

Summary
Human cells are able to sense and adapt to variations in oxygen levels. Historically, much research in this field has focused on hypoxia inducible factor (HIF) signaling and reactive oxygen species (ROS). Here, we perform genome-wide CRISPR growth screens at 21%, 5%, and 1% oxygen to systematically identify gene knockouts with relative fitness defects in high oxygen (213 genes) or low oxygen (109 genes), most without known connection to HIF/ROS. Knockouts of many mitochondrial pathways thought to be essential, including complex I and enzymes in Fe-S biosynthesis, grow relatively well at low oxygen and thus are buffered by hypoxia. In contrast, in certain cell types, knockout of lipid biosynthetic and peroxisomal genes cause fitness defects only in low oxygen. Our resource nominates genetic diseases whose severity may be modulated by oxygen and links hundreds of genes to oxygen homeostasis.
Introduction
Oxygen is the most commonly used substrate across all known biochemical reactions, surpassing even ATP and NADH (Raymond and Segre, 2006). The evolution of photosynthesis nearly two billion years ago led to a critical turning point for the earth’s atmosphere – the rise of oxygen. The availability of oxygen facilitated new types of chemistry (e.g. sterol synthesis) while simultaneously boosting energy yields by providing a thermodynamically favorable terminal electron acceptor. However, the ”great oxygenation event” also resulted in the increased production of toxic free radicals. This balance between the beneficial and deleterious effects of oxygen imparted a strong selective pressure on the evolution of different lifeforms.
Humans are regularly exposed to different oxygen tensions. We live at altitudes ranging from sea-level (21% O2) to 4,500 m (11% O2), and can even visit the peak of Mt. Everest (5% O2) while breathing atmospheric air. Acutely, humans can adapt to rapid O2 fluctuations via changes in ventilation and an increase in erythropoiesis. Over evolutionary timescales, there has been genetic selection for adaptive pathways in high altitude populations. These studies have identified polymorphisms in the canonical vHL-PHD-HIF O2 sensing pathways (Beall et al., 2010; Peng et al., 2011; Simonson et al., 2010; Xu et al., 2011; Yang et al., 2017; Yi et al., 2010), representing the most rapid known example of recent natural selection. Even at a fixed altitude, most metabolites are maintained at a relatively stable steady state in the human body (Ivanisevic et al., 2015). However, oxygen levels vary drastically from the arterial (100mmHg) to venous circulation (40 mmHg), and even within a given tissue can span a wide range of tensions (Ast and Mootha, 2019; Carreau et al., 2011). It follows that humans must have cellular programs to cope with fluctuating oxygen levels.
Extreme fluctuations in oxygen availability can exceed our body’s adaptive capacity, resulting in organ damage. Oxygen limitation contributes to some of the leading causes of death in developed countries, including stroke, ischemic heart disease, and kidney injury (Semenza, 2014). At the opposite extreme, excess oxygen can lead to retinopathy of prematurity (Hartnett and Penn, 2012), seizures, and acute respiratory distress (Wertlake and Winter, 1965). Recent epidemiological studies are beginning to demonstrate detrimental effects of excess supplemental oxygen in critically-ill patients (Chu et al., 2018; Girardis et al., 2016). We recently demonstrated that the neurological disease of a mouse model of mitochondrial Leigh syndrome is dramatically alleviated by chronic hypoxia and exacerbated at high ambient oxygen tensions (Ferrari et al., 2017; Jain et al., 2019; Jain et al., 2016). We also showed that neurological symptoms in a mouse model of Friedreich’s Ataxia can be prevented with hypoxia (Ast et al., 2019).
Despite the fundamental importance of oxygen in evolutionary biology, human physiology, and disease, we currently lack a complete understanding of human genes and pathways that are sensitive to fluctuations in oxygen tensions. In recent times, this field has primarily focused on the role of the vHL-PHD-HIF system in hypoxic adaptation. In contrast, a systematic investigation may highlight other pathways that inform our understanding of oxygen homeostasis as well as clinical conditions of ischemia, hypoxia and oxygen toxicity. Importantly, such work will help prioritize genetic diseases that may benefit from modulating oxygen availability.
Here, we use CRISPR/Cas9 technology to systematically identify human genes and pathways that exhibit different fitness in high and low oxygen environments, namely 21% O2 vs. 5% and 1% O2. This contrasts with all recent databases of essential genes in proliferating cell lines, which were performed in standard tissue culture conditions of 21% O2 (Blomen et al., 2015; Wang et al., 2017) even though most tissues are exposed to less than 10% O2 (Carreau et al., 2011). Our resource identifies pathways differentially essential in high and low oxygen and nominates genes likely to be involved in oxygen sensing, signaling, and metabolism.
Results
Genome-wide Screen Identifies Genes with Differential Fitness in High versus Low Oxygen
Tissue oxygen levels vary from less than 1% O2 in the lumen of the large intestine to just under 21% O2 in trachea (Carreau et al., 2011) (Figure 1A). Ultimately, a balance between oxygen delivery and oxygen consumption sets the local partial pressure of oxygen (PO2). To span the full spectrum of physiologically-relevant oxygen tensions, we focused on 1%, 5% and 21% O2 exposure. We generated a genome-wide pool of K562 knockout (KO) cells using the Brunello library, targeting approximately 20,000 genes using >76,000 guide RNAs (Figure 1B) (Doench, 2016). Following puromycin selection, cells were split into the three different oxygen tensions. Samples were collected at days 9 and 15 post-infection to track the gradual depletion or accumulation of a given gene KO in the various oxygen tensions. Of note, all experiments were performed in magnetic spinner flasks to ensure rapid gas equilibration and to ensure a near-equal oxygenation across cells in each condition. A doubling time of ~24h was maintained at 21% and 5% O2, whereas 1% O2 exposure resulted in a doubling time of ~36h (Figure 1C).
Figure 1. Genome-wide screen identifies genes that are selectively essential as a function of oxygen levels.
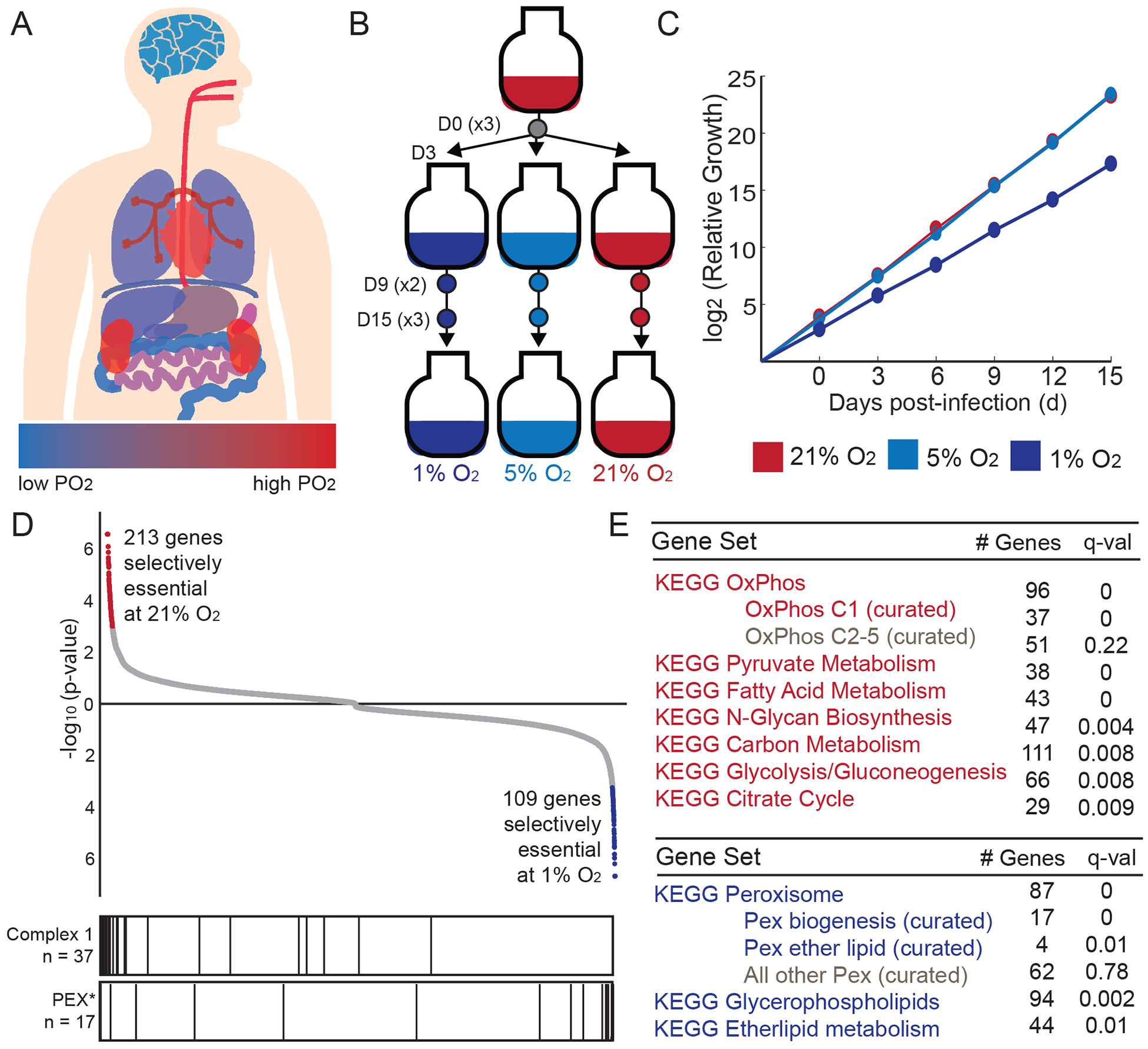
(A) Approximate tissue PO2 values across the human body (Carreau et al., 2011); blue for low PO2 and red for high PO2. (B) Experimental design for genome-wide screen. Three environmental oxygen tensions were tested at Day 0, 9 and 15 of treatment (n = 3 per D15 and D0 time points, n = 2 for D9 time points). (C) Cumulative relative growth throughout screens for cells exposed to 1, 5, 21% O2 (average shown for all technical replicates, curves at 21% and 5% overlap). (D) All 20,112 genes are ranked by their selective essentiality in 21% vs. 1% O2. The two sides of the list are compiled independently by running MAGeCK analysis on 21% vs. 1% O2 and then in 1% vs. 21% O2. Red and blue color indicates genes selectively essential at 10% FDR. Bar plot vertical lines indicate position of genes in complex I or peroxisome biogenesis (PEX*) gene sets. (E) KEGG pathways that are selectively essential in 1% O2 (blue) or 21% O2 (red).
To identify metabolic pathways that exhibit differential fitness as a function of oxygen tension, we used the MAGeCK algorithm to find gene KOs that are differentially abundant in 21% versus 1% O2 at the 15 day time point (Li et al., 2014) (Table S1). We identified 213 gene knockouts that showed significant depletion in 21% vs. 1% O2, and 109 gene knockouts that showed significant depletion at 1% vs. 21% O2, at a 0.1 false discovery rate (FDR) (Figure 1D, Table S1). The vast majority of these differentially abundant knockouts were significantly depleted compared to the early time points, and therefore we refer to hits as being “selectively essential” in 21% or 1% oxygen. However, a small subset are not truly essential at either oxygen tension but rather exhibit a growth advantage in one oxygen tension and hence are high scoring (see Table S1). Although we use the term “selectively essential” in this resource, careful comparison to initial guide counts, and follow-up experiments, are required to formally determine the nature of the interaction (synthetic lethal, buffering, etc) between the gene knockout and ambient oxygen.
We sought to determine if any biological pathways emerge from the screening hits. Interestingly, oxygen-consuming enzymes as defined by the KEGG reactions database, were not selectively essential in high or low oxygen (Table S1). We performed gene set enrichment analysis (GSEA) (Subramanian et al., 2005) on the rank-ordered lists using a custom database of 92 metabolism gene sets defined from KEGG Metabolism and Cellular Process (Kanehisa et al., 2016) (see Methods, Table S2). The mitochondrial oxidative phosphorylation (OXPHOS) gene set was the most enriched for being selectively essential at high oxygen tensions (Figure 1D–E). We further subdivided this gene set into complex I versus complex II-V, finding that the signal arose primarily from complex I subunits. At the opposite side of the analysis, peroxisome gene sets were the most selectively essential at low oxygen tensions. Subdividing this category further demonstrated that the signal arose primarily from peroxisome biogenesis genes and peroxisomal ether lipid synthesis.
Genes Related to High Altitude Adaptation and the Hypoxia Transcriptional Response are Not Selectively Essential in Hypoxia
We asked whether the canonical hypoxia adaptation pathways are enriched at either extreme in our CRISPR screen. We examined genes that are under recent natural selection in humans adapted to high altitude (Yang et al., 2017). Interestingly, these genes (Table S3) do not appear to be selectively essential in high or low oxygen in our screening dataset (Figure 2A). The most reproducibly identified SNPs associated with high altitude are found in the canonical cellular oxygen sensing pathway of vHL-PHD-HIF (Yang et al., 2017). These variants of PHD2 and HIF2α are believed to dampen the endogenous hypoxia transcriptional program and limit extreme polycythemia. Interestingly, while vHL knockouts show a fitness advantage in 21% oxygen, other key members of the hypoxia transcriptional response (HIF, PHD1–3, ARNT) do not appear to be selectively essential in our screen (Table S1).
Figure 2. High-altitude SNPs and hypoxia-inducible transcripts are not selectively essential as a function of oxygen levels.
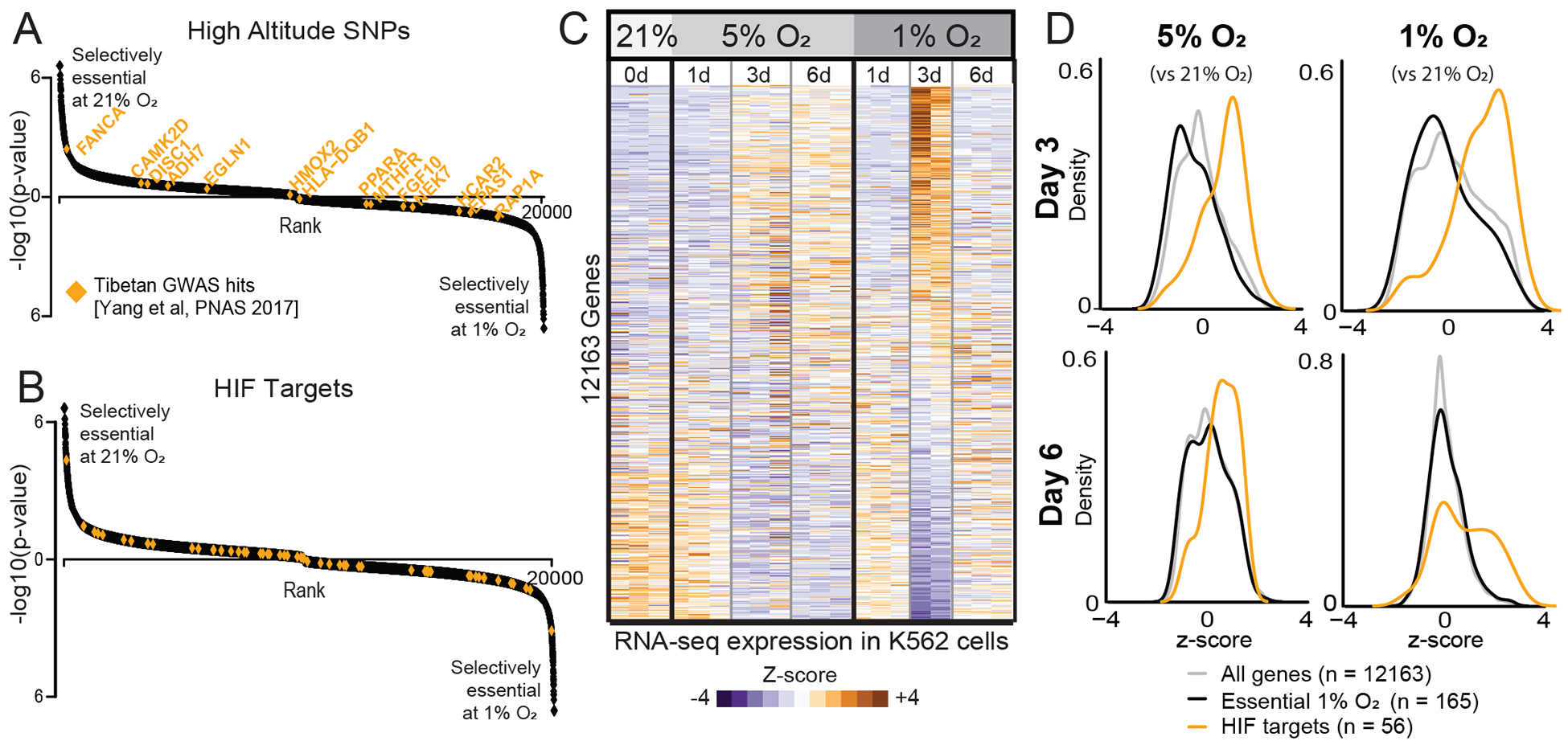
(A) High-altitude SNPs identified in Tibetan high-altitude natives (Yang et al., 2017) shown in orange on genes ranked as in Figure 1D. (B) Canonical HIF targets (Benita et al., 2009) shown in orange. (C) Heatmap of normalized transcript levels in 21%, 5% and 1% O2 performed in triplicate. Relative expression (Z-score) indicates genes down-regulated (blue) or up-regulated (orange) in hypoxia. (D) Distribution of gene expression Z-scores at two time points at 5% and 1% O2 relative to 21% O2. All genes shown in light gray. Genes selectively essential in 1% vs. 21% O2 from CRISPR screen (FDR<0.3) shown in black. Experimentally defined HIF targets (Benita et al., 2009) shown in orange.
We next asked whether targets of the vHL-PHD-HIF are selectively essential in high vs. low oxygen. Using previously curated lists of HIF targets (Benita et al., 2009), we did not find any enrichment for HIF targets being selectively essential in our dataset (Figure 2B). Notably, glycolytic enzymes are key targets of the HIF program, however the core glycolytic machinery is essential at all oxygen tensions, whereas other glycolytic components have paralogs and thus are insensitive to knockout at any oxygen tension.
To better compare the transcriptional response in our experimental system, we performed a matched RNA-seq time-course to accompany the CRISPR screening conditions. As expected, known HIF targets (including the glycolysis pathway) were upregulated at 1% and 5% O2 at the 3d time point (Figure 2C–D). However, due to a known regulatory negative feedback loop (Henze and Acker, 2010), some key HIF targets returned to baseline expression after 6d at 1% O2. As our genetic screen was performed over a 15d timeframe, such transcriptional dynamics were collectively integrated into our final readout of gene essentiality. Although key HIF targets were transcriptionally upregulated during acute hypoxia (Figure 2D), the genes that we found to be selectively essential as a function of oxygen tension were not transcriptionally regulated and were indistinguishable from the background distribution of all expressed genes, even when employing a more less conservative FDR of <0.3 (Figure 2D, black vs. gray curves). The incongruence between gene essentiality and the canonical HIF transcriptional program may result from cell autonomous vs. non-cell autonomous components of the hypoxia response (e.g. control of red blood cell mass, catecholamine release, changes in capillary density, etc.) or differences in timescales (e.g. acute and chronic hypoxia adaptations).
Dozens of Mitochondrial Disease Genes are Selectively Essential in High Oxygen
We previously demonstrated the benefit of breathing 11% O2 in a mouse model of complex I deficiency (Ferrari et al., 2017; Jain et al., 2016) as well as in preclinical models of Friedreich’s ataxia due to frataxin (FXN) deficiency (Ast et al., 2019). A key motivation for the current study was to understand which genetic disorders may be buffered or exacerbated by variations in oxygen levels. Of the 213 genes that are selectively essential in 21% versus 1% O2 (Table S1), 76 underlie Mendelian disorders (Figure 3A). Strikingly, 52 of these genes are associated with primary or secondary mitochondrial disease (Frazier et al., 2019) (Figure 3B). Indeed, 103 of the 213 genes encode mitochondrial proteins, reflecting a near 10-fold enrichment compared to non-mitochondrial genes (Figure 3A). Notably, our screen recovers the majority of core complex I subunits and FXN as selectively essential in 21% O2 (Figure 3B and C). However, NDUFS4 (the exact complex I disease gene whose loss we previously rescued in mice with hypoxia therapy) was not a hit in this screen, as this accessory subunit is not essential for K562 cell proliferation. Dozens of additional human mitochondrial disorders are now predicted to be sensitive to oxygen (Figure 3B), including pyruvate dehydrogenase (PDH) deficiency, autosomal dominant optic atrophy (OPA1), Charcot-Marie Tooth (MFN2), sideroblastic anemia (GLRX5) and Barth syndrome (TAZ). Our screen prioritizes other disease genes including monogenic forms of Parkinson’s Disease (HTRA2) and several non-mitochondrial genes underlying porphyrias (PPOX, UROD). Of course, these screening results only generate hypotheses that would require extensive additional preclinical studies to further substantiate the beneficial effects of hypoxia.
Figure 3. Mitochondrial genes are selectively essential at high oxygen levels.
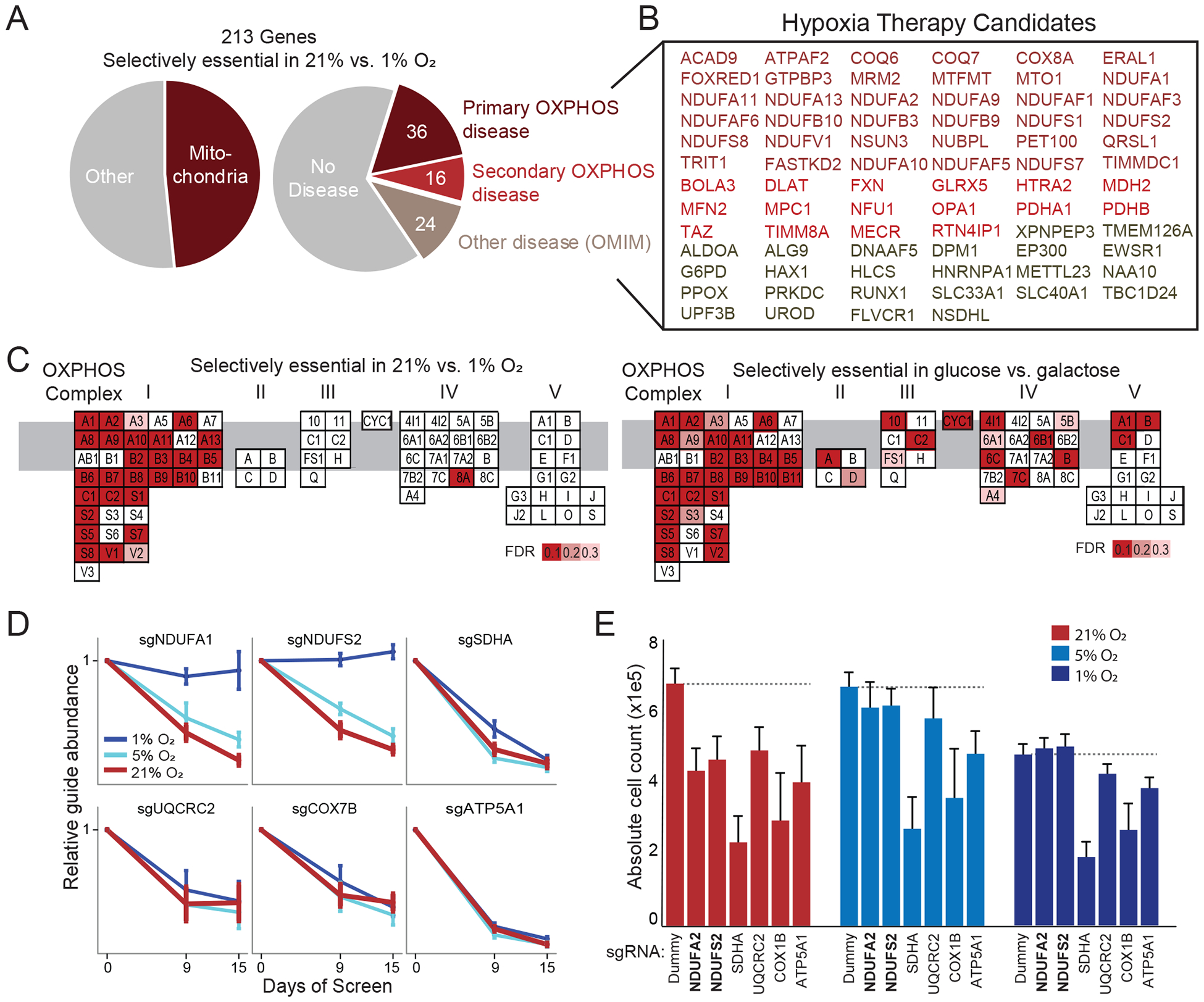
(A) Percentage of screening hits that encode mitochondrial proteins (left) or underlie human Mendelian disease (right) (Frazier et al., 2019; Hamosh et al., 2005). (B) Disease genes whose loss is buffered by hypoxia in cell culture. Different colors correspond to primary or secondary mitochondrial disease (Frazier et al., 2019), or non-mitochondrial diseases. (C) OXPHOS genes are organized by complex with red indicating genes that are hits in current screen (selectively essential in 21% vs. 1% O2, left panel) or in the glucose-galactose screen (unable to survive in galactose, right panel) (Arroyo et al., 2016). Genes are ordered alphabetically within complex using complex-specific prefixes (NDUF, SDH, UQCR, COX, ATP5) (e.g. A1 in CI refers to NDUFA1 whereas A in CII refers to SDHA). (D) sgRNA abundance at different time points of screen and oxygen tensions for CI-CV genes. Mean relative abundance (+/− SEM) shown across 4 guides per gene across all screen replicates. (E) Experimental validation in HEK293T knockout cell lines shows three-day growth as a function of oxygen tension (mean n=6 replicates +/− SD, Dummy indicates non-coding control). While all knockouts showed reduced growth compared to Dummy in 21% O2 and 5% O2, only complex I knockouts (bold text) show similar or increased growth in 1% O2.
Complex I, but not other ETC Complexes, is Selectively Essential in High Oxygen
Our screen shows a striking enrichment for certain mitochondrial pathways being selectively essential at 21% vs. 1% O2: complex I of the electron transport chain, the pyruvate dehydrogenase (PDH) complex, the mitochondrial pyruvate transporter (MPC), type II fatty acid synthesis (FASII), CoQ biosynthesis, and iron-sulfur (Fe-S) cluster biosynthesis (Figure 3C, Figure S1). Specifically, five Fe-S cluster biosynthesis genes were high-scoring (FXN, HSCB, NARFL, NFU1, NUBPL). NFS1 shows oxygen sensitivity in other contexts (Alvarez et al., 2017) but was not significant in our screen as its knockout led to a fitness defect in all oxygen tensions. Intriguingly, not all mitochondrial pathways were selectively essential in high oxygen. Using the classic experimental paradigm of glucose versus galactose media (in which cells rely primarily on glycolysis and OXPHOS metabolism, respectively), we previously showed that all OXPHOS complexes are essential for survival in glucose-free media (Arroyo et al., 2016), as well as many genes required for mitochondrial genome maintenance and expression. In contrast, we observe that only genes encoding OXPHOS complex I (and not other complexes) are selectively essential in 21% vs. 1% O2 (Figure 3C). Notably, ~70% (25 of 37) of nuclear-encoded complex I subunit deficiencies were buffered by hypoxia, i.e., the combined growth defect of gene loss and hypoxia was less than expected in an additive model. But almost none of complex II-V subunit deficiencies were buffered at 1% O2 (Figure 3C,D). This effect was validated in follow-up experiments in a second cell type (HEK293T) where lentivirus-infected pools of individual subunit KOs were tested for growth in 21%, 5% and 1% O2 (Figure 3E). Even after only three days in 1% O2, complex I mutants grew at a similar rate as WT cells in hypoxia, whereas the other mutants displayed impaired growth (Figure 3E).
Genes Involved in Lipid Metabolism are Selectively Essential in Low Oxygen
We then turned to the opposite side of the screen to explore the 109 genes whose loss causes a growth defect in 1% vs. 21% O2 (Figure 1 and 4). This side of the list contains a large number of genes related to lipid metabolism and peroxisomal metabolism. The former pointed us to literature linking oxygen availability to membrane fluidity (Ackerman and Simon, 2014; Ackerman et al., 2018; Sormendi and Wielockx, 2018). Membrane fluidity is a dynamic parameter known to be influenced by variables such as lipid composition, temperature and oxygen. In normoxia, membrane fluidity is maintained by the production of unsaturated fatty acids. However, desaturase enzymes are oxygen-dependent, and hypoxic inhibition of stearoyl-CoA desaturase (SCD) can decrease the ratio of unsaturated to saturated fatty acids. This leads to impaired cell growth, likely via increasing membrane rigidity (Kamphorst et al., 2013; Volmer et al., 2013). Certain tumor cell lines can adapt to this membrane stress by scavenging unsaturated lipids from the media (Kamphorst et al., 2013) or by sequestering toxic saturated fatty acids into lipid droplets (Ackerman et al., 2018; Piccolis et al., 2019).
Figure 4. Peroxisomal genes are selectively essential in low oxygen.
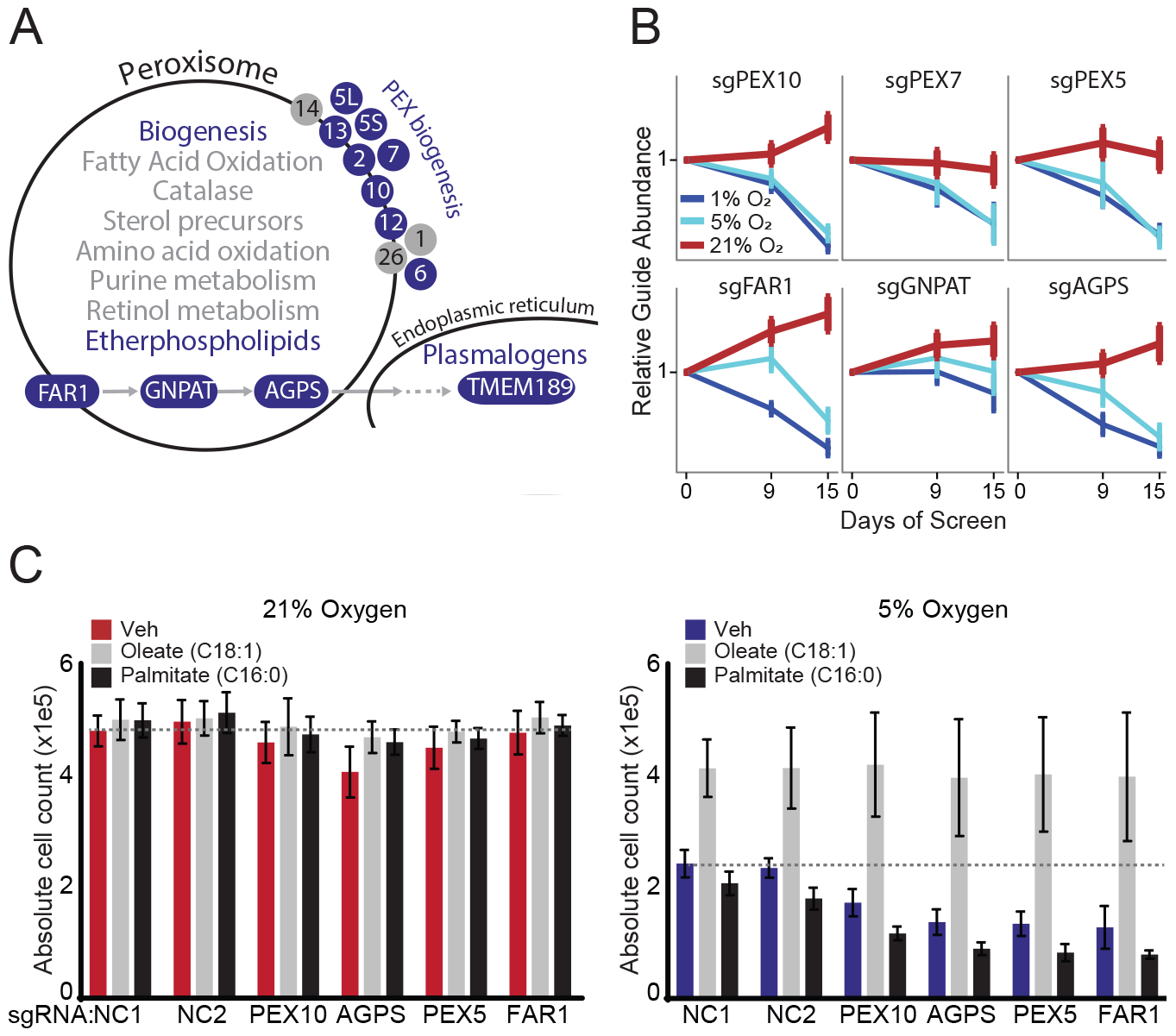
(A) Schematic of peroxisomal pathways with blue color indicating pathways and genes selectively essential in 1% vs. 21% O2 (FDR≤0.3). Subunits refer to PEX biosynthesis genes (e.g. 10 refers to PEX10). (B) sgRNA abundance corresponding to ether phospholipid synthesis genes and peroxisome biogenesis genes. Mean sgRNA relative abundance (+/− SEM) shown across 4 guides per gene across all screen replicates. (C) Cell counts for HEK293T non-coding (NC) control and knockout cell lines grown in low serum (3% FBS) for 2 days at different oxygen tensions and treated with vehicle, 10μM oleic or 10μM palmitic acid. Bars indicate averages (+/−SD) for triplicate experiments each with technical duplicates (n = 6). Dotted line indicates level of NC1 vehicle-treated control cells.
Many of the genes we observe to be selectively essential at 1% vs. 21% O2 are logical hits given their established roles in regulating production of unsaturated lipids and membrane fluidity. AMFR (rank #1) and SREBF1 (rank #2) are key regulators of lipid metabolism (Liu et al., 2012) and were recently identified to guard against saturated fatty acid (palmitate) toxicity (Piccolis et al., 2019). ADIPOR2 (rank #4) was reported to be a regulator of membrane fluidity in worms and in human cell lines (Ruiz et al., 2019). ACSL4 (rank #13) encodes an acyl-CoA synthase with a preference for polyunsaturated fatty acids (Klett et al., 2017), while its paralog ACSL3 prefers saturated fatty acids and scored on the opposite side of our screen.
We more closely examined hits related to the peroxisome (Figure 4A). Peroxisomes are single membrane organelles that house approximately 100 proteins responsible for ether lipid synthesis, very long chain and branched fatty acid oxidation, and H2O2 metabolism (Schluter et al., 2007; Waterham and Ebberink, 2012; de Vet et al., 1999). Defects in any of the 14 peroxisomal biogenesis proteins abolish or attenuate nearly all peroxisomal pathways (Waterham and Ebberink, 2012). Amongst 87 KEGG-annotated peroxisomal genes, eleven genes were selectively essential (FDR≤0.3) in low oxygen: seven genes involved in peroxisomal biogenesis (PEX*), ACSL4, and enzymes involved in ether phospholipid biosynthesis (FAR1, GNPAT, AGPS) (Figure 4 A,B). Peroxisome-derived ether lipids have a signature ether linkage at the sn-1 position. A subset of ether lipids are plasmalogens, which have a vinyl-ether linkage at the sn-1 position (Braverman and Moser, 2012) introduced by an endoplasmic reticulum protein TMEM189 (Gallego-Garcia et al., 2019), which is also selectively essential in 1% vs. 21% O2 (rank #6). We did not observe any high scoring peroxisomal pathways related to amino acid metabolism, fatty acid oxidation, or catalase.
Hypoxic Growth Defects are Rescued by Unsaturated Fatty Acids
We hypothesized that the differential essentiality of peroxisomes and ether phospholipid biosynthesis in 1% vs. 21% O2 might be acting through hypoxia’s known effects on membrane fluidity. Indeed, previous studies have shown that peroxisome mutants at 21% O2 have reduced membrane fluidity (Nagura et al., 2004). To test this hypothesis, and to assess a second cell type, we exposed HEK293T peroxisomal KO cell lines to high levels of either saturated (palmitic acid) or unsaturated (oleic acid) fatty acids at 21% and 5% O2. As in our screen, HEK293T cells also showed selective essentiality of peroxisomal genes in low oxygen (Figure 4C). WT and KO cells both show a baseline growth defect in low oxygen that is worsened with addition of palmitic acid, while oleic acid rescued growth defects in hypoxia (Figure 4C). Impaired growth of peroxisomal KO cells in hypoxia is not related to impaired formation of lipid droplets, which is grossly intact in FAR1 KO cell lines (Figure S2). We also note that although previous studies have shown that hypoxia can induce pexophagy in animals (Walter et al., 2014), peroxisomes are intact in HEK293T cells exposed to hypoxia (Figure S3).
We propose that peroxisome metabolism becomes important in low oxygen secondary to the saturated fatty acid toxicity present in hypoxia, but we note that this phenomenon is likely to be highly cell type specific and sensitive to cell culture conditions (e.g. lipid composition of media, seeding density). The growth defects of peroxisome knockouts that we observed in our screen (K562 cells) was confirmed by growth assays in follow-up studies in HEK293T cells, but not in HeLa cells, MCH58 fibroblasts cells, or in K562 cells outside of the screening conditions. Similar to experiments investigating SCD inhibition (Kamphorst et al., 2013), the selective essentiality of PEX genes is masked by high serum conditions (10% FBS), and only becomes apparent in low-lipid conditions (3% FBS).
Biosynthesis of Peroxisome-Derived Lipids is Increased in Hypoxia
Given these screening results, we asked whether the production of any peroxisome-derived lipids is increased in hypoxia. Applying lipidomics in K562 cells, we measured 454 annotated lipid species in WT and PEX KO cells as a function of oxygen tension (in low serum conditions) (Table S4). As expected, cells exposed to 24h hypoxia (5% O2) had increased levels of saturated lipids (Figure 5A). Of the 21 measured lipid classes, lyso-phosphatidylethanolamines (LPEs) and lyso-phosphatidylcholines (LPCs) were particularly increased (Figure 5B), consistent with previous reports showing they can be scavenged by hypoxic cells (Kamphorst et al., 2013). Of the 44 lipids annotated to be peroxisome-derived, eight were significantly elevated in hypoxia (p<0.05 after Bonferroni correction, Figure 5C) – the most significant being saturated lyso-ether-phospholipids: LPE(O-18:0), LPE(O-16:0), LPC(O-18:0) and LPC(O-16:0), where ‘O’ denotes an ether linkage (Figure 5D). These eight ether lipids were elevated in hypoxia only in cells with intact PEX genes, indicating they were synthesized by the peroxisome during hypoxia and not scavenged from the media (Figure 5D). Using a complementary approach (see Methods), we replicated these results using HEK293T cells (Figure 5E,F), and again, the elevation of saturated, peroxisome-derived lyso-ether-phospholipids only occurred in cells in which peroxisome genes were intact (Figure S4). We then asked whether the observed changes in peroxisome-derived lipids could be attributable to oxygen limitation of SCD action during hypoxia. Exposing WT HEK293T cells grown at 21% O2 to a potent small molecule inhibitor of SCD (SCDi) recapitulated key features of the hypoxic profile, notably a striking increase in saturated lyso-phospholipids, including saturated lyso-ether-phospholipids (Figure 5G). We also observed a similar pattern for di-saturated diacylglycerols (DAGs) (Figure 5H), recently identified to be effectors of saturated lipid toxicity (Piccolis et al., 2019).
Figure 5: Peroxisomal lipids are synthesized in hypoxia.
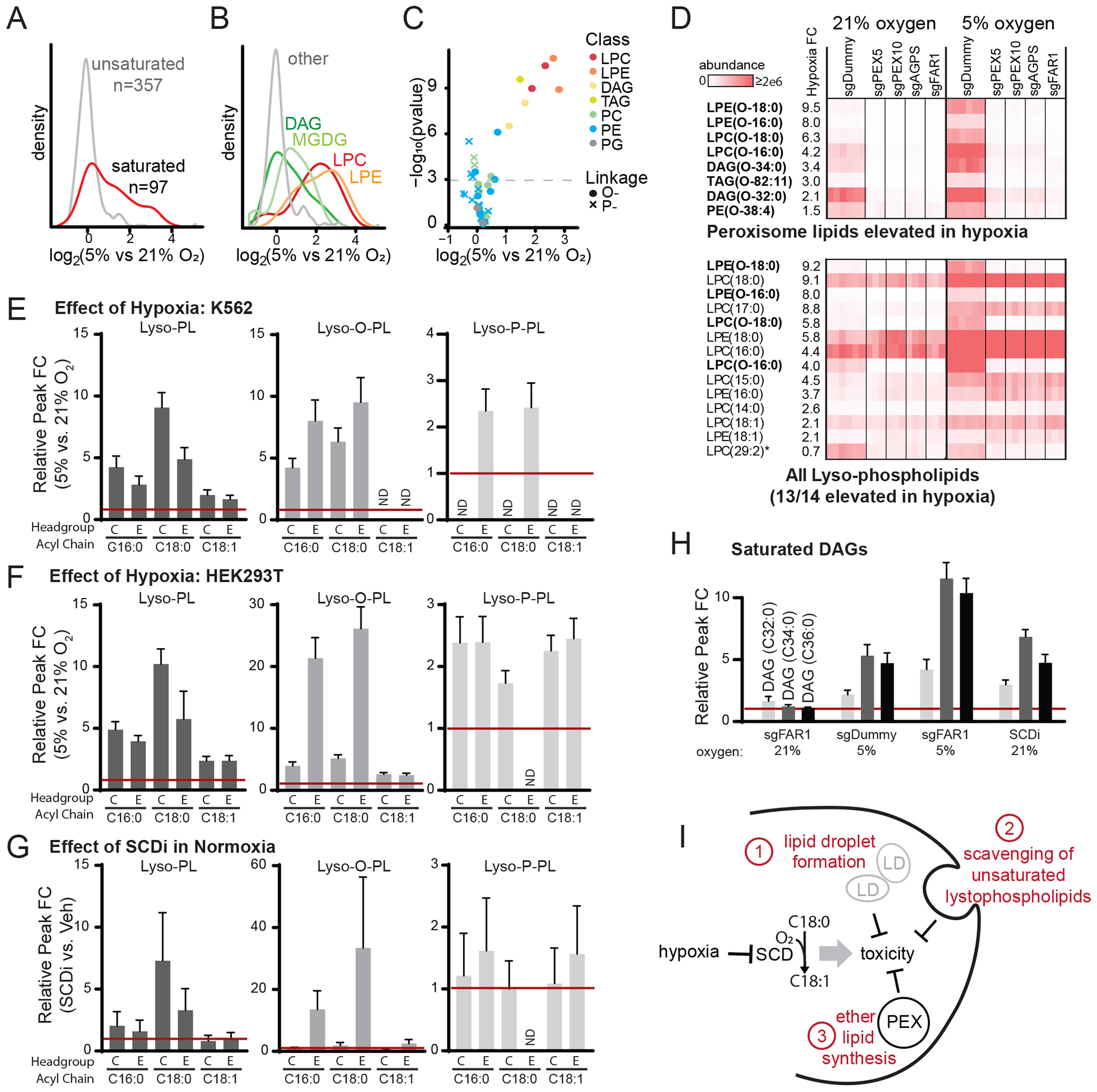
(A) Density plot shows saturated lipid levels increased after 24 hr exposure to hypoxia (5% oxygen) in K562 control cells treated with sgDummy (non-coding control). (B) Density plot shows lipid classes that are increased during hypoxia in K562 control cells treated with sgDummy. (C) Volcano plot shows all confirmed peroxisome-derived lipids, where color indicates lipid class and shape indicates linkage (circle denotes ether, cross denotes vinyl-ether). (D) Heatmaps show all eight peroxisome-derived lipids that are significantly increased in hypoxia in K562 cells treated with sgDummy (non-coding control) (top) and all lyso-phospholipids (bottom). Heatmaps show relative lipid abundance in sgDummy and CRISPR knockout cell lines, with lipids ordered by fold change (FC) between mean of control cells in 5% vs. 21% oxygen. Bold indicates peroxisome-derived lipids and asterisk indicates lipid not elevated in hypoxia. (E-G) Fold change of selected lyso-phospholipid species in hypoxia (5% vs. 21%) in K562 cells (panel E) and HEK293T cells (panel F) and in HEK293T cells with SCDi treatment (SCDi vs. Veh, panel G). (H) Fold change of saturated DAG species in different conditions (compared to sgDummy at 21% O2) shows changes due to FAR1 knockout, hypoxia, and FAR1 knockout and hypoxia. (I) Schematic model shows hypoxia inhibits SCD which induces toxicity that can be abrogated by three known mechanisms: lipid droplet formation (Piccolis et al., 2019), lyso-phospholipid scavenging (Kamphorst et al., 2013), or peroxisomal ether lipid synthesis shown here.
Collectively, our screening results and follow-up studies suggest a role for peroxisomal metabolism in maintaining membrane fluidity. Previous studies have suggested that in the setting of hypoxia, the toxicity that arises from inhibition of SCD can be buffered either by the formation of lipid droplets or by scavenging of unsaturated fatty acids from the media. Our work proposes a third strategy – peroxisomal ether lipid synthesis – that may represent an additional strategy to alleviate membrane rigidity stress (Figure 5I).
Essentiality of Peroxisomes Correlates with Lipid Saturation across Hundreds of Cell Lines
We next wondered if the fitness contributions of peroxisomes under hypoxia might extend to other cell states characterized by high levels of saturated lipids. Therefore, we searched for a correlation between gene essentiality and saturated lipid levels using the Cancer Dependency Map which performed CRISPR essentiality screens and metabolomics across 447 cancer cell lines (Li et al., 2019; Meyers et al., 2017; Tsherniak et al., 2017). For each cell line and each lipid class, we defined a metric of lipid desaturation (defined as mean number of double bonds, weighted by lipid abundance within the individual lipids in the class). We then asked which genes’ essentially scores (CERES scores) correlated with lipid desaturation. Following correction for multiple hypothesis testing, 9 genes emerged (Figure 6A, Figure S5): SCD itself, regulators of SCD (SREBF1, SCAP), four genes we show are selectively essential at 1% vs. 21% O2 (PEX2, PEX6, SREBF1, TMEM189), and three additional genes (CEP44, CTNNB1, and MBTPS1). Specifically, there is a clear correlation between the number of double bonds and the dependency of SCD and certain peroxisomal genes (e.g. PEX2) in three different lipid classes (Figure 6B, Figure S5). Across hundreds of cell lines, cells with high levels of saturated lipids tend to require both SCD and certain peroxisomal genes for growth, extending the relationship between peroxisomes and lipid saturation to other conditions besides hypoxia.
Figure 6. Essentiality of peroxisome genes correlates with lipid saturation across hundreds of cell lines.
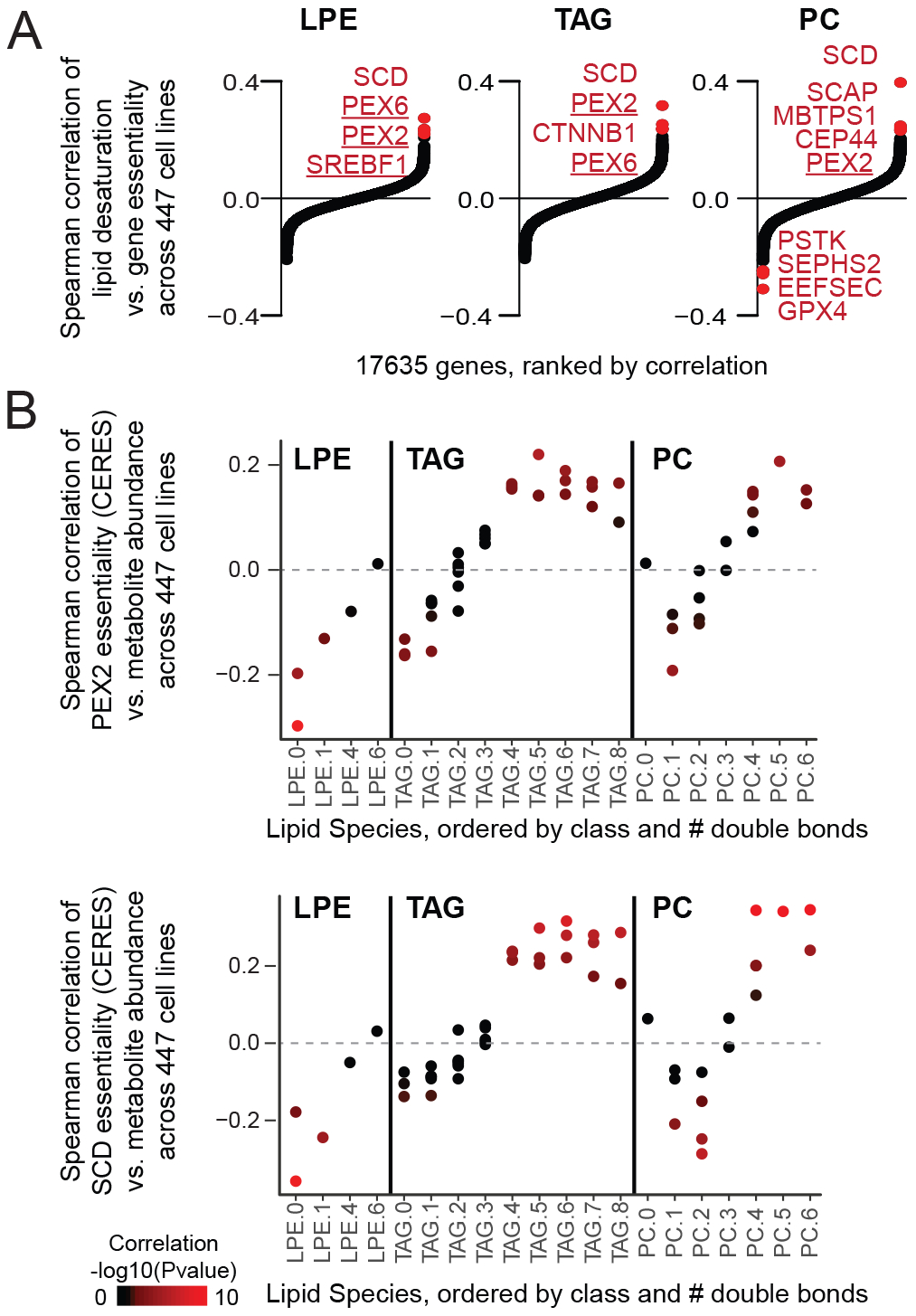
(A) For each of 17635 genes, correlation between growth of CRISPR knockout (CERES score) and lipid desaturation metric (weighted mean number double bonds) are shown for three lipid classes: lyso-phosphatidylethanolamine (LPE), triacylglyerol (TAG), and phosphatidylcholine (PC). Significant hits shown in red (Bonferroni corrected p-value < 0.05). Positive correlation indicates genes are essential in cells with high levels of saturated lipids. (B) For two top-scoring genes (PEX2 and SCD), Spearman correlations between individual lipid abundance and growth of CRISPR knockout (CERES score, where lower score indicates lower growth, i.e. greater essentiality) are shown for all measured lipids in each class. Lipids are grouped by the number of double bonds (e.g. LPE(16:0) and LPE(18:0) grouped under LPE.0), and individual lipids are colored by significance of correlation. Figure S5 shows results for all lipids.
Discussion
Cellular and organismal health is based on interactions between genetic background and external factors such as environment, nutrition and behavior. It seems logical that the optimal oxygen environment for a cell, tissue or organism differs across genetic backgrounds. We recently found that defects in mitochondrial complex I or frataxin are buffered by reduced environmental oxygen tensions. This led us to propose hypoxia as an experimental therapy for mitochondrial diseases (Ferrari et al., 2017; Jain et al., 2019; Jain et al., 2016). Here, we ask the converse question – which genetic backgrounds induce fitness defects that can be ameliorated by increasing or decreasing oxygen. Our screen yields a resource of hundreds of genes that show differential fitness as a function of ambient oxygen. We envision that our screening data will serve as a valuable resource for a number of different applications.
Our screen yields a resource of hundreds of genes and pathways that should be of interest in studying the basic biology of oxygen. Notably, the majority of the genes we have identified fall outside of extensively studied areas (e.g. HIF signaling and reactive oxygen species), indicating that new oxygen-related biology awaits discovery. Recent publications help to credential the quality of our screening hits. For example, the histone demethylase KDM6A (9th most selectively essential gene in 1% vs. 21% O2) was recently shown to directly sense oxygen and relay this information to chromatin modifications and cell fate (Chakraborty et al., 2019). TMEM189 (5th most selectively essential gene in 1% vs. 21% O2) was recently shown to encode an oxygen-regulated fatty acid desaturase in the synthesis of vinyl-ether bond containing plasmalogens (Gallego-Garcia et al., 2019). We recently demonstrated that GLRX5, FBXL5, and FXN (ranking 1st, 23rd and 33rd in the 21% side, respectively) KO cells exhibit a severe growth defect in 21% O2, however show comparable growth to WT cells at low O2 (Ast et al., 2019). These contemporary studies provide strong independent validation of hits on both sides of our screen. Of course, the precise set of genes that exhibit oxygen-sensitive fitness defects will be influenced by cell type, media conditions, and the type of genetic interaction (e.g., buffering, suppressor, synthetic lethal). Future work is needed to validate and mechanistically characterize these screen hits.
Our inventory is also likely to be of use in translational research. For example, tumor hypoxia is one of the worst prognosis factors for survival (Martin et al., 2016). The cores of solid tumors are typically deprived of nutrients and oxygen. By identifying the genetic dependencies of cell proliferation in a hypoxic environment (Table S1), including lipid and peroxisomal metabolism, we are also identifying potential targets for novel chemotherapeutics. Additionally, we have identified a list of essential genes at the more physiological 5% oxygen level (Table S1). This will provide clues as to which small molecule inhibitors can be safely administered without affecting cellular health. Moreover, we have highlighted additional inborn errors of metabolism which may benefit from modulating oxygen tensions and delivery (Figure 3B).
We find a striking enrichment for several mitochondrial pathways, including complex I deficiency and iron sulfur biogenesis, as being buffered by lowering oxygen tensions. In addition, other mitochondrial pathways are also high scoring, including the pyruvate dehydrogenase complex (PDH) and the pyruvate transporter (MPC), type II fatty acid synthase (FASII), amongst others. Our screen is consistent with our previous work in which we showed that a mouse model of complex I deficiency could be alleviated by breathing of low inspired oxygen (Jain et al., 2016), and that frataxin deficiency can be rescued in low oxygen in yeast, human cells, and worms (Ast et al., 2019). An important question is why knockouts of certain mitochondrial pathways, most notably complex I, but not complexes II-V, are buffered by low oxygen in our screen. We previously showed that complex I-V deficiencies all result in an ATP crisis in low glucose conditions in a classic “glucose/galactose” screen (Arroyo et al., 2016), consistent with a key role of all of these genes in ATP production via OXPHOS in aerobic conditions. The difference between these results suggests that the ability of hypoxia to rescue complex I deficiency is unlikely to be simply related to its contribution to ATP production by OXPHOS. Our recent in vivo work on hypoxia in the mouse model of Ndufs4 deficiency suggests a key role for high unused oxygen itself as a potential culprit (Jain et al., 2019). Future work is required to determine the detailed mechanism by which hypoxia alleviates complex I deficiency.
On the opposite end of the spectrum, we find that many pathways related to lipid metabolism and peroxisomal ether lipid metabolism are selectively essential at lower oxygen tensions. Among the most essential in 1% vs. 21% oxygen are AMFR, SREBF1, ACSL4, ADIPOR2, while on the reverse side we observe ACSL3, CHP1, AGPAT as high scoring. Prior literature has identified these genes as important regulators of membrane fluidity, and in fact, this pattern of hits is reminiscent of two recent genetic screens assaying saturated fatty acid (palmitate) toxicity (Zhu et al., 2019; Piccolis et al., 2019). Given the known effects of hypoxia in limiting fatty acid and lipid desaturases, this leads us to speculate that peroxisomal metabolism may also contribute to the maintenance of membrane fluidity. Supporting this, we find that across hundreds of cancer cell lines, cells with high saturated lipids require certain peroxisome genes for growth. Interestingly, C. elegans worm mutants deficient in peroxisomal ether lipid synthesis are extremely sensitive to low temperatures, an environmental pressure that decreases membrane fluidity (Los and Murata, 2004); (Shi et al., 2016).
Future work is required to follow-up on our screening hits to dissect the exact fitness contributions of peroxisomes in hypoxia, but several possibilities exist. First, it is possible that peroxisome-derived ether lipids, notably plasmalogens, are themselves beneficial in hypoxia. Previous studies showed the vinyl-ether linkage in plasmalogens can prevent propagation of lipid peroxidation (Chen and Yu, 1994; Sindelar et al., 1999; Zoeller et al., 2002; Zoeller et al., 1999). A second possibility is that the production of peroxisomal lipids is beneficial by sequestering saturated fatty acids that would otherwise be incorporated into di-saturated glycerolipids that are believed to be toxic (Piccolis et al., 2019). A third possibility is that peroxisomal derived lipids are sensed and monitored as a part of a homeostatic loop, and the loss of this regulation becomes detrimental in low oxygen (Honsho et al., 2015; Garcia-Bermudez et al., 2019). Future work is needed to determine to what extent these or other mechanisms are at play.
The growth deficits of peroxisome gene knockouts in hypoxia is extremely cell type specific and media dependent. This was a robust finding in the context of the pooled screen, but follow-up studies showed this result to be limited to 293T cells and also dependent on media lipid content and cell seeding conditions. These features are reminiscent of studies of saturated fatty acid toxicity – which also tend to be very cell type specific and also sensitive to the media conditions. A better understanding of the molecular mechanisms linking peroxisomes to low oxygen is required to explain the source of this variability and also predict which cell types will exhibit this sensitivity in hypoxia.
Finally, we anticipate that the resource of genes we have identified may useful in understanding the evolution of aerobic metabolism. Life on our planet began under anaerobic conditions, and with the rise in atmospheric oxygen hundreds of genetic pathways co-evolved. The pathways revealed by our screen may provide hypotheses into the underlying mechanisms of evolution. For example, it is known that Fe-S clusters can form spontaneously under anaerobic conditions, but in the presence of oxygen, require enzymatic synthesis (Imlay, 2006). These pathways were high scoring on the high oxygen side of our screen. Mitochondrial oxygen metabolism has been proposed to be a key driver in metazoan evolution at a time when earth’s oxygen rose to levels higher than today (Nursall, 1959). And it is notable that plasmalogen synthesis is reported to have appeared and disappeared in bacterial evolution, in a way that may be related to oxygen (Goldfine, 2010). The hundreds of other genes identified in our resource may similarly provide additional clues about the adaptations required to cope with rising oxygen on our planet.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vamsi K. Mootha (vamsi@hms.harvard.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
K562 (ATCC CCL-243) and 293T (ATCC CRL-3216) cells were obtained from ATCC. All experiments with wild-type cells or CRISPR-Cas9 mediated knockouts were performed within 30 passages upon receipt from ATCC. Cells were always maintained in 37C incubators.
METHOD DETAILS
Genome-wide CRISPR screen
The genome-wide CRISPR screen was performed in the leukemic cell line, K562 (ATCC CCL-243). Cells were maintained in culture in magnetic spinner flasks (Corning) throughout the duration of the screen. ~1.5e8 cells were infected with the Brunello library virus (Doench, 2016) at a target MOI of 0.3, allowing for a representation of ~500 cells infected with a given sgRNA. Briefly, ~1.5e8 cells were pelleted and resuspended in DMEM Media (Gibco/Life Technologies, Ref. #11995–065) with 10% FBS (Sigma Aldrich, Catalog #F6178). Cells were diluted to a concentration of 1.5e6/ml and 4ug/ml polybrene. The cell suspension was then placed in 24-well plates with 2ml of cells per well. 300ul of virus was added to each well. Cells were spinfected at 1000g for 2h. 24h after spinfection, cells were resuspended in DMEM media containing puromycin (2ug/ml), placed in magnetic spinner flasks and incubated in tissue culture incubators set to 21% O2. Puromycin selection continued for 3d (cells were passaged as needed when nearing confluency: ~1e6/ml). On day 5 post-infection, infected cells were split into three new flasks at 5e4/ml and placed at three different oxygen tensions – 21%, 5% and 1% O2. Tissue culture incubators were adapted to maintain different oxygen tensions. A liquid nitrogen tank was used to pulse N2 into incubators with a feedback loop to maintain the specified oxygen tension. Cell suspensions were passaged every 3d with a seeding density of 0.7–1e5/ml at the beginning of each passage. All tissue culture handling was done outside of hypoxia incubators (at environmental oxygen tensions of 21% O2). The screen was performed in triplicate independent experimental set-ups.
Cells were pelleted and stored at each passage. Day 0, Day 9 and Day 15 samples were used for sequencing. Genomic DNA was extracted using the Qiagen Blood and Cell Culture DNA Maxi Kit. One set of replicates from the Day 9 samples was lost due to technical error at this stage. Library prep and sequencing was performed as previously described (Arroyo et al., 2016; Doench, 2016).
RNA-Seq
K562 cells were seeded at a density of 1e5/ml in 3ml of DMEM media (same conditions as screen) per well of 6-well plate. Plates were maintained in 1%, 5% or 21% O2 incubators and passaged at 3d of growth. All experiments were performed in triplicate for each oxygen tension. Samples were collected separately for the following time points after seeding: 0h, 6h, 24h, 3d and 6d (after an intermediate passage at 3d). At each time point, cells were pelleted, rinsed with PBS and then store at −80C. RNA extraction was performed using the Qiagen RNAeasy kit. NOTE: Unlike the screen, which was performed in spinner flasks, samples used for RNASeq were maintained in 6-well plates. Therefore, oxygen diffusion was more limited and may have been affected by parameters such as cell density and media levels over time.
RNASeq libraries were generated at the Broad Institute Technology Labs using the Smart-seq2 protocol which includes polyA selection, as described (Picelli et al., 2014; Trapnell et al., 2012). RNA was isolated using RNASPRI beads. Following reverse transcription, cDNA was fragmented and barcoded. Samples were subjected to paired-end sequencing of 2×25bp on an Illumina NextSeq500 instrument.
Mitochondrial KO experimental follow-up
Knockouts were made from HEK293T cells (ATCC CRL-3216) using media conditions analogous to the CRISPR screen conditions: DMEM, 10% FBS, Pen/Strep, Glutamax. As hypoxic cells rely on anaerobic glycolysis, all experiments were performed in starting conditions of 25mM glucose. Spinfections for lenti-Cas9 follow-up experiments were performed as described in (Jain et al., 2016). Infected 293T cells were seeded at 0.5e5/ml in 1ml of media (in wells of 24-well plates). Counts were performed ~72h after initial seeding at different oxygen tensions. Cells were manually counted using a Beckman coulter counter. Noncoding guide RNAs (see above) or GFP-targeting guide RNA constricts were used as controls.
Timing of lentivirus infections:
Lentivirus pools of knockout cells are mixed populations of cells with different genomic edits in the region-of-interest. Thus, it is likely that a combination of in-frame and out-of-frame edits are generated after infection, leading different types of mutations: hypomorph, loss-of-function, etc. Therefore, all growth curves are performed within three weeks of spinfection. With longer time points, it is possible that hypomorph or unedited cells will out-compete loss-of-function mutations. Thus, the exact growth defect of OXPHOS mutants may vary based on the time after spinfection/selection. However, the overall effect of C1 rescue should be maintained if these technical considerations are kept in mind.
Oxygen diffusion considerations:
Various aspects of experimental design can affect local oxygen tensions that cells are experiencing. The “environmental oxygen tension” present in hypoxia chambers may be different from the local oxygen tension that cells are experiencing. The following parameters were considered in experimental design: cell density, oxygen consumption of given cell line, media levels, glucose concentrations.
Interpreting relative vs. absolute growth rescues:
Severe hypoxia slows growth in proliferating cells. OXPHOS mutants likely have impaired oxygen consumption. Therefore, local oxygen tensions in OXPHOS mutants may be higher than in WT controls. This should be kept in mind when interpreting the “relative rescue” of KO cells compared to WT cells in a given oxygen tension.
Peroxisomal KO experimental follow-up
Knockouts for peroxisome genes were performed in HEK293T cells. Cells were seeded at 1e5/ml in a 1ml of media in 24-well plates. Counts were performed 2d after plating into different oxygen tensions. Peroxisome-related follow-up experiments were performed in the following media: 25mM glucose DMEM, 3% FBS. Cells were incubated for 2 days with 10μM oleic or 10μM palmitic acid (Cayman Chemicals).
Since hypoxic cells can scavenge lipids from media, low-serum conditions must be used to see peroxisome KO growth defects. The exact FBS concentration and lipid composition may affect the extent of growth defect in hypoxia.
Lipid droplets
For Figure S2, K562 or 293T cells expressing Cas9 and sgRNA guides to FAR1 or noncutting control (sgDummy) were incubated in 21% or 5% or 1% O2 (2–3 days) in delipidated media and then treated for 24h with 0.04 mM BSA, 0.25 mM palmitate, or 0.25mM palmitate plus 0.25 mM oleate. Cells were fixed with 10% formalin and stained with 0.5 μM BODIPY 493/503 (Invitrogen) as described in (Piccolis et al., 2019) and co-stained with Hoechst 33342 (Invitrogen) at a concentration of 0.5 μg/mL, and analyzed by confocal microscopy on a spinning disk confocal system (Yokogawa CSU-22 spinning disk confocal scan head with Andor Borealis modification) using a 63X objective (1.4 N.A.). Image analysis was performed with ImageJ (NIH).
Peroxisome number
For Figure S3, HEK293T cells were lentivirally infected with a GFP-PTS1 expressing vector and grown for 5 days in 21% O2 or 1% O2. A day prior to imaging, 1×106 cells were plated on 35mm poly-D-lysine coated glassbottom dishes (MatTek Corporation). Immediately prior to imaging, cells were co-stained with Hoechst 33342 (Invitrogen) at a concentration of 0.1ug/ml. Imaging was conducted on a spinning disk confocal system (Yokogawa CSU-22 spinning disk confocal scan head with Andor Borealis modification) using a 63X objective (1.4 N.A.), with MetaMorph acquisition software and a Hamamatsu Orca-ER cooled CCD camera. Images were analyzed using the CellProfiler software 3.1.8. At least 130 cells were analyzed per condition.
Lipidomics in K562 cells (Figure 5A–D)
K562 KO cells lines were generated for non-coding guideRNAs or peroxisome KO cell lines using guides listed in Table S5. Cells were plated in 6-well plates at a seeding density of 0.4e6/ml (3ml per well) in media containing 3% FBS. Plates were then incubated at 21%, 5% or 1% O2 for 24h. Cells were collected, pelleted, rinsed with PBS and then used for lipidomics analyses. Lipids were extracted using 225μL MeOH mixed with an internal standard (Avanti SPLASH Lipidomix) and 750 μL MTBE (methyl tert-butyl ether). Samples were sonicated for 1min, placed on ice for 15 min, and then added to 300ul ddH2O. Samples were then centrifuged at 14,000 g for 10 min. The organic phase was vacuum-dried. Samples were then reconstituted in 100 μL ACN:H2O:IPA (1:1:2) + 0.1% formic acid (Fisher Scientific) and used for LC-MS. LC-MS was performed as described in (Simcox et al., 2017). We note that the addition of formic acid may break down vinyl ether lipids present in our samples.
Lipidomics in HEK293T Cells (Figure 5E–H)
HEK293T cells were plated in 6-well plates at a seeding density of 0.4e6/ml (3ml per well) in media containing 3% FBS. Plates were then incubated at 21% or 5% O2 for 24h. Cells were collected, pelleted, rinsed with PBS and then used for lipidomic analyses. Cell extracts were prepared using 300 μl of Buffer B (100% methanol with 0.15% formic acid, Fisher Scientific). Samples were mixed by pipetting, vortexed, and centrifuged for 5 min @ 21,000g. For LC/MS experiments, 20 μL of cell extract was injected on to a 30 mm × 2 mm Phenomenex Luna 3 μM C8(2) 100Å column. Buffer A was 97% water, 3% methanol with 0.15% formic acid, and Buffer B was 100% methanol with 0.15% formic acid. The LC flowrate was 0.2 mL/min and initially set to 85% B for sample injection and a 1.5 min wash. The solvent was linearly increased to 99% B from 1.5–12 mins, held at 99% B from 12–17 mins, decreased to 85% B from 17–18 mins, and held at 85% for 18–29 mins to re-equilibrate. A Thermo QExactive Plus instrument equipped with an Ion Max heated electrospray ionization source was used to monitor the eluent. Samples were run with a sheath gas flow of 30, auxiliary gas flow of 7, and sweep gas flow of 2, and at 2.8 kV spray voltage. Most experiments were run in polarity switching mode at resolving power 70,000 (at m/z 200), with a scan range of 70 – 1000 m/z. Again, we note that the addition of formic acid may break down vinyl ether lipids present in our samples
MS data analysis was performed using the Xcalibur QuanBrowser software (Thermo Scientific). Peak areas were quantified +/− 10ppm, and normalized by dividing by the total area of the positive-ion TIC for each run. To confirm peak identity, standards were purchased (Avanti Polar Lipids) for LPC-P-18:0, LPC-P-16:0, LPC-O-18:0, LPC-O-18:1, and LPC-O-16:0. Of the samples for which no standards were available, LPC-P-18:1, LPE-P-18:1, LPE-P-16:0, LPE-O-18:0, LPE-O-16:0 eluted in one peak and have unambiguous masses, so their identification was made by mass alone. LPE-P-18:0 and LPE-O-18:1 are isomeric and are the same mass, however, the order of their peak elution was used to identify them. In that case as well, LPE-O-18:1 was a very low-abundance species and was not regularly detected.
For SCD inhibitor treatment (Figure 5G), cells were grown overnight in 6-well plates, and seeded at 3 ml per well, 0.4e6 cells/ml in 3% FBS growth media. Samples were treated +/− 20nM SCD inhibitor (ApexBio A939572).
QUANTIFICATION AND STATISTICAL ANALYSIS
Genome-wide CRISPR screen
MAGeCK software version 0.5.4 (Li et al., 2014) was used to collapse guide RNA data to gene-level data (parameters: gene-test-fdr-threshold 0.1; sort-criteria pos) (Table S1).
Genes were ranked for GSEA analysis (Figure 1D–E). Genes essential in 21% vs. 1% oxygen were determined based on MAGeCK comparing the 3 replicates in 21% O2 (day 15) to the 3 replicates in 1% O2 (day 15). All 20112 genes were ranked (Figure 1D) by first categorizing genes as either “Depleted in 21% vs. 1% O2” (MAGeCK pval.pos<pval.neg) or “Depleted in 1% vs. 21% O2” (otherwise), and then each category was ranked based on –log10(pval) from MAGeCK. Using this ranking, GSEA (Subramanian et al., 2005) was performed with default parameters using a customized geneset database containing all KEGG Metabolism or Cellular Process pathways with 15–500 genes (data downloaded 5/30/2017), yielding a total of 92 pathways. The KEGG Oxidative Phosphorylation pathway was manually reviewed to remove the lysosomal ATPase complex (ATP6*) and the Peroxisome pathway was reviewed to remove mitochondrial contaminant MPV17. All pathways with GSEA FDR qval<0.05 were considered significant. For GO enrichment analysis (Table S2), genes selectively essential in 21% vs. 1% oxygen (FDR≤0.1) were searched for enriched Gene Ontology biological processes using PANTHER Overrepresentation Test (Release 20190711) (Mi et al., 2013) using Fisher’s Exact Test with FDR correction.
Disease genes and comparison to Glu-Gal screen
The list of primary and secondary mitochondrial disease genes was obtained from (Frazier et al., 2019) and the OMIM database was used to identify genes underlying additional non-mitochondrial disorders. The glu-gal analysis was taken from (Arroyo et al., 2016) using a FDR of 0.3.
Comparison to High Altitude SNPs and HIF targets
HIF targets were obtained from (Benita et al., 2009) and the list of high altitude SNPs was generated using genome-wide significant hits and previous study hits that replicated across datasets (Yang et al., 2017).
RNA-Seq analysis
RNASeq data was processed using the Cufflinks suite (tophat, cuffquant, cuffnorm) as previously described (Trapnell et al., 2012) and deposited to GEO (accession GSE144527). We excluded poorly expressed genes (maximum FPKM<1), transcripts not coding for proteins, and one replicate that failed quality control metrics. The remaining 12163 transcripts were normalized across samples (Z-score) and sorted by mean expression on day 3 in 1% oxygen (Figure 2C).
Lipidomics in K562 cells
Lipid data from K562 cells were filtered to remove unidentified metabolites (n=176), internal standards (n=16), and duplicate measurements (n=15) yielding 642 metabolites annotated into 21 lipid classes. All adducts corresponding to the same lipid species (same class, number carbons, number double bonds) were summed, yielding 454 unique lipids. Each of 21 lipid classes were assessed for enrichment within the 86 hypoxia-increased lipids via Fisher’s Exact test. Lipids increased during hypoxia were defined using a t-test, comparing 6 measurements in 21% oxygen (two different non-coding controls, each with three technical replicates) versus 6 measurements in 5% oxygen. Annotated peroxisome-derived lipids were considered (O- or P- linkages, n=63). In order to exclude any potential misannotations, a subset of 44 confirmed peroxisome-derived lipids was defined as those also showing significant decrease in any of the peroxisome derived knockout cell lines (PEX5, PEX10, AGPS, FAR1) versus non-coding controls in 21% oxygen. Some peroxisome-derived lipids were ambiguous as to presence of an ether or vinyl-ether bond, and for display purposes in Figure 6C, 8 ambiguous lipids were considered vinyl-ether linkages (none of these was increased in hypoxia).
Lipid desaturation within Cancer Dependency Map (Figure 6)
447 cancer cell lines with CERES dependency scores (DepMap version 19Q2) (Meyers et al., 2017) that also had metabolomics profiling (Li et al., 2019) were analyzed. Lower CERES scores indicate lower cell growth, or greater gene dependence. For each cell line, a lipid desaturation metric was computed as follows: DesaturationC = Σi € C (Di * Ai) / Σ i € C (Ai) where C indicates lipid class { LPE, TAG, PC, LPC, CE, SM, DAG}, i indicates the specific lipid within the class, Di indicates the number of double bonds, and Ai indicates the observed abundance. While it can be problematic to compare MS/MS abundances across different metabolite species, since the different species in the same lipid class have such similar chemical properties we believed it appropriate to average abundances within the same class. However for robustness, we ensured that our lipid desaturation metric correlated strongly with other measures of lipid class desaturation such as percent_unsaturated (sum of unsaturated species / sum of all species) and also with metrics that relied only on relative abundance for each lipid class (e.g. first compute Z-score for each lipid across 447 cell lines, then measure mean(Z-score for lipids with ≥4 double bonds)- mean(Z-scores for lipids with <4 double bonds). The results for SCD, PEX2, and PEX6 were detected using these three different metrics of lipid desaturation. We note that the 89 lipids measured were not comprehensive, and some lipid classes were uninformative. For example, no saturated DAG species were reported, likely explaining the lower correlation of desaturation metrics between DAGs and other lipid classes.
For each of seven lipid classes, Spearman correlations were performed for all 17635 genes: comparing gene CERES score (lower score indicates lower growth or greater dependency) versus lipid desaturation metric across 447 cell lines. For two genes (PEX2 and SCD), Spearman correlations were performed for each of 225 metabolites: comparing CERES score versus metabolite abundance across 447 cell lines.
DATA AND CODE AVAILABILITY
RNA-seq data (Figure 2) is available from the NCBI Gene Expression Omnibus (GSE144527). This study did not generate new unique reagents.
Supplementary Material
Table S2, Related to Figure 1. Enrichment of gene sets including custom KEGG gene sets used for GSEA, and GO biological process analysis of genes selectively essential at 21% and 1% oxygen.
Table S4, Related to Figure 5. Lipidomics data from K562 knockout cell lines grown at 21% and 5% oxygen.
Table S3, Related to Figure 2. High-altitude SNPs and ranking in oxygen screen.
Table S5, Related to STAR Methods. sgRNA guide sequences for gene-specific CRISPR knockouts.
Figure S1. Gene ontology biological processes selectively essential at 21% vs. 1% oxygen, related to Figure 3. All genes within Gene Ontology biological process categories that were enriched in the 213 genes selectively essential at 21% vs. 1% oxygen, based on PANTHER analysis. See also Table S2.
Figure S3. Peroxisome number in 21% and 1% oxygen, related to Figure 4. GFP-PTS1 labeled peroxisomes were imaged (left) and quantified (right) in HEK293T cells cultured for 5 days under 21% O2 or 1% O2. Cells were co-stained with the nuclear dye Hoechst. Peroxisomes were quantified via CellProfiler version 3.1.8 using minimum 130 cells per condition. No differences were observed in peroxisome abundance or gross appearance between the two conditions. Scale bar- 5μm.
Figure S2. K562 and HEK293T cells are capable of forming lipid droplets, related to Figure 4. (A) K562 cells expressing Cas9 and sgRNA guides to FAR1 or noncutting control (sgDummy) were incubated in 21% or 1% O2 (2 days) in delipidated media (supplemented with 0.5% fatty acid free BSA) and then treated for 24h with 0.04 mM BSA, 0.25 mM palmitate, or 0.25mM palmitate plus 0.25 mM oleate. Cells were fixed and stained with 0.5μM BODIPY 493/503 (Piccolis et al., 2019), and analyzed by confocal microscopy. Blue scale bar = 5 μm. (B) HEK293T cells expressing Cas9 and sgRNA guides to FAR1 or noncutting control (sgDummy) or FAR1 were incubated in 21% or 5% O2 (3 days) in delipidated media (supplemented with 10% charcoal stripped FBS) and then treated for 24h with 0.04 mM BSA, 0.25 mM palmitate, or 0.25mM palmitate plus 0.25 mM oleate. Cells were fixed and stained with 0.5μM BODIPY 493/503 (Piccolis et al., 2019), and analyzed by confocal microscopy. Blue scale bar = 5 μm. (C) Abundance of TAG species in K562 cells in cells transiently transfected with sgRNA guides to a non-coding region (sgDummy) or peroxisomal genes (PEX5, PEX10, AGPS, FAR1) in 21% or 1% O2 shows similar abundance levels across most species and conditions, with C50.0.TAG showing nearly twofold elevation in hypoxia (regardless of peroxisome knockout).
Figure S5. Metrics of lipid desaturation across seven lipid classes, related to Figure 6. (A) Density plot shows distribution of lipid desaturation metric across 447 cancer cell lines, for all seven lipid classes (LPE: lyso-phosphatidylethanolamine; TAG: triacylglycerol; PC: phosphatidylcholine; LPC: lyso-phosphatidylcholine; CE: cholesteryl ester; SM: sphingomyelin; DAG: diacylglycerol). (B) Pearson correlation between desaturation metrics for seven lipid classes, across the same 447 cancer cell lines. (C) For each of 17635 genes, Spearman correlation is calculated for growth of CRISPR knockout (CERES score, negative indicates lower growth, i.e. greater essentiality) and lipid desaturation metric across 447 cell lines. Significant hits shown in red (Bonferroni corrected p-value < 0.05). (D) For two top-scoring genes (PEX2 and SCD) and all 89 measured lipids, Spearman correlations between CERES score and lipid abundance are shown. Lipids are grouped by the number of double bonds (e.g. C16.0.LPE and C18.0.LPE grouped under LPE.0), and individual lipids are colored by significance of correlation. Lipid data derived from (Li et al., 2019) and CRISPR dependency data derived from DepMap version 19Q2 (Meyers et al., 2017; Tsherniak et al., 2017).
Figure S4. Hypoxia-induced lyso-phospholipid changes in cells lacking ether lipid biosynthesis, related to Figure 5. The fold change in peak area of lyso-phospholipids with hypoxia and FAR1 KO vs. control cells (sgDummy non-coding control cells grown at 21% oxygen).
Table S1, Related to Figures 1–4. (Tab 1) sgRNA raw counts for all conditions; (Tab 2) Lists of genes selectively essential in each oxygen level based on MAGeCK comparisons; (Tab 3) Lists of genes generally essential in each oxygen level based on MAGeCK analysis on day 15 vs. day 0; (Tab 4) MAGeCK analysis comparing 21% and 1% oxygen at day 15; (Tab 5) MAGeCK analysis comparing 21% and 5% oxygen at day 15; (Tab 6) Summaries of all pairwise MAGeCK comparisons.
Acknowledgements
We would like to thank John Doench and David Root of the Genetic Perturbation Platform at Broad Institute for advice on the CRISPR screen. We would like to thank James Cox and John Alan Maschek of the University of Utah Metabolomics Core and Rohit Sharma (MGH) for assistance with mass spectrometry experiments, Bob Farese, Jean Vance, and Raphael Zoeller for thoughtful feedback. The Broad Technology Labs generated the Smart-Seq2 libraries, which were then sequenced by the Broad Genomics Platform. I.H.J was supported by the Department of Energy Computational Science Graduate Fellowship Program (grant DE-FG02-97ER25308). O.S.S was supported by an NIH F32 fellowship (1F32GM133047-01). This work was supported by a gift from the Marriott Foundation and a grant R35GM122455 from the National Institutes of Health (V.K.M). V.K.M. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Declaration of Interests
V.K.M. is a paid scientific advisor to 5AM Ventures and Janssen Pharmaceuticals. O.S.S. is a paid consultant for Proteinaceous, Inc. V.K.M. and I.H.J are listed as inventors on a patent application filed by Massachusetts General on the use of hypoxia as a therapeutic strategy.
REFERENCES
- Ackerman D, and Simon MC (2014). Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell Biol 24, 472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman D, Tumanov S, Qiu B, Michalopoulou E, Spata M, Azzam A, Xie H, Simon MC, and Kamphorst JJ (2018). Triglycerides Promote Lipid Homeostasis during Hypoxic Stress by Balancing Fatty Acid Saturation. Cell Rep 24, 2596–2605 e2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, Sabatini DM, Birsoy K, and Possemato R (2017). NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo JD, Jourdain AA, Calvo SE, Ballarano CA, Doench JG, Root DE, and Mootha VK (2016). A Genome-wide CRISPR Death Screen Identifies Genes Essential for Oxidative Phosphorylation. Cell Metab 24, 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ast T, Meisel JD, Patra S, Wang H, Grange RMH, Kim SH, Calvo SE, Orefice LL, Nagashima F, Ichinose F, et al. (2019). Hypoxia Rescues Frataxin Loss by Restoring Iron Sulfur Cluster Biogenesis. Cell 177, 1507–1521 e1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ast T, and Mootha VK (2019). Oxygen and mammalian cell culture: are we repeating the experiment of Dr. Ox? Nature Metabolism, 858–860. [DOI] [PubMed] [Google Scholar]
- Beall CM, Cavalleri GL, Deng L, Elston RC, Gao Y, Knight J, Li C, Li JC, Liang Y, McCormack M, et al. (2010). Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci U S A 107, 11459–11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, and Xavier RJ (2009). An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res 37, 4587–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomen VA, Majek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A, et al. (2015). Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096. [DOI] [PubMed] [Google Scholar]
- Braverman NE, and Moser AB (2012). Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta 1822, 1442–1452. [DOI] [PubMed] [Google Scholar]
- Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, and Kieda C (2011). Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med 15, 1239–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty AA, Laukka T, Myllykoski M, Ringel AE, Booker MA, Tolstorukov MY, Meng YJ, Meier SR, Jennings RB, Creech AL, et al. (2019). Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 363, 1217–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, and Yu BP (1994). Alterations in mitochondrial membrane fluidity by lipid peroxidation products. Free Radic Biol Med 17, 411–418. [DOI] [PubMed] [Google Scholar]
- Chu DK, Kim LH, Young PJ, Zamiri N, Almenawer SA, Jaeschke R, Szczeklik W, Schunemann HJ, Neary JD, and Alhazzani W (2018). Mortality and morbidity in acutely ill adults treated with liberal versus conservative oxygen therapy (IOTA): a systematic review and meta-analysis. Lancet 391, 1693–1705. [DOI] [PubMed] [Google Scholar]
- de Vet EC, Ijlst L, Oostheim W, Dekker C, Moser HW, van Den Bosch H, and Wanders RJ (1999). Ether lipid biosynthesis: alkyl-dihydroxyacetonephosphate synthase protein deficiency leads to reduced dihydroxyacetonephosphate acyltransferase activities. J Lipid Res 40, 1998–2003. [PubMed] [Google Scholar]
- Doench JG (2016). CRISPR/Cas9 gene editing special issue. FEBS J 283, 3160–3161. [DOI] [PubMed] [Google Scholar]
- Ferrari M, Jain IH, Goldberger O, Rezoagli E, Thoonen R, Cheng KH, Sosnovik DE, Scherrer-Crosbie M, Mootha VK, and Zapol WM (2017). Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc Natl Acad Sci U S A 114, E4241–E4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier AE, Thorburn DR, and Compton AG (2019). Mitochondrial energy generation disorders: genes, mechanisms, and clues to pathology. J Biol Chem 294, 5386–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego-Garcia A, Monera-Girona AJ, Pajares-Martinez E, Bastida-Martinez E, Perez-Castano R, Iniesta AA, Fontes M, Padmanabhan S, and Elias-Arnanz M (2019). A bacterial light response reveals an orphan desaturase for human plasmalogen synthesis. Science 366, 128–132. [DOI] [PubMed] [Google Scholar]
- Garcia-Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, Yucel B, Fiore D, Tavora B, Freinkman E, et al. (2019). Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 567, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardis M, Busani S, Damiani E, Donati A, Rinaldi L, Marudi A, Morelli A, Antonelli M, and Singer M (2016). Effect of Conservative vs Conventional Oxygen Therapy on Mortality Among Patients in an Intensive Care Unit: The Oxygen-ICU Randomized Clinical Trial. JAMA 316, 1583–1589. [DOI] [PubMed] [Google Scholar]
- Goldfine H (2010). The appearance, disappearance and reappearance of plasmalogens in evolution. Prog Lipid Res 49, 493–498. [DOI] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, Bocchini CA, and McKusick VA (2005). Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 33, D514–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartnett ME, and Penn JS (2012). Mechanisms and management of retinopathy of prematurity. N Engl J Med 367, 2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze AT, and Acker T (2010). Feedback regulators of hypoxia-inducible factors and their role in cancer biology. Cell Cycle 9, 2749–2763. [DOI] [PubMed] [Google Scholar]
- Honsho M, Abe Y, and Fujiki Y (2015). Dysregulation of Plasmalogen Homeostasis Impairs Cholesterol Biosynthesis. J Biol Chem 290, 28822–28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay JA (2006). Iron-sulphur clusters and the problem with oxygen. Mol Microbiol 59, 1073–1082. [DOI] [PubMed] [Google Scholar]
- Ivanisevic J, Elias D, Deguchi H, Averell PM, Kurczy M, Johnson CH, Tautenhahn R, Zhu Z, Watrous J, Jain M, et al. (2015). Arteriovenous Blood Metabolomics: A Readout of Intra-Tissue Metabostasis. Sci Rep 5, 12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain IH, Zazzeron L, Goldberger O, Marutani E, Wojtkiewicz GR, Ast T, Wang H, Schleifer G, Stepanova A, Brepoels K, et al. (2019). Leigh Syndrome Mouse Model Can Be Rescued by Interventions that Normalize Brain Hyperoxia, but Not HIF Activation. Cell Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, Goldberger O, Peng J, Shalem O, Sanjana NE, et al. (2016). Hypoxia as a therapy for mitochondrial disease. Science 352, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DC, Dean DR, Smith AD, and Johnson MK (2005). Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem 74, 247–281. [DOI] [PubMed] [Google Scholar]
- Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB, and Rabinowitz JD (2013). Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci U S A 110, 8882–8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Sato Y, Kawashima M, Furumichi M, and Tanabe M (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44, D457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klett EL, Chen S, Yechoor A, Lih FB, and Coleman RA (2017). Long-chain acyl-CoA synthetase isoforms differ in preferences for eicosanoid species and long-chain fatty acids. J Lipid Res 58, 884–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ning S, Ghandi M, Kryukov GV, Gopal S, Deik A, Souza A, Pierce K, Keskula P, Hernandez D, et al. (2019). The landscape of cancer cell line metabolism. Nat Med 25, 850–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TF, Tang JJ, Li PS, Shen Y, Li JG, Miao HH, Li BL, and Song BL (2012). Ablation of gp78 in liver improves hyperlipidemia and insulin resistance by inhibiting SREBP to decrease lipid biosynthesis. Cell Metab 16, 213–225. [DOI] [PubMed] [Google Scholar]
- Los DA, and Murata N (2004). Membrane fluidity and its roles in the perception of environmental signals. Biochim Biophys Acta 1666, 142–157. [DOI] [PubMed] [Google Scholar]
- Martin JD, Fukumura D, Duda DG, Boucher Y, and Jain RK (2016). Reengineering the Tumor Microenvironment to Alleviate Hypoxia and Overcome Cancer Heterogeneity. Cold Spring Harb Perspect Med 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, and Thomas PD (2013). PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res 41, D377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagura M, Saito M, Iwamori M, Sakakihara Y, and Igarashi T (2004). Alterations of fatty acid metabolism and membrane fluidity in peroxisome-defective mutant ZP102 cells. Lipids 39, 43–50. [DOI] [PubMed] [Google Scholar]
- Nursall J (1959). Oxygen as a Prerequisite to the Origin of the Metazoa. Nature 183, 1170–1172. [Google Scholar]
- Peng Y, Yang Z, Zhang H, Cui C, Qi X, Luo X, Tao X, Wu T, Ouzhuluobu Basang, et al. (2011). Genetic variations in Tibetan populations and high-altitude adaptation at the Himalayas. Mol Biol Evol 28, 1075–1081. [DOI] [PubMed] [Google Scholar]
- Piccolis M, Bond LM, Kampmann M, Pulimeno P, Chitraju C, Jayson CBK, Vaites LP, Boland S, Lai ZW, Gabriel KR, et al. (2019). Probing the Global Cellular Responses to Lipotoxicity Caused by Saturated Fatty Acids. Mol Cell 74, 32–44 e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Raymond J, and Segre D (2006). The effect of oxygen on biochemical networks and the evolution of complex life. Science 311, 1764–1767. [DOI] [PubMed] [Google Scholar]
- Ruiz M, Stahlman M, Boren J, and Pilon M (2019). AdipoR1 and AdipoR2 maintain membrane fluidity in most human cell types and independently of adiponectin. J Lipid Res 60, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter A, Fourcade S, Domenech-Estevez E, Gabaldon T, Huerta-Cepas J, Berthommier G, Ripp R, Wanders RJ, Poch O, and Pujol A (2007). PeroxisomeDB: a database for the peroxisomal proteome, functional genomics and disease. Nucleic Acids Res 35, D815–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2014). Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol 9, 47–71. [DOI] [PubMed] [Google Scholar]
- Shi X, Tarazona P, Brock TJ, Browse J, Feussner I, and Watts JL (2016). A Caenorhabditis elegans model for ether lipid biosynthesis and function. J Lipid Res 57, 265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simcox J, Geoghegan G, Maschek JA, Bensard CL, Pasquali M, Miao R, Lee S, Jiang L, Huck I, Kershaw EE, et al. (2017). Global Analysis of Plasma Lipids Identifies Liver-Derived Acylcarnitines as a Fuel Source for Brown Fat Thermogenesis. Cell Metab 26, 509–522 e506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson TS, Yang Y, Huff CD, Yun H, Qin G, Witherspoon DJ, Bai Z, Lorenzo FR, Xing J, Jorde LB, et al. (2010). Genetic evidence for high-altitude adaptation in Tibet. Science 329, 72–75. [DOI] [PubMed] [Google Scholar]
- Sindelar PJ, Guan Z, Dallner G, and Ernster L (1999). The protective role of plasmalogens in iron-induced lipid peroxidation. Free Radic Biol Med 26, 318–324. [DOI] [PubMed] [Google Scholar]
- Sormendi S, and Wielockx B (2018). Hypoxia Pathway Proteins As Central Mediators of Metabolism in the Tumor Cells and Their Microenvironment. Front Immunol 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal VJ (2009). Oxygen in the evolution of complex life and the price we pay. Am J Respir Cell Mol Biol 40, 507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volmer R, van der Ploeg K, and Ron D (2013). Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci U S A 110, 4628–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter KM, Schonenberger MJ, Trotzmuller M, Horn M, Elsasser HP, Moser AB, Lucas MS, Schwarz T, Gerber PA, Faust PL, et al. (2014). Hif-2alpha promotes degradation of mammalian peroxisomes by selective autophagy. Cell Metab 20, 882–897. [DOI] [PubMed] [Google Scholar]
- Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, and Sabatini DM (2017). Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 168, 890–903 e815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, and Ebberink MS (2012). Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim Biophys Acta 1822, 1430–1441. [DOI] [PubMed] [Google Scholar]
- Wertlake PT, and Winter TS (1965). Oxygen Poisoning. N Engl J Med 273, 448–449. [DOI] [PubMed] [Google Scholar]
- Xu S, Li S, Yang Y, Tan J, Lou H, Jin W, Yang L, Pan X, Wang J, Shen Y, et al. (2011). A genome-wide search for signals of high-altitude adaptation in Tibetans. Mol Biol Evol 28, 1003–1011. [DOI] [PubMed] [Google Scholar]
- Yang J, Jin ZB, Chen J, Huang XF, Li XM, Liang YB, Mao JY, Chen X, Zheng Z, Bakshi A, et al. (2017). Genetic signatures of high-altitude adaptation in Tibetans. Proc Natl Acad Sci U S A 114, 4189–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZX, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS, et al. (2010). Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329, 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XG, Nicholson Puthenveedu S, Shen Y, La K, Ozlu C, Wang T, Klompstra D, Gultekin Y, Chi J, Fidelin J, et al. (2019). CHP1 Regulates Compartmentalized Glycerolipid Synthesis by Activating GPAT4. Mol Cell 74, 45–58 e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoeller RA, Grazia TJ, LaCamera P, Park J, Gaposchkin DP, and Farber HW (2002). Increasing plasmalogen levels protects human endothelial cells during hypoxia. Am J Physiol Heart Circ Physiol 283, H671–679. [DOI] [PubMed] [Google Scholar]
- Zoeller RA, Lake AC, Nagan N, Gaposchkin DP, Legner MA, and Lieberthal W (1999). Plasmalogens as endogenous antioxidants: somatic cell mutants reveal the importance of the vinyl ether. Biochem J 338 (Pt 3), 769–776. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2, Related to Figure 1. Enrichment of gene sets including custom KEGG gene sets used for GSEA, and GO biological process analysis of genes selectively essential at 21% and 1% oxygen.
Table S4, Related to Figure 5. Lipidomics data from K562 knockout cell lines grown at 21% and 5% oxygen.
Table S3, Related to Figure 2. High-altitude SNPs and ranking in oxygen screen.
Table S5, Related to STAR Methods. sgRNA guide sequences for gene-specific CRISPR knockouts.
Figure S1. Gene ontology biological processes selectively essential at 21% vs. 1% oxygen, related to Figure 3. All genes within Gene Ontology biological process categories that were enriched in the 213 genes selectively essential at 21% vs. 1% oxygen, based on PANTHER analysis. See also Table S2.
Figure S3. Peroxisome number in 21% and 1% oxygen, related to Figure 4. GFP-PTS1 labeled peroxisomes were imaged (left) and quantified (right) in HEK293T cells cultured for 5 days under 21% O2 or 1% O2. Cells were co-stained with the nuclear dye Hoechst. Peroxisomes were quantified via CellProfiler version 3.1.8 using minimum 130 cells per condition. No differences were observed in peroxisome abundance or gross appearance between the two conditions. Scale bar- 5μm.
Figure S2. K562 and HEK293T cells are capable of forming lipid droplets, related to Figure 4. (A) K562 cells expressing Cas9 and sgRNA guides to FAR1 or noncutting control (sgDummy) were incubated in 21% or 1% O2 (2 days) in delipidated media (supplemented with 0.5% fatty acid free BSA) and then treated for 24h with 0.04 mM BSA, 0.25 mM palmitate, or 0.25mM palmitate plus 0.25 mM oleate. Cells were fixed and stained with 0.5μM BODIPY 493/503 (Piccolis et al., 2019), and analyzed by confocal microscopy. Blue scale bar = 5 μm. (B) HEK293T cells expressing Cas9 and sgRNA guides to FAR1 or noncutting control (sgDummy) or FAR1 were incubated in 21% or 5% O2 (3 days) in delipidated media (supplemented with 10% charcoal stripped FBS) and then treated for 24h with 0.04 mM BSA, 0.25 mM palmitate, or 0.25mM palmitate plus 0.25 mM oleate. Cells were fixed and stained with 0.5μM BODIPY 493/503 (Piccolis et al., 2019), and analyzed by confocal microscopy. Blue scale bar = 5 μm. (C) Abundance of TAG species in K562 cells in cells transiently transfected with sgRNA guides to a non-coding region (sgDummy) or peroxisomal genes (PEX5, PEX10, AGPS, FAR1) in 21% or 1% O2 shows similar abundance levels across most species and conditions, with C50.0.TAG showing nearly twofold elevation in hypoxia (regardless of peroxisome knockout).
Figure S5. Metrics of lipid desaturation across seven lipid classes, related to Figure 6. (A) Density plot shows distribution of lipid desaturation metric across 447 cancer cell lines, for all seven lipid classes (LPE: lyso-phosphatidylethanolamine; TAG: triacylglycerol; PC: phosphatidylcholine; LPC: lyso-phosphatidylcholine; CE: cholesteryl ester; SM: sphingomyelin; DAG: diacylglycerol). (B) Pearson correlation between desaturation metrics for seven lipid classes, across the same 447 cancer cell lines. (C) For each of 17635 genes, Spearman correlation is calculated for growth of CRISPR knockout (CERES score, negative indicates lower growth, i.e. greater essentiality) and lipid desaturation metric across 447 cell lines. Significant hits shown in red (Bonferroni corrected p-value < 0.05). (D) For two top-scoring genes (PEX2 and SCD) and all 89 measured lipids, Spearman correlations between CERES score and lipid abundance are shown. Lipids are grouped by the number of double bonds (e.g. C16.0.LPE and C18.0.LPE grouped under LPE.0), and individual lipids are colored by significance of correlation. Lipid data derived from (Li et al., 2019) and CRISPR dependency data derived from DepMap version 19Q2 (Meyers et al., 2017; Tsherniak et al., 2017).
Figure S4. Hypoxia-induced lyso-phospholipid changes in cells lacking ether lipid biosynthesis, related to Figure 5. The fold change in peak area of lyso-phospholipids with hypoxia and FAR1 KO vs. control cells (sgDummy non-coding control cells grown at 21% oxygen).
Table S1, Related to Figures 1–4. (Tab 1) sgRNA raw counts for all conditions; (Tab 2) Lists of genes selectively essential in each oxygen level based on MAGeCK comparisons; (Tab 3) Lists of genes generally essential in each oxygen level based on MAGeCK analysis on day 15 vs. day 0; (Tab 4) MAGeCK analysis comparing 21% and 1% oxygen at day 15; (Tab 5) MAGeCK analysis comparing 21% and 5% oxygen at day 15; (Tab 6) Summaries of all pairwise MAGeCK comparisons.
Data Availability Statement
RNA-seq data (Figure 2) is available from the NCBI Gene Expression Omnibus (GSE144527). This study did not generate new unique reagents.