

Abstract
Background/Aims:
Cell migration and extracellular matrix remodeling underlie normal mammalian development and growth as well as pathologic tumor invasion. Skeletal muscle is no exception, where satellite cell migration replenishes nuclear content in damaged tissue and extracellular matrix reforms during regeneration. A key set of enzymes that regulate these processes are matrix metalloproteinases (MMP)s. The collagenase MMP-13 is transiently upregulated during muscle regeneration, but its contribution to damage resolution is unknown. The purpose of this work was to examine the importance of MMP-13 in muscle regeneration and growth in vivo and to delineate a satellite cell specific role for this collagenase.
Methods:
Mice with total and satellite cell specific Mmp13 deletion were utilized to determine the importance of MMP-13 for postnatal growth, regeneration after acute injury, and in chronic injury from a genetic cross with dystrophic (mdx) mice. We also evaluated insulin-like growth factor 1 (IGF-1) mediated hypertrophy in the presence and absence of MMP-13. We employed live-cell imaging and 3D migration measurements on primary myoblasts obtained from these animals. Outcome measures included muscle morphology and function.
Results:
Under basal conditions, Mmp13−/− mice did not exhibit histological or functional deficits in muscle. However, following acute injury, regeneration was impaired at 11 and 14 days post injury. Muscle hypertrophy caused by increased IGF-1 was blunted with minimal satellite cell incorporation in the absence of MMP-13. Mmp13−/− primary myoblasts displayed reduced migratory capacity in 2D and 3D, while maintaining normal proliferation and differentiation. Satellite cell specific deletion of MMP-13 recapitulated the effects of global MMP-13 ablation on muscle regeneration, growth and myoblast movement.
Conclusion:
These results show that satellite cells provide an essential autocrine source of MMP-13, which not only regulates their migration, but also supports postnatal growth and resolution of acute damage.
Keywords: Satellite cells, Extracellular matrix, Myoblast migration
Introduction
Tissue repair and development are facilitated by well-coordinated remodeling of the extracellular matrix (ECM), as well as migration of cells through the ECM to form new tissues. Cell movement through the ECM is aided by the ability of the cells to digest macromolecules in the matrix, and proteolytic enzymes that degrade ECM proteins are essential. These processes are important for successful structural formation of tissues and wound healing [1, 2]. Alternatively, disease processes such as tumor invasion and metastasis take advantage of the same mechanisms to enable cancer cells to escape through biological barriers [3]. Similar mechanisms are used by skeletal muscle, which has a renowned ability to repair and regenerate following injury. Much of the regenerative capacity of skeletal muscle is attributable to the resident muscle stem cell population, the satellite cell [4, 5]. Satellite cells act in coordination with a milieu of inflammatory and stromal cells present in damaged muscle to resolve the injury and restore skeletal muscle structure. This heterogeneous cell population within the muscle tissue secretes many factors to help guide cell maturation, degrade and re-construct ECM, and re-form muscle fibers. Of these factors, the matrix metalloproteinases (MMP) family of proteinases is important for the ECM remodeling process, where their primary function is to degrade components of the ECM [6]. In addition to actions on the ECM, MMPs have also been shown to regulate satellite cell migration [7, 8], in part by altering the matrix and lowering the barriers for cell movement.
The tight regulation of MMPs occurs at many levels including transcription, activation, and inhibition by the tissue inhibitors of metalloproteinases (TIMPs) [9]. During development, repair, and regeneration, MMPs are upregulated and activated to remodel the ECM. However, without regulation over-active MMPs can cause several pathological conditions [10–12]. Alternatively, loss of MMP activity leads to pathologies including stalled development [13], unresolved bone repair [14], diminished wound healing [15], and impaired skeletal muscle repair [16]. Hence, regulation of MMP activity is a significant factor for successful skeletal muscle regeneration. Of the more than 2 dozen MMPs identified, the gelatinases (MMP-2 and −9), collagenases (MMP-1 and −13), and the membrane anchored MT1-MMP/MMP-14 have been examined in skeletal muscle [7, 17–21]. These MMPs are upregulated to enable regeneration with distinct time courses. For instance, during the resolution of acute damage, MMP-9 is quickly activated in the injured muscle and then disappears, whereas MMP-2, MMP-14, and MMP-13 appear in the later phases of the repair process [7, 20]. By extension, the continual cycles of degeneration and regeneration associated with several muscular dystrophies is accompanied by sustained elevation of MMP levels [7]. As the dystrophic muscles are progressively replaced by fibrosis [22], heightened MMP activity may be compensatory to counter fibrosis, or part of the initiation of ECM remodeling. Loss of MMP-2 from the dystrophic mdx mouse resulted in a more pronounced phenotype with lack of proper regeneration and angiogenesis [23]. The role of MMP-9 in the mdx mice is more complex, with loss of MMP-9 and overexpression of MMP-9 both resulting in an improved phenotype [24, 25] as well as reports of a beneficial impact early followed by a detrimental effect later in the lifespan [25]. While the effects of MMPs on the muscle ECM integrity have been studied, little attention on how they contribute to or alter satellite cell function in the regenerating environment has been addressed.
A role for MMP-13 in myoblast migration was originally noted in a wound healing study that observed high expression of MMP-13 in migrating myoblasts [26]. This is consistent with the ability of MMP-13 to modulate C2C12 myoblast migration in vitro [7]. In addition to its actions on cell migration, MMP-13 is a collagenase capable of cleaving native interstitial collagens [27], providing a counter weight to the abundant interstitial collagen present in fibrosis. MMP-13 is a potent ECM degrading enzyme with activity against multiple collagens, proteoglycans, and fibronectin as well as activating other MMPs, including MMP-2 and MMP-9 [28]. MMP-13 can also support regeneration through its role in releasing vascular endothelial growth factor (VEGF) from the ECM to accommodate angiogenesis [29]. In comparison to its other family members, MMP-13 levels are very low, suggesting that its actions on overall ECM proteolysis may be less than more abundant MMPs. However, the local concentration of MMP-13 in the vicinity of a cell that secretes it may be sufficient to provide the ECM remodeling necessary for that particular cell. No previous studies have specifically manipulated MMP-13 in primary muscle cells or in muscle to examine its necessity during phases of matrix remodeling. The purpose of this work is to examine the importance of MMP-13 in muscle regeneration and growth in vivo using global genetic ablation of MMP-13 [13], and to delineate a satellite cell specific role for this collagenase. We hypothesized that myoblasts lacking MMP-13 would have impaired migration, resulting in impaired regenerative capacity and matrix remodeling in vivo.
Materials and Methods
Animals
The Animal Care and Use Committees at the University of Pennsylvania and the University of Florida approved all experiments in this study. Male and female mice were used for all studies, unless otherwise stated. Mice were maintained in the animal facility under a 12-hour light/dark cycle. Animals had ad libitum access to food and water. Mouse lines included those with whole body ablation of Mmp13 (Mmp13−/−) [13, 30] and C57BL/6 strain matched wildtype (WT) controls. Mmp13−/− mice were crossed with mdx mice, a model for DMD. Experiments on male Mmp13−/−:mdx and mdx mice were carried out in mice at 12-weeks of age and a small subset at 1 year of age. In addition, mice with satellite cell specific deletion of Mmp13 (scMmp13fl/fl) were generated. Mice harboring floxed exons 3–5 of Mmp13 (Mmp13fl/fl) [30] (005710, Jackson Laboratory) were crossed with mice carrying tamoxifen-inducible Cre recombinase under the Pax7 promoter (Pax7CE) [31, 32] (012476, Jackson Laboratory), and also crossed with the ROSAmT/mG reporter mouse (007676, Jackson Laboratory) [33], which widely expresses membrane localized TdTomato prior to Cre exposure and expresses GFP in Cre expressing cells, to monitor deletion. Intraperitoneal injection of 0.2 mg/g bodyweight of tamoxifen (TAM) dissolved in corn oil was administered for five consecutive days prior to the experimental timepoint to induce satellite cell deletion of Mmp13 (+scMmp13fl/fl). Controls included scMmp13fl/fl mice that received intraperitoneal corn oil (vehicle) injections (−scMmp13fl/fl), and scMmp13+/+ mice injected with TAM (+scMmp13+/+). Genotypes of mouse lines were determined by PCR with oligonucleotides listed in Supplementary Table 1 (for all supplemental material see www.cellphysiolbiochem.com). Experiments were conducted on age- and sex-matched mice.
Muscle Acute Injury
For muscle injury experiments mice were injected in the anterior hind limb, targeting the TA muscle, with 50 ul of 10 μM CTX (Sigma-Aldrich). This instigates acute muscle damage with a well-documented time course of regeneration from which muscles were harvested after 7, 11, 14, or 21 days. Initial injection of CTX took place in young adult mice aged 10–15 weeks.
Viral Delivery of IGF-1
IGF-1 mediated hypertrophy was induced by injection of adeno-associated virus (AAV) harboring Igf1a cDNA as previously described [34]. Specifically, 2.5 × 1010 copies self-complementing AAV 2/8 in 50 μL PBS was injected into the anterior compartment of the lower hindlimb, with the contralateral limb receiving PBS only. Mice were injected at 2 months of age, and were then sacrificed one month after injection. Following this, both TA and EDL muscles were dissected and weighed. TA muscles were flash frozen for IGF-1 quantification, and the EDL muscles embedded in OCT and frozen in melting isopentane muscles and stored at −80°C for subsequent morphological analysis. Tissue IGF-1 levels were determined in the TA muscles using the Rat/Mouse IGF-1 Enzyme-linked Immunosorbent ELISA-Kit (R&D Systems, Cat#MG100) as described previously [35] and according to the manufacturer’s instructions. Data are expressed as ng/g tissue.
Muscle Mechanics
Mice were anesthetized with a ketamine/xylazine cocktail. Muscles were dissected and incubated in a bath of modified Ringer’s solution (119 mM NaCl, 4.74 mM KCl, 3.36 mM CaCl2, 1.18 mM KH2PO4, 1.18 mM MgSO4, 25 mM HEPES, and 2.75 mM glucose) gas equilibrated with 95% O2-5% CO2. The proximal and distal tendons of the EDL were tied with 6–0 silk suture, and for diaphragm suture was tied to the central tendon on one end and two adjacent ribs on the other. The mechanical measurements were made on an apparatus previously described (Aurora Scientific, Ontario, Canada) [36], on muscles maintained in modified Ringers solution at 25°C. Optimum muscle length (Lo) was determined with iterative manual length extensions to reach maximal force production from a supramaximal stimulation to induce twitch. Muscle physiological cross-sectional area (PCSA) was determined as previously described using Lf/Lo of 0.45 for EDL and 1.0 for diaphragm. Maximum isometric twitch force was measured and then after 20 s maximum isometric tetanus was measured during a 500 ms stimulation. The twitch and tetanus forces were measured a total of three times with a 5-minute interval between each set of stimulations. For eccentric contraction (ECC) induced injury an isometric maximal force was produced by supramaximal stimulation for 500 ms with a 10% strain during an additional 200 ms of supramaximal stimulation. The eccentric contraction was repeated 5 times with a period of 5 minutes between each contraction. An index of ECC induced damage was calculated by the isometric force following the ECC contractions compared to the isometric force prior to the ECC contractions [37].
Passive muscle mechanics were measured as previously described [38]. Briefly, each muscle underwent strains of 2.5, 5, 7.5, 10, and 12.5%. Muscles were preconditioned for each strain with a cyclical strain at 1 Hz for 5 s. The muscle then underwent a ramp strain at 1 Lo /s and allowed to stress relax for 2 minutes. The stress at 2 minutes was used as the elastic stress. The muscles were allowed to relax for 20 s prior to the following induced strain. The elastic stiffness was calculated from the tangent modulus of the elastic stress at 10% strain.
Muscle Histochemistry
Muscle cryosections were cut in cross section of 10 μm from the mid belly of the muscle. For immunofluorescence, sections were washed in PBS, blocked for 1 hour at room temperature in 5% bovine serum albumin, and incubated in primary antibody at 4°C overnight in a humidified chamber. Primary antibodies used include laminin (Thermo FB-082A) and CD31 (eBiosciences). The following day sections were washed with PBS, incubated with fluorescently labeled secondary antibodies (Alexa Fluor 488 goat antirat IgG (H+L) (Invitrogen A-11006), and Alexa Fluor 568 goat anti-rabbit IgG (H+L) (Invitrogen A-11011), washed again in PBS, and mounted with Vectashield containing DAPI (VectorLabs). In addition, slides were exposed to mouse IgG alone (Invitrogen A-11029) to assess fiber damage. Negative control slides, incubated only with the secondary antibodies, were included in each immunostaining. Samples were visualized in a Leica DMR Fluorescent Phase Contrast Microscope (Leica), equipped with a DFC7000T Microscope camera (Leica). Images were acquired and processed using the Leica Suite Microscope Imaging software (Leica). The images of TA and EDL muscles were reconstituted by merging images using Photoshop to contain the whole cross-section. Analysis of fiber cross-sectional area, proportion of centrally nucleated fibers, and capillary density were conducted using SMASH software for analysis of skeletal muscle histology [39].
Picrosirius red staining was conducted similarly to previous studies [38]. Briefly, sections were fixed in 4% paraformaldehyde for 10 min, rinsed, air dried, and stained for 1 h in 0.1% (wt/vol) sirius red (Sigma-Aldrich) dissolved in saturated aqueous picric acid (Sigma-Aldrich). Sections were then washed in two changes of 0.5% acetic acid, dehydrated in three changes of 100% ethanol, cleared with Citra Solv, and mounted with Cytoseal. A series of micrographs from each muscle were captured using a ×10 objective on a Leica DMLP (Leica) scope with a Micropublisher 5.0 (Q Imaging) camera. A custom script was written in MATLAB (Mathworks) to determine number of pixels stained red and total number of pixels stained.
Collagen packing density was evaluated using picrosirius red-stained muscle sections viewed under circularly polarized light using the Leica DMLP (Leica) scope, rotating polarizer, rotating analyzer, and dual quarter wave plates as previously described [38]. A custom script was written in MATLAB to determine the number of pixels stained above an intensity threshold in bins determined by 8-bit hue values (245–255 and 0–34 for red, 35–49 for yellow, and 50–115 for green) where high wavelengths represent more densely packed collagen.
Hydroxyproline content was determined as described in previous studies by our group [38, 40]. Frozen muscle tissue was pulverized on dry ice with careful attention to remove any attached tendon. The pulverized muscle was weighed, and hydrolyzed overnight in 1 ml of 6 M hydrochloric acid at 105°C. 10 μl of hydrolysate was mixed with 150 μl isopropanol followed by 75 μl of 1.4% chloramine-T (Sigma, St. Louis, MO) in citrate buffer and oxidized at room temperature for 10 min. The samples were then mixed with 1 ml of a 3:13 solution of Ehrlich’s reagent [1.5 g of 4-(dimethylamino) benzaldehyde (Sigma); 5 ml ethanol; 337 μl sulfuric acid] to isopropanol and incubated for 45 min at 55°C. Quantification was determined by extinction measurement of the resulting solution at 558 nm with the inclusion of a standard (0–1,000 μM trans-4-hydroxy-l-proline; Sigma). Results are reported as micrograms of hydroxyproline per milligrams of tissue wet weight.
MMP-13 Enzyme Activity
Frozen muscles were lysed with zymogen extraction buffer (100 mM Tris·HCl, 200 mM NaCl, 100 mM CaCl2, and 1% Triton X-100, pH 7.6) to measure MMP-13 activity as previously described [7]. Protein concentrations were measured by Bradford method (Bio-Rad). Samples were diluted to 100 μg/ml in assay buffer (50 mM Tris·HCl, 10 mM CaCl2). Trypsin (10 μg/ml) was added for cleavage of the inhibiting proregion of recombinant MMP-13. All samples were incubated at 37°C for 20 min to facilitate MMP activation. Soybean inhibitor (80 μM) was added to inactivate trypsin and to prevent nonspecific cleavage. Activity was assessed using fluorogenic MMP-13--specific substrate (EMD Millipore, Billerica, MA) dissolved in DMSO. Fluorescence was measured with 325 nm (excitation) and 393 nm (emission) every 10 min for a total of 120 min using a fluorescence plate reader (SpectraMax M5, Molecular Devices). Each sample was measured in triplicate. For calculation of MMP activity, each measurement was background-corrected to the average of the controls containing substrate only at the same time point. The slope of background-corrected fluorescence vs. time was calculated as a measure of MMP activity for each sample.
Single Fiber Satellite Cell isolation for Live Cell Microscopy
To directly visualize myoblast migration, single fiber cultures were used as a source of satellite cells, modified from [41]. Briefly, freshly dissected EDL muscles were rinsed in PBS and placed in DMEM containing 2 mg/ml collagenase and incubated at 37°C for 90 minutes with agitation. Collagenase was then rinsed with DMEM and muscles placed in DMEM + 10% FBS. Muscle was dissociated first by passing through a wide-bore Pasteur pipette followed by further mechanical separation of fibers with Pasteur pipette. Isolated single fibers were carefully transferred to another dish with DMEM + 10% FBS and kept at 37°C as fibers were collected. Single fibers were then transferred again into 4-chamber glass bottom 35 mm dishes coated in 2 mg/ml matrigel (Corning Life Sciences). Fibers were allowed to attach for approximately 1 hour prior to addition of myoblast media (F-10 base, 20% FBS, 0.5 μg/ml bFGF, and PBS). Fiber cultures were left undisturbed for 48 hours and then media was changed with fresh myoblast media or reduced serum myoblast media (2% FBS). The samples were brought to the School of Dental Medicine Imaging Core (University of Pennsylvania). DIC and fluorescent signals were acquired with a Nikon A1R confocal microscope equipped with an automated stage. Cells were maintained at 37°C under 5% CO2 for the duration of the imaging via a stage-top incubator. Images were captured with a 10x (NA 0.45) objective data analyzed using Nikon Elements AR 4.30.01 software. Images of the hypercontracted myofiber and surrounding myoblasts were taken every 15 minutes for a total of 16 hours. Cells were tracked using ImageJ cell tracker plugin [42] and the velocity, total displacement, and divisions were calculated.
Bulk Satellite Cell Isolation
Satellite cells were isolated as described in [43]. Briefly, freshly dissected TA and gastrocnemius muscles were rinsed in pre-warmed DMEM and under a dissecting scope and tendon, fat, vessels, and connective tissue were removed. The muscle was then cut into small fragments and remaining pieces of connective tissue were removed. Fragments were transferred to DMEM containing 2 mg/ml collagenase (Sigma) and incubated with agitation at 37°C for 1 hour. Fragments were spun down and washed in DMEM before being placed in DMEM supplemented with 10% horse serum. Enzymatically digested muscle fragments were then mechanically titrated with a wide-bore pasture pipette and a 9” glass pasture pipette. The supernatant was filtered through a 40 μm cell filter, centrifuged, and suspended in DMEM with 10% horse serum. Cells were counted using a hemocytometer and used in the transwell migration assay or fusion assay, or differentiated.
Transwell invasion assay
Transwell membranes were used according to the manufactures instructions (Trevigen). The day prior to satellite cell isolation, 1X basement membrane extract (BME, Trevigen) and 1.5 mg/ml collagen I (Trevigen) solutions were made and 100 μl was placed on top of a transwell membrane containing 8 μm pores. Transwells were incubated overnight at 37°C to polymerize the substrate. The following day any liquid was carefully aspirated from the substrate and 1 × 105 freshly isolated satellite cells from the muscle bulk in DMEM + 10% horse serum were added to the top of the chamber. On the bottom of the chamber 500 μl of growth media (DMEM with 20% fetal bovine serum, 10% horse serum, and 1% chick embryo extract) was added. Assembled transwells were incubated for 24 hrs at 37°C with 5% CO2. Following incubation, nonmigrated cells on the upper side of the filter were wiped with Q-tips; the migrated cells attached to the lower side of the filter were fixed with 4% formaldehyde for 10 min, washed three times with PBS, stained with crystal violet solution for 30 min, and rinsed with PBS. The filters were cut out from the transwell plates, mounted onto glass slides with mounting medium, and viewed under a Leica DFC300 light microscope. For each membrane, 3 fields were randomly chosen and imaged at 20X, and the total number of cells per field was determined. Each condition was performed in triplicate.
Fusion assay
Satellite cells freshly isolated as described above were centrifuged and resuspended with growth media. Cell suspension was placed on 24 mm coverslips coated with BME or collagen I within a 6 well plate at a density of 1 × 105 cells/well. Cells were cultured for 3 days undisturbed and then carefully washed with DMEM to remove debris and cultured in growth media an additional 3 days. On the 6th day the medium was changed to DMEM with 10% horse serum to induce differentiation. At 0, 1, and 2 days post differentiation the cells were fixed with 4% paraformaldehyde for 10 minutes. The cells were then permeabilized with 1% Triton X-100 and blocked in 5% BSA. Coverslips were incubated with MF20 antibody to label sarcomeric myosin (MF20, Iowa Developmental Studies Hybridoma Bank) overnight at 4°C. Coverslips were washed and incubated with secondary antibody (Alexa Fluor 568 goat anti-mouse IgG (H+L) (Invitrogen A-11004), and phalloidin conjugated to Alexa Fluor 488 (Thermo) for 1 hour at room temperature. Coverslips were washed and mounted on slides with Vectashield containting DAPI. From each coverslip, 3 images were obtained at 10X magnification. Nuclei were counted within cells that expressed sarcomeric myosin and total nuclei. The ratio of nuclei within myotubes expressing sarcomeric myosin to total nuclei is used as the fusion index.
Gene expression analysis
Total RNA was isolated from TA muscles using Trizol ® Reagent (Life Technologies, #15596018) and DNAse treated with DNase I recombinant, RNase- free (Roche, #04716728001). cDNA was prepared using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, #4368814) according to the manufacturer’s instructions. Duplicates of cDNA samples were amplified on Step One Plus Real Time PCR System (Applied Biosystems) using Power SYBR Green PCR Master Mix (Applied Biosystems, #4367659). The expression level of Mmp2 was determined and normalized to 18S and plotted as mean ± SEM in fold-changes. The oligonucleotide primers used are listed in Supplementary Table 1.
Statistical methods
Analysis of results included two-tailed unpaired students t-test, one-way, or two-way ANOVA followed by Bonferroni’s post hoc analysis. For two-way ANOVAs, genotype was a main effect along with one of the following: serum concentration, substrate, days post differentiation, days post cardiotoxin, or muscle.
Results
Mmp-13 is dispensable in homeostatic muscle
Mice with total ablation of Mmp13 were previously evaluated for the effects on bone formation, but no role in skeletal muscle had been addressed [30]. We examined mice at 4 and 11 weeks of age to capture a period of active growth. At post-weaning age, both male and female Mmp13−/− mice were smaller than their age and sex-matched controls, similar to previous reports ascribing lower body mass to disruption of skeletal development at the growth plate [30]. By 11 weeks of age, there was no appreciable difference in body weight (Fig. 1A). Mmp13−/− mice age 11–13 weeks displayed divergent phenotypes with respect to muscle mass, where males had smaller extensor digitorum longus (EDL) and larger tibialis anterior (TA) mass, while female muscle masses were similar to wildtype (WT) cohorts (Fig. 1B). Normalized muscle mass of male TA muscles was also larger, while female EDL muscles were smaller in Mmp13−/− mice (Fig. 1C). Muscles from Mmp13−/− mice maintained normal function in terms of force normalized to cross sectional area (specific force) (Fig. 1D) and passive stiffness (Supplementary Fig. 1), in both the EDL and diaphragm. Cross sections from TA muscles were immunostained for laminin and CD31 to determine if Mmp13 ablation led to changes in fiber dimensions or vascularization, respectively (Fig. 1E), as MMP-13 plays a role in angiogenesis that could overlap with defects in muscle growth [44]. The cumulative distribution of fiber sizes showed no significant difference between the Mmp13−/− and WT TA muscles for unchallenged adult mice (Fig. 1F). Similarly, there was not a significant decrease in capillary density of the Mmp13−/− TA muscles as measured by CD31 labeled capillaries per muscle fiber (Fig. 1G). As MMP-13 is an archetypal collagenase, we anticipated higher collagen levels in its absence. However, the percent area of ECM in TA muscles was unchanged in Mmp13−/− compared to WT muscles (Fig. 1H/I), indicating no major changes in collagen balance. Further, collagen packing density was also similar between strains (Fig. 1J). Quantification of hydroxyproline performed on quadriceps muscles were consistent with the morphological assessments in TA muscles, showing no apparent differences between strains (Fig 1K). Thus, in unchallenged adult mice, the absence of MMP-13 did not manifest in deficiencies in muscle function, fiber size, angiogenesis, or ECM content. However, homeostatic muscle has virtually undetectable levels of MMP-13 [7], and compared to the expression of other MMPs in muscle (Supplementary Fig. 2), suggesting that it might not play a critical role in healthy adult muscle.
Fig. 1.
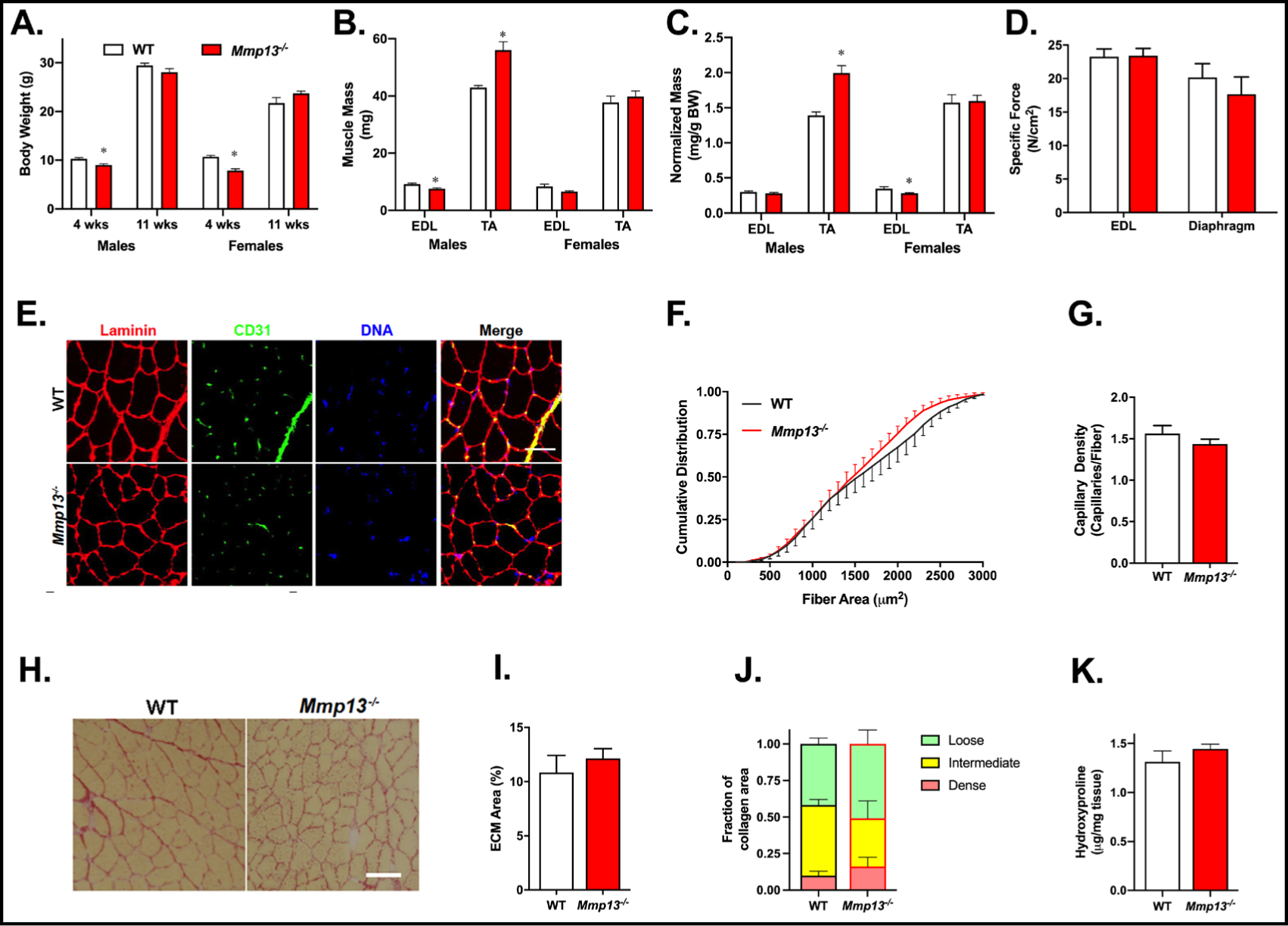
Mmp13−/− muscles exhibit differential size with normal function, vasculature, and ECM. (A) Male and female Mmp13−/− mice at 4 weeks of age are smaller than age-and sex-matched WT mice (N=3–5 per group), but by 11 weeks of age, no body mass differences are apparent. (B) Muscle masses in 11-week old male Mmp13−/− mice have smaller absolute EDL mass but larger TA mass when compared to WT mice (N=5 each). Female mice have no differences in muscle masses between genotypes. (C) When normalized to the body mass, Mmp13−/− male mice had larger TA but females had smaller EDL muscles. (D) The specific force in muscles from Mmp13−/− mice (N=5) are similar to WT (N=5) for EDL and diaphragm. (E) WT and Mmp13−/− were visualized with immunofluorescent staining of laminin for fiber border, CD31 for endothelial cells, and DAPI to visualize DNA from the TA muscle. Scale bar 50 μm. (F) There was no significant change in the cumulative distribution of fiber areas of Mmp13−/− (N=5) compared to WT (N=6) TA muscles. (G) There was no difference in the capillary density (the number of capillaries normalized to fiber number) of the same TA muscles. (H) Picrosirius red staining of TA muscles showed areas occupied by ECM from WT and Mmp13−/− muscles. Scale bar 50 μm. (I) The area fraction of ECM was similar for WT (N=6) and Mmp13−/− (N=8) TA muscles. (J) The proportion of loose (green), intermediate (yellow), and dense (red) collagen was similar for WT (N=4) and Mmp13−/− (N=4) TA muscles. (K) The amount of hydroxyproline was similar for WT (N=4) and Mmp13−/− (N=4) quadriceps muscles. The * represents a significant (p<0.05) difference between genotype.
MMP-13 is required for efficient muscle regeneration
Muscles that undergo injury activate satellite cells, which differentiate and then migrate and fuse to damaged fibers or to each other to form new muscle fibers, and this is accompanied by a transient increase in MMP-13 activity [5]. To determine if loss of MMP-13 altered muscle regeneration, young adult WT and Mmp13−/− mice received CTX injections into the TA muscles, which were then dissected after 7, 11, 14, and 21 days, and immunostained for laminin and CD31 (Fig. 2A). At 7 days post CTX injection, fiber sizes from both Mmp13−/− and WT muscles were significantly smaller than WT uninjured muscle fiber size, but they were not significantly different from each other (Fig. 2B/C). By 11 days post CTX injection the WT muscle fiber sizes were similar to uninjured sizes, but the Mmp13−/− fibers were significantly smaller. The differences in fiber size persisted to 14 days post CTX injection, but strains exhibited similar sizes by day 21. Hence, there was a delayed recovery of fiber size during muscle regeneration following CTX injury in Mmp13−/− TA muscles. The smaller fiber size at 14 days post CTX was accompanied by a reduced capillary density in Mmp13−/− TA muscles (Fig. 2D). However, the number of regenerating fibers as marked by centrally nucleated fibers was not significantly different from Mmp13−/− muscles at any time point (Fig. 2E). Fibers may undergo necrosis when regeneration is not optimal, with necrotic fibers being marked by positive IgG staining (Fig. 2F) [45, 46]. Necrotic fibers were observed in clusters in a subset of Mmp13−/− TA muscles at each of 7, 11, and 14 days post CTX injection, while WT muscles lacked appreciable necrotic fibers at any time point, a difference that was significant for a main effect in Mmp13−/− muscles (Fig. 2G). It may again be presumed that a lack of MMP-13 would lead to an imbalance between ECM production and degradation, favoring ECM accumulation. However, there was no significant difference in ECM area between the strains at any time point after CTX injection (Fig. 2H/I). Because several MMPs share common substrates, compensatory expression of other MMP family members could occur in the absence of MMP-13. MMP-2 is the likely candidate as it has a similar timecourse to MMP-13 during muscle regeneration [7]. mRNA levels of Mmp2 were transiently elevated in TA muscles following CTX injection, but with no alteration in the timecourse in Mmp13−/− TA muscles (Fig. 2J). Taken together, the absence of MMP-13 resulted in delayed resolution of acute muscle damage.
Fig. 2.
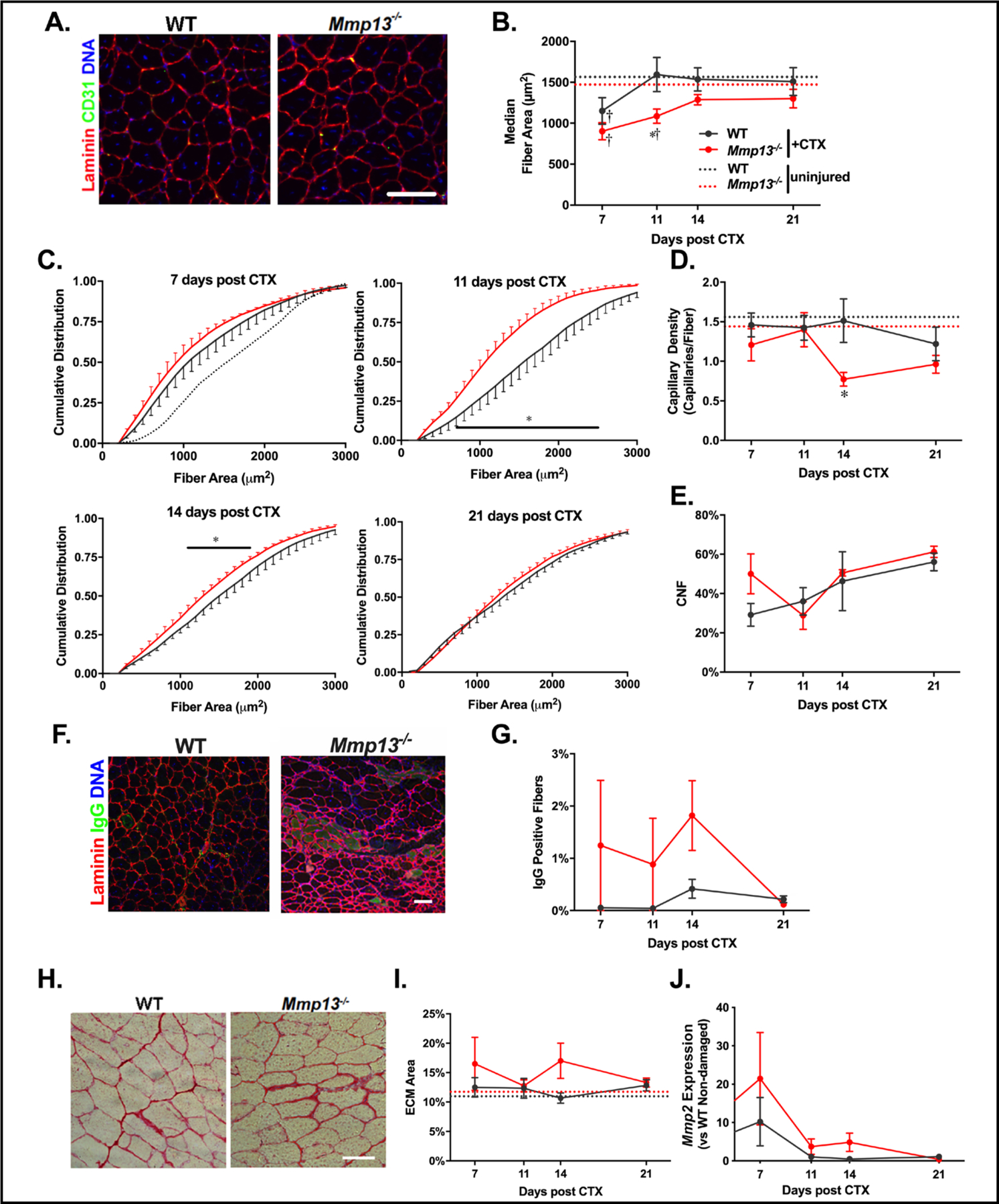
Mmp13−/− muscles have impaired regeneration following cardiotoxin injury. (A) Examples of WT and Mmp13−/− TA muscles with staining of laminin, CD31, and DNA on TA muscles 14 days after cardiotoxin (CTX) injection. Scale bar 50 μm. (B) The median fiber area is decreased for both genotypes 7 days after CTX and recovers to baseline by day 11 for WT, but recovery is significantly delayed in Mmp13−/− TA muscles. (C) The cumulative distributions of fiber areas from WT and Mmp13−/− TA muscles 7 days (WT N=8;Mmp13−/− N=4), 11 days (WT N=8;Mmp13−/− N=7), 14 days (WT N=6;Mmp13−/− N=6), and 21 days (WT N=10;Mmp13−/− N=12) following CTX injections. The uninjured WT cumulative distribution (black dotted line) is shown for reference with day 7. (D) The capillary density is significantly reduced at 14 days after CTX in the Mmp13−/− TA muscles. (E) There was no significant difference in the percentage of centrally nucleated fibers (CNF)s between WT and Mmp13−/− TA muscles throughout the injury recovery period. (F) Examples of WT and Mmp13−/− TA muscle 14 and 7 days post CTX, respectively, with staining of laminin, IgG to label damaged or necrotic fibers, and DNA. WT Day 14 was used so that rare IgG positive fibers could be shown. Scale bar 50 μm. (G) There is a significant main effect of Mmp13−/− compared to WT across the recovery timeframe. (H, I) The area fraction of ECM was similar for WT and Mmp13−/− TA muscles following CTX injury. (J) There was a significant main effect of increased expression of Mmp2 dependent on time post CTX injury, but no differences with respect to strain (all conditions N=4 per group). The * represents a significant (p<0.05) difference between WT and Mmp13−/−, with the bar representing the range of fiber areas that have a significant difference in the cumulative distribution. The † represents significant (p<0.05) difference between injured and uninjured muscles.
The most commonly used mouse model of muscular dystrophy, the mdx mouse, lacks dystrophin expression and consequently undergoes chronic muscle degeneration and regeneration cycles, and has elevated levels of MMP-13 [7]. Due to the impaired regeneration observed following acute CTX injury, mdx mice were crossed with Mmp13−/− mice to determine if the loss of MMP-13 exacerbated the dystrophic phenotype. Surprisingly, no significant defects were observed in muscle function for adult male Mmp13−/−:mdx mice compared to mdx mice in terms of maximum specific force, passive stiffness, or eccentric contraction induced force loss in the EDL or diaphragm, which is known as the most severely affected muscle [47], although there were apparent deficits in these measures compared to WT muscles (Supplementary Fig. 1A–C). The lack of functional changes between mdx and Mmp13−/−:mdx mice persisted out to 1 year of age, which was the longest time studied (data not shown). Consistent with the lack of functional defect, the fiber size distribution was not significantly shifted in Mmp13−/−:mdx mice relative to mdx mice in EDL muscles (Supplementary Fig. 1D/E). Correspondingly, the percentage of centrally nucleated fibers and the capillary density were unchanged between Mmp13−/−:mdx and mdx EDL and diaphragm muscles (Supplementary Fig. 1F/G). Muscles undergoing chronic degeneration and regeneration are known to have a subset of necrotic fibers, shown by positive IgG staining (Supplementary Fig. 1H). While a small population was observed in mdx muscles, there was a significant main effect across muscle for an increase in the population of necrotic fibers in Mmp13−/−:mdx, indicating a small defect in the muscle fiber survival. The chronic damage in the mdx mice leads to elevated ECM deposition and eventually fibrosis, yet the loss of MMP-13 in mdx mice did not result in an increase in ECM area of the muscle. Thus, the small increase in necrotic fibers that results from loss of MMP-13 is not sufficient to drive functional or morphological defects of the muscle on the mdx background, where chronic injury and ECM remodeling occur in the presence of high levels of several MMPs [7, 48].
Absence of MMP-13 alters the hypertrophic response to IGF-1
In additional to rapid growth during postnatal development, mature muscles are also capable of undergoing profound hypertrophy in response to mechanical overload [49], reduction of muscle growth inhibitors [49], or overexpression of hypertrophic signaling molecules, such as Insulin-Like Growth Factor-1 (IGF-1) [50]. With Mmp13 being one of the most upregulated genes in response to local IGF-1 overexpression [51], we anticipated that MMP-13 contributed to IGF-1 induced hypertrophy. To determine if MMP-13 was necessary for IGF-1 mediated hypertrophy, viral mediated gene transfer of IGF-1 was performed in the anterior hindlimbs of young adult Mmp13−/− and WT mice, causing a 7–10 fold increase in IGF-I levels (Fig. 3A). To ensure that IGF-1 led to an increase in MMP-13, enzyme activity assays were performed on muscle lysates, and confirmed a significant increase in MMP13 activity in WT muscles injected with AAV-Igf1, whereas, muscles from Mmp13−/− mice displayed no apparent MMP-13 activity in response to increased IGF-1 levels (Fig. 3B). Consistent with previous studies, viral production of IGF-1 resulted in a 10–15% increase in muscle mass relative to the contralateral limb muscles 4 weeks after injection, but this effect was blunted in the Mmp13−/− muscles (Fig. 3C). Hypertrophy was driven primarily by increased fiber size, which was evident in the EDL muscles of both mouse strains (Fig. 3D). Further, WT EDL muscles also displayed a significant increase in centrally nucleated fibers following increased IGF-1 expression, as with previous studies [52] (Fig. 3E). In contrast, EDL muscles from Mmp13−/− mice lacked IGF-1 dependent increases in central nuclei. Hence, hypertrophy was possible in the absence of MMP-13, but it was less robust and occurred through a satellite cell independent mechanism.
Fig. 3.
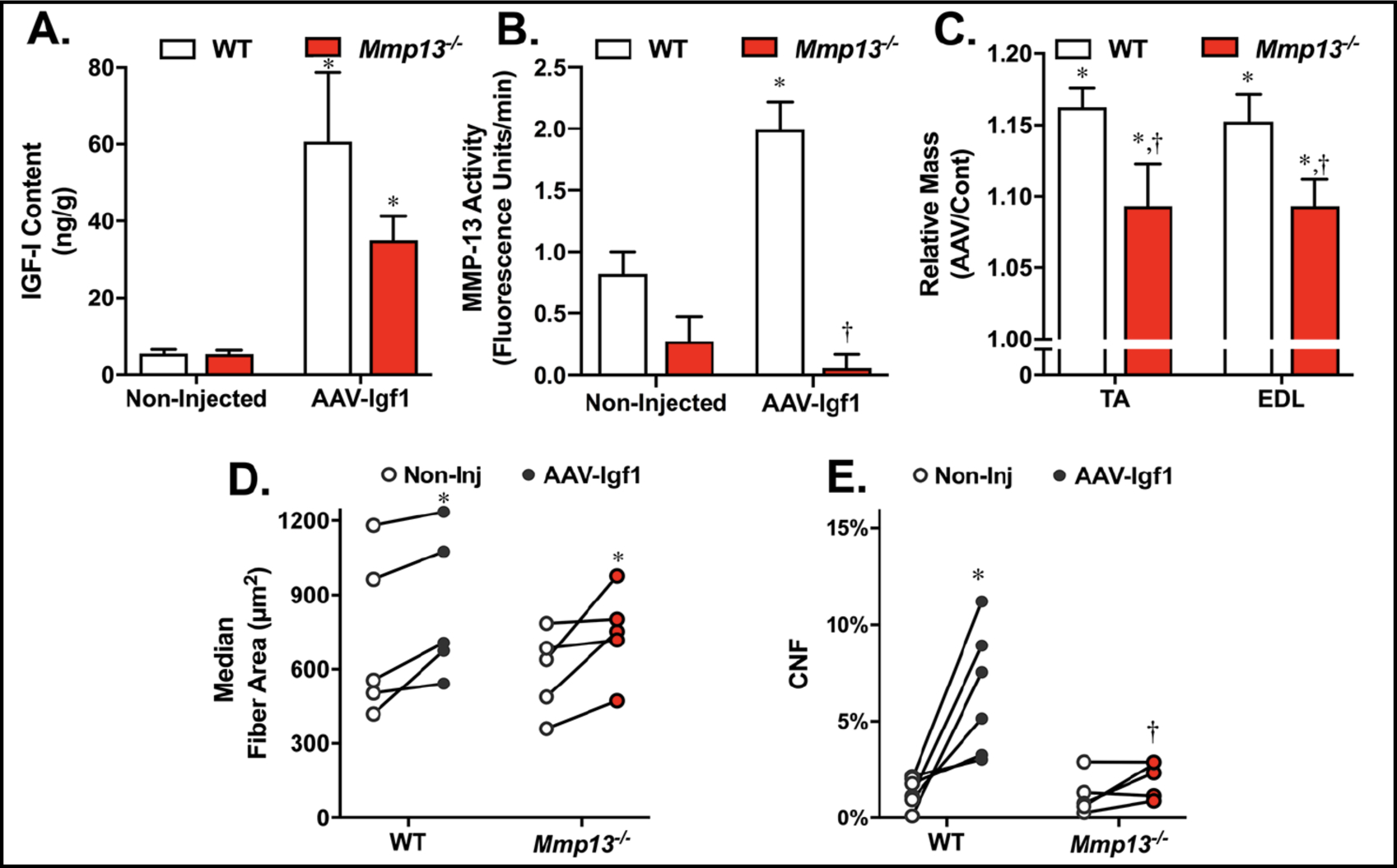
Mmp13−/− muscles exhibit hypertrophy in response to IGF-1 but without satellite cell involvement. (A) IGF-I content significantly increases in TA muscles of both WT and Mmp13−/− mice following injection with AAV-Igf1 (N=3 muscle pairs). (B) MMP-13 activity increased in response to IGF1 AAV in WT muscles, but not in muscles from Mmp13−/− mice (N=3 muscles pairs). (C) Relative muscle mass increased following AAV-Igf1 injection in TA and EDL muscles compared to contralateral control muscles regardless of genotype (N=6 muscle pairs), but muscles from Mmp13−/− mice displayed a blunted hypertrophic response. (D) Median fiber areas significantly increased in response to AAV-Igf1 injection in EDL muscles. (E) The percent of CNF increased in WT but not in EDL muscles from Mmp13−/− mice in response to AAV-Igf1 injection. The * represents p<0.05 between +AAV and non-injected muscles. The † represents p<0.05 between genotypes.
MMP-13 is critical for myoblast migration
Previous studies have demonstrated that MMP-13 is expressed by myoblasts in vitro and modulates their migration [7]. To extend these findings, we examined muscle progenitor cells migrating from single muscle fibers using live cell microscopy. Muscle progenitors were tracked for migration and cell division following their exit from isolated fibers, 48 – 64 hours following fiber isolation (Fig. 4A; Supplementary Videos for Fig. 4). Under standard primary culture conditions containing 20% serum, no difference in migration velocity or proliferation rate between WT and Mmp13−/− myoblasts was observed (Fig. 4B/C). Because serum contains many MMPs that may mask effects of MMP-13 deficiency, serum levels were reduced to 2%. In 2% serum, WT myoblast migration was minimally impacted, yet the Mmp13−/− myoblasts migrated significantly more slowly when compared to WT myoblasts in 2% or Mmp13−/− myoblasts in 20% serum conditions. Qualitatively, the Mmp13−/− myoblasts also showed less directional persistence than the WT myoblasts. While the 2D migration of myoblasts was impaired, other properties were unaffected by MMP-13 absence, including proliferation rate (Fig. 4C), although reduced serum conditions generally lowered proliferation.
Fig. 4.
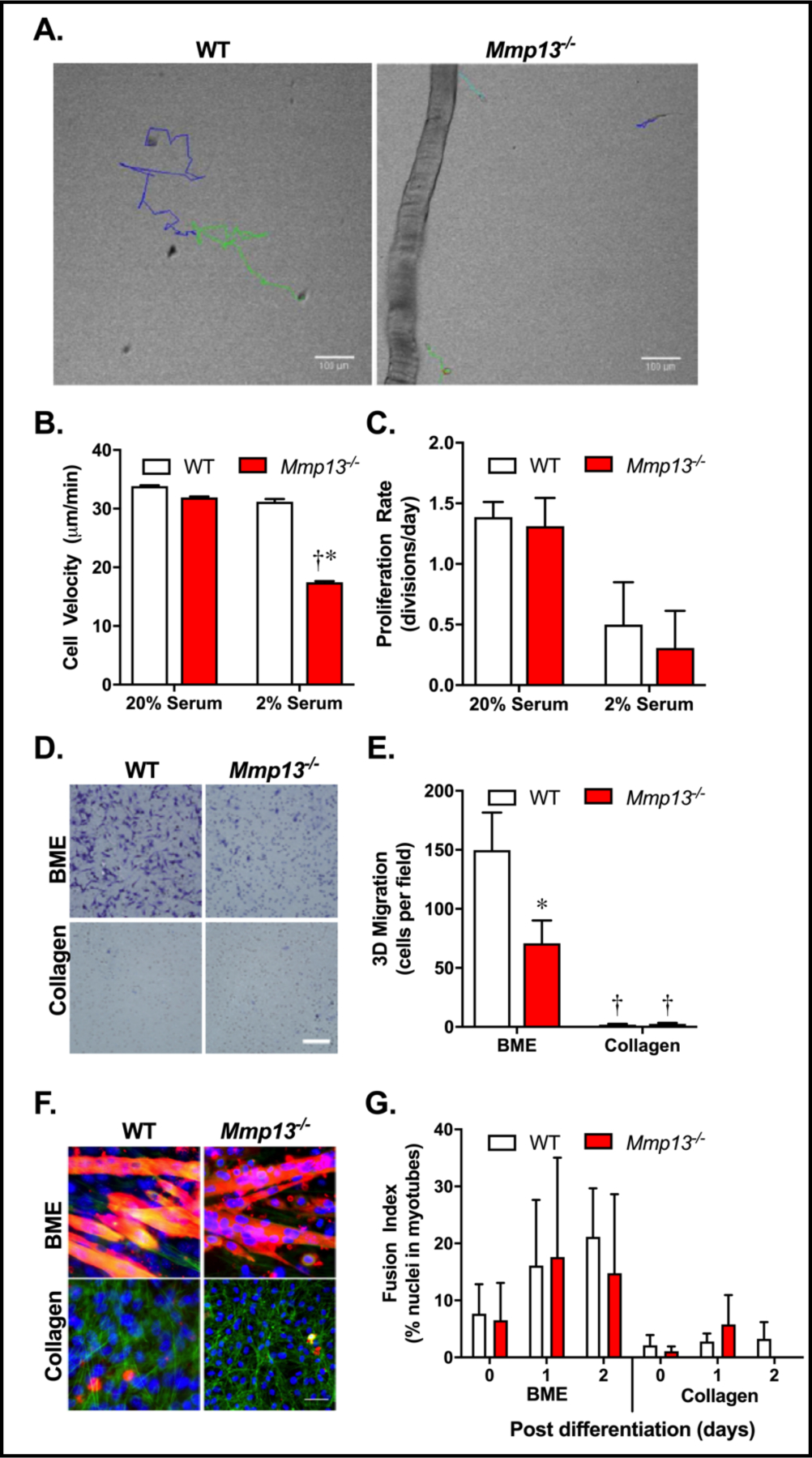
Mmp13−/− myoblasts in vitro have impaired migration. (A) A brightfield frame of live cell microscopy with multicolored fiber tracks showing the course of migration over 14 hours in WT and Mmp13−/− primary satellite cells. A fiber is shown on the left side of the Mmp13−/− frame. Scale bar is 100 μm. Representative videos are included in the supplement. (B) Average velocity of migrating cells in 2D from live cell experiments were similar in WT cells (N=307) and Mmp13−/− cells (N=215) at 20% serum and significantly different between WT cells (N=27) and Mmp13−/− cells (N=35) in 2% serum. Numbers of cells analyzed are shown on each column of the graph. (C) Proliferation rate measured as cells are migrating in 2D obtained from the same experiments as in B, which consisted of cells from WT (N=9) fibers and Mmp13−/− (N=7) fibers in 20% serum and WT (N=6) fibers and Mmp13−/− (N=6) fibers in 2% serum were not significantly different between genotypes. (D) Examples of 3D migration of WT and Mmp13−/− cells showing bottom side of membrane with cells invading the designated substrate (basement membrane extract (BME) or collagen) over the course of 24 hours. Scale bar = 50 μm. (E) 3D migration of Mmp13−/− cells through BME is impaired compared to WT cells, and almost non-existent through collagen by cells of both genotypes. 3 images per membrane (N=3 membranes for each genotype and substrate). (F) Examples of images of myotube formation at day 2 of differentiation from WT and Mmp13−/− myoblasts on BME and collagen. Sarcomeric myosin heavy chain (sMyHC) in red, phalloidin labeling actin in green, and DAPI labeling DNA. Scale bar = 50 μm. (G) The fusion index (percentage of nuclei in myotubes of all nuclei) is not changed between genotypes at any timepoint or substrate. There is a main effect of collagen substrates with less differentiation than BME substrates (N=3 wells per genotype per substrate per day). The * represents p<0.05 difference between WT and Mmp13−/−;† represents a significant difference within each genotype of serum conditions or substrate.
Migration in 2D does not necessarily require the cleavage of any MMP substrate, but 3D invasion assays require cells to penetrate ECM components in order to facilitate directed migration. When basement membrane extract (BME) was used as a substrate in transwell invasion assays for myoblasts isolated from muscle in bulk, there was robust invasion by WT myoblasts, but the invasion was reduced by ~50% for the Mmp13−/− myoblasts (Fig. 4D/E). Notably, neither WT nor Mmp13−/− myoblasts were able to penetrate a collagen I gel over a 24-hour period. Myoblasts from bulk isolation were also differentiated on top of either BME or collagen substrates to examine the role of MMP-13 in differentiation and myoblast fusion using immunostaining for myosin by MF20 (in red) and actin labeling by phalloidin (in green) (Fig. 4F). There was no significant difference in the fusion index between myoblasts from WT and Mmp13−/− mice for either substrate (Fig. 4G). However, in both genotypes BME served as a better substrate than collagen for myosin positive myotube formation, with a significant main effect of the substrate. These data demonstrate that Mmp13−/− myoblasts are impaired in 2D and 3D migration, but loss of MMP-13 does not have a dramatic impact on their ability to proliferate, differentiate or fuse.
Targeted loss of MMP-13 from myoblasts replicates impairments of total MMP-13 ablation
While the global Mmp13−/− mouse has defects in muscle regeneration, it is not known if cells other than myoblasts produce MMP-13 needed for this process. A satellite cell specific knockout of MMP-13, +scMmp13fl/fl, was used to elucidate the role of muscle progenitors in expressing MMP-13 (Fig. 5A). Pilot studies in +scMmp13+/+ cells showed that 83±15% of the cells were GFP positive indicating efficient TAM induction of the ROSA reporter (2 biological replicates). A second strategy of model validation used bulk primary cultures from muscles of +scMmp13fl/fl mice treated with TAM or vehicle, allowed to differentiate, and then subjected to immunostaining for MMP-13. GFP positive myotubes had no evidence of MMP-13 staining, even though contaminating fibroblasts had strong signal for MMP-13 (Supplementary Fig. 3). Thus, the intended deletion of Mmp13 from satellite cells was efficient and specific for myogenic cells.
Fig. 5.
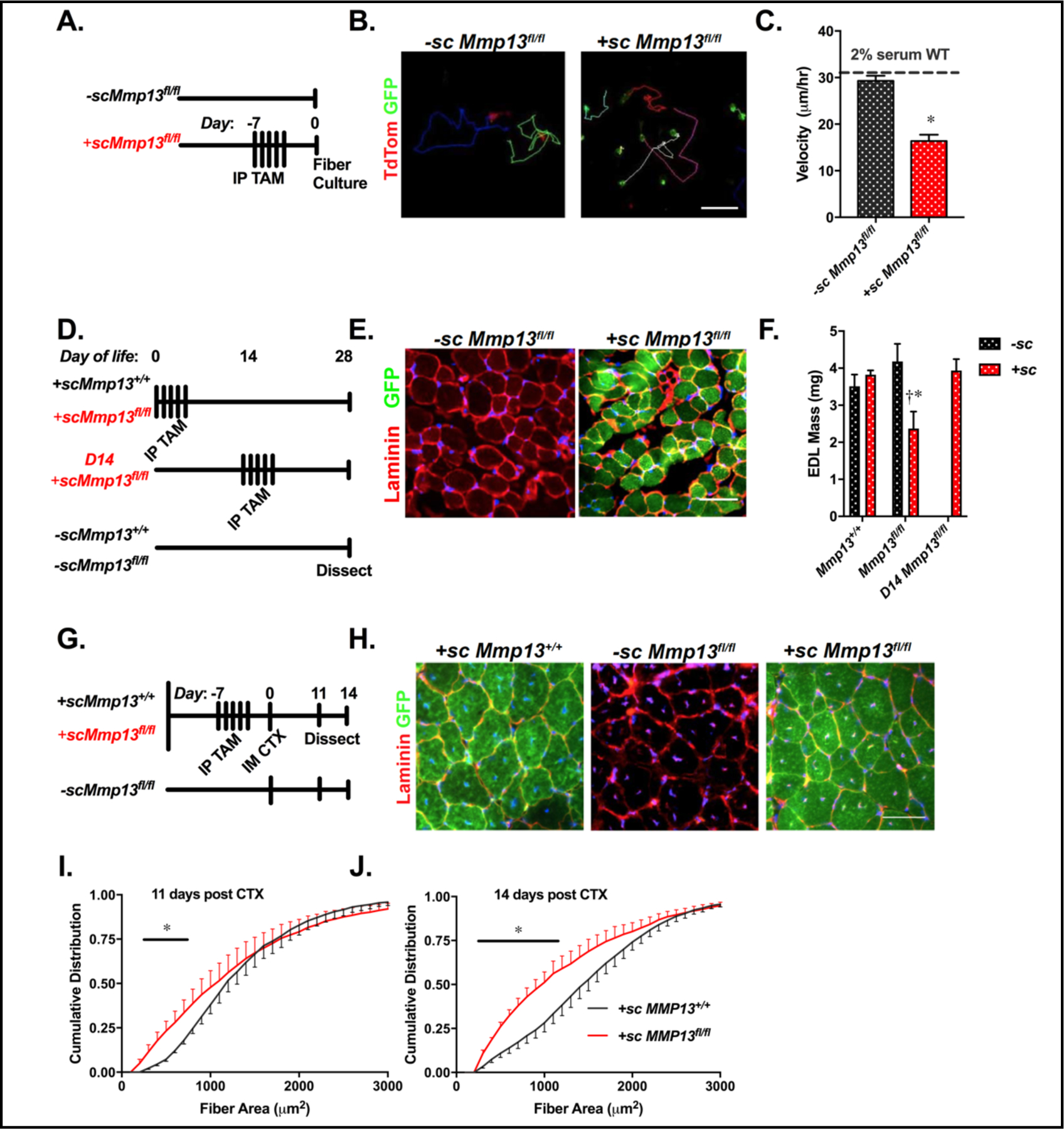
Satellite cell expression of MMP-13 underlies the Mmp13−/− phenotype. (A) Adult mice with Mmp13fl/fl Pax7CreER mT/mG received intraperitoneal injections of tamoxifen (TAM) (+sc) or corn oil (−sc) for 5 consecutive days 1 week prior to tissue harvest for single fiber culture. (B) A frame of live cell microscopy with multicolored fiber tracks showing the course of migration over 14 hours of cells from −sc/+scMmp13fl/fl mice (Sup Videos Overlay_RG-Control.avi and Overlay_RG-ko.avi). The +sc cells express GFP in satellite cells due to recombination of the mT/mG reporter element that expresses TdTomato in −sc cells. Scale bar = 100 μm. Representative videos are included in the supplement. (C) There is a significant reduction in migration velocity of +sc cells (N=114) that lack MMP-13 compared to −sc cells (N=103) expressing MMP-13 at 2% serum. In 20% serum +sc cells (N=73) do not exhibit impaired migration. (D) Similar schematic to (A), with TAM injections beginning on Day 1 or 14 of life, and mice being dissected at day 28 of life. (E) Examples of +sc/−scMmp13fl/fl TA muscles stained with laminin and expressing GFP indicating derivation from Pax7 cells recombined to ablate Mmp13. Scale bar 50 μm. (F) Muscle mass is significantly reduced in EDLs from +sc (N=5) compared to −scMmp13fl/fl (N=7) EDL muscles when TAM treatment occurred at Days 0–4. EDL mass was not reduced when TAM treatment occurred starting at Day 14 (N=3). In scMmp13+/+ mice, TAM treatment starting at Day 0 did not alter EDL mass (N=3 −sc, and N=4 +sc). (G) Similar schematic as (A) except at day 0 adult mice are injected with CTX and dissected at day 11 and day 14 of regeneration. Additionally, a mouse with Mmp13+/+ Pax7CreER mT/mG (+scMmp13+/+) was used as a further control for TAM injection. (H) Examples of +sc/−scMmp13fl/fl and +scMmp13+/+ TA muscles stained with laminin and expressing GFP indicating derivation from Pax7 cells recombined to express GFP from 14 days post CTX injection. Scale bar 50 μm. The cumulative distributions from (I) 11 days (Mmp13+/+ N=6;Mmp13fl/fl N=6) and (J) 14 days (Mmp13+/+ N=7;Mmp13fl/fl N=3) following CTX injections show significantly smaller fibers in +scMmp13fl/fl compared to +scMmp13+/+ muscles, similar to global Mmp13−/−. The * represents a significant (p<0.05) difference between satellite cells which express and do not express MMP-13, with the bar representing the range of fiber areas that have a significant difference in the cumulative distribution. The † represents a significant difference between each genotype with TAM treatment.
To confirm that the in vitro phenotype of +scMmp13fl/fl cells could replicate the global knockout model, the migration velocity was measured as previously shown (Fig. 5B; Supplementary Videos for Fig. 5). Consistent with Mmp13−/− myoblasts, tamoxifen (TAM) treated +scMmp13fl/fl cells had a similar velocity to WT cells at 20% serum and cells of the same genotype that were not exposed to TAM (−scMmp13fl/fl). However, while −scMmp13fl/fl maintained a consistent velocity in 2% serum, +scMmp13fl/fl cells displayed a significant decrease in migration velocity (Fig. 5C), similar to Mmp13−/− myoblasts in the same condition. Taken together, the impairment of myoblast migration was dependent predominantly on autocrine sources of MMP-13, rather than that secreted from fibers.
Muscles in +scMmp13fl/fl mice were examined early in life when satellite cell incorporation into growing muscle fibers is most dramatic [53], through removal of MMP-13 from the Pax7 lineage +scMmp13fl/fl starting postnatal Day 1 (Fig. 5D). The presence of GFP positive fibers in +scMmp13fl/fl EDL muscles indicated that Mmp13−/− satellite cells fused to growing fibers (Fig. 5E). However, EDL muscle mass was significantly smaller compared to muscles from littermate and sex-matched −scMmp13fl/fl mice (Fig. 5F). This effect was not attributable to the early TAM injection as Mmp13+/+ mice with and without TAM injection showed no changes in muscle mass. Importantly, the growth impairment occurred only when TAM treatment began early in postnatal life, as the same treatment regimen employed at Day 14 of life did not exhibit growth impairments (Fig. 5F). Hence, MMP-13 expressed by satellite cells is important for normal post-natal growth.
Our initial observation of regeneration defects used mice with complete ablation of Mmp13. To clarify if MMP-13 production by satellite cells contributed to regeneration, we performed CTX injection into muscles from +scMmp13fl/f mice, mice with the same genotype but without TAM injection (−scMmp13fl/fl), and mice treated with TAM but lacking floxed alleles of Mmp13 (+scMmp13+/+) (Fig. 5G). GFP positive fibers were evident in regenerating muscles from the TAM injected animals (Fig. 5H), and centrally nucleated fibers were apparent in all CTX treated muscles, indicating robust satellite cell fusion. Analysis of GFP positive fibers in +scMmp13fl/fl and +scMmp13+/+ mice revealed significantly smaller muscles fibers in +scMmp13fl/fl mice at 11 and 14 days after CTX injection (Fig. 5I/J). This difference recapitulated much of the defect observed in Mmp13−/− muscles injected with CTX. Together these results show that the impaired regenerative capacity associated with Mmp13−/− muscles is present when MMP-13 is removed only from muscle progenitors, implicating muscle progenitors as a critical source of MMP-13 in regenerating muscle, despite the likely production of MMP-13 by a multitude of cell types present during the repair process.
Discussion
The goal of this study was to determine the role of MMP-13 in muscle homeostasis, regeneration, and growth. Several studies have taken advantage of the Mmp13−/− mouse line, and other than abnormal growth plates [13, 54], no other phenotypes have been described [55–57]. While healthy adult skeletal muscle has minimal MMP-13 activity, our findings place MMP-13 among the family of MMPs with important roles in skeletal muscle repair and growth. Even though MMP-13 may be provided by other cells and by fibers within the muscle bed, defects in growth and regeneration persist in the absence of satellite cell expression of MMP-13. Thus, a critical partner and source of MMP-13 activity is the satellite cell, which relies on MMP-13 for migration.
As skeletal growth and muscle growth are linked, the impact of MMP-13 on bone growth may have indirectly affected muscle size in young mice. However, the levels of MMP-13 are negligible in healthy adult muscle, which may underly the minimal impact of this collagenase in homeostatic muscle. Similar results have been reported for MMP-2 deficient mice, which have unchanged fiber size and capillary density at baseline conditions [23]. A more relevant situation to examine MMP-13 actions in muscle is during regeneration, given MMP-13’s known role in wound healing [54], and previous demonstrations of MMP-13 up-regulation 7–14 days after CTX injection (GDS234) [7, 58]. Our current findings reflect this timecourse, with delayed regeneration in the intermediate timepoints, but the possible functional overlap within the MMP family may have enabled resolution of damage by day 21. Despite the clear phenotype during regeneration in mice lacking MMP-13, the collagenase’s absence had no consistent impact on collagen area of the muscle. In part, this points to the extensive overlap in target substrates by the MMP family [59], as well as the several-fold lower MMP-13 levels with respect to its other family members. However, the lack of an MMP-13 dependent impact on collagen content is distinct from other clinical situations, such as idiopathic pulmonary fibrosis. Absence of MMP-13 in a bleomycin murine idiopathic pulmonary fibrosis model results in excessive ECM deposition, supporting the need for MMP-13 for collagenolytic activity and to counter inflammatory-mediated ECM production [60]. Thus, in muscle, despite the requirement for MMP-13 in the efficient resolution of damage, it does not appear to be essential as a counterbalance to progressive fibrosis associated with inflammation.
This situation is exemplified in the clinically relevant model of DMD, the mdx mouse, which undergoes persistent injury and regeneration and progressive fibrosis, which is, in part, due to heightened chronic inflammation. By crossing mdx and Mmp13−/− mouse lines, we could then determine the role of MMP-13 in chronically damaged muscle. We evaluated multiple functional muscle parameters known to be disrupted in the mdx mouse; specific force, passive stiffness [38], and susceptibility to ECC injury displayed no significant changes caused by lack of MMP-13, although all of these parameters were different than WT muscles. Likewise, muscles were histologically similar, which is in contrast to the effects observed when other MMPs are altered in mdx muscle. MMP-2 ablation in mdx mice has shown a decrease in both centrally and peripherally nucleated fiber sizes in TA muscle with a similar effect in capillary density linked to reduced VEGF availability [23]. Ablating MMP-9 from the mdx mouse has protective effects, with increased specific force associated with a decrease in inflammatory factors [24]. However, a confounding observation is that increased MMP-9 expression protected mdx muscle with fiber hypertrophy and minimized fibrosis [25]. These previous studies highlight the multifunctional roles of MMPs in muscle disease, which can promote inflammation, inhibit fibrosis, support angiogenesis, and increase damage. The effect of an MMP likely depends on timing and location of MMP activity. These factors could neutralize each other in the Mmp13−/−:mdx muscle to maintain the same muscle function. The lack of histological phenotype suggests that MMP-13 is compensated for by other proteases or is not required in mdx muscle. As observed in the high serum 2D migration experiments, high levels of MMPs can mask a role for MMP-13; similarly, the high overall MMP levels persistent in mdx muscle [20] could hide a more severe phenotype from Mmp13−/−:mdx mice. As myoblasts appear unable to migrate through a dense collagen network, myoblast migration through the fibrotic mdx muscles may be severely impaired regardless of the ability to express MMP-13. MMP-13 can cleave fibrillar collagen type II much faster than collagen I or III [59], and as the ECM of muscle is predominantly comprised collagens types I and III, with little type II [61] MMP-13 may serve little role in satellite cell migration through the interstitial matrix. Alternatively, the progressive crosslinking within the dystrophic ECM may prevent the ability for MMP-13 to cleave interstitial fibrillar collagens [40]. This would be in contrast to the denatured collagen substrates for MMP-2/9, which may still persist in the dystrophic muscle.
Despite the lack of phenotype observed in Mmp13−/−:mdx mice, MMP-13 as a collagenase may well limit collagen accumulation if expressed in fibrotic muscle at supraphysiological levels. The determination of what levels would be therapeutic could be minimal given the relatively low abundance of this MMP. Activation of MMPs with relaxin has been shown to enhance muscle regeneration and limit fibrosis [62]. It is intriguing that, as opposed to limb muscle, the diaphragm of mdx mice (GDS641) [63] and muscles from DMD patients (GDS3027) [64] do not have elevated Mmp13 expression. This is despite dramatic upregulation of fibrillar collagen and TIMPs, suggesting that MMP-13 overexpression may help muscles restore collagen balance. Alternatively, supplementing MMP-13 may enhance migration even in the fibrotic environment, as a similar collagenase, MMP-1, which enhances myoblast engraftment in skeletal muscle [65]. The potential of MMP-13 to serve as a supplementary therapy in muscle disease may exist, but this was not evaluated in this study.
Hypertrophy of muscle fibers necessitates the remodeling of the ECM that surrounds them to afford their expansion, and thus notionally, MMP activity would be required. In line with this premise, Mmp13 expression is elevated from day 7 to day 14 synergist ablation model of hypertrophy (GDS4932) [66]. MMP-2 ablation has been shown to limit muscle hypertrophy in a synergistic ablation model as well [67], supporting the role for MMPs in muscle growth. Because our initial indication that MMP-13 was expressed in muscle arose following increased IGF-1 levels, we reasoned that elevated MMP-13 contributed to IGF-1 dependent actions in skeletal muscle [51], and it also provided a way to validate the ablation of MMP-13 in the knock out mice. In the absence of MMP-13, IGF-1 mediated hypertrophy still occurred; however, the surprising reduction of centrally nucleated muscle fibers suggested an inhibition on satellite cell actions led to the blunted hypertrophic response. Previous work removing satellite cell activity through irradiation demonstrated an approximate 50% decrease in muscle hypertrophy in response to IGF-1 [50], and more recently, satellite cell ablation prevented hypertrophy in muscle overload experiments [68]. However, a limitation of this former study was the elimination of many cells within the muscle in addition to satellite cells. Indeed, the current observation is more reflective of the ability of muscle to grow following genetic ablation of satellite cells dependent on age [49]. While we did not pursue longer term evaluation of hypertrophy in this model, it is clear that MMP-13 is not required for hypertrophy per se, but it is important for the incorporation of satellite cells to growing fibers.
The above experiments suggested that satellite cells suffered the primary consequences of MMP-13 loss. As such, satellite cell migration in vitro was also impaired, even when only satellite cell expression of Mmp13 was targeted. From a broader perspective, MMP13 mediated migration is a phenomenon shared with other cell types in physiological and pathological conditions, including keratinocytes [54] and melanocytes [69], where myoblasts appear to have adopted a similar use to ensure efficient muscle growth and repair. Previous work had shown that in the C2C12 myogenic cell line a small molecular inhibitor of MMP-13 decreased migration and transcriptional markers of myogenic differentiation [7], with no change in proliferation or fusion. Our current work is consistent with this study in that the principal effect of MMP-13 on myogenic cells is on migration. As expected, Mmp13−/− primary myoblasts were not able to migrate through a fibrillar collagen I substrate in 3D, given collagen is MMP-13’s primary substrate. However, fibrillar collagen also presented a virtually impenetrable barrier to WT cells, which was recently observed of mouse myoblasts in a related study [70]. This indicates that endogenous levels of MMP-13 are not sufficient to migrate through a collagen network similar to the interstitial matrix, or dense networks in fibrotic muscle. The impact of MMP-13 was readily observed in cells migrating through BME, which is a condition that mimics the basement membrane juxtaposed to muscle fibers [71]. While BME does not contain fibrillar collagen, it does contain many MMP-13 substrates, including fibronectin [59]. MMP-13’s role in 2D is less readily apparent, and required low serum levels to unmask the impact that loss of MMP-13 had on migration. Estimations of MMP-13 levels in serum are extremely low in comparison to other MMPs, where in human serum it is 4 orders of magnitude lower than MMP-2 and −9 [72], supporting our speculation that other MMPs can substitute for the absence of MMP-13 at high concentrations. For myoblasts, the local MMP-13 source provides sufficient activity in their microenvironment to move. In addition, myoblasts could secrete their own matrix components they can utilize as scaffolds on which to migrate, or a role for MMP-13 in the migration machinery in the cell. MMP-13 has been observed in myoblasts intracellularly, especially during proliferation and later phases of myoblast maturation [7].
Many cells in addition to myoblasts within the muscle milieu may secrete MMP-13, including fibroblasts [73] and macrophages [44] that are present in muscle tissue. In order to selectively remove MMP-13 from myogenic cells in a manner that did not interfere with development, a tamoxifen inducible system was used [31]. Despite the expression of MMP-13 by other cell types, removing MMP-13 from satellite cells largely mirrored the results of the global Mmp13−/− mice in muscle regeneration. This result establishes myogenic cells as a critical source of MMP-13 in skeletal muscle regeneration. MMPs are also known to be active during development, as is the case in skeletal muscle [74]. While adult mice had no overt phenotype, investigation of 4-week-old mice revealed a delayed increase in muscle mass when MMP-13 is removed specifically from satellite cells, and a general deficit in growth at the same age with total MMP-13 ablation. This coincides with a period in which Pax7+ satellite cells are highly active in providing new myonuclei [32], which may continue into pre-puberty [75]. While we can establish that MMP-13 is important in satellite cells in vivo, we cannot definitively conclude if myoblast migration is altered in vivo in Mmp13−/− cells. However, recent intravital imaging of myoblasts in regenerating muscle does confirm that mammalian myoblasts are highly migratory cells [76], further supporting evidence for MMP-13’s role in muscle regeneration to be a consequence of satellite cell migration.
The satellite cell specific knockout of MMP-13 eliminates inflammatory cells and interstitial cells as being critical sources of MMP-13 during regeneration, however it remains to be determined if the satellite cells themselves are a critical source, or if newly regenerated fibers derived from the satellite cells lacking Mmp13 are the limitation. In our previous studies, we noted that Mmp13 expression occurred late in the differentiation process as nascent myotubes matured, as well as later in regenerating muscle [7]. Hence, this suggested that muscle fibers were the primary source of MMP-13. ECM remodeling around the forming muscle fiber could then enable the fiber to expand in girth or digest the ECM to afford satellite cell incorporation. In addition to the muscle fibers, we also observed that satellite cells exhibited positive MMP-13 staining particularly as they proliferated, where the intensity of MMP-13 signal was greatest on the side opposing the cleavage furrow. This suggested that a second source of MMP-13 might complement that from the muscle fibers and enable satellite cells to digest the ECM as they moved through it. Indeed, the live cell imaging analysis of satellite cells lacking MMP-13 move more slowly, regardless of whether the cells are from mice with total MMP-13 ablation or satellite specific deletion. Our interpretation of these findings must now acknowledge that the source of MMP-13 is critical. Even though the satellite cell production of MMP-13 may be small with respect to that from the muscle fibers or other cell types, the satellite cell source appears to be necessary to enable satellite cell migration, and their contribution to regeneration and muscle growth.
Supplementary Material
Acknowledgements
This work was supported by NIH AR057363 to ERB, and the Wellstone MDCRC Physiological Assessment Core (NIH U54 AR052646). LRS was supported, in part, by NIH K99/R00 AR067867. K. Rakoczy was supported by the Undergraduate Scholars Program, University of Florida.
The authors are grateful to Drs. D. Leong and B. Wheeler for assistance with primary cultures and image analysis, and to the Penn Dental Medicine Live Cell Imaging Core for their expertise.
Abbreviations
- BME
basement membrane extract
- CTX
cardiotoxin
- DMD
Duchenne Muscular Dystrophy
- ECC
eccentric contraction
- EDL
extensor digitorum longus
- IGF-1
InsulinLike Growth Factor-1
- MMP
matrix metalloproteinase
- +scMmp13fl/fl
satellite cell specific knockout of MMP-13
- TA
tibialis anterior
- TAM
tamoxifen
- TIMPs
tissue inhibitors of metalloproteinases
- VEGF
vascular endothelial growth factor
Footnotes
Ethics Statement
The research does not include human studies, and all animal studies were approved by institutional animal care and use committees.
Disclosure Statement
The authors have no conflicts of interest to declare.
References
- 1.Rohani MG, Parks WC: Matrix remodeling by MMPs during wound repair. Matrix Biol 2015;44–46:113–121. [DOI] [PubMed] [Google Scholar]
- 2.Mittal R, Patel AP, Debs LH, Nguyen D, Patel K, Grati M, Mittal J, Yan D, Chapagain P, Liu XZ: Intricate Functions of Matrix Metalloproteinases in Physiological and Pathological Conditions. J Cell Physiol 2016;231:2599–2621. [DOI] [PubMed] [Google Scholar]
- 3.Kessenbrock K, Plaks V, Werb Z: Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 2010;141:52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moss FP, Leblond CP: Nature of dividing nuclei in skeletal muscle of growing rats. J Cell Biol 1970;44:459–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yin H, Price F, Rudnicki MA: Satellite cells and the muscle stem cell niche. Physiol Rev 2013;93:23–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giannandrea M, Parks WC: Diverse functions of matrix metalloproteinases during fibrosis. Dis Model Mech 2014;7:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lei H, Leong D, Smith LR, Barton ER: Matrix metalloproteinase 13 is a new contributor to skeletal muscle regeneration and critical for myoblast migration. Am J Physiol Cell Physiol 2013;305:C529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishimura T, Nakamura K, Kishioka Y, Kato-Mori Y, Wakamatsu J, Hattori A: Inhibition of matrix metalloproteinases suppresses the migration of skeletal muscle cells. J Muscle Res Cell Motil 2008;29:37–44. [DOI] [PubMed] [Google Scholar]
- 9.Brew K, Nagase H: The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta 2010;1803:55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller MC, Manning HB, Jain A, Troeberg L, Dudhia J, Essex D, Sandison A, Seiki M, Nanchahal J, Nagase H, Itoh Y: Membrane type 1 matrix metalloproteinase is a crucial promoter of synovial invasion in human rheumatoid arthritis. Arthritis Rheum 2009;60:686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nistico P, Bissell MJ, Radisky DC: Epithelial-mesenchymal transition: general principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb Perspect Biol 2012;4:pii:a011908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deryugina EI, Quigley JP: Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev 2006;25:9–34. [DOI] [PubMed] [Google Scholar]
- 13.Inada M, Wang Y, Byrne MH, Rahman MU, Miyaura C, Lopez-Otin C, Krane SM: Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc Natl Acad Sci U S A 2004;101:17192–17197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kosaki N, Takaishi H, Kamekura S, Kimura T, Okada Y, Minqi L, Amizuka N, Chung UI, Nakamura K, Kawaguchi H, Toyama Y, D’Armiento J: Impaired bone fracture healing in matrix metalloproteinase-13 deficient mice. Biochem Biophys Res Commun 2007;354:846–851. [DOI] [PubMed] [Google Scholar]
- 15.Gill SE, Parks WC: Metalloproteinases and their inhibitors: regulators of wound healing. Int J Biochem Cell Biol 2008;40:1334–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bellayr I, Holden K, Mu X, Pan H, Li Y: Matrix metalloproteinase inhibition negatively affects muscle stem cell behavior. Int J Clin Exp Pathol 2013;6:124–141. [PMC free article] [PubMed] [Google Scholar]
- 17.Snyman C, Niesler CU: MMP-14 in skeletal muscle repair. J Muscle Res Cell Motil 2015;36:215–225. [DOI] [PubMed] [Google Scholar]
- 18.Zimowska M, Brzoska E, Swierczynska M, Streminska W, Moraczewski J: Distinct patterns of MMP-9 and MMP-2 activity in slow and fast twitch skeletal muscle regeneration in vivo. Int J Dev Biol 2008;52:307–314. [DOI] [PubMed] [Google Scholar]
- 19.Bedair H, Liu TT, Kaar JL, Badlani S, Russell AJ, Li Y, Huard J: Matrix metalloproteinase-1 therapy improves muscle healing. J Appl Physiol 2007;102:2338–2345. [DOI] [PubMed] [Google Scholar]
- 20.Kherif S, Lafuma C, Dehaupas M, Lachkar S, Fournier JG, Verdiere-Sahuque M, Fardeau M, Alameddine HS: Expression of matrix metalloproteinases 2 and 9 in regenerating skeletal muscle: a study in experimentally injured and mdx muscles. Dev Biol 1999;205:158–170. [DOI] [PubMed] [Google Scholar]
- 21.Ohtake Y, Tojo H, Seiki M: Multifunctional roles of MT1-MMP in myofiber formation and morphostatic maintenance of skeletal muscle. J Cell Sci 2006;119:3822–3832. [DOI] [PubMed] [Google Scholar]
- 22.Smith LR, Barton ER: Regulation of fibrosis in muscular dystrophy. Matrix Biol 2018;68–69:602–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyazaki D, Nakamura A, Fukushima K, Yoshida K, Takeda S, Ikeda S: Matrix metalloproteinase-2 ablation in dystrophin-deficient mdx muscles reduces angiogenesis resulting in impaired growth of regenerated muscle fibers. Hum Mol Genet 2011;20:1787–1799. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Mittal A, Makonchuk DY, Bhatnagar S, Kumar A: Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum Mol Genet 2009;18:2584–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dahiya S, Bhatnagar S, Hindi SM, Jiang C, Paul PK, Kuang S, Kumar A: Elevated levels of active matrix metalloproteinase-9 cause hypertrophy in skeletal muscle of normal and dystrophin-deficient mdx mice. Hum Mol Genet 2011;20:4345–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu N, Jansen ED, Davidson JM: Comparison of mouse matrix metalloproteinase 13 expression in freeelectron laser and scalpel incisions during wound healing. J Invest Dermatol 2003;121:926–932. [DOI] [PubMed] [Google Scholar]
- 27.Balbin M, Fueyo A, Knauper V, Lopez JM, Alvarez J, Sanchez LM, Quesada V, Bordallo J, Murphy G, LopezOtin C: Identification and enzymatic characterization of two diverging murine counterparts of human interstitial collagenase (MMP-1) expressed at sites of embryo implantation. J Biol Chem 2001;276:10253–10262. [DOI] [PubMed] [Google Scholar]
- 28.Leeman MF, Curran S, Murray GI: The structure, regulation, and function of human matrix metalloproteinase-13. Crit Rev Biochem Mol Biol 2002;37:149–166. [DOI] [PubMed] [Google Scholar]
- 29.Lederle W, Hartenstein B, Meides A, Kunzelmann H, Werb Z, Angel P, Mueller MM: MMP13 as a stromal mediator in controlling persistent angiogenesis in skin carcinoma. Carcinogenesis 2010;31:1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stickens D, Behonick DJ, Ortega N, Heyer B, Hartenstein B, Yu Y, Fosang AJ, Schorpp-Kistner M, Angel P, Werb Z: Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development 2004;131:5883–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G: Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 2011;138:3625–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lepper C, Conway SJ, Fan CM: Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature 2009;460:627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L: A global double-fluorescent Cre reporter mouse. Genesis 2007;45:593–605. [DOI] [PubMed] [Google Scholar]
- 34.Park S, Brisson BK, Liu M, Spinazzola JM, Barton ER: Mature IGF-I excels in promoting functional muscle recovery from disuse atrophy compared with pro-IGF-IA. J Appl Physiol (1985) 2014;116:797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Durzynska J, Philippou A, Brisson BK, Nguyen-McCarty M, Barton ER: The pro-forms of insulin-like growth factor I (IGF-I) are predominant in skeletal muscle and alter IGF-I receptor activation. Endocrinology 2013;154:1215–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barton ER, Morris L, Kawana M, Bish LT, Toursel T: Systemic administration of L-arginine benefits mdx skeletal muscle function. Muscle Nerve 2005;32:751–760. [DOI] [PubMed] [Google Scholar]
- 37.Moorwood C, Liu M, Tian Z, Barton ER: Isometric and eccentric force generation assessment of skeletal muscles isolated from murine models of muscular dystrophies. J Vis Exp 2013:e50036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith LR, Barton ER: Collagen content does not alter the passive mechanical properties of fibrotic skeletal muscle in mdx mice. Am J Physiol Cell Physiol 2014;306:C889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith LR, Barton ER: SMASH - semi-automatic muscle analysis using segmentation of histology: a MATLAB application. Skelet Muscle 2014;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith LR, Hammers DW, Sweeney HL, Barton ER: Increased collagen cross-linking is a signature of dystrophin-deficient muscle. Muscle Nerve 2016;54:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shefer G, Yablonka-Reuveni Z: Isolation and culture of skeletal muscle myofibers as a means to analyze satellite cells. Methods Mol Biol 2005;290:281–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schindelin J, Rueden CT, Hiner MC, Eliceiri KW: The ImageJ ecosystem: An open platform for biomedical image analysis. Mol Reprod Dev 2015;82:518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Danoviz ME, Yablonka-Reuveni Z: Skeletal muscle satellite cells: background and methods for isolation and analysis in a primary culture system. Methods Mol Biol 2012;798:21–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kudo Y, Iizuka S, Yoshida M, Tsunematsu T, Kondo T, Subarnbhesaj A, Deraz EM, Siriwardena SB, Tahara H, Ishimaru N, Ogawa I, Takata T: Matrix metalloproteinase-13 (MMP-13) directly and indirectly promotes tumor angiogenesis. J Biol Chem 2012;287:38716–38728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Straub V, Rafael JA, Chamberlain JS, Campbell KP: Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol 1997;139:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hindi SM, Sato S, Choi Y, Kumar A: Distinct roles of TRAF6 at early and late stages of muscle pathology in the mdx model of Duchenne muscular dystrophy. Hum Mol Genet 2014;23:1492–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, Narusawa M, Leferovich JM, Sladky JT,Kelly AM: The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 1991;352:536–539. [DOI] [PubMed] [Google Scholar]
- 48.Bani C, Lagrota-Candido J, Pinheiro DF, Leite PE, Salimena MC, Henriques-Pons A, Quirico-Santos T: Pattern of metalloprotease activity and myofiber regeneration in skeletal muscles of mdx mice. Muscle Nerve 2008;37:583–592. [DOI] [PubMed] [Google Scholar]
- 49.Murach KA, White SH, Wen Y, Ho A, Dupont-Versteegden EE, McCarthy JJ, Peterson CA: Differential requirement for satellite cells during overload-induced muscle hypertrophy in growing versus mature mice. Skelet Muscle 2017;7:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barton-Davis ER, Shoturma DI, Sweeney HL: Contribution of satellite cells to IGF-I induced hypertrophy of skeletal muscle. Acta Physiol Scand 1999;167:301–305. [DOI] [PubMed] [Google Scholar]
- 51.Barton ER, DeMeo J, Lei H: The insulin-like growth factor (IGF)-I E-peptides are required for isoformspecific gene expression and muscle hypertrophy after local IGF-I production. J Appl Physiol (1985) 2010;108:1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL: Viral mediated expression of insulinlike growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci U S A 1998;95:15603–15607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.White RB, Bierinx AS, Gnocchi VF, Zammit PS: Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev Biol 2010;10:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hattori N, Mochizuki S, Kishi K, Nakajima T, Takaishi H, D’Armiento J, Okada Y: MMP-13 plays a role in keratinocyte migration, angiogenesis, and contraction in mouse skin wound healing. Am J Pathol 2009;175:533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fukuda H, Mochizuki S, Abe H, Okano HJ, Hara-Miyauchi C, Okano H, Yamaguchi N, Nakayama M, D’Armiento J, Okada Y: Host-derived MMP-13 exhibits a protective role in lung metastasis of melanoma cells by local endostatin production. Br J Cancer 2011;105:1615–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takaishi H, Kimura T, Dalal S, Okada Y, D’Armiento J: Joint diseases and matrix metalloproteinases: a role for MMP-13. Curr Pharm Biotechnol 2008;9:47–54. [DOI] [PubMed] [Google Scholar]
- 57.Singh A, Rajasekaran N, Hartenstein B, Szabowski S, Gajda M, Angel P, Brauer R, Illges H: Collagenase-3 (MMP-13) deficiency protects C57BL/6 mice from antibody-induced arthritis. Arthritis Res Ther 2013;15:R222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao P, Iezzi S, Carver E, Dressman D, Gridley T, Sartorelli V, Hoffman EP: Slug is a novel downstream target of MyoD. Temporal profiling in muscle regeneration. J Biol Chem 2002;277:30091–30101. [DOI] [PubMed] [Google Scholar]
- 59.Knauper V, Lopez-Otin C, Smith B, Knight G, Murphy G: Biochemical characterization of human collagenase-3. J Biol Chem 1996;271:1544–1550. [DOI] [PubMed] [Google Scholar]
- 60.Nkyimbeng T, Ruppert C, Shiomi T, Dahal B, Lang G, Seeger W, Okada Y, D’Armiento J, Gunther A: Pivotal role of matrix metalloproteinase 13 in extracellular matrix turnover in idiopathic pulmonary fibrosis. PLoS One 2013;8:e73279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gillies AR, Lieber RL: Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 2011;44:318–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mu X, Urso ML, Murray K, Fu F, Li Y: Relaxin regulates MMP expression and promotes satellite cell mobilization during muscle healing in both young and aged mice. Am J Pathol 2010;177:2399–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haslett JN, Kang PB, Han M, Kho AT, Sanoudou D, Volinski JM, Beggs AH, Kohane IS, Kunkel LM: The influence of muscle type and dystrophin deficiency on murine expression profiles. Mamm Genome 2005;16:739–748. [DOI] [PubMed] [Google Scholar]
- 64.Pescatori M, Broccolini A, Minetti C, Bertini E, Bruno C, D’Amico A, Bernardini C, Mirabella M, Silvestri G, Giglio V, Modoni A, Pedemonte M, Tasca G, Galluzzi G, Mercuri E, Tonali PA, Ricci E: Gene expression profiling in the early phases of DMD: a constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J 2007;21:1210–1226. [DOI] [PubMed] [Google Scholar]
- 65.Pan H, Vojnits K, Liu TT, Meng F, Yang L, Wang Y, Huard J, Cox CS, Lally KP, Li Y: MMP1 gene expression enhances myoblast migration and engraftment following implanting into mdx/SCID mice. Cell Adh Migr 2015;9:283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chaillou T, Lee JD, England JH, Esser KA, McCarthy JJ: Time course of gene expression during mouse skeletal muscle hypertrophy. J Appl Physiol (1985) 2013;115:1065–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Q, Joshi SK, Lovett DH, Zhang B, Bodine S, Kim HT, Liu X: Matrix metalloproteinase-2 plays a critical role in overload induced skeletal muscle hypertrophy. Muscles Ligaments Tendons J 2014;4:446–454. [PMC free article] [PubMed] [Google Scholar]
- 68.Egner IM, Bruusgaard JC, Gundersen K: Satellite cell depletion prevents fiber hypertrophy in skeletal muscle. Development 2016;143:2898–2906. [DOI] [PubMed] [Google Scholar]
- 69.Gunn S, Yeh IT, Lytvak I, Tirtorahardjo B, Dzidic N, Zadeh S, Kim J, McCaskill C, Lim L, Gorre M, Mohammed M: Clinical array-based karyotyping of breast cancer with equivocal HER2 status resolves gene copy number and reveals chromosome 17 complexity. BMC Cancer 2010;10:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lund DK, Mouly V, Cornelison DD: MMP-14 is necessary but not sufficient for invasion of three-dimensional collagen by human muscle satellite cells. Am J Physiol Cell Physiol 2014;307:C140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gutierrez JM, Escalante T, Rucavado A, Herrera C, Fox JW: A Comprehensive View of the Structural and Functional Alterations of Extracellular Matrix by Snake Venom Metalloproteinases (SVMPs): Novel Perspectives on the Pathophysiology of Envenoming. Toxins (Basel) 2016;8:pii:E304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jonsson A, Hjalmarsson C, Falk P, Ivarsson ML: Levels of matrix metalloproteinases differ in plasma and serum - aspects regarding analysis of biological markers in cancer. Br J Cancer 2016;115:703–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, Iredale JP: Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol 2007;178:5288–5295. [DOI] [PubMed] [Google Scholar]
- 74.Chin JR, Werb Z: Matrix metalloproteinases regulate morphogenesis, migration and remodeling of epithelium, tongue skeletal muscle and cartilage in the mandibular arch. Development 1997;124:1519–1530. [DOI] [PubMed] [Google Scholar]
- 75.Bachman JF, Klose A, Liu W, Paris ND, Blanc RS, Schmalz M, Knapp E, Chakkalakal JV: Prepubertal skeletal muscle growth requires Pax7-expressing satellite cell-derived myonuclear contribution. Development 2018;145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Webster MT, Manor U, Lippincott-Schwartz J, Fan CM: Intravital Imaging Reveals Ghost Fibers as Architectural Units Guiding Myogenic Progenitors during Regeneration. Cell Stem Cell 2016;18:243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.