

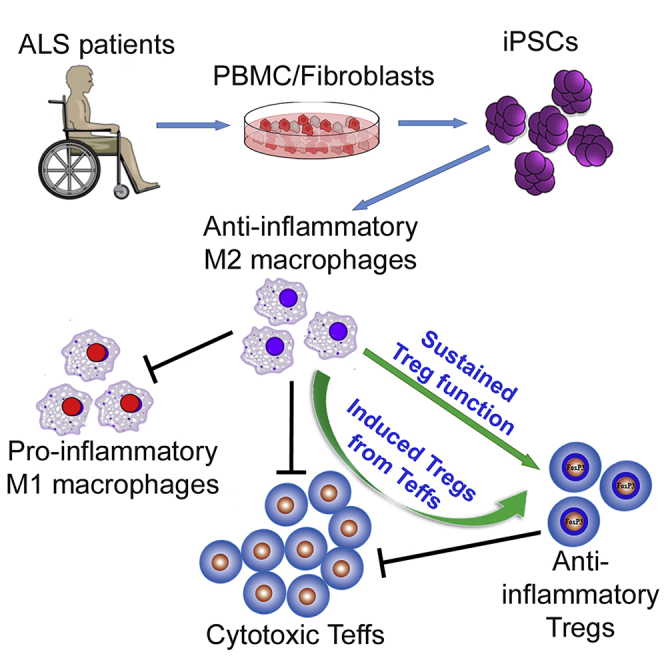
Summary
Amyotrophic lateral sclerosis (ALS) is a disorder with immune alterations that augment disease severity. M2 macrophages benefit diabetic and nephrotic mice by suppressing the pro-inflammatory state. However, neither have M2 cells been investigated in ALS nor have human induced pluripotent stem cell (iPSC)-derived M2 cells been thoroughly studied for immunosuppressive potentials. Here, iPSCs of C9orf72 mutated or sporadic ALS patients were differentiated into M2 macrophages, which suppressed activation of pro-inflammatory M1 macrophages as well as proliferation of ALS CD4+CD25- Tc (Teffs). M2 cells converted ALS Teffs into CD4+CD25+Foxp3+ regulatory T cells (Tregs) and rescued Tregs of ALS patients from losing CD25 and Foxp3. Furthermore, Tregs induced or rescued by iPSC-derived M2 had strong suppressive functions. ALS iPSC-derived M2 cells including those with C9orf72 mutation had similar immunomodulatory activity as control iPSC-derived M2 cells. This study demonstrates that M2 cells differentiated from iPSCs of ALS patients are immunosuppressive, boost ALS Tregs, and may serve as a candidate for immune-cell-based therapy to mitigate inflammation in ALS.
Subject Areas: Cellular Neuroscience, Immunology
Graphical Abstract
Highlights
-
•
ALS iPSC-derived M2 inhibit M1 activation and proliferation of ALS effector T cells
-
•
ALS iPSC-derived M2 induce functional Tregs from ALS effector T cells
-
•
ALS iPSC-derived M2 sustain ALS Tregs' function and phenotypic expression
-
•
Functions of sporadic ALS, C9 mutant ALS, and control iPSC-derived M2 are comparable
Cellular Neuroscience; Immunology
Introduction
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig disease, is a common, devastating, and invariably fatal adult neurodegenerative disease. In addition to the loss of upper and lower motor neurons, ALS is now regarded as a disorder with immune dysregulation, which is characterized by alterations/activation of inflammatory cells that augment disease burdens and rates of disease progression. Earlier studies documented the presence of activated pro-inflammatory microglia/macrophages and lymphocytes in the central nervous system (CNS) of patients with ALS (Appel et al., 2010, Appel et al., 2011, Beers and Appel, 2019, Butovsky et al., 2012, Keizman et al., 2009, Zhao et al., 2017). Furthermore, a meta-analysis reported that cytokine levels of interleukin (IL)-6, IL-1β, and tumor necrosis factor (TNF)-α were increased in the blood of patients, implicating a peripheral systemic pro-inflammatory immune response (Hu et al., 2017). Similar CNS and peripheral inflammatory responses have been reported in transgenic animal models of ALS (Beers et al., 2008; Chiu et al., 2008). Unfortunately, no treatments are presently available to arrest or substantially delay these inexorable inflammatory responses in patients with ALS.
Monocytes are myeloid cells circulating in the blood. When monocytes enter tissues, they become macrophages and the tissue milieu can induce changes in these cells. Therefore, monocytes and macrophages are closely related. Microglia are the innate immune myeloid cells within the CNS. Although microglia originate from the yolk sac and enter the CNS during embryonic development, microglia and macrophages have similar functions, such as patrolling for pathogens, responding to damage signals, performing phagocytosis of microbes and cellular debris, and acting as professional antigen-presenting cells to stimulate T cell responses. During chronic neuroinflammation, CNS-infiltrating macrophages acquire microglial markers (Grassivaro et al., 2020) and also retain significant differences (Ajami et al., 2018). Based on different activation stimuli, both microglia and macrophages can be polarized to M1 or M2 cells that produce pro-inflammatory or anti-inflammatory cytokines.
Polarization of CNS microglia and macrophages is a spectrum categorized by the subjective nomenclature M1 and M2 cells that represent the two extreme pro- and anti-inflammatory phenotypes, respectively (Ransohoff, 2016). Classical activation of M1 cells can be generated in response to interferon (IFN)-γ and lipopolysaccharide (LPS). They possess anti-microbial and cytotoxic properties due to secretion of reactive oxygen species and pro-inflammation cytokines (Nathan and Shiloh, 2000). The persistence or escalation of the inflammatory processes mediated by M1 macrophages can result in cytotoxicity (Laskin et al., 2011). In animal models of ALS, activated microglia are pro-inflammatory and neurotoxic during the rapidly progressing phase of disease (Beers et al., 2006, Boillee et al., 2006, Liao et al., 2012, Meissner et al., 2010, Zhao et al., 2010). In the periphery, monocytes of patients with ALS are also skewed toward a pro-inflammatory M1 state and potentially contribute to disease progression (Zhao et al., 2017). In contrast, M2 cells can be established after exposure to IL-4/IL-10/IL-13, TGF-β, or glucocorticoids. M2 cells secrete high levels of anti-inflammatory cytokines and neurotrophic factors, and have high endocytic clearance capacities (Tiemessen et al., 2007). M2 cells have been reported to promote blockade of inflammatory responses and enhance tissue repair (Bai et al., 2017, Cherry et al., 2014).
During the early stage of motor neuron injury in ALS models, the surveying microglia exhibit an M2 phenotype and react to the signals (possibly CD200 and fractalkine) with release of cytokines and trophic factors to promote repair and regeneration (Appel et al., 2010, Beers et al., 2011a, Liao et al., 2012). However, as disease progresses, the injured motor neurons release “danger signals,” possibly misfolded oxidized proteins (Zhao et al., 2010), or ATP (D'Ambrosi et al., 2009), which induce microglia to release reactive oxygen species and pro-inflammatory cytokines and display an M1 phenotype. Activated M1 microglia promote further motor neuron injury, enhancing the release of pro-inflammatory signals, which activate microglia to an even greater extent, and induce a self-propagating cycle of a pro-inflammatory injurious dialog between motor neurons and microglia (Zhao et al., 2013).
The responses of the adaptive immune system also play a pivotal role regulating the rate of disease progression and survival in patients with ALS (Appel et al., 2010, Appel et al., 2011, Beers and Appel, 2019). It has been reported that regulatory T cells (Tregs), an adaptive immune response, had a protective role in patients with ALS; reduced Treg numbers and FOXP3 expression were associated with a more rapid disease progression and increased mortality (Henkel et al., 2013). In addition, Tregs from patients with ALS were less suppressive and the function of their Tregs correlated with disease progression rate and severity (Beers et al., 2017). Thus, the current working hypothesis is that as disease progresses, the initial supportive anti-inflammatory M2/Treg immune response is transformed to an injurious pro-inflammatory M1/Th1 response.
M2 cells derived from human blood have been shown to decrease the Th1 response to bacteria (Verreck et al., 2004). These blood-derived M2 cells also have the ability to convert CD4+ T cells to Tregs with a strong suppressive function (Savage et al., 2008, Schmidt et al., 2016). However, peripheral monocytes have limited proliferation potential and do not yield sufficient numbers of M2 cells to study. Human iPSCs (induced pluripotent stem cells) can provide a source of unlimited M2 cells. Furthermore, the functions of iPSC-derived M2 have not been fully studied. In the present study, we generated M2 cells from iPSCs of patients with ALS and healthy controls and compared their immune-suppressive functions. The purpose of this study is to determine whether ALS iPSCs could be used to generate unlimited immunosuppressive M2 cells in vitro as an autologous cell-based therapy for patients with ALS.
Results
M1 and M2 Macrophages Are Differentiated from iPSCs of Patients with ALS and Healthy Controls
Human fibroblasts or blood cells from six healthy controls (CTR) and seven patients with ALS (three patients with C9orf72 (C9) mutation, four patients with sporadic disease with either fast or slow disease progression) were reprogrammed to iPSCs and subsequently differentiated into monocytes. Flow cytometry revealed that more than 90% CD14+ cells co-expressed CD115 and HLA-DR (Figures 1A and 1B), indicating that these CD14+ cells were monocytes.
Figure 1.

Cells Differentiated from Human iPSCs Express Monocyte Markers
After differentiation of human iPSCs, floating cells were collected and subjected to flow cytometry. More than 90% of CD14+ cells (shown in blue boxes) express CD115 (A) and HLA-DR (B).
iPSC-derived CD14+ monocytes were further differentiated to M1 macrophages using granulocyte-macrophage colony-stimulating factor (GM-CSF) plus LPS + IFN-γ, and in separate cultures, differentiated into M2 macrophages using macrophage-CSF (M-CSF) plus IL-4 + IL-10 + TGF-β. iPSC-derived M1 cells up-regulated the pro-inflammatory cytokines IL-6, TNF-α, and IL-8 (Figures 2A–2C). iPSC-derived M2 cells expressed the macrophage lineage marker, CD68 (Figure 3A), and typical M2 markers, CD163 and CD206 (Figures 3B, 3C, and S9). Furthermore, the production of anti-inflammatory cytokines was measured in the supernatants of M2 cultures. As the TGF-β ELISA kit also recognized the latent TGF-β complex present in culture media, enhanced TGF-β secreted by M2 macrophages was shown as subtracted levels from M0 cells cultured at the same time (M2-M0) (Figure 3E). M2 cells from CTR iPSCs, ALS C9 iPSCs, or sporadic ALS iPSCs produced more IL-10 and TGF-β than their corresponding resting M0 macrophages; the levels of IL-10 and TGF-β were not different among M2 cells derived from CTR iPSCs, ALS C9 iPSCs, and sporadic ALS iPSCs (Figures 3D and 3E).
Figure 2.

M1 Macrophages Differentiated from iPSC-Derived Monocytes
M0 macrophages were differentiated from monocytes derived from control (CTR) iPSCs (n = 5) and ALS iPSCs (n = 7) in the presence of GM-CSF (50ng/ml) for 7 days. To generate M1 macrophages, resting M0 cells were activated with LPS (1ng/ml) and IFN-γ (2 ng/ml) for different time periods as indicated. Both CTR and ALS M1 cells (1 x 105/well) expressed up-regulated mRNA and protein of IL-6 (A), TNF-α (B), and IL-8 (C). The mRNA level of M0 were set as 1 arbitrary unit. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 vs. CTR M0 for mRNA; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. ALS M0 for mRNA; &p < 0.05, &&p < 0.01, &&&p < 0.001 vs. CTR 1hr for protein; ∆∆p < 0.01, ∆∆∆p < 0.001 vs. ALS 1 h for protein.
Figure 3.

M2 Macrophages Differentiated from iPSC-Derived Monocytes
(A–C) To differentiate M2 cells, iPSC-derived monocytes were cultured with M-CSF (100ng/ml) for 7 days, then with the addition of IL-4, IL-10, and TGF-β (20ng/ml each) for 5-16 hours. M2 cells derived from iPSCs expressed CD68 (A), CD206 (B), and CD163 (C).
(D and E) M2 cells derived from control (CTR) iPSCs (n = 5), ALS C9orf72 (C9) iPSCs (n = 3), and sporadic ALS iPSCs (n = 4) released more IL-10 (D) and TGF-β (E) than their resting M0 cells. Enhanced TGF-β secreted by M2 cells were shown as subtracted levels from M0 cells cultured at the same time (M2-M0) to remove the amount of TGF-β existing in the culture serum. ∗∗∗p < 0.001 vs. their corresponding M0. The levels of IL-10 and TGFβ were not different among CTR M2, ALS C9 M2 and ALS sporadic M2. ns = no significant difference.
ALS iPSC-Derived M2 Macrophages Inhibit M1 Activation
M2 cells were cultured on coverslips so that they could be removed after co-cultured with M1 cells at different time points , and M1 and M2 cells could be investigated separately. After co-culturing with M2 cells derived from an ALS iPSC line with C9 mutation, M1 cells expressed less IL-6 and TNF-α mRNA than M1 cultures alone; M2 in the co-cultures only express minimal IL-6 and TNF-α mRNA (Figures 4A and 4C). IL-6 and TNF-α protein levels were also suppressed in M1+M2 co-cultures compared with M1 alone (Figures 4B and 4D). M2 cells suppressed M1 IL-8 mRNA at early time points (3–7 h). However, M2 cells upregulated IL-8 mRNA at 24-h time point (Figure 4E). IL-8 protein levels in M1+M2 co-cultures were not lower than those in M1 cells alone (Figure 4F).
Figure 4.

ALS iPSC-Derived M2 Macrophages Inhibit M1 Activation
(A–F) Representative time curves of M1 cells alone, M1 cells after being co-cultured with M2 cells, and M2 cells after being co-cultured with M1 cells. M1 and M2 cells in this graph were differentiated from a representative ALS28 C9 iPSC. IL-6 mRNA (A), TNFα mRNA (C) and IL-8 mRNA (E) were detected in M1 alone, or M1 or M2 cells after co-cultures. IL-6 protein (B), TNFα protein (D) and IL-8 protein (F) were measured in the supernatants of M1 alone, or M1+M2 co-cultures.
(G) IL-6, TNF-α and IL-8 mRNA levels in M1 cells after being co-cultured with resting M0 cells from ALS iPSCs (n=3), M2 cells from CTR iPSCs (n=3), and M2 cells from ALS iPSCs (n=6) for 5hr. &p < 0.05, &&p < 0.01 vs ALS M0+M1.
(H) IL-6, TNF-α and IL-8 proteins in CTR M1 cells or ALS M1 cells alone or co-cultures with CTR M2 cells, ALS M2 cells, or ALS resting M0 for 7hrs. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 vs. their corresponding M1 cell alone cultures; ###p < 0.001 vs. ALS M1+M0.
M1 and M2 co-cultures were then performed using three CTR iPSCs and five additional ALS iPSCs from three patients with sporadic ALS and two ALS patients with C9 mutation. M2 from each of these iPSCs suppressed IL-6 and TNF-α expression, but did not change IL-8 protein of M1 in a similar pattern (Figures S1–S8). To determine whether the function of M2 cells is specific, resting M0 cells derived from same ALS iPSCs were used as controls. M2 differentiated from CTR iPSCs were also used as controls to study the influence of C9 mutation and ALS disease insults. The suppression of ALS M2 cells on IL-6, TNF-α, and IL-8 mRNA of M1 cells was significantly higher than that of resting ALS M0 cells, but similar to the suppressive function of CTR M2 cells (Figure 4G). IL-6 protein levels were reduced in ALS M1+M2 co-cultures compared with M1 alone or ALS M1+M0 cultures; M0 cells did not change IL-6 protein after co-culturing with M1 cells (Figure 4H). TNF-α protein levels increased in ALS M1+M0 co-cultures compared with M1 alone cultures, whereas TNF-α protein levels were lower in ALS M1+M2 co-cultures than ALS M1 alone or ALS M1+M0 cultures (Figure 4H). No differences were observed on IL-6 and TNF-α production between CTR M1+M2 and ALS M1+M2 co-cultures (Figure 4H), indicating that the suppressive abilities of ALS M2 were not reduced due to C9 mutation or ALS-related changes, and at same levels as control M2 on M1. However, IL-8 protein was increased in M1+M2 co-cultures compared with M1 cultures (Figure 4H).
ALS iPSC-Derived M2 Macrophages Suppress Proliferation of ALS Teffs
After being co-cultured with increased number of M2 cells derived from three ALS iPSC lines, less proliferation of ALS Teffs was observed in a dose-dependent manner (Figure 5B). Representative flow cytometry graphs were shown in Figure 5A. ALS iPSC-derived M2 cells behaved differently from M0 cells, which enhanced proliferation of ALS Teffs at lower doses (Figure 5B). M2 cells from seven ALS iPSCs (including three ALS C9 iPSCs) were able to suppress ALS Teff proliferation as efficiently as M2 from four CTR iPSCs (Figure 5C).
Figure 5.

ALS iPSC-Derived M2 Macrophages Suppress Proliferation of ALS Teffs
(A) Representative proliferation plots examined by flow cytometry: ALS Teffs had fewer proliferative divisions after being co-cultured with an increased number of M2 cells derived from a representative ALS iPSC line. (B) Proliferation of ALS Teffs was lower when co-cultured with M2 cells from ALS iPSCs than co-cultured with resting M0 cells from same ALS iPSCs at 1:1/8 and 1:1/4 ratios of Teffs:M2/M0. ∗p < 0.05 vs. ALS Teffs+ALS M0 at same ratio. (C) M2 cells from CTR iPSCs (n = 4) and ALS iPSCs (n = 7) suppressed ALS Teffs’ proliferation in a dose-dependent manner. The suppressive activity of ALS M2 cells on ALS Teffs was not different from CTR M2 cells.
ALS iPSC-Derived M2 Macrophages Induce Functional Tregs from ALS Teffs
The expression of CD25 and Foxp3 (Tregs functional markers) were examined in ALS Teffs co-cultured with iPSC-derived M2 cells. The percentages of CD4+CD25+Foxp3+ Tregs were increased in M2 and ALS Teffs co-cultures compared with cultures of ALS Teffs alone (Figure 6A), indicating that iPSC M2 can induce Tregs from ALS Teffs. We then compared Treg induction ability of M2 with resting M0 and M1 cells derived from same ALS iPSCs. ALS M2 and M0 cells induced increased numbers of Tregs from ALS Teffs than ALS M1 cells or ALS Teffs alone; ALS M1 cells did not increase the percentage of Tregs compared with ALS Teffs alone (Figure 6B). M2 cells from three ALS C9 and four sporadic iPSCs had similar Treg induction capabilities as three CTR M2 cells (Figure 6C). In addition, M2-induced Tregs were functionally suppressive; Tregs induced by five ALS iPSC-derived M2 cells all suppressed Teff proliferation in a dose-dependent manner, as strongly as Tregs induced by three CTR M2 (Figure 6D).
Figure 6.

ALS iPSC-Derived M2 Macrophages Induce Functional Tregs from ALS Teffs
M2, M0, or M1 cells derived from iPSCs were co-cultured with ALS Teffs at a ratio of 1:1/4 (Teffs:M2) for 2 days. Cells were then subjected for flow cytometric analyses.
(A) Representative plots showed CD25 and Foxp3 expression of CD4+ T-cells in ALS Teffs either cultured alone or co-cultured with M2 cells derived from one CTR25 iPSC, one ALS29 C9 iPSC.
(B) The percentages of CD4+CD25+Foxp3+ Tregs in ALS M2+ALS Teffs (n = 3) and ALS M0+ALS Teffs (n = 3) were higher than ALS M1+ALS Teffs (n = 3) or ALS Teffs alone (n = 3). No differences were observed between ALS M1+ALS Teffs and ALS Teffs alone. ∗∗∗p<0.001 vs. ALS Teffs alone; ##p<0.01, #p<0.05 vs. ALS M1+ALS Teffs.
(C) M2 cells from ALS C9 iPSCs (n = 3) and sporadic ALS iPSCs (n = 4) increased the percentage of CD4+CD25+Foxp3+ Tregs from ALS Teffs compared with ALS Teffs alone cultures; Treg induction capacity of M2 cells from both ALS C9 and sporadic iPSCs were not different from CTR M2 cells (n = 3). ∗∗∗p<0.001 vs. ALS Teffs alone.
(D) M2-induced CD4+CD25+ Tregs were isolated from M2+ALS Teffs co-cultures. These M2-induced Tregs (iTregs(M2)) were then co-cultured with CFSE-stained responder Teffs from ALS patients for 5 days at different Teffs:Tregs(M2) ratio (1:1/8, 1:1/4, 1:1/2, 1:1). Tregs induced by CTR M2 (n = 3) and ALS iPSC M2 (n = 5) suppressed proliferation of ALS Teffs similarly in a dose-dependent manner.
ALS iPSC-Derived M2 Macrophages Sustain CD25 and Foxp3 Expression and Suppressive Function of ALS Tregs
To determine the effects of iPSC-derived M2 cells on Tregs, M2 cells were co-cultured with Tregs isolated from the blood of patients with ALS. After culturing ALS Tregs alone for 2 days, the percentage of CD4+CD25+Foxp3+ Tregs was decreased from 32.6% ± 0.9% to 11.8% ± 1.6%. When ALS Tregs were co-cultured with M2 cells derived from a representative ALS C9 iPSC, the percentage of CD4+CD25+Foxp3+ Tregs increased in a dose-dependent manner (Figure 7A). M2 cells from both ALS C9 and sporadic ALS iPSCs had similar effects as CRT M2 cells and increased the percentage of CD4+CD25+Foxp3+ Tregs (Figure 7B). In addition, there were no differences in viability and CD4+ T cell numbers among culture groups (Figure S10), which argues against the possibility that M2 cells selectively supported the survival of Tregs, or that the increased percentage of CD4+CD25+Foxp3+ Tregs was due to loss of CD4+Foxp3- T cells. M2-rescued Tregs were further isolated from ALS Tregs and ALS M2 co-cultures. As low numbers of Tregs were initially obtained from blood samples, fewer M2-rescued Tregs were isolated for their functional assay. Even at a ratio of 1:1/16 (responder Teffs:M2-rescued Tregs), significant reduction of Teff proliferation was noted (from 92.3% ± 0.42% in Teff alone to 50.9% ± 9.95% in Teffs + M2-rescued Tregs, Figures 7C and 7D).
Figure 7.

ALS iPSC-Derived M2 Macrophages Sustain the Expression of Treg Functional Markers CD25 and Foxp3
Tregs purified from blood of ALS patients were either cultured alone or co-cultured with iPSC-derived M2 cells at a ratio of 1:1/2 (Tregs:M2). After 2 days, T-cells were collected for flow cytometric analyses.
(A) Representative plots showed that the percentage of CD4+CD25+Foxp3+ Tregs increased in a dose-dependent fashion after being co-cultured with M2 cells derived from a representative ALS C9 iPSC.
(B) Similar to CRT M2 cells (n = 3), M2 cells from ALS C9 iPSCs (n = 3) and sporadic ALS iPSCs (n = 4) increased the percentage of CD4+CD25+Foxp3+ Tregs. ∗∗∗p < 0.001 vs. ALS Tregs alone.
(C and D) M2-rescued CD4+CD25+ Tregs were isolated from M2 and ALS Tregs co-cultures. These M2-rescued Tregs (rTregs(M2)) were then co-cultured with CFSE-stained responder Teffs from ALS patients for 5 days at a ratio of 1:1/16 (Tresp : M2-rescued Tregs). After co-cultured with Tregs rescued by ALS M2 cells, proliferation of ALS Tresp was lower than ALS Tresp alone (D) (#p < 0.05, n = 3). Representative proliferation plots were shown in C.
Discussion
Accumulating evidence suggests that tissue inflammatory responses promote increased burden of disease and rate of progression in ALS (Appel et al., 2010, Appel et al., 2011, Beers et al., 2006, Beers et al., 2011b, Beers et al., 2017); ALS is now regarded as a disorder with immune alterations and dysregulation (Beers and Appel, 2019). The current study demonstrated for the first time that M2 cells derived from iPSCs of patients with ALS suppressed toxic M1 cell responses and inhibited ALS Tresp proliferation. Furthermore, these iPSC-derived M2 cells converted Teffs of patients with ALS into functionally suppressive Tregs and sustained the immunomodulatory functions of ALS Tregs. ALS iPSC-derived M2 cells were functionally different when compared with resting M0 cells or M1 cells derived from same ALS iPSCs, indicating that the effects of M2 cells are phenotype specific. This study also demonstrates that the immunomodulatory effects of ALS iPSC-derived M2 cells are as strong as those of control iPSC-derived M2 cells, suggesting that the capacity of ALS iPSCs to become functional M2 cells is not influenced by the ongoing disease process or by the expression of the C9 mutation in patients with ALS.
A recent meta-analysis reported that cytokine levels of IL-6, IL-1β, and TNF-α were increased in the blood of patients with ALS, implicating a peripheral systemic pro-inflammatory immune response (Hu et al., 2017). These cytokines not only contribute to the innate immune system's pro-inflammatory milieu in patients with ALS but also contribute to the alteration/education of the adaptive immune system in these patients. IL-6 has been shown to induce differentiation of pathogenic Th17 and inhibit Tregs generation induced by TGF-β (Bettelli et al., 2006). TNF-α is not only known as a modulator of inflammation and is upregulated in patients with ALS (Brohawn et al., 2016) but also inhibits Treg-suppressive function by down-regulating Foxp3 expression and phosphorylation, and their adaptive immune response (Nie et al., 2013, Valencia et al., 2006). Thus, both the innate and adaptive immune systems play a pivotal and intertwined role in the ALS disease process; as disease burden accumulates and progression rates accelerates, the initial supportive anti-inflammatory M2/Tregs immune response is transformed into an injurious pro-inflammatory M1/Th1 response (Beers et al., 2011a, Henkel et al., 2013, Liao et al., 2012). The current study demonstrated that M2 cells derived from ALS iPSC lines suppressed M1 cell inflammatory responses by reducing IL-6 and TNF-α production, inhibited the proliferation of ALS Teffs, and induced and rescued ALS Tregs. These data suggest that iPSC-derived M2 cells may reverse the imbalance between Tregs/M2 and Th1/M1 reactivity and reduce the pro-inflammatory milieu promoted by M1 and Th1.
iPSC-derived M2 cells expressed low levels of IL-6 and TNF-α in the M1 and M2 co-cultures and suppressed IL-6 and TNF-α produced by M1 cells. However, M2 cells upregulated IL-8 expression 24 h after being co-cultured with M1 cells, and IL-8 protein levels in M1 + M2 co-cultures were higher than M1 cultures alone. IL-8 is a chemokine responsible for attracting neutrophils and T cells to inflamed sites. A previous study demonstrated that IL-8 expression was upregulated in peripheral monocytes of patients with ALS (Zhao et al., 2017). Another study reported that IL-8 stimulated the M2 polarization of tumor-associated macrophages (Xiao et al., 2018); IL-8 mediated migration of CD4+ T cells including Tregs (Eikawa et al., 2010). These cumulative data suggest that IL-8 may attract pro-inflammatory as well as anti-inflammatory cells to lesions. Thus the role of IL-8 in ALS remains unclear, and further investigations are required.
The current data also suggest that iPSC-derived M2 cells enhanced ALS Tregs by sustaining their number and converting Teffs into Tregs. Tregs are potent modulators of the immune system, suppressing injurious pro-inflammatory responses; Tregs can directly suppress the toxic properties of microglia (Zhao et al., 2012). The passive transfer of Tregs prolonged the slowly progressing phase of disease in ALS mice, augmented survival, increased M2 markers, and decreased M1 markers and their pro-inflammatory cytokines (Beers et al., 2011a). Furthermore, in vivo expansion of Tregs using an IL-2/IL-2Ab complex prolonged survival of ALS mice with reduced microglial immunoreactivity (Sheean et al., 2018). In patients with ALS, both number and function of Tregs were decreased as disease advanced from a slowly progressing stage to a rapidly progressing disease (Beers et al., 2017, Henkel et al., 2013). More importantly, autologous infusions of expanded Treg cells in patients with ALS were shown to be safe and slowed the patient's disease progression (Thonhoff et al., 2018). Thus, the demonstration that iPSC-derived M2 cells induce and rescue suppressive Tregs provides a potentially novel approach to maintain suppressive and anti-inflammatory effects of Treg-related therapy; M2 and Tregs may have a synergistic potential to quench the pro-inflammatory milieu.
The mechanism of how iPSC-derived M2 cells suppress cytokine production and secretion from M1 cells is unclear. TGF-β and IL-10 have been reported to regulate microglia, macrophages, and Teffs (Butovsky et al., 2014, Fiorentino et al., 1991, Kelly et al., 2018, Zhao et al., 2012, Zheng et al., 2007) and play an important role in the induction of Tregs (Hsu et al., 2015, Zheng et al., 2007). However, in our experiments, blocking antibodies (abs) against IL-10 and TGF-β did not reverse the effects of iPSC-derived M2 cell on M1 cell activation, Teff proliferation, and induction of Tregs from Teffs (data not shown). We could not document that either IL-10 or TGF-β contributed to the suppressive mechanisms in vitro. Further studies are clearly required to define the specific suppressive mechanisms.
The blood-brain barrier (BBB) is impaired in patients with ALS and in ALS animal models (Garbuzova-Davis et al., 2012, Henkel et al., 2009, Nicaise et al., 2009, Winkler et al., 2013, Zhong et al., 2008). As a result, peripheral monocytes can invade the spinal cord parenchyma (Zondler et al., 2016). Monocyte-derived macrophages have a distinct transcriptional profile, and when they enter the CNS, they maintain their transcriptional profile, which differs from the resident microglial transcriptional profile (Cronk et al., 2018). However, it has also been reported that when monocytes enter the CNS in pathological conditions and become macrophages, many of the genes expressed in macrophages are similar to the ones expressed in microglia (Friedman et al., 2018, Grassivaro et al., 2020). Further studies are needed to determine whether activated ALS macrophages remain distinct from activated microglia.
Blood-born macrophages have been shown to contribute to disease progression in ALS mice (Butovsky et al., 2012) and could similarly induce either neuroinflammation or neuroprotection in ALS. It is clear that extensive neuro-immune cross talk occurs between the brain and peripheral immune systems, especially in the presence of an altered BBB. Thus, peripheral myeloid M2 cells could modulate disease progression by suppressing M1-mediated inflammation in the periphery or in the CNS. The demonstration that iPSC-derived M2 cells were able to suppress the activation of M1 cells suggests that iPSC-derived M2 cells have the functional ability to protect motor neurons from injury mediated by M1 cells, and thereby slow ALS disease progression.
Our data indicate that C9 mutation and some ALS-related changes in iPSCs did not compromise their in vitro development to M2 cells with strong immunosuppressive function. However, we could not rule out the fact that ALS iPSCs-derived M2 may differ from control iPSC-derived M2 cells in other functions (such as phagocytosis) or following diverse differentiation conditions. It may also be possible that microglia/macrophages in patients with ALS contain genetic and disease-specific changes, which could promote M1 activation of ALS macrophages in vivo. In our recent study, monocytes from patients with ALS were more readily differentiated to a pro-inflammatory M1 phenotype than healthy control monocytes (Du et al., 2020). We had too few iPSC specimens to document whether different regimens that promote the M1 phenotype from iPSC cells could differentiate ALS M1 from control M1 cells. It is also possible that exposure to in vivo ALS-related signals as well as genetic differences may yield different responses in ALS and healthy control monocytes. More studies are clearly required to explore the underlining mechanisms.
Our previous studies demonstrated that M2 microglia in spinal cords of ALS mice lost their markers and a shift of M2 to M1 phenotype occurred during rapid progressing stage (Beers et al., 2011a, Liao et al., 2012). Macrophages infiltrating to CNS or along peripheral nerves may undergo this shift from M2 to M1 cells in a similar way. However, it is still unknown which exact signals in ALS drive this M2 to M1 transition. It is also unclear whether iPSC-derived M2 cells would maintain their phenotype after transfer in vivo. We used M-CSF and IL-4/IL-10/TGF-β to differentiate M2 macrophages. This optimized protocol has been shown to induce human M2 cells to release more anti-inflammatory cytokines with higher suppressive function, especially more resistant to pro-inflammatory stimulation than other M2 differentiation protocols (Mia et al., 2014).
Although the in vivo protective anti-inflammatory functions of M2 have not been reported in ALS, mouse M2 cells have been shown to have protective effects in mouse models of diabetes and nephrosis; transferred M2 macrophages were shown to maintain their anti-inflammatory phenotypes in vivo in these models (Cao et al., 2010, Parsa et al., 2012). One single injection of mouse M2 cells before disease onset protected >80% of treated mice against type 1 diabetes for at least 3 months and suppressed T cell proliferation in pancreatic lymph nodes of treated mice; injected M2 cells were found to migrate predominantly into the inflamed pancreas and associated lymph nodes (Parsa et al., 2012). M2 transfer also reduced renal inflammation in murine adriamycin nephrosis; accumulation of injected M2 was also observed in inflamed kidney and renal draining lymph nodes after adriamycin administration (Cao et al., 2010). Thus, these reports indicate that M2 macrophages have in vivo immunosuppressive properties, are able to enter inflamed lesion sites, and provide clinical benefit.
The current study demonstrates that ALS does not cause deficits in iPSC-derived M2 cells; M2 cells differentiated from iPSCs of patients with ALS suppress inflammation as strongly as M2 derived from healthy controls' iPSCs. Our data provide critical evidence for the autologous treatment of patients with ALS using their own iPSC-derived M2 cells. It remains to be determined whether iPSC-derived M2 cells can effectively ameliorate the pathogenesis of disease in ALS.
Limitations of the Study
The first limitation of this study is that therapeutic mechanisms of suppressive action and induction of Tregs from Teffs mediated by iPSC-derived M2 macrophages are presently unclear. Second, we do not have definitive evidence that the ex vivo immunomodulatory activity of iPSC-derived M2 macrophages will be translated into in vivo therapy. Third, how to maintain the suppressive function of M2 cells and prevent them from shifting to M1 cells in pro-inflammatory environment is not addressed in this study. The answer to these questions would be valuable for further in vivo therapeutic investigation.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Stanley H. Appel (sappel@houstonmethodist.org).
Materials Availability
iPSC lines generated in this study will be made available upon request for a minimal fee to cover production of iPSC Cryovials after completing a Materials Transfer Agreement with the Cedars-Sinai Biomanufacturing Center (CBC). The iPSC lines are readily accessible from the CBC Cell Line Catalog. This study did not generate any other unique reagents.
Data and Code Availability
This study did not generate datasets or codes. There is no any unpublished custom code, software, or algorithm that is central to supporting the main claims of the paper.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We are grateful to Yuanfang Liu, Hui Xuan, and Douglas Casey for their technical assistance. This work was supported by The ALS Association (# 0001442744) and ALS Finding a Cure Foundation (ALSFAC-ACT-807).
Author Contributions
W.Z., J.W., and S.W. performed the research and collected data. L.O. and D.S. generated iPSC lines. W.Z. analyzed data. S.H.A. and W.Z. designed the project and interpreted the data. S.H.A. and C.N.S. organized patient recruitment and provide clinical information. C.N.S., H.S.G., D.R.B., J.R.T., A.D.T., and A.F. conceptualized the study, performed further data interpretation, and provided critical experimental suggestions. W.Z., S.H.A., D.R.B., and A.F. wrote and revised the manuscript.
Declaration of Interests
The authors declare no conflict of interest.
Published: June 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101192.
Supplemental Information
References
- Ajami B., Samusik N., Wieghofer P., Ho P.P., Crotti A., Bjornson Z., Prinz M., Fantl W.J., Nolan G.P., Steinman L. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat. Neurosci. 2018;21:541–551. doi: 10.1038/s41593-018-0100-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel S.H., Beers D.R., Henkel J.S. T cell-microglial dialogue in Parkinson's disease and amyotrophic lateral sclerosis: are we listening? Trends Immunol. 2010;31:7–17. doi: 10.1016/j.it.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel S.H., Zhao W., Beers D.R., Henkel J.S. The microglial-motoneuron dialogue in ALS. Acta Myol. 2011;30:4–8. [PMC free article] [PubMed] [Google Scholar]
- Bai L., Liu X., Zheng Q., Kong M., Zhang X., Hu R., Lou J., Ren F., Chen Y., Zheng S. M2-like macrophages in the fibrotic liver protect mice against lethal insults through conferring apoptosis resistance to hepatocytes. Sci. Rep. 2017;7:10518. doi: 10.1038/s41598-017-11303-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers D.R., Appel S.H. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol. 2019;18:211–220. doi: 10.1016/S1474-4422(18)30394-6. [DOI] [PubMed] [Google Scholar]
- Beers D.R., Henkel J.S., Xiao Q., Zhao W., Wang J., Yen A.A., Siklos L., McKercher S.R., Appel S.H. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U S A. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers D.R., Henkel J.S., Zhao W., Wang J., Appel S.H. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc. Natl. Acad. Sci. U S A. 2008;105:15558–15563. doi: 10.1073/pnas.0807419105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers D.R., Henkel J.S., Zhao W., Wang J., Huang A., Wen S., Liao B., Appel S.H. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain. 2011;134:1293–1314. doi: 10.1093/brain/awr074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers D.R., Zhao W., Liao B., Kano O., Wang J., Huang A., Appel S.H., Henkel J.S. Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain Behav. Immun. 2011;25:1025–1035. doi: 10.1016/j.bbi.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers D.R., Zhao W., Wang J., Zhang X., Wen S., Neal D., Thonhoff J.R., Alsuliman A.S., Shpall E.J., Rezvani K. ALS patients' regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight. 2017;2:e89530. doi: 10.1172/jci.insight.89530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E., Carrier Y., Gao W., Korn T., Strom T.B., Oukka M., Weiner H.L., Kuchroo V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Boillee S., Yamanaka K., Lobsiger C.S., Copeland N.G., Jenkins N.A., Kassiotis G., Kollias G., Cleveland D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Brohawn D.G., O'Brien L.C., Bennett J.P., Jr. RNAseq analyses identify tumor necrosis factor-mediated inflammation as a major abnormality in ALS spinal cord. PLoS One. 2016;11:e0160520. doi: 10.1371/journal.pone.0160520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O., Siddiqui S., Gabriely G., Lanser A.J., Dake B., Murugaiyan G., Doykan C.E., Wu P.M., Gali R.R., Iyer L.K. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J. Clin. Invest. 2012;122:3063–3087. doi: 10.1172/JCI62636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O., Jedrychowski M.P., Moore C.S., Cialic R., Lanser A.J., Gabriely G., Koeglsperger T., Dake B., Wu P.M., Doykan C.E. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q., Wang Y., Zheng D., Sun Y., Wang Y., Lee V.W., Zheng G., Tan T.K., Ince J., Alexander S.I. IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J. Am. Soc. Nephrol. 2010;21:933–942. doi: 10.1681/ASN.2009060592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry J.D., Olschowka J.A., O'Banion M.K. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J. Neuroinflammation. 2014;11:98. doi: 10.1186/1742-2094-11-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu I.M., Chen A., Zheng Y., Kosaras B., Tsiftsoglou S.A., Vartanian T.K., Brown R.H., Jr., Carroll M.C. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc. Natl. Acad. Sci. U S A. 2008;105:17913–17918. doi: 10.1073/pnas.0804610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronk J.C., Filiano A.J., Louveau A., Marin I., Marsh R., Ji E., Goldman D.H., Smirnov I., Geraci N., Acton S. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J. Exp. Med. 2018;215:1627–1647. doi: 10.1084/jem.20180247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosi N., Finocchi P., Apolloni S., Cozzolino M., Ferri A., Padovano V., Pietrini G., Carri M.T., Volonte C. The proinflammatory action of microglial P2 receptors is enhanced in SOD1 models for amyotrophic lateral sclerosis. J. Immunol. 2009;183:4648–4656. doi: 10.4049/jimmunol.0901212. [DOI] [PubMed] [Google Scholar]
- Du Y., Zhao W., Thonhoff J.R., Wang J., Wen S., Appel S.H. Increased activation ability of monocytes from ALS patients. Exp. Neurol. 2020;328:113259. doi: 10.1016/j.expneurol.2020.113259. [DOI] [PubMed] [Google Scholar]
- Eikawa S., Ohue Y., Kitaoka K., Aji T., Uenaka A., Oka M., Nakayama E. Enrichment of Foxp3+ CD4 regulatory T cells in migrated T cells to IL-6- and IL-8-expressing tumors through predominant induction of CXCR1 by IL-6. J. Immunol. 2010;185:6734–6740. doi: 10.4049/jimmunol.1000225. [DOI] [PubMed] [Google Scholar]
- Fiorentino D.F., Zlotnik A., Mosmann T.R., Howard M., O'Garra A. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 1991;147:3815–3822. [PubMed] [Google Scholar]
- Friedman B.A., Srinivasan K., Ayalon G., Meilandt W.J., Lin H., Huntley M.A., Cao Y., Lee S.H., Haddick P.C.G., Ngu H. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of alzheimer's disease not evident in mouse models. Cell Rep. 2018;22:832–847. doi: 10.1016/j.celrep.2017.12.066. [DOI] [PubMed] [Google Scholar]
- Garbuzova-Davis S., Hernandez-Ontiveros D.G., Rodrigues M.C., Haller E., Frisina-Deyo A., Mirtyl S., Sallot S., Saporta S., Borlongan C.V., Sanberg P.R. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 2012;1469:114–128. doi: 10.1016/j.brainres.2012.05.056. [DOI] [PubMed] [Google Scholar]
- Grassivaro F., Menon R., Acquaviva M., Ottoboni L., Ruffini F., Bergamaschi A., Muzio L., Farina C., Martino G. Convergence between microglia and peripheral macrophages phenotype during development and neuroinflammation. J. Neurosci. 2020;40:784–795. doi: 10.1523/JNEUROSCI.1523-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel J.S., Beers D.R., Wen S., Bowser R., Appel S.H. Decreased mRNA expression of tight junction proteins in lumbar spinal cords of patients with ALS. Neurology. 2009;72:1614–1616. doi: 10.1212/WNL.0b013e3181a41228. [DOI] [PubMed] [Google Scholar]
- Henkel J.S., Beers D.R., Wen S., Rivera A.L., Toennis K.M., Appel J.E., Zhao W., Moore D.H., Powell S.Z., Appel S.H. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol. Med. 2013;5:64–79. doi: 10.1002/emmm.201201544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P., Santner-Nanan B., Hu M., Skarratt K., Lee C.H., Stormon M., Wong M., Fuller S.J., Nanan R. IL-10 potentiates differentiation of human induced regulatory T cells via STAT3 and Foxo1. J. Immunol. 2015;195:3665–3674. doi: 10.4049/jimmunol.1402898. [DOI] [PubMed] [Google Scholar]
- Hu Y., Cao C., Qin X.Y., Yu Y., Yuan J., Zhao Y., Cheng Y. Increased peripheral blood inflammatory cytokine levels in amyotrophic lateral sclerosis: a meta-analysis study. Sci. Rep. 2017;7:9094. doi: 10.1038/s41598-017-09097-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keizman D., Rogowski O., Berliner S., Ish-Shalom M., Maimon N., Nefussy B., Artamonov I., Drory V.E. Low-grade systemic inflammation in patients with amyotrophic lateral sclerosis. Acta Neurol. Scand. 2009;119:383–389. doi: 10.1111/j.1600-0404.2008.01112.x. [DOI] [PubMed] [Google Scholar]
- Kelly A., Gunaltay S., McEntee C.P., Shuttleworth E.E., Smedley C., Houston S.A., Fenton T.M., Levison S., Mann E.R., Travis M.A. Human monocytes and macrophages regulate immune tolerance via integrin alphavbeta8-mediated TGFbeta activation. J. Exp. Med. 2018;215:2725–2736. doi: 10.1084/jem.20171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin D.L., Sunil V.R., Gardner C.R., Laskin J.D. Macrophages and tissue injury: agents of defense or destruction? Annu. Rev. Pharmacol. Toxicol. 2011;51:267–288. doi: 10.1146/annurev.pharmtox.010909.105812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao B., Zhao W., Beers D.R., Henkel J.S., Appel S.H. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 2012;237:147–152. doi: 10.1016/j.expneurol.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner F., Molawi K., Zychlinsky A. Mutant superoxide dismutase 1-induced IL-1beta accelerates ALS pathogenesis. Proc. Natl. Acad. Sci. U S A. 2010;107:13046–13050. doi: 10.1073/pnas.1002396107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mia S., Warnecke A., Zhang X.M., Malmstrom V., Harris R.A. An optimized protocol for human M2 macrophages using M-CSF and IL-4/IL-10/TGF-beta yields a dominant immunosuppressive phenotype. Scand. J. Immunol. 2014;79:305–314. doi: 10.1111/sji.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C., Shiloh M.U. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. U S A. 2000;97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicaise C., Mitrecic D., Demetter P., De Decker R., Authelet M., Boom A., Pochet R. Impaired blood-brain and blood-spinal cord barriers in mutant SOD1-linked ALS rat. Brain Res. 2009;1301:152–162. doi: 10.1016/j.brainres.2009.09.018. [DOI] [PubMed] [Google Scholar]
- Nie H., Zheng Y., Li R., Guo T.B., He D., Fang L., Liu X., Xiao L., Chen X., Wan B. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat. Med. 2013;19:322–328. doi: 10.1038/nm.3085. [DOI] [PubMed] [Google Scholar]
- Parsa R., Andresen P., Gillett A., Mia S., Zhang X.M., Mayans S., Holmberg D., Harris R.A. Adoptive transfer of immunomodulatory M2 macrophages prevents type 1 diabetes in NOD mice. Diabetes. 2012;61:2881–2892. doi: 10.2337/db11-1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff R.M. A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 2016;19:987–991. doi: 10.1038/nn.4338. [DOI] [PubMed] [Google Scholar]
- Savage N.D., de Boer T., Walburg K.V., Joosten S.A., van Meijgaarden K., Geluk A., Ottenhoff T.H. Human anti-inflammatory macrophages induce Foxp3+ GITR+ CD25+ regulatory T cells, which suppress via membrane-bound TGFbeta-1. J. Immunol. 2008;181:2220–2226. doi: 10.4049/jimmunol.181.3.2220. [DOI] [PubMed] [Google Scholar]
- Schmidt A., Zhang X.M., Joshi R.N., Iqbal S., Wahlund C., Gabrielsson S., Harris R.A., Tegner J. Human macrophages induce CD4(+)Foxp3(+) regulatory T cells via binding and re-release of TGF-beta. Immunol. Cell Biol. 2016;94:747–762. doi: 10.1038/icb.2016.34. [DOI] [PubMed] [Google Scholar]
- Sheean R.K., McKay F.C., Cretney E., Bye C.R., Perera N.D., Tomas D., Weston R.A., Scheller K.J., Djouma E., Menon P. Association of regulatory T-cell expansion with progression of amyotrophic lateral sclerosis: a study of humans and a transgenic mouse model. JAMA Neurol. 2018;75:681–689. doi: 10.1001/jamaneurol.2018.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thonhoff J.R., Beers D.R., Zhao W., Pleitez M., Simpson E.P., Berry J.D., Cudkowicz M.E., Appel S.H. Expanded autologous regulatory T-lymphocyte infusions in ALS: a phase I, first-in-human study. Neurol. Neuroimmunol. Neuroinflamm. 2018;5:e465. doi: 10.1212/NXI.0000000000000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiemessen M.M., Jagger A.L., Evans H.G., van Herwijnen M.J., John S., Taams L.S. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl. Acad. Sci. U S A. 2007;104:19446–19451. doi: 10.1073/pnas.0706832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia X., Stephens G., Goldbach-Mansky R., Wilson M., Shevach E.M., Lipsky P.E. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006;108:253–261. doi: 10.1182/blood-2005-11-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verreck F.A., de Boer T., Langenberg D.M., Hoeve M.A., Kramer M., Vaisberg E., Kastelein R., Kolk A., de Waal-Malefyt R., Ottenhoff T.H. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc. Natl. Acad. Sci. U S A. 2004;101:4560–4565. doi: 10.1073/pnas.0400983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler E.A., Sengillo J.D., Sullivan J.S., Henkel J.S., Appel S.H., Zlokovic B.V. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125:111–120. doi: 10.1007/s00401-012-1039-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao P., Long X., Zhang L., Ye Y., Guo J., Liu P., Zhang R., Ning J., Yu W., Wei F. Neurotensin/IL-8 pathway orchestrates local inflammatory response and tumor invasion by inducing M2 polarization of Tumor-Associated macrophages and epithelial-mesenchymal transition of hepatocellular carcinoma cells. Oncoimmunology. 2018;7:e1440166. doi: 10.1080/2162402X.2018.1440166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W., Beers D.R., Henkel J.S., Zhang W., Urushitani M., Julien J.P., Appel S.H. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia. 2010;58:231–243. doi: 10.1002/glia.20919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W., Beers D.R., Liao B., Henkel J.S., Appel S.H. Regulatory T lymphocytes from ALS mice suppress microglia and effector T lymphocytes through different cytokine-mediated mechanisms. Neurobiol. Dis. 2012;48:418–428. doi: 10.1016/j.nbd.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W., Beers D.R., Appel S.H. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J. Neuroimmune Pharmacol. 2013;8:888–899. doi: 10.1007/s11481-013-9489-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W., Beers D.R., Hooten K.G., Sieglaff D.H., Zhang A., Kalyana-Sundaram S., Traini C.M., Halsey W.S., Hughes A.M., Sathe G.M. Characterization of gene expression phenotype in amyotrophic lateral sclerosis monocytes. JAMA Neurol. 2017;74:677–685. doi: 10.1001/jamaneurol.2017.0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S.G., Wang J., Wang P., Gray J.D., Horwitz D.A. IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J. Immunol. 2007;178:2018–2027. doi: 10.4049/jimmunol.178.4.2018. [DOI] [PubMed] [Google Scholar]
- Zhong Z., Deane R., Ali Z., Parisi M., Shapovalov Y., O'Banion M.K., Stojanovic K., Sagare A., Boillee S., Cleveland D.W. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat. Neurosci. 2008;11:420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zondler L., Muller K., Khalaji S., Bliederhauser C., Ruf W.P., Grozdanov V., Thiemann M., Fundel-Clemes K., Freischmidt A., Holzmann K. Peripheral monocytes are functionally altered and invade the CNS in ALS patients. Acta Neuropathol. 2016;132:391–411. doi: 10.1007/s00401-016-1548-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets or codes. There is no any unpublished custom code, software, or algorithm that is central to supporting the main claims of the paper.