

Abstract

Chagas disease is a parasitic infection affecting millions of people across Latin America, imposing a dramatic socioeconomic burden. Despite the availability of drugs, nifurtimox and benznidazole, lack of efficacy and incidence of side-effects prompt the identification of novel, efficient, and affordable drug candidates. To address this issue, one strategy could be probing the susceptibility of Trypanosoma parasites toward NADP-dependent enzyme inhibitors. Recently, steroids of the androstane group have been described as highly potent but nonselective inhibitors of parasitic glucose-6-phosphate dehydrogenase (G6PDH). In order to promote selectivity, we have synthesized and evaluated 26 steroid derivatives of epiandrosterone in enzymatic assays, whereby 17 compounds were shown to display moderate to high selectivity for T. cruzi over the human G6PDH. In addition, three compounds were effective in killing intracellular T. cruzi forms infecting rat cardiomyocytes. Altogether, this study provides new SAR data around G6PDH and further supports this target for treating Chagas disease.
Keywords: Chagas disease, Trypanosoma cruzi, G6PDH, selective inhibitors, steroids
Discovered in the early 20th century by the Brazilian physician Carlos Chagas, Chagas disease is attributed to the infection by the flagellate protozoan Trypanosoma cruzi (T. cruzi). According to the World Health Organization, it is estimated that about 6 to 7 million people worldwide are at risk of infection by T. cruzi.1 Currently there are two licensed drugs for the treatment of Chagas disease: nifurtimox (NFX) and benznidazole (BNZ). Both have serious side effects such as neurological and digestive disorders, hypersensitivity in the form of rashes, in addition to having low efficacy and efficiency during the chronic stage.2,3 Given this current scenario, it is necessary to develop more effective and efficient drugs for the treatment of all stages of infection. The enzyme glucose-6-phosphate dehydrogenase (G6PDH) is the first of the oxidative branch of the pentose-phosphate pathway (PPP). This pathway is key to cellular metabolism because it is a major producer of dihydronicotinamide adenine dinucleotide phosphate (NADPH), a molecule necessary for biosynthetic reductions and important in the maintenance of cellular redox homeostasis, by counteracting the deleterious effects of oxygen radicals.4 The PPP is especially important for parasites, since mammalian hosts use oxidative burst as the first line of defense against infection via the formation of reactive oxygen species, which are counterbalanced in the parasite by NADPH-dependent reactions. This assertion is corroborated by in vitro experiments performed in T. cruzi by Maugeri and co-workers, which points out the increased flow through the PPP in response to oxidative stress.5 G6PDH is of central importance for PPP, since it often presents a high control coefficient, being considered a potential target for the development of drugs.6 In addition, the genetic validation of this enzyme as a target was demonstrated in T. brucei by Cordeiro and co-workers through the partial depletion of this enzyme using tetracycline inducible RNA interference.7
Steroids such as dehydroepiandrostone (DHEA, 1) and epiandrosterone (EA, 2) have long been known for inhibiting human glucose-6-phosphate dehydrogenase (HsG6PDH).8 Recently, HsG6PDH inhibition by DHEA in erythrocytes was shown to promote resistance toward infection by Plasmodium falciparum, the causative agent of malaria.9 DHEA is further believed to act as an immunomodulating agent by increasing the host’s immunity toward infections by T. cruzi.10−12 Epiandrosterone (EA) analogues have been studied in the context of their potential anticancer activity in phenotypic assays, with modifications occurring mainly at the C3 or the C16/17 of the androstane core (Figure 1).13,14 Hamilton and co-workers, in particular, have developed a series of EA-derivatized inhibitors of HsG6PDH, allowing for the identification of key features that promote or disfavor binding to this enzyme.15 16α-Bromoepiandrosterone (BrEA, 3), which is a potent inhibitor of HsG6PDH, was also shown to strongly inhibit T. cruzi G6PDH (TcG6PDH),16 and its parasiticidal effects have been reported for Taenia and Entamoeba.17 In particular, the safety of BrEA (also registered as HE2000) has been confirmed in clinical trials in HIV and malaria patients while its immune-boosting properties were assessed.18−20 Although the mode of inhibition (uncompetitive mechanism) has long been elucidated,21 the exact mode of binding of steroid inhibitors to the enzyme remains unknown, despite a combination of computational and biochemical efforts suggesting it lies near the binding site of its substrate, G6P, and cofactor, NADP.22,23 So far, attempts by Mercaldi and co-workers have yielded the crystal structure of TcG6PDH obtained in a ternary complex with the substrate G6P and reduced cofactor NADPH; however, the desired quaternary complex with a steroid inhibitor, for instance, has not yet been achieved.24 Despite BrEA displaying high affinity for the parasitic enzyme TcG6PDH, it is only twice less active against the human homologous enzyme. Likewise, already known 16α-halogenated analogues (4, 6, and 7) have also proven strong inhibitors of HsG6PDH, which could potentially hamper their use for the treatment of this parasitic disease, on the basis of undesired drug–host interactions.
Figure 1.

Endogenous steroid hormone DHEA (1) and synthetic analogues (2–7).
According to Hamilton and co-workers, the presence of an H-bond donor at C3β substituents is essential for human G6PDH inhibition,15 but this has not yet been reported for TcG6PDH. Thus, the derivatization of the C3β–OH group would allow for the removal of the key H-bonding feature, while introducing chemical diversity to the androstane core, potentially yielding TcG6PDH selective inhibitors. As such, we herein describe the synthesis and biochemical evaluation of a series of C3β-O-modified analogues of EA and BrEA, composed of carboxylic and sulfonic esters, as well as ethers, where aliphatic and heterocyclic groups are introduced. To the best of our knowledge, these compounds have not been described in the literature in the context of G6PDH inhibition and would thus provide the scientific community with novel data in this field.
The first set of compounds (8–24) was composed of carboxylic and sulfonic esters of EA and BrEA25 containing either lipophilic or heterocyclic moieties. Compounds 8–15 represented esters of increasing size, as well as the simple methyl sulfonic esters, and were prepared by reacting steroids 2 and 3 with the desired acyl or sulfonyl chlorides (Scheme 1A). Intermediates 16–18 were synthesized using the same conditions with the appropriate reagent and further treated with either morpholine or N-methylpiperazine to yield heterocyclic steroid analogues 19–24. Although the 16α-brominated equivalents of compounds 16–18 (Scheme 1A, X = Br) were successfully synthesized (data not shown), their reaction with the two heterocyclic amines led to partial elimination of the bromine atom, and attempts to brominate compounds 20 and 24 with copper(II) bromide in methanol at reflux led to the elimination of the carboxylic or sulfone ester moieties at C3.
Scheme 1. Synthesis of EA (2) and BrEA (3) Derivatives Comprising: (A) Aliphatic and Heterocycle-Containing Esters; (B) Aliphatic and Heterocycle-Containing Ethers; (C) Nonhydrolyzable Moieties.

Target compounds are highlighted in blue rectangles.
Because of the potential lability of esters in vivo, we sought to produce C3-ether derivatives, thus demanding the use of harsher conditions to perform O-alkylations at C3. As a consequence, the protection of the C17-ketone as a ketal (25) was required and was performed under conditions described by Hitchin and co-workers (Scheme 1B).26 We chose the simple methyl ethers of EA and BrEA, as well as the equivalent of compounds 10 and 14, that is, the isobutyl ethers. Ketal (25) was thus treated with sodium hydride in methanol under reflux, followed by the addition of methyl iodide to form the desired methyl ether (26).27 Deprotection of the ketal group under acidic condition delivered the target methyl ether of EA (27), and further treatment with copper(II) bromide generated the 16α-brominated analogue 28. In a similar fashion, isobutyl tosylate (29) was reacted with ketal 25 and sodium hydride in the presence of sodium iodide to yield the desired isobutyl ether (30). After deprotection of the ketal, ketone 31 was treated with copper(II) bromide, thus producing its 16α-brominated analogue 32. In order to introduce polar groups similar to the heterocycle-containing compounds 19–24, we focused on analogues of derivatives 22 and 24, where the carbonyl/sulfonyl group and the methylene adjacent to the nitrogen atom would be inverted, producing amide 33 and sulfonamide 34, respectively. From a synthetic point of view, these new compounds would be formed by an SN2 or a Michael addition-type O-alkylation of the corresponding α-bromoacetamide (35) and vinyl sulfone (36) derivatives, which were prepared by known procedures.28,29 Treating ketal 25 with n-butyllithium, compound 35 and tetrabutylammonium iodide (TBAI) gave the desired ether (37) in modest yield,30 and further deprotection of the ketal delivered the target compound (33). For the alkylation of 25 with vinyl sulfone 36, sodium hydroxide and tetrabutylammonium bromide (TBAB) were used, giving the desired ether (38) in 66% yield.31 Likewise, removal of the ketal group yielded the proposed compound (34). Pleasingly, we were able to introduce the 16α-bromine atom under the usual conditions, delivering target compounds 39 and 40.
Finally, we designed a series of compounds deprived of hydrolyzable groups, where the attachment to the steroid and the introduction of heterocycles would be facilitated by using the reactivity of an epoxide (Scheme 1C). Seeking to produce both epimers of the resulting secondary alcohols, we used the adequate glycidyl tosylate surrogates (R)-41 and (S)-42, obtained from the commercially available (S)-(+)-glycidol (43) and (R)-(−)-glycidol (44), respectively. Substitution of the tosylate group by the C3-alcohol of 25 occurred best using an excess of sodium hydride at room temperature, despite the modest yields observed for the glycidyl ethers 45 and 46. Next, the desired target compounds 47–50 were achieved by aminolysis of the epoxides with either morpholine or N-methylpiperazine under gentle heating, subsequent treatment of the crude reactions mixtures with aqueous hydrochloric acid to remove the ketal group, and purification by recrystallization.
All 26 target compounds were evaluated in biochemical assays against Tc and HsG6PDH to calculate the selectivity of the inhibitors and perform a structure–activity relationship (SAR) analysis (Supporting Information, Table S1). Reference compounds DHEA (1), EA (2), and their C3-deoxygenated and 16α-halogenated analogues (3–7) were also assayed. The potency of the inhibitors toward both enzymes is illustrated in Figure 2, facilitating the SAR analysis.
Figure 2.
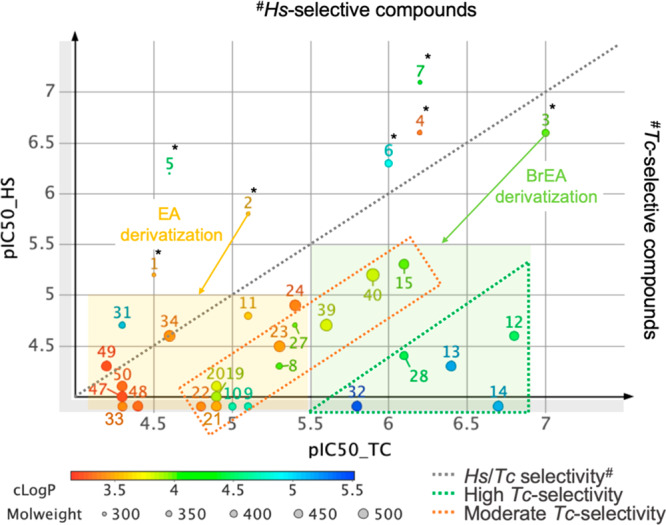
SAR analysis of EA (2) and BrEA (3) derivatives against the parasitic and human G6PDHs. Affinities are shown as pIC50 values, where IC50 is expressed in μM (pIC50 = −log(IC50.10–6). The green dotted triangle represents highly Tc-selective compounds, defined by an IC50_Hs/IC50_Tc ratio >40. Moderate Tc-selectivity (5 < ratio <30) is represented by a dotted orange rectangle. The light green filled rectangle covers BrEA derivatives, whereas the light orange filled rectangle represents EA derivatives. The chart was generated with the DataWarrior software (Osiris).33
As mentioned in the introduction, all reference compounds (1–7)* are HsG6PDH-selective (Figure 2, above the gray dotted line), to the exception of BrEA (3), which is, however, only twice more potent for TcG6PDH. These compounds all have in common the free C3β-alcohol (1–4) or its absence (5–7), further supporting our strategy to produce C3β-derivatized steroids to favor the inhibition of TcG6PDH. Indeed, most of the synthesized EA-analogues are found below the dotted gray line. Special attention should be given to the ester derivatives of BrEA (12–14) in the dotted green triangle, which showed highest potency (0.15 < IC50 (μM) < 0.38) and selectivity toward TcG6PDH, especially compound 14, for which no activity was detected for HsG6PDH even at the highest assayed concentration of 80 μM. Further, it appears that increasing the bulkiness of the ester group contributes to increased selectivity, as can be noted by comparing the acetyl (12, 160-fold) and the isobutyryl (14, >400-fold) esters of BrEA. This same trend is observed for the nonhalogenated analogues (8–10), in the dotted orange rectangle, which display moderate selectivity, due to lower potencies for TcG6PDH (IC50 > 3 μM; pIC50 < 5.5, Figure 2). Methyl sulfones 11 and 15 showed weak (2-fold) and moderate (6-fold) selectivity, respectively.
More polar EA ester analogues containing morpholine and N-methylpiperazine moieties (19–22) were weak inhibitors (IC50 = 10 μM), while the heterocyclic sulfone esters (23–24) performed better (IC50 = ∼5 μM), with only 23 showing moderate selectivity. In the C3β-ether series, 16α-brominated ethers 28 and 32 achieved high selectivities (50-fold range), while morpholine-containing analogues 39 and 40 showed moderate selectivity (5- to 10-fold). Nonhalogenated methyl ether 27 achieved some selectivity toward TcG6PDH, while unexpectedly, isobutyl ether 31 proved 3-fold HsG6PDH-selective, but also a weak inhibitor. Compounds 47–50 showed almost no activity (IC50 > 40 μM) toward either of the G6PDH homologues, noting that they all contain a hydrogen-bond donating group in their propanolamine moiety. Similarly, compound 33, which bears a hydrogen-bond acceptor (amide) at the same position, was also one of the weakest compounds of the series, although the addition of the 16α-bromine somewhat restored potency toward TcG6PDH, as shown with compound 39. Compound 34 was also a weak inhibitor (IC50 = 20 μM) and nonselective.
Finally, looking at the distribution of the compounds according to their calculated partition coefficient (cLogP), compounds with values higher than 3.5 tend to make the more potent inhibitors, while polar compounds (cLogP < 3.5) are poor inhibitors (Figure 2). Clearly, the most potent inhibitors owe their high affinities to the presence of the 16α-bromine atom.
In summary, our strategy to remove a H-bond donating feature at epiandrosterone’s C3β substituents, which is key for HsG6PDH binding, proved successful for producing TcG6PDH selective inhibitors. Despite a recent comparative study highlighting structural features unique to TcG6PDH,32 the exact mechanism underlying high selectivities for 16αBrEA-O-acetylated inhibitors, for instance, still cannot be rationalized.
Next, we evaluated the efficacy of TcG6PDH inhibitors (with pIC50_TC > 5) in killing intracellular T. cruzi (Y strain) amastigotes infecting H9C2 rat cardiomyocytes by an image-based cellular assay. Such multiparametric assay enables us to assess compound efficacy against parasites and toxicity toward host cells, simultaneously. Initially, compounds were assayed at the single concentration of 20 μM. At this concentration, no compounds were able to reduce infection without showing significant host cell toxicity (Figure 3), as being the case for compounds 3, 6, 12, 13, 15, 32, 39, and 40, all derived from BrEA. Nonhalogenated steroids 2, 8, 9, 10, 11, 23, 24, and 27, on the other hand, were not cytotoxic toward host cells, but neither were they effective against T. cruzi. These observations reinforce the idea that bromine addition at C-16 is important for potent TcG6PDH inhibition but may also be responsible for the pronounced toxicity in cell assays. Concerning fluorine-derivatives 4 and 7, and BrEA derivative 14, which were not active in the cell assays despite showing potent enzyme inhibition, one ought to be reminded that several factors such as solubility and membrane permeability (which needs to occur twice to reach intracellular parasites), as well as metabolization, can impact on the general bioavailability of drug candidates. Complex cellular pharmacokinetics have also been suggested by Khare and co-workers for the intracellular T. cruzi forms in particular, while assessing proteasome inhibitors of kinetoplastids.34
Figure 3.

Anti-T. cruzi intracellular assay at single concentration of 20 μM. Black and gray bars represent mean and standard deviation of normalized total host and infected cells, respectively. DMSO values were used for data normalization.
Due to the toxicity observed for a set of compounds at 20 μM, these were selected to be retested at lower concentrations in a dose–response assay format for EC50 and selectivity index (S.I.) determination. The results are presented in Table 1.
Table 1. EC50 Values (μM) for Host Cells and Intracellular T. cruzi Forms.
| Sample | EC50_T. cruzi | EC50_host | S.I.a |
|---|---|---|---|
| BNZ | 2.2 ± 0.4 | >40 | >18 |
| 40 | 2.4 ± 0.4 | 8.5 ± 1.9 | 3.5 |
| 15 | 2.4 ± 0.3 | 7.5 ± 0.6 | 3.1 |
| 39 | 3.2 ± 1 | 5.6 ± 1.6 | 1.8 |
| 6 | 3.5 ± 0.4 | 8.4 ± 1.8 | 2.4 |
| 32 | 4.2 ± 0.5 | 12.3 ± 1.8 | 2.9 |
| 3 | 6.4 ± 2 | 13 ± 2 | 2.0 |
| 12 | 8.5 ± 1.5 | 14.7 ± 4.2 | 1.7 |
| 13 | 16.7 ± 4.2 | 54 ± 3.3 | 3.2 |
S.I. = selectivity index calculated as EC50_host/EC50_T. cruzi. Mean and standard deviation for 3 independent experiments.
At first glance, the Tc/HsG6PDH selectivity and potencies observed in enzymatic assays were not reflected in cell assays. On one hand, highly Tc-selective compounds 32, 12, and 13 showed higher EC50 values for host cells than compounds with no or low Tc-selectivity such as 40, 15, 39, and 6 (Table 1), which indicates that a certain degree of selectivity in cell assays was achieved, at least in terms of toxicity. However, it is the least Tc-selective compounds, which despite having lower potencies for the parasitic enzyme, showed the highest efficacies toward T. cruzi in cell assays. On the other hand, BrEA (3), which is only twice more potent for TcG6PDH, still has a higher EC50_host value and is thus less toxic than its moderately selective congeners. One could propose that possible metabolic pathways could be involved. In fact, BrEA possesses a free C3-OH group which could be prone to sulfatation, as is the case in vivo for DHEA, which renders it inactive toward HsG6PDH.35 In a similar fashion, compounds 12 and 13, which are highly Tc-selective but show lower efficacies could also easily be metabolized. Being esters, they could indeed suffer hydrolysis by esterases and further undergo conjugation. As such, these transformations occurring in the host cells’ cytosol would lower the concentration of inhibitor entering the parasites, which would account for their higher EC50 values in T. cruzi. Conversely, this could explain why nonhydrolyzable ethers 39 and 40 were more active in cell assays. Still, this is merely speculative, since T. cruzi pharmacokinetics are reported to be quite complex.34
Nevertheless, we were pleased to observe that compounds 40, 15, and 32 showed EC50 values for intracellular T. cruzi equivalent to BNZ and at least three times higher for host cells (Table 1 and Figure 4).
Figure 4.

Dose–response curves for normalized host (black) and infected cells (gray) for BNZ, compounds 32, 15, and 40. Plots prepared with GraphPad Prism.
Inspection of cell assay images for these compounds acquired at 10 μM (Figure 5), the concentration closest to the EC50 for host cells, shows a significative reduction in the number of infected cells and infection ratio, equivalent to BNZ. Despite the observed decrease in total host cells, a significative reduction in parasitemia was clearly observed in the remaining cells, supporting the notion that the parasites are more susceptible to the compounds’ action than the host cells.
Figure 5.
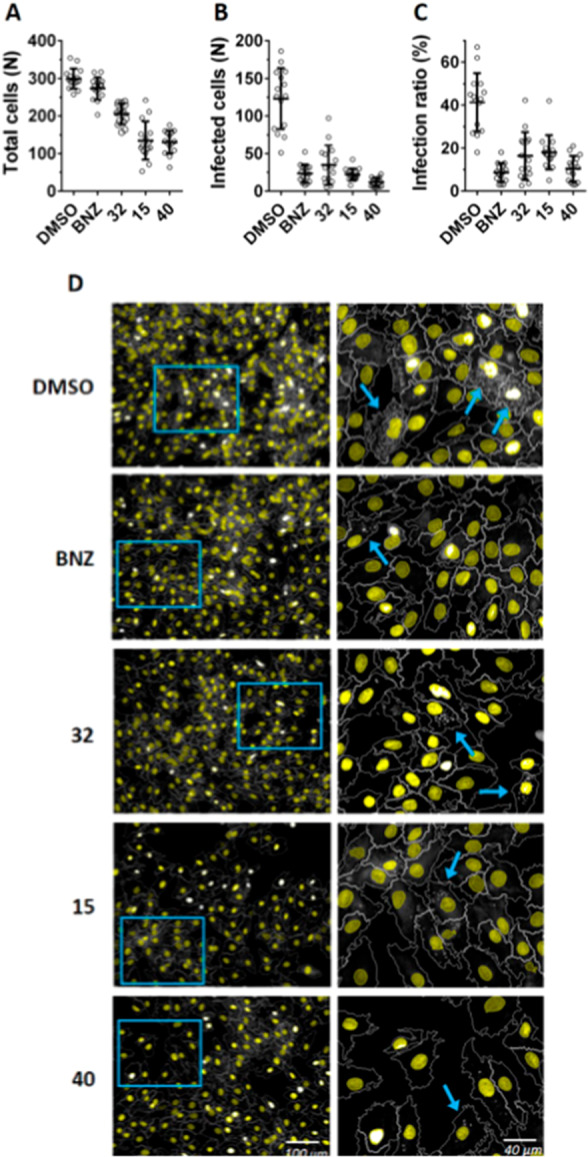
Image analysis of anti-T. cruzi intracellular assay of compounds 32, 15, and 40 (10 μM). Mean and standard deviation for total host cells (A), infected cells (B), and infection ratio (C) per image (N = 15). (D) Representative images (left) and zoom from blue boxes (right) for DMSO, BNZ, and compounds 32, 15, and 40. Host cell nuclei are colored in yellow, and T. cruzi amastigotes appear as white spots in the cytoplasm of infected cells (indicated by blue arrows).
In conclusion, for the first time, selective inhibitors of the parasitic G6PDH enzyme, with respect to the human isoform, have been described. Our data shows that esters of the C3β-OH group of epiandrosterone (2) abolish activity toward the human enzyme, while maintaining high affinity for TcG6PDH, especially in the presence of the 16α-bromine atom, as supported by consistent SAR analyses. On assessing the efficacy of these compounds in cell-based assays, however, most of the halogenated compounds proved cytotoxic at 20 μM. On performing dose–response experiments, isobutyl ether 32, sulfone 15, and sulfonamide 40 displayed EC50 values close to BNZ’s but with narrow therapeutic windows. New chemical scaffolds are thus pursued by our group in order to achieve safer drug candidates.36 Altogether, these are encouraging results showing for the first time that uncompetitive G6PDH inhibitors are able to kill intracellular T. cruzi amastigotes, the parasite form associated with the chronic stage of Chagas disease.
Acknowledgments
The authors wish to thank Prof. Ronaldo Pilli and the Chemistry Institute/UNICAMP for their support, as well as the Nuclear Magnetic Resonance Laboratory at LNBio/CNPEM.
Glossary
Abbreviations
- NADP
nicotinamide adenine dinucleotide phosphate
- G6PDH
glucose-6-phosphate dehydrogenase
- NFX
nifurtimox
- BNZ
benznidazole
- PPP
pentose-phosphate pathway
- NADPH
dihydronicotinamide adenine dinucleotide phosphate
- DHEA
dehydroepiandrosterone
- EA
epiandrosterone
- BrEA
16α-bromoepiandrosterone
- DMSO
dimethyl sulfoxide
- HIV
human immunodeficiency virus.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00106.
Author Contributions
‡ F.F.N. and J.N.F. contributed equally. F.F.N. and M.B. performed the synthesis; J.N.F. produced the purified enzymes and performed the enzymatic assays; A.G.E. performed the cell-assays; A.T.C. designed and supervised the biological assays; M.B. planned the target compounds; A.T.C. and M.B. jointly designed the study and analyzed the results. The manuscript was written through contributions of all authors.
This research was funded by the São Paulo Research Foundation [FAPESP grants for M.B. (2013/16534-4) and A.T.C. (2016/14271-4 and 2018/22202-8)] and by the Coordination for the Improvement of Higher Education Personnel (CAPES 7775/2014-98). The Brazilian Ministry of Science, Technology, Innovation and Communication sponsored F.F.N. (PCI-DB 313424/2015-2).
The authors declare no competing financial interest.
Supplementary Material
References
- WHO . Chagas disease (American trypanosomiasis) fact sheet. https://www.who.int/en/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (accessed 2020-04-08).
- Pérez-Molina J. A.; Pérez-Ayala A.; Moreno S.; Fernández-González M. C.; Zamora J.; López-Velez R. Use of Benznidazole to Treat Chronic Chagas’ Disease: A Systematic Review with a Meta-Analysis. J. Antimicrob. Chemother. 2009, 64 (6), 1139–1147. 10.1093/jac/dkp357. [DOI] [PubMed] [Google Scholar]
- Maya J. D.; Cassels B. K.; Iturriaga-Vásquez P.; Ferreira J.; Faúndez M.; Galanti N.; Ferreira A.; Morello A. Mode of Action of Natural and Synthetic Drugs against Trypanosoma Cruzi and Their Interaction with the Mammalian Host. Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol. 2007, 146 (4), 601–620. 10.1016/j.cbpa.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Babior B. M. Phagocytes and Oxidative Stress. Am. J. Med. 2000, 109 (1), 33–44. 10.1016/S0002-9343(00)00481-2. [DOI] [PubMed] [Google Scholar]
- Maugeri D. A.; Cazzulo J. J. The Pentose Phosphate Pathway in Trypanosoma Cruzi. FEMS Microbiol. Lett. 2004, 234 (1), 117–123. 10.1111/j.1574-6968.2004.tb09522.x. [DOI] [PubMed] [Google Scholar]
- Kovářová J.; Barrett M. P. The Pentose Phosphate Pathway in Parasitic Trypanosomatids. Trends Parasitol. 2016, 32 (8), 622–634. 10.1016/j.pt.2016.04.010. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Cordeiro A. T.; Michels P. A. M. Glucose-6-Phosphate Dehydrogenase Is the Target for the Trypanocidal Action of Human Steroids. Mol. Biochem. Parasitol. 2011, 176 (2), 112–115. 10.1016/j.molbiopara.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Marks P. A.; Banks J. Inhibition of Mammalian Glucose-6-Phosphate Dehydrogenase by Steroids. Proc. Natl. Acad. Sci. U. S. A. 1960, 46 (4), 447–452. 10.1073/pnas.46.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Chen X.; Jiang C.; Fang Z.; Feng Y.; Jiang W. The Effect and Mechanism of Inhibiting Glucose-6-Phosphate Dehydrogenase Activity on the Proliferation of Plasmodium Falciparum. Biochim. Biophys. Acta, Mol. Cell Res. 2017, 1864 (5), 771–781. 10.1016/j.bbamcr.2017.02.010. [DOI] [PubMed] [Google Scholar]
- Santos C. D.; Toldo M. P. A.; Santello F. H.; Filipin M. D. V.; Brazão V.; do Prado Júnior J. C. Dehydroepiandrosterone Increases Resistance to Experimental Infection by Trypanosoma Cruzi. Vet. Parasitol. 2008, 153 (3–4), 238–243. 10.1016/j.vetpar.2008.01.039. [DOI] [PubMed] [Google Scholar]
- Domingues Santos C.; Loria R. M.; Rodrigues Oliveira L. G.; Collins Kuehn C.; Alonso Toldo M. P.; Albuquerque S.; do Prado Júnior J. C. Effects of Dehydroepiandrosterone-Sulfate (DHEA-S) and Benznidazole Treatments during Acute Infection of Two Different Trypanosoma Cruzi Strains. Immunobiology 2010, 215 (12), 980–986. 10.1016/j.imbio.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Kuehn C. C.; Oliveira L. G. R.; Santos C. D.; Augusto M. B.; Toldo M. P. A.; Do Prado J. C. Prior and Concomitant Dehydroepiandrosterone Treatment Affects Immunologic Response of Cultured Macrophages Infected with Trypanosoma Cruzi in Vitro?. Vet. Parasitol. 2011, 177 (3–4), 242–246. 10.1016/j.vetpar.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Romero-Hernández L. L.; Merino-Montiel P.; Meza-Reyes S.; Vega-Baez J. L.; López Ó.; Padrón J. M.; Montiel-Smith S. Synthesis of Unprecedented Steroidal Spiro Heterocycles as Potential Antiproliferative Drugs. Eur. J. Med. Chem. 2018, 143, 21–32. 10.1016/j.ejmech.2017.10.063. [DOI] [PubMed] [Google Scholar]
- Cui H. W.; Peng S.; Gu X. Z.; Chen H.; He Y.; Gao W.; Lv F.; Wang J. H.; Wang Y.; Xie J.; Liu M. Y.; Yi Z.; Qiu W. W. Synthesis and Biological Evaluation of D-Ring Fused 1,2,3-Thiadiazole Dehydroepiandrosterone Derivatives as Antitumor Agents. Eur. J. Med. Chem. 2016, 111, 126–137. 10.1016/j.ejmech.2016.01.058. [DOI] [PubMed] [Google Scholar]
- Hamilton N. M.; Dawson M.; Fairweather E. E.; Hamilton N. S.; Hitchin J. R.; James D. I.; Jones S. D.; Jordan A. M.; Lyons A. J.; Small H. F.; Thomson G. J.; Waddell I. D.; Ogilvie D. J. Novel Steroid Inhibitors of Glucose 6-Phosphate Dehydrogenase. J. Med. Chem. 2012, 55 (9), 4431–4445. 10.1021/jm300317k. [DOI] [PubMed] [Google Scholar]
- Cordeiro A. T.; Thiemann O. H. 16-Bromoepiandrosterone, an Activator of the Mammalian Immune System, Inhibits Glucose 6-Phosphate Dehydrogenase from Trypanosoma Cruzi and Is Toxic to These Parasites Grown in Culture. Bioorg. Med. Chem. 2010, 18 (13), 4762–4768. 10.1016/j.bmc.2010.05.008. [DOI] [PubMed] [Google Scholar]
- Carrero J. C.; Cervantes-Rebolledo C.; Vargas-Villavicencio J. A.; Hernández-Bello R.; Dowding C.; Frincke J.; Reading C.; Morales-Montor J. Parasiticidal Effect of 16α-Bromoepiandrosterone (EpiBr) in Amoebiasis and Cysticercosis. Microbes Infect. 2010, 12 (8–9), 677–682. 10.1016/j.micinf.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Stickney D. R.; Noveljic Z.; Garsd A.; Destiche D. A.; Frincke J. M. Safety and Activity of the Immune Modulator HE2000 on the Incidence of Tuberculosis and Other Opportunistic Infections in AIDS Patients. Antimicrob. Agents Chemother. 2007, 51 (7), 2639–2641. 10.1128/AAC.01446-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frincke J. M.; Stickney D. R.; Onizuka-Handa N.; Garsd A.; Reading C.; Krudsood S.; Wilairatana P.; Looareesuwan S. Reduction of Parasite Levels in Patients with Uncomplicated Malaria by Treatment with HE2000. Am. J. Trop. Med. Hyg. 2007, 76 (2), 232–236. 10.4269/ajtmh.2007.76.232. [DOI] [PubMed] [Google Scholar]
- Reading C.; Dowding C.; Schramm B.; Garsd A.; Onizuka-Handa N.; Stickney D.; Frincke J. Improvement in Immune Parameters and Human Immunodeficiency Virus-1 Viral Response in Individuals Treated with 16α-Bromoepiandrosterone (HE2000). Clin. Microbiol. Infect. 2006, 12 (11), 1082–1088. 10.1111/j.1469-0691.2006.01520.x. [DOI] [PubMed] [Google Scholar]
- Gordon G.; Mackow M. C.; Levy H. R. On the Mechanism of Interaction of Steroids with Human Glucose 6-Phosphate Dehydrogenase. Arch. Biochem. Biophys. 1995, 318 (1), 25–29. 10.1006/abbi.1995.1199. [DOI] [PubMed] [Google Scholar]
- Zhao Z. B.; Liu Y.; Yao Y. Computational Determination of Binding Structures and Free Energies of Glucose 6-Phosphate Dehydrogenase with Novel Steroid Inhibitors. J. Mol. Graphics Modell. 2014, 51, 168–172. 10.1016/j.jmgm.2014.05.009. [DOI] [PubMed] [Google Scholar]
- Ortiz C.; Moraca F.; Medeiros A.; Botta M.; Hamilton N.; Comini M. A.; Schmidt T. J. Binding Mode and Selectivity of Steroids towards Glucose-6-Phosphate Dehydrogenase from the Pathogen Trypanosoma Cruzi. Molecules 2016, 21 (3), 1–14. 10.3390/molecules21030368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercaldi G. F.; Dawson A.; Hunter W. N.; Cordeiro A. T. The Structure of a Trypanosoma Cruzi Glucose-6-Phosphate Dehydrogenase Reveals Differences from the Mammalian Enzyme. FEBS Lett. 2016, 590, 2776–2786. 10.1002/1873-3468.12276. [DOI] [PubMed] [Google Scholar]
- Numazawa M.; Nagaoka M.; Osawa Y. Stereospecific Synthesis of 16α-Hydroxy-17-Oxo Steroids by Controlled Alkaline Hydrolysis of Corresponding 16-Bromo 17-Ketones and Its Reaction Mechanism. J. Org. Chem. 1982, 47 (21), 4024–4029. 10.1021/jo00142a004. [DOI] [Google Scholar]
- Hitchin J. R.; Hamilton N. M.; Jordan A. M.; Lyons A. J.; Ogilvie D. J. A Novel Scalable and Stereospecific Synthesis of 3α- and 3β-Amino-5α-Androstan-17-Ones and 3α- and 3β-Amino- 5α-Pregnan-20-Ones. Tetrahedron Lett. 2012, 53 (23), 2868–2872. 10.1016/j.tetlet.2012.03.124. [DOI] [Google Scholar]
- Tchédam Ngatcha B.; Luu-The V.; Labrie F.; Poirier D. Androsterone 3α-Ether-3β-Substituted and Androsterone 3β-Substituted Derivatives as Inhibitors of Type 3 17β-Hydroxysteroid Dehydrogenase: Chemical Synthesis and Structure–Activity Relationship. J. Med. Chem. 2005, 48 (16), 5257–5268. 10.1021/jm058179h. [DOI] [PubMed] [Google Scholar]
- Roiser L.; Waser M. Enantioselective Spirocyclopropanation of Para-Quinone Methides Using Ammonium Ylides. Org. Lett. 2017, 19 (9), 2338–2341. 10.1021/acs.orglett.7b00869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Wang Y.; Qi X.; Xia G.; Tong K.; Tu J.; Pittman C. U.; Zhou A. Selective Synthesis of Seven- and Eight-Membered Ring Sultams via Two Tandem Reaction Protocols from One Starting Material.. Org. Lett. 2012, 14 (14), 3700–3703. 10.1021/ol301535j. [DOI] [PubMed] [Google Scholar]
- Ohgane K.; Karaki F.; Dodo K.; Hashimoto Y. Discovery of Oxysterol-Derived Pharmacological Chaperones for NPC1: Implication for the Existence of Second Sterol-Binding Site. Chem. Biol. 2013, 20 (3), 391–402. 10.1016/j.chembiol.2013.02.009. [DOI] [PubMed] [Google Scholar]
- Giardinà A.; Giovannini R.; Petrini M. Radical Induced Allylations of Functionalized α-Haloalkylphenyl Sulfones. Tetrahedron Lett. 1997, 38 (11), 1995–1998. 10.1016/S0040-4039(97)00213-X. [DOI] [Google Scholar]
- Ortíz C.; Botti H.; Buschiazzo A.; Comini M. A. Glucose-6-Phosphate Dehydrogenase from the Human Pathogen Trypanosoma Cruzi Evolved Unique Structural Features to Support Efficient Product Formation. J. Mol. Biol. 2019, 431 (11), 2143–2162. 10.1016/j.jmb.2019.03.023. [DOI] [PubMed] [Google Scholar]
- Sander T.; Freyss J.; von Korff M.; Rufener C. DataWarrior: An Open-Source Program For Chemistry Aware Data Visualization And Analysis. J. Chem. Inf. Model. 2015, 55 (2), 460–473. 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- Khare S.; Nagle A. S.; Biggart A.; Lai Y. H.; Liang F.; Davis L. C.; Barnes S. W.; Mathison C. J. N.; Myburgh E.; Gao M. Y.; Gillespie J. R.; Liu X.; Tan J. L.; Stinson M.; Rivera I. C.; Ballard J.; Yeh V.; Groessl T.; Federe G.; Koh H. X. Y.; Venable J. D.; Bursulaya B.; Shapiro M.; Mishra P. K.; Spraggon G.; Brock A.; Mottram J. C.; Buckner F. S.; Rao S. P. S.; Wen B. G.; Walker J. R.; Tuntland T.; Molteni V.; Glynne R. J.; Supek F. Proteasome Inhibition for Treatment of Leishmaniasis, Chagas Disease and Sleeping Sickness. Nature 2016, 537 (7619), 229–233. 10.1038/nature19339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel G. W.; Benes P. Inhibition of Glucose-6-Phosphate Dehydrogenase by Steroids. J. Steroid Biochem. 1972, 3 (6), 903–905. 10.1016/0022-4731(72)90019-2. [DOI] [PubMed] [Google Scholar]
- Mercaldi G. F.; Ranzani A. T.; Cordeiro A. T. Discovery of New Uncompetitive Inhibitors of Glucose-6-Phosphate Dehydrogenase. J. Biomol. Screening 2014, 19 (10), 1362–1371. 10.1177/1087057114546896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.