSD and CSF is generally considered to have a lower sensitivity than serum for AQP4-IgG detection.25 Serum titer may not strongly correlate with CSF titer as both Dujmovic et al. and Sato et al. showed correlation of CSF but not serum AQP4-IgG titer with disease course.7,21
In regard to the lack of clear association between AQP4-IgG titers and clinical course or outcome, additional explanations may be related to the basis of interindividual and intraindividual variability with respect to (1) avidity/affinity20; it is possible that measures of affinity (strength of interaction between the epitope and the antibody's antigen binding site) and avidity (overall strength of the antigen-antibody complex) could affect functional pathogenicity of antibody-antigen interaction. Studies investigating avidity and affinity in individual patient sera and CSF would be of interest for further research; (2) epitope specificity; (3) complement regulatory protein function4; and (4) blood-brain barrier permeability and coexistence of glucose-regulated protein 78 autoantibody.26 Given the relapsing nature of AQP4-IgG + NMOSD and the fact that antibody titers may be high during periods of remission indicate that a multitude of immunologic steps are likely required in addition to the presence of the antibody. These steps remain elusive to date.
In a separate study, we reported that AQP4-IgG titer seroconverted to negative after autologous nonmyeloablative hematopoietic stem cell transplantation (HSCT) in 7 of 9 patients who remained relapse-free 5 years post-HSCT. Complement-mediated cell killing ability of patient serum was switched off in 6 of 7 patients after treatment. This study showed that seroconversion from positive to negative and loss of complement killing activity after HSCT in patients with NMOSD may be predictive of relapse freedom.
This study has 3 potentially significant limitations. First, most of the samples were collected many years before the study (median 7.1 years, range 3.1–14.2 years), and although they were kept frozen at −80°C, some had multiple freeze-thaw cycles, which may have reduced titers. The second limitation is the potential effect of acute attack immunotherapies given before collection of attack sera. Of 83 attack samples with known acute treatment, 42% were drawn after treatments and 5% were drawn before treatments, while timing of immunotherapy with respect of blood draw was unclear in 53% of these samples. Importantly, however, none of the preattack samples were exposed to acute immunotherapies. The third limitation is that samples tested for AQP4-IgG using AQP4-M1–transfected cells were screened at 1:5 dilution and, if positive, titrated further in 10-fold dilution steps. Given such a 10-fold dilution approach, it is possible that small differences in titers could have been missed. Future studies may avoid such limitations. For example, testing placebo arm patient serial sera from drug trials would mitigate any potential impact of acute or chronic immunotherapies.
Acknowledgment
This work was supported by the NIH (R01-NS065829), Mayo Clinic's Department of Laboratory Medicine and Pathology, and the Mayo Clinic Center for Multiple Sclerosis and Autoimmune Neurology.
Glossary
- AChR
acetylcholine receptor
- AQP4
aquaporin-4
- EDSS
Expanded Disability Status Scale
- GEE
generalized estimating equation
- HSCT
hematopoietic stem cell transplantation
- IgG
immunoglobulin G
- IVMP
IV methylprednisolone
- NMDAR
NMDA receptor
- NMOSD
neuromyelitis optica spectrum disorder
- ON
optic neuritis
- PI
propidium iodide
- PLEX
plasma exchange
- TM
transverse myelitis
Appendix. Authors

Footnotes
Editorial, page e759
Study funding
This study was funded by the Department of Laboratory Medicine; Center for MS and Autoimmune Neurology, Mayo Clinic; and NIH (R01-NS065829 Sean J. Pittock).
Disclosure
J. Jitprapaikulsan, J.P. Fryer, C.Y. Smith, S.M. Jenkins, P. Cabre, S.R. Hinson, B.G. Weinshenker, J. Mandrekar, J.J. Chen, C.F. Lucchinetti, Y. Jiao, J. Sagen, J.E. Schmeling, E.P. Flanagan, A. McKeon, and S.J. Pittock report no disclosures relevant to the manuscript. Go to Neurology.org/NN for full disclosures.
References
- 1.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489. [DOI] [PubMed] [Google Scholar]
- 2.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805–815. [DOI] [PubMed] [Google Scholar]
- 3.Collongues N, Cabre P, Marignier R, et al. . A benign form of neuromyelitis optica: does it exist? Arch Neurol 2011;68:918–924. [DOI] [PubMed] [Google Scholar]
- 4.Pittock SJ, Lucchinetti CF. Neuromyelitis optica and the evolving spectrum of autoimmune aquaporin-4 channelopathies: a decade later. Ann NY Acad Sci 2016;1366:20–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aouad P, Li J, Arthur C, Burt R, Fernando S, Parratt J. Resolution of aquaporin-4 antibodies in a woman with neuromyelitis optica treated with human autologous stem cell transplant. J Clin Neurosci 2015;22:1215–1217. [DOI] [PubMed] [Google Scholar]
- 6.Pisani F, Sparaneo A, Tortorella C, et al. . Aquaporin-4 autoantibodies in neuromyelitis optica: AQP4 isoform-dependent sensitivity and specificity. PLoS One 2013;8:e79185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dujmovic I, Mader S, Schanda K, et al. . Temporal dynamics of cerebrospinal fluid anti-aquaporin-4 antibodies in patients with neuromyelitis optica spectrum disorders. J Neuroimmunol 2011;234:124–130. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi T, Miyazawa I, Misu T, et al. . Intractable hiccup and nausea in neuromyelitis optica with anti-aquaporin-4 antibody: a herald of acute exacerbations. J Neurol Neurosurg Psychiatry 2008;79:1075–1078. [DOI] [PubMed] [Google Scholar]
- 9.Jarius S, Aboul-Enein F, Waters P, et al. . Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 2008;131:3072–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi T, Fujihara K, Nakashima I, et al. . Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 2007;130:1235–1243. [DOI] [PubMed] [Google Scholar]
- 11.Hinson SR, Pittock SJ, Lucchinetti CF, et al. . Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007;69:2221–2231. [DOI] [PubMed] [Google Scholar]
- 12.Hinson SR, Roemer SF, Lucchinetti CF, et al. . Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down-regulating EAAT2. J Exp Med 2008;205:2473–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinoshita M, Nakatsuji Y, Kimura T, et al. . Neuromyelitis optica: passive transfer to rats by human immunoglobulin. Biochem Biophys Res Commun 2009;386:623–627. [DOI] [PubMed] [Google Scholar]
- 14.Nishiyama S, Misu T, Nuriya M, et al. . Complement-dependent and -independent aquaporin 4-antibody-mediated cytotoxicity in human astrocytes: pathogenetic implications in neuromyelitis optica. Biochem Biophys Rep 2016;7:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinson SR, McKeon A, Fryer JP, Apiwattanakul M, Lennon VA, Pittock SJ. Prediction of neuromyelitis optica attack severity by quantitation of complement-mediated injury to aquaporin-4-expressing cells. Arch Neurol 2009;66:1164–1167. [DOI] [PubMed] [Google Scholar]
- 16.Pittock SJ, Lennon VA, McKeon A, et al. . Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol 2013;12:554–562. [DOI] [PubMed] [Google Scholar]
- 17.Fryer JP, Lennon VA, Pittock SJ, et al. . AQP4 autoantibody assay performance in clinical laboratory service. Neurol Neuroimmunol Neuroinflamm 2014;1:e11 doi: 10.1212/NXI.0000000000000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waters PJ, McKeon A, Leite MI, et al. . Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 2012;78:665–671; discussion 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burt RK, Balabanov R, Han X, et al. . Autologous nonmyeloablative hematopoietic stem cell transplantation for neuromyelitis optica. Neurology 2019;93:e1732–e1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitley J, Woodhall M, Leite MI, Palace J, Vincent A, Waters P. Aquaporin-4 antibody isoform binding specificities do not explain clinical variations in NMO. Neurol Neuroimmunol Neuroinflamm 2015;2:e121 doi: 10.1212/NXI.0000000000000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato DK, Callegaro D, de Haidar Jorge FM, et al. . Cerebrospinal fluid aquaporin-4 antibody levels in neuromyelitis optica attacks. Ann Neurol 2014;76:305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindstrom JM, Seybold ME, Lennon VA, Whittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis: prevalence, clinical correlates, and diagnostic value. Neurology 1976;26:1054–1059. [DOI] [PubMed] [Google Scholar]
- 23.Drachman DB. Myasthenia gravis. N Engl J Med 1994;330:1797–1810. [DOI] [PubMed] [Google Scholar]
- 24.Gresa-Arribas N, Titulaer MJ, Torrents A, et al. . Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol 2014;13:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Majed M, Fryer JP, McKeon A, Lennon VA, Pittock SJ. Clinical utility of testing AQP4-IgG in CSF: guidance for physicians. Neurol Neuroimmunol Neuroinflamm 2016;3:e231 doi: 10.1212/NXI.0000000000000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimizu F, Schaller KL, Owens GP, et al. . Glucose-regulated protein 78 autoantibody associates with blood-brain barrier disruption in neuromyelitis optica. Sci Transl Med 2017;9:eaai9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All authors have full access to all data sets and take full responsibility for the integrity of the data and accuracy of the data analysis. All data pertaining to this article are contained within or published as online supplement.