

Summary
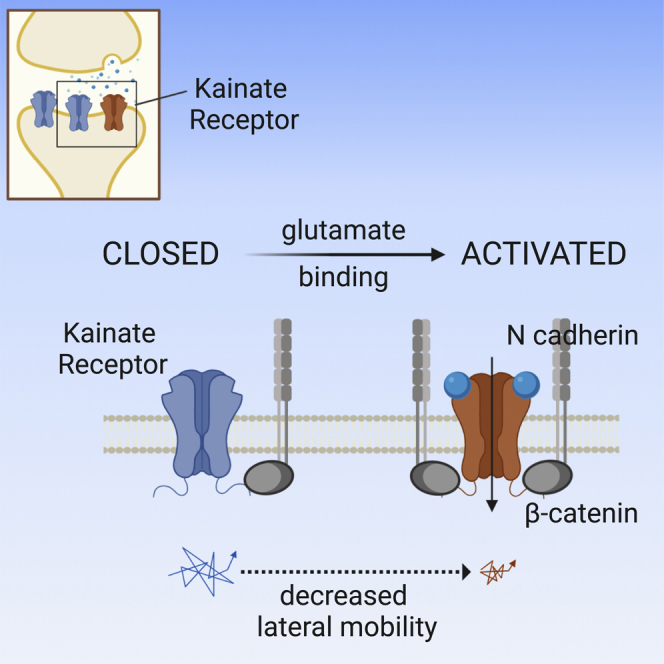
Kainate receptors (KARs) mediate postsynaptic currents with a key impact on neuronal excitability. However, the molecular determinants controlling KAR postsynaptic localization and stabilization are poorly understood. Here, we exploit optogenetic and single-particle tracking approaches to study the role of KAR conformational states induced by glutamate binding on KAR lateral mobility at synapses. We report that following glutamate binding, KARs are readily and reversibly trapped at glutamatergic synapses through increased interaction with the β-catenin/N-cadherin complex. We demonstrate that such activation-dependent synaptic immobilization of KARs is crucial for the modulation of short-term plasticity of glutamatergic synapses. Thus, the present study unveils the crosstalk between conformational states and lateral mobility of KARs, a mechanism regulating glutamatergic signaling, particularly in conditions of sustained synaptic activity.
Keywords: kainate receptors, glutamatergic synapses, short-term synaptic plasticity, lateral diffusion, single-particle tracking, glutamatergic PSD, glutamate uncaging, cadherin, catenin, model simulations
Graphical Abstract
Highlights
-
•
Anchoring of KARs at glutamatergic synapses depends on receptor-glutamate binding
-
•
KARs activation/desensitization promotes receptors trapping at glutamatergic synapses
-
•
N-cadherins mediate the KAR activation/desensitization-dependent anchoring at synapses
-
•
Synaptic trapping of desensitized KARs affects short-term synaptic plasticity
Polenghi et al. report that following their activation, kainate receptors (KARs) are readily trapped at glutamatergic synapses in the open/desensitized state. This mechanism defines the number of ready-to-be-activated KARs at synapses, with direct impacts on short-term synaptic plasticity.
Introduction
Kainate receptors (KARs) are glutamate-gated ion channels that play an important role in the modulation of both inhibitory and excitatory synaptic transmission. They exert depolarizing actions both pre- and postsynaptically and mediate metabotropic and ionotropic signaling (Contractor et al., 2011, Lerma and Marques, 2013). Among their many roles, KARs mediate slow-decaying postsynaptic currents (KAR-EPSCs) that tune the summation of excitatory inputs (Frerking and Ohliger-Frerking, 2002, Goldin et al., 2007) and modulate the induction of spike-timing-dependent Hebbian long-term potentiation (Sachidhanandam et al., 2009). Synaptic localization of KARs, in analogy with other neurotransmitter receptors, is assumed to rely on the receptor anchoring to synaptic scaffold proteins that act as “diffusion traps” (Choquet and Triller, 2013). One of the major subunits of postsynaptic KARs, GluK2, interacts with specific scaffold/adhesion proteins at the excitatory postsynaptic density (PSD) that provide the molecular basis for KAR accumulation at the postsynaptic level (Coussen et al., 2002, Garcia et al., 1998, Hirbec et al., 2003, Fièvre et al., 2016). Analysis of KARs’ lateral diffusion revealed that the dispersion of KARs from the synapse is due to reduced interactions of KARs with PSD-95, which is responsible for the expression of long-term depression of KAR-mediated responses. Thus, the dynamic regulation of KAR-scaffold protein interactions strongly modulates KAR-EPSC strength (Carta et al., 2013). However, the KAR lateral diffusion in relation to the KAR activation by glutamate binding has never been investigated. To address this issue, we exploited an engineered light-gated GluK2 (LiGluK2) receptor that can be activated by light and controlled in the open/desensitized or closed states by illumination with UV or visible light, respectively (Volgraf et al., 2006). This optogenetic tool, in combination with single-particle tracking (SPT), allows the study of LiGluK2 receptor lateral mobility in specific receptor light-induced conformational states at unprecedented temporal resolution. In addition, the use of LiGluK2 allows the selective activation of GluK2 receptors without application of exogenous glutamate, thus preventing potential confounders due to the activation of AMPA and/or NMDA glutamate receptors. We find that LiGluK2 activation (followed by fast desensitization) reversibly immobilizes KARs at glutamatergic synapses. Immobilization of wild-type (WT) GluK2 was also observed in response to GluK2 activation by glutamate bath applications, indicating that this is not a specific feature of LiGluK2 activated by light. KAR activation-dependent stabilization at postsynaptic sites was mediated by the interaction between the GluK2-C terminus and the adhesion protein N-cadherin. This mechanism is responsible for the activity-dependent fast regulation of KARs availability at glutamatergic synapses and the short-term regulation of excitatory synaptic signaling.
Results
LiGluK2 Receptors Are Accumulated at Glutamatergic Synapses
Transfection of LiGluK2 in hippocampal cultured neurons induced the formation of functional KARs. These receptors mediated light-evoked responses (Figure S1A, left panel) with kinetics comparable to those of light- and glutamate-evoked currents mediated by homomeric GluK2 receptors in heterologous systems (Bowie and Mayer, 1995, Reiner and Isacoff, 2014). Moreover, similarly to native GluK2 receptors activated by glutamate, the lectin Concanavalin-A (Con-A) increased the size and abolished the desensitization of LiGluK2 light-activated currents (Bowie et al., 2003, Mayer and Vyklicky, 1989, Paternain et al., 1998) (Figure S1A, right panel). Next, we considered the possibility that heteromerization of LiGluK2 with non-photoswitchable endogenous GluK5 could lead to heteromeric LiGluK2/GluK5 receptors only partially activated by light. However, the K5 subunit was unlikely to be incorporated in surface KARs upon LiGluK2 neuronal transfection, given the markedly distinct kinetics of our light-evoked responses (Figure S1A, left panel) from light-evoked LiGluK2/GluK5-mediated currents (Reiner and Isacoff, 2014) and the lack of response to focal application of the GluK5 subunit selective agonist 5-iodowillardiine (5-IW; Figure S1B). This is in line with previous studies showing that cultured hippocampal neurons do not express the GluK5 subunit (Palacios-Filardo et al., 2016, Ruano et al., 1995). Subsequently, we investigated the surface distribution of LiGluK2 receptors by quantifying the immunoreactivity of hemagglutinin (HA)-tagged LiGluK2 at glutamatergic synapses identified either by overexpression of the postsynaptic marker Homer1c-DsRed or by immunolabeling the presynaptic marker VGLUT1 (Hayashi et al., 2009). The integrated fluorescence intensity of HA-LiGluK2 at synapses (i.e., that co-localized with Homer1c) was significantly higher than that at extrasynaptic sites, suggesting LiGluK2 accumulates at glutamatergic synapses (121.3 ± 15.1 a.u. and 71.3 ± 9.5 a.u. for synaptic and extrasynaptic HA-LiGluK2, respectively; n = 48, from 3 cultures; p < 0.01, Student’s t test; Figures S1C and S1D). The percentage of synaptic HA-LiGluK2 clusters co-localizing with Homer1c was 23.3% ± 1.4% of total HA-LiGluK2 clusters (Figure S1D), in line with previous studies addressing the synaptic localization of both overexpressed and native GluK2 receptors (Martin et al., 2008, Salinas et al., 2006). Comparable values (18.0% ± 1.3%) were obtained when synaptic LiGluK2 receptors were identified by their close proximity to the presynaptic marker VGLUT1 (Figure S1D). Taken together, these data indicate that the transfection of the LiGluK2 subunit in hippocampal cultured neurons leads to the surface expression of functional homomeric LiGluK2 receptors that accumulate at glutamatergic synapses.
Desensitized LiGluK2 Receptors Are Reversibly Immobilized at Glutamatergic Synapses
In a second set of experiments, we studied the relationship between the conformational state and the lateral mobility of GluK2 receptors by combining the use of photoswitchable LiGluK2 with the SPT technique. We tracked the diffusion of individual LiGluK2 receptors coupled to quantum dots (QDs) while simultaneously maintaining receptor conformation in a closed or open/desensitized state by shedding light at 488 nm or 378 nm, respectively (Figures 1A and S1A). Considering that profound desensitization occurs within a few tens of ms after the receptor activation, hereafter we will define the receptors as “desensitized” when illuminated with 378 nm light, although we cannot exclude that a small fraction of receptors might be in the open state. The mobility of synaptic and extrasynaptic LiGluK2 was measured at glutamatergic synapses identified by transfection of Homer1c-DsRed. Similar to other neurotransmitter receptors (Borgdorff and Choquet, 2002, Dahan et al., 2003, Groc et al., 2006, Jacob et al., 2005, Petrini et al., 2014), LiGluK2 freely diffused in the extrasynaptic area but was more confined at synaptic sites (Figures 1C and 1D). Interestingly, LiGluK2 activation/desensitization by 378 nm light reversibly immobilized receptors at synapses (Figure 1B, Videos S1 and S2), as indicated by the reduced receptor diffusion coefficient (closed state = 0.016 μm2s−1, interquartile range (IQR): 0.004–0.033 μm2s−1; desensitized state = 0.008 μm2s−1, IQR: 0.002–0.016 μm2s−1; ntrajectories = 140; p < 0.001, Mann-Whitney U test; Figure 1C, left panel) and by the increased receptor immobile fraction (closed state = 0.27 ± 0.03; desensitized state = 0.44 ± 0.04; p < 0.001, Student’s t test; Figure 1C, middle panel). Moreover, light-induced LiGluK2 desensitization increased receptor confinement at synapses, as shown by the reduction of mean square displacement (MSD) versus time curve plateau (p < 0.001, t test at steady state; Figure 1C, right panel), the increased fraction of time spent at synapses (closed state = 58% ± 0.02%; desensitized state = 0.67 ± 0.02; p < 0.01, Student’s t test; Figure S1E), and the decreased number of transitions between extrasynaptic and synaptic compartments (closed state = 2.39 ± 0.09; desensitized state = 1.95 ± 0.08; p < 0.0005, respectively, Student’s t test; Figure S1E). Notably, LiGluK2 photoactivation did not change the lateral mobility of extrasynaptic LiGluK2, suggesting that the light-evoked changes of LiGluK2 lateral diffusion were specific for synaptic receptors (median diffusion coefficient: closed state = 0.020 μm2s−1, IQR: 0.006–0.051 μm2s−1, desensitized state = 0.021 μm2s−1, IQR: 0.005–0.055 μm2s−1, ntrajectories = 250; ns, Mann–Whitney U test; Figure 1D, left panel; immobile fraction: closed state = 0.27 ± 0.01, desensitized state = 0.29 ± 0.01; ns, Student’s t test; Figure 1D, middle panel; MSD versus time curve plateau; ns, Student’s t test at steady state; Figure 1D, right panel). These results indicate that LiGluK2 receptor transition into the desensitized state is sufficient for its reversible trapping at glutamatergic synapses.
Figure 1.

Desensitized LiGluK2 Receptors Are Reversibly Trapped at Glutamatergic Synapses
(A) Schematic of SPT experiments: 488 nm and 380 nm light illumination was used to track receptors in the closed (blue) and desensitized (purple) states, respectively. A second 488-nm pulse induced recovery in the closed state (gray). The protocol was repeated five times every 2 min.
(B) Example trajectories (yellow) of the same individual LiGluK2 receptor diffusing at synapses (Homer1c, red) in the states described in (A). Scale bar, 1 μM.
(C and D) Summary of median diffusion coefficient (±IQR), immobile fraction, and MSD versus time curves of synaptic LiGluK2 (C) (ntrajectories = 140, in 10 neurons from three independent cultures) and extrasynaptic LiGluK2 (D) (ntrajectories = 250, in 10 neurons from three independent cultures) in the closed, desensitized, and recovery closed states.
(E) Schematic of SPT experiments as in (A), in the continued presence of VGCC blockers (2-APB, D-APV, ω-conotoxin MVIIC, GYKI 53655, nifedipine, and ryanodine; black) delivered after the initial 488-nm illumination (blue).
(F) Example trajectories (yellow) of an individual LiGluK2 receptor diffusing over a portion of dendrite (green) in the indicated states. Homer1c indicates synapses (red). Scale bar, 1 μM.
(G and H) Summary of median diffusion coefficient, immobile fraction, and MSD versus time curve of synaptic (G) and extrasynaptic (H) LiGluK2 in different states as indicated in (E) (ntrajectories: synaptic = 100; extrasyaptic = 206).
Unless otherwise stated, data are presented as mean ± SEM, ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.005; ns, non-significant. See also Figures S1 and S2.
KARs’ Desensitization-Dependent Synaptic Immobilization Is Not Calcium Dependent
The lateral mobility of excitatory and inhibitory fast ligand-gated receptors critically depends upon intracellular calcium concentration (Bannai et al., 2009, Borgdorff and Choquet, 2002). Thus, LiGluK2 synaptic immobilization could be caused by the increase of cytosolic calcium from several different pathways: (1) voltage-gated calcium channels (VGCCs) activated by LiGluK2-induced depolarization; (2) NMDA receptors (NMDARs) activated by spontaneous glutamate release concomitant to LiGluK2-induced depolarization; (3) calcium-permeable AMPA receptors (AMPARs) activated by spontaneous glutamate release; and (4) activated LiGluK2 receptor. To assess each pathway, we sequentially applied nifedipine/ω-conotoxin MVIIC, D-APV, and GYKI 53655 to block L-/P-Q-/N-type VGCCs, NMDARs, and AMPARs, respectively (Figures 1E and S1F). In addition, we administered 2-APB and ryanodine to block intracellular IP3Rs and RyRs calcium channels, respectively (Figure S1F). Notably, neither of these blockers reduced light-induced LiGluK2 immobilization at synapses (Figure 1F) (median diffusion coefficient: closed state = 0.014 μm2s1, IQR: 0.004–0.026 μm2s–1; desensitized state = 0.003 μm2s–1, IQR: 0.001–0.018 μm2s–1; ntrajectories = 100; p < 0.001, Mann-Whitney U test; Figures 1G, left panel, and S1F; immobile fraction: closed state = 0.33 ± 0.02; desensitized state = 0.62 ± 0.04; p < 0.001, Student’s t test; Figure 1G, middle panel). Consistently, the inhibition of any of the four pathways did not affect light-induced LiGluK2 synaptic confinement (MSD versus time curve plateau; p < 0.01, Student’s t test at steady state; Figure 1G, right panel; fraction of time spent at synapses: closed state, 68% ± 0.02%; desensitized state, 85% ± 0.02%; p < 0.01, Student’s t test; number of transitions between extrasynaptic and synaptic compartments: closed state = 2.39 ± 0.09; desensitized state = 1.95 ± 0.08; p < 0.001, Student’s t test; Figure S1G). Moreover, such VGCC blockers failed to change the diffusion properties of extrasynaptic LiGluK2 in the different conformational states (median diffusion coefficient: closed state = 0.021 μm2s1, IQR: 0.006–0.056 μm2s−1; desensitized state = 0.022 μm2s−1, IQR: 0.003–0.06 μm2s−1; ntrajectories = 320; ns, Mann-Whitney U test; Figure 1H, left panel; immobile fraction: closed state = 0.27 ± 0.01; desensitized state = 0.33 ± 0.01; ns, Student’s t test; Figure 1H, middle panel; MSD versus time curve plateau; ns, Student’s t test at steady state; Figure 1H, right panel).
To exclude direct calcium entry through LiGluK2 receptors (and/or through T- and R-type VGCCs), we removed calcium from the extracellular solution. In extracellular nominal zero calcium, UV light illumination caused similar synaptic LiGluK2 immobilization (Figure S1F). In contrast, in neurons expressing LiGluK2 not tethered with the photoswitch maleimide-azobenzene-glutamate (MAG; see STAR Methods), hence unable to be activated by light, the same illumination protocol previously used to photoactivate LiGluK2 receptors failed to change the diffusion of synaptic and extrasynaptic LiGluK2, indicating that the reduction of LiGluK2 synaptic receptor lateral mobility was not caused by UV-induced oxidative stress, which can alter calcium homeostasis (Csordás and Hajnóczky, 2009) (diffusion coefficient synaptic LiGluK2: closed state = 0.015 μm2s−1, IQR: 0.005–0.036 μm2s−1; desensitized state = 0.016 μm2s−1, IQR: 0.005–0.034 μm2s−1; extrasynaptic LiGluK2: closed state = 0.021 μm2s−1, IQR: 0.005–0.059 μm2s−1; desensitized state = 0.020 μm2s−1, IQR: 0.005–0.050 μm2s−1, ntrajectories = 100; ns, Mann-Whitney U test; Figure S1H).
Although the use of LiGluK2 receptors allows rapid and reversible control of the KAR’s conformational state, it cannot be excluded that receptor activation following UV illumination may differ from that induced by the conventional binding of free glutamate molecules. To address this point, we studied the diffusion properties of LiGluK2 receptors during bath application of glutamate (100 μM). Similarly to UV light exposure, bath application of glutamate was able to reversibly immobilize LiGluK2 receptors at synapses and increase their confinement in the synaptic compartment (median diffusion coefficient synaptic LiGluK2: closed state = 0.011 μm2s−1, IQR: 0.004–0.032 μm2s−1; desensitized state = 0.006 μm2s−1, IQR: 0.002–0.013 μm2s−1; ntrajectories = 60; p < 0.01, Mann-Whitney U test; Figure S2A, left; MSD versus time curve plateau; p < 0.01, Student’s t test at steady state; Figure S2A, right) without affecting the mobility of extrasynaptic receptors (median diffusion coefficient extrasynaptic LiGluK2: closed state = 0.018 μm2s−1, IQR: 0.007–0.037 μm2s−1; desensitized state = 0.016 μm2s−1, IQR: 0.006–0.039 μm2s−1; ntrajectories = 270; ns, Mann-Whitney U test; Figure S2B, left; MSD versus time curve plateau; ns, Student’s t test at steady state; Figure S2B, right). Since LiGluK2 is a mutated channel harboring the L439C amino acid (aa) substitution for the tethering of the photoswitch MAG, the functional properties of LiGluK2 may potentially differ from that of WT GluK2 receptors. To exclude this possibility, we studied the surface mobility of overexpressed WT GluK2 receptors in response to activation by glutamate. We found that the effects of bath applications of glutamate on GluK2 synaptic lateral diffusion were indistinguishable from those observed in LiGluK2 (median diffusion coefficient synaptic GluK2: closed state = 0.012 μm2s−1, IQR: 0.003–0.028 μm2s−1; desensitized state = 0.006 μm2s−1, IQR: 0.001–0.015 μm2s−1; ntrajectories = 64; p < 0.01, Mann-Whitney U test; Figure S2C, left; MSD versus time curve plateau; p < 0.01, Student’s t test at steady state; Figure S2C, right). In line with the previous results obtained with LiGluK2, glutamate activation did not change the mobility of extrasynaptic GluK2 receptors (median diffusion coefficient extrasynaptic GluK2: closed state = 0.018 μm2s−1, IQR: 0.005–0.045 μm2s−1; desensitized state = 0.019 μm2s−1, IQR: 0.005–0.046 μm2s−1, ntrajectories = 360; ns, Mann-Whitney U test; Figure S2D, left; MSD versus time curve plateau; ns, Student’s t test at steady state; Figure S2D, right). These data demonstrate that LiGluK2 and GluK2 are similarly trapped at glutamatergic synapses after activation and that their immobilization at synapses does not depend on intracellular calcium.
Desensitization-Dependent LiGluK2 Synaptic Immobilization Is Mediated by Interactions between GluK2 C Terminus and N-Cadherins
GluK2 subunits bind the scaffold protein PSD-95 through a PDZ binding motif at the GluK2 C-terminal domain (CTD) (Garcia et al., 1998). Since scaffold proteins tune the lateral diffusion of neurotransmitter receptors (Choquet and Triller, 2013), we hypothesized that conformational changes induced by glutamate binding may strengthen the interaction between LiGluK2 and PSD-95, thus promoting LiGluK2 clustering at synapses. To test this, we generated a mutated LiGluK2 (LiGluK2Δ4) lacking the last four C-terminal residues (aa 905–908, ETMA), corresponding to the PDZ binding site domain. Immunocytochemistry experiments showed comparable intensity of HA-tagged LiGluK2Δ4 and LiGluK2 synaptic clusters, indicating comparable expression for these two receptors at excitatory synapses (LiGluK2 = 134.2 ± 10.5 a.u., n = 20 cells, LiGluK2Δ4 = 138.2 ± 7.8 a.u., n = 24 cells; ns, Mann-Whitney U test) and similar dendritic cluster density (LiGluK2 = 0.072 ± 0.005 cluster/μm2, LiGluK2Δ4 = 0.066 ± 0.008 cluster/μm2; ns, Mann-Whitney U test; Figures S3A and S3B). The ETMA deletion did not affect the expression of the postsynaptic scaffold Homer1c (LiGluK2 = 0.23 ± 0.04 cluster/μm2, LiGluK2Δ4 = 0.20 ± 0.04 cluster/μm2; ns, Mann-Whitney U test; Figure S3B). In line with these observations, LiGluK2Δ4 and LiGluK2 receptors showed similar co-localization with Homer1c (Figures S3A and S3B). In addition, LiGluK2Δ4 receptor-mediated light-evoked currents were also comparable to those of LiGluK2, indicating that the ETMA deletion does not interfere with receptor gating properties (Figure S3C; cf. Figure S1A). SPT experiments revealed that the lateral diffusion of closed LiGluK2Δ4 at synapses matched those of LiGluK2 (compare Figures 1B and 2A) (median diffusion coefficients in the close state: LiGluK2 = 0.016 μm2s−1, IQR: 0.004–0.033 μm2s−1; LiGluK2Δ4 = 0.014 μm2s−1, IQR: 0.006–0.030 μm2s−1; immobile fraction: LiGluK2 = 0.27 ± 0.03, LiGluK2Δ4 = 0.30 ± 0.02; see Figures 1C and 2B). Consistent with the ETMA deletion not affecting the diffusion properties of LiGluK2, the receptor immobilization produced by light-induced desensitization of the LiGluK2Δ4 receptor was indistinguishable with respect to that of WT LiGluK2 receptors (closed state = 0.014 μm2s−1, IQR: 0.006–0.030 μm2s−1, desensitized state = 0.009 μm2s−1, IQR: 0.002–0.017 μm2s−1; ntrajectories = 94 from 7 neurons; p < 0.0001, Mann-Whitney U test; MSD versus time curve plateau; p < 0.01, Student’s t test at steady state; Figures 2A and 2B). The ETMA deletion also left unchanged the mobility of extrasynaptic LiGluK2Δ4 receptors (median diffusion coefficient extrasynaptic LiGluK2Δ4: closed state = 0.02 μm2s−1, IQR: 0.009–0.046 μm2s−1; desensitized state = 0.02 μm2s−1, IQR: 0.009–0.048 μm2s−1; ntrajectories = 200; ns, Mann-Whitney U test; Figure S3D; MSD versus time curve plateau; ns, Student’s t test at steady state; Figure S3D).
Figure 2.

The LiGluK2 C-Terminal Domain Mediates the Desensitization-Induced Synaptic Immobilization of LiGluK2 Receptors
(A) Representative trajectories (yellow) of LiGluK2Δ4 in the closed, desensitized, and recovery closed states diffusing at glutamatergic synapses (red). Scale bar, 1 μM.
(B) Desensitization-induced LiGluK2Δ4 immobilization, similar to that of LiGluK2. Summary of median diffusion coefficient (±IQR) (left), immobile fraction (middle), and the MSD versus time curve (right) of synaptic LiGluK2Δ4 in different activation states, similar to LiGluK2 (ntrajectories = 94, from 7 neurons; cf. Figure 1C).
(C) Representative trajectories (yellow) of surface LiGluK2Δ16 dynamics at synapses (red) as in (A). Scale bar, 1 μM.
(D) The deletion of the last 16 residues of LiGluK2 C-terminal domain prevents desensitization-induced receptor immobilization at synapses (cf. A and B) (ntrajectories = 102, from 7 neurons).
(E) Trajectories (yellow) of an individual synaptic LiGluK2 in the indicated activation states upon overexpression of N-cadΔE. Scale bar, 1 μM.
(F) Quantification of LiGluK2 surface dynamics as in (B), in the presence of N-cadΔE (ntrajectories = 72, from 5 neurons).
Unless otherwise stated, data are presented as mean ± SEM, ∗p < 0.05; ∗∗∗p < 0.005; ns, non-significant. See also Figure S3.
To further investigate the role of the LiGluK2 C-terminal interactions with the glutamatergic PSD in the light-induced LiGluK2 synaptic immobilization, we generated a second mutant LiGluK2 receptor lacking aa 893–908 (LiGluK2Δ16; previously reported to be unable to interact with N-cadherin), an adhesion protein that, together with β-catenin, forms stable complexes at synapses (Coussen et al., 2002, Fièvre et al., 2016). HA-tagged LiGluK2Δ16 receptors showed reduced synaptic accumulation compared to LiGluK2 (LiGluK2 = 101.5 ± 6.4 a.u., n = 48 cells, LiGluK2Δ16 = 75.1 ± 7.1 a.u., n = 39 cells; p < 0.001, Mann-Whitney U test; Figures S3E and S3F), though with similar (1) dendritic density of the synaptic clusters (LiGluK2 = 0.045 ± 0.003 cluster/μm2, LiGluK2Δ16 = 0.048 ± 0.004 cluster/μm2; ns, Mann-Whitney U test; Figure S3F), (2) kinetics of light-evoked currents (Figure S3G), and (3) intensity and density of Homer1c puncta (integrated intensity: LiGluK2 = 102.3 ± 38.1 a.u., LiGluK2Δ16 = 109.1 ± 42.2 a.u.; cluster density: LiGluK2 = 0.11 ± 0.006 cluster/μm2, LiGluK2Δ16 = 0.12 ± 0.007 cluster/μm2; ns, Mann-Whitney U test; Figure S3F). Importantly, the aa 893–908 (Δ16) deletion abolished the desensitization-induced LiGluK2 immobilization at synapses (LiGluK2Δ16: closed state = 0.020 μm2s−1, IQR: 0.012–0.034 μm2s−1; desensitized state = 0.020 μm2s−1, IQR: 0.010–0.039 μm2s−1; ntrajectories = 102 from 7 neurons; ns, Mann-Whitney U test; Figures 2C and 2D). The mobility of extrasynaptic LiGluK2Δ16 receptors was not affected by the Δ16 deletion (median diffusion coefficient extrasynaptic LiGluK2Δ16: closed state = 0.02 μm2s−1, IQR: 0.008–0.043 μm2s−1; desensitized state = 0.022 μm2s−1, IQR: 0.006–0.051 μm2s−1; ntrajectories = 250; ns, Mann-Whitney U test; Figure S3H; MSD versus time curve plateau; ns, Student’s t test at steady state; Figure S3H). These data indicate that the interaction of aa 893–908 of the GluK2 subunit CTD with the glutamatergic PSD is crucial the for the desensitization-induced immobilization of LiGluK2 receptors at glutamatergic synapses. Next, we considered the possibility that the functional interaction between LiGluK2 and the postsynaptic glutamatergic scaffold could be mediated by N-cadherins. To test this hypothesis, we took advantage of N-cadΔE, a dominant-negative mutant that impairs endogenous N-cadherin function (Garcia et al., 2015, Togashi et al., 2002) by preventing the binding between GluK2 and N-cadherins. Immunocytochemical experiments in neurons overexpressing N-cadΔE revealed a decreased N-cadherin immunoreactivity at glutamatergic synapses, as compared to control neurons in which only endogenous N-cadherin was present (integrated fluorescence intensity: control = 522.6 ± 5.1 a.u.; N-cadΔE = 380 ± 6.1.2 a.u.; p < 0.05, Mann-Whitney U test; percentage of co-localization with Homer1c: WT = 19.5 ± 1.2, n = 50 cells; N-cadΔE = 13.8 ± 1.1, n = 40 cells; p < 0.005, Mann-Whitney U test; Figures S3I and S3J), thus confirming that in our experimental conditions, N-cadΔE was indeed acting as a dominant negative. Next, we probed the effect of N-cadherin dominant negative on LiGluK2 surface expression by immunocytochemistry. As shown in Figure S3K, N-cadΔE overexpression decreased the accumulation of LiGluK2 receptors at synapses as compared to control conditions (synaptic LiGluK2 integrated intensity: control = 82.5 ± 5.3 a.u., n = 46 cells; N-CadΔE = 52.8 ± 5.3 a.u., n = 31; p < 0.001, Mann-Whitney U test; Figure S3L) without affecting the dendritic density of LiGluK2 clusters and the intensity and dendritic density of Homer1c clusters (Homer integrated fluorescence intensity: control = 98.7 ± 2.8 a.u.; N-cadΔE = 99.8 ± 4.4 a.u.; ns, Mann-Whitney U test; density of Homer clusters: control = 0.22 ± 0.01 clusters/μm2; N-cadΔE = 0.24 ± 0.01 clusters/μm2; ns, Mann-Whitney U test; Figure S3L) and the kinetics of the light-activated current (Figure S3M).
In a following set of experiments, we aimed at assessing LiGluK2 surface mobility upon impairment of N-cadherin activity. SPT experiments showed that the N-cadΔE overexpression prevented the synaptic immobilization of LiGluK2 induced by receptor desensitization (closed state = 0.019 μm2s−1, IQR: 0.009–0.042 μm2s−1; desensitized state = 0.020 μm2s−1, IQR: 0.010–0.031 μm2s−1; ns, Mann-Whitney U test; ntrajectories = 70 from 5 neurons; Figures 2E and 2F). In line with previous results, the N-cadΔE mutant did not alter the diffusion of extrasynaptic LiGluK2 receptors (median diffusion coefficient extrasynaptic LiGluK2: closed state = 0.023 μm2s−1, IQR: 0.008–0.054 μm2s−1; desensitized state = 0.022 μm2s−1, IQR: 0.008–0.058 μm2s−1; ntrajectories = 195; ns, Mann-Whitney U test; MSD versus time curve plateau; ns, Student’s t test at steady state; Figure S3N). Thus, the LiGluK2 C terminus (aa 893–908) and N-cadherins play an important role in the activation-dependent trapping of LiGluK2 receptors at postsynaptic sites.
In subsequent coimmunoprecipitation experiments, we investigated the molecular interaction between LiGluK2 and the N-cadherin/β-catenin complex. Cadherins are a large family of adhesion proteins showing highly variable synapse-specific expression, typically directly binding β-catenin (Jou et al., 1995). In order to benefit from high antibody specificity, we focused on tagged proteins (i.e., HA-tagged LiGluK2 and HA-GluK2, along with N-cadΔE-myc). We found that HA-LiGluK2 receptors coimmunoprecipitated with β-catenin, whereas no interaction was observed between HA-LiGluK2 and β-catenin in conditions of N-cadΔE-myc overexpression or between HA-LiGluK2Δ16 and β-catenin (Figure 3A). In line with the results obtained with HA-LiGluK2, the WT HA-tagged GluK2 receptor also coimmunoprecipitated with β-catenin (as already demonstrated by Coussen et al., 2002) and failed to interact with β-catenin upon N-cadΔE-myc overexpression (Figure 3A). Next, we assessed whether the interaction of GluK2 with the N-cadherin/β-catenin complex could be modulated by the induction of receptor activation/desensitization by glutamate binding. In the presence of glutamate 100 μM, the amount of HA-GluK2 coimmunoprecipitated with β-catenin was significantly increased with respect to control conditions, both in the presence and the absence of calcium (Figures 3B and 3C). These biochemical data nicely corroborate the results obtained with the SPT experiment and reinforce the hypothesis that the LiGluK2 synaptic immobilization upon activation/desensitization is mediated by receptor state-dependent molecular interactions with the N-cadherin/β-catenin complex expressed at glutamatergic synaptic sites. Furthermore, we demonstrate that LiGluK2 and GluK2 similarly interact with the key proteins of the glutamatergic PSD, N-cadherin/β-catenin.
Figure 3.

N-Cadherin/β-Catenin Complex Associates with the C-Terminal Domain of LiGluK2
(A) Left: coimmunoprecipitation experiment showing the interaction between HA-LiGluK2 and the β-catenin/N-cadherin complex. Cell lysates from HEK293 cells transfected with Li-GluK2 alone, LiGluK2 along with N-cadΔE-myc, or HA-LiGluK2Δ16 were immunoprecipitated with the anti-HA antibody and immunoblotted with anti-HA, anti-β-catenin, and anti-myc antibodies. β-catenin co-precipitates with HA-LiGluK2 transfected alone, but not with HA-LiGluK2 cotransfected along with the dominant-negative N-cadΔE-myc. β-catenin fails to interact with the mutant receptor HA-LiGluK2Δ16. Right: coimmunoprecipitation experiment showing the interaction between HA-GluK2 and the β-catenin/N-cadherin complex. Cell lysates from HEK293 cells transfected with GluK2 alone or GluK2 along with N-cadΔE-myc were immunoprecipitated and immunoblotted. β-catenin co-precipitates with HA-GluK2, similarly to full-length HA-LiGluK2. The dominant-negative N-cadΔE-myc prevents the interaction of β-catenin with HA-GluK2. Note that the presence of N-cadΔE-myc in the supernatants (S) indicates that it does not interact with full-length LiGluK2 and GluK2 receptors.
(B) Glutamate enhances the coprecipitation of HA-LiGluK2 with β-catenin. Anti-HA immunoprecipitation (IP) in the control condition (ctrl) and upon receptor desensitization with glutamate 100 μM (glu) in the presence or absence of calcium. The “input” represents 10% of the total extract before the IP.
(C) Quantification of the co-precipitation experiments described in (B). Effect of glutamate on β-catenin co-IP with LiGluK2 in the presence (top) or absence (bottom) of calcium. Bar plots represent IP β-catenin signal normalized to HA-LiGluK2 signal (left) or to β-catenin input (right). n = 6.
Data are presented as mean ± SEM, ∗p < 0.05.
Desensitization-Dependent LiGluK2 Synaptic Immobilization Affects KAR-Mediated Responses
Previous works have demonstrated that the mobility of synaptic glutamate receptors significantly impacts the amplitude of glutamatergic synaptic currents (Constals et al., 2015, Heine et al., 2008). Indeed, the fast exchange of desensitized AMPARs with a pool of naive extrasynaptic/perisynaptic receptors significantly reduces the accumulation of desensitization during repetitive activation of synaptic receptors. We reasoned that while the immobilization of KARs at synapses by glutamate binding would determine a higher susceptibility to desensitization of KAR-mediated components of EPSCs, conversely, the lack of LiGluK2 immobilization after receptor activation/desensitization in LiGluK2Δ16 and N-cadΔE conditions could reduce the synaptic KARs’ desensitization. To investigate this, we performed laser photolysis of caged glutamate (MNI-Glu) in diffraction-limited spots to elicit uncaging currents (uEPSCs) at individual glutamatergic synapses identified by Homer1c-DsRed overexpression. With this approach, we elicited six consecutive uEPSCs (at 20 Hz) in hippocampal neurons expressing LiGluK2Δ16 or N-cadΔE/LiGluK2 and compared the current amplitudes with those obtained from LiGluK2-expressing neurons with the same stimulation protocol (Figures 4A–4C). We found that uEPSCs in both LiGluK2Δ16- and N-cadΔE/LiGluK2-expressing neurons exhibited less desensitization with respect to control neurons expressing LiGluK2 (at pulse 2: LiGluK2: 62.9% ± 1.7%, n = 21 cells; LiGluK2Δ16: 40.9% ± 4.0%, n = 12 cells; N-cadΔE/LiGluK2: 40.3% ± 4.0%, n = 16 cells; p < 0.0001, Mann-Whitney U test; Figure 4D, left panel). These results are in agreement with the hypothesis that in LiGluK2Δ16- and N-cadΔE-expressing neurons, the higher mobility of synaptic LiGluK2 favors the dispersal of desensitized receptors and the insertion of ready-to-be-activated receptors at the synapse. To confirm this hypothesis, we repeated the same experiments upon the LiGluK2 receptor cross-link (X-link) procedure, where receptors are immobilized by binding a molecular mesh of primary and secondary antibodies (Ashby et al., 2006, Bats et al., 2007, Heine et al., 2008). In LiGluK2Δ16- and N-cadΔE-expressing neurons, the X-link protocol reverted the extent of uEPSCs’ desensitization to values observed in LiGluK2-expressing neurons (Figures 4A–4C, red traces; at pulse 2: 60.0% ± 1.9%, n = 14; 60.6% ± 2.9%, n = 13; 63.3% ± 2.8%, n = 16, for LiGluK2, LiGluK2Δ16, and N-cadΔE, respectively; ns, Mann-Whitney U test; Figure 4D, right panel). Of note, in control LiGluK2-transfected neurons, the X-link protocol did not affect the extent of desensitization of uEPSCs (Figures 4A and 4D, black bars).
Figure 4.

Increased Synaptic LiGluK2 Receptor Mobility Affects Glutamatergic Synaptic Currents
(A) Top: stimulating protocol: train of 6 brief (0.8 ms) consecutive UV light pulses at 20 Hz to uncage MNI-glutamate. Bottom: representative traces of LiGluK2-mediated uEPSCs in control (left) and X-link conditions (right).
(B) Representative traces of uEPSCs mediated by LiGluK2Δ16 in control (left) and X-link conditions (right).
(C) Representative traces of uEPSCs evoked in N-cadΔE/LiGluK2-expressing neurons in control (left) and X-link conditions (right).
(D) Left: the percentage of desensitization of uEPSCs is larger in LiGluK2- (black) expressing neurons than in LiGluK2Δ16- (gray) and N-cadΔE/LiGluK2- (cyan) expressing neurons. Right: percentage of desensitization of synaptic uEPSCs as on the left, upon receptor X-link. Receptor immobilization by X-link did not change the control (LiGluK2) currents but reverted the desensitization of currents evoked in LiGluK2Δ16- and N-cadΔE/LiGluK2-expressing neurons to control values.
Data are presented as mean ± SEM, ∗p < 0.05; ns, non-significant.
Activity-Dependent LiGluK2 Synaptic Immobilization Affects the Kinetics of Mixed AMPAR/KAR-Mediated Synaptic Responses
In a previous study, glutamate binding has been shown to increase AMPAR lateral mobility (Constals et al., 2015), an opposite paradigm with respect to that reported here for KARs. Since the presence of both AMPARs and KARs at glutamatergic synapses leads to the generation of mixed AMPAR-KAR EPSCs (Castillo et al., 1997, Vignes and Collingridge, 1997, Bureau et al., 2000, Cossart et al., 2002, Frerking et al., 1998, Kidd and Isaac, 1999), we hypothesized that during repetitive stimulations, the fraction of KAR-mediated components would be more susceptible to desensitization with respect to the AMPAR-mediated one due to the KARs’ immobilization at synapses after glutamate binding. In particular, taking into account that AMPARs and KARs mediate fast and slow EPSCs, respectively (Frerking and Ohliger-Frerking, 2002), such an increase of the AMPAR-KAR ratio should lead to faster kinetics of mixed AMPA-KA EPSCs. To address this issue, in order to avoid possible distortions of uEPSC kinetics due to slow glutamate clearance, we studied synaptic AMPAR-KAR currents evoked by extracellular stimulation of hippocampal principal cells in the presence of 50 μM D-APV and 10 μM bicuculline. As expected, in untransfected neurons, evoked EPSCs (eEPSCs) were purely AMPAR-mediated currents with fast decay kinetics (τ = 1.3 ± 0.1 ms; Figures 5A, black, and S4A, black). On the other hand, in LiGluK2-expressing neurons, mixed AMPAR-KAR eEPSCs exhibiting fast and slow decay kinetics were best fitted with a double exponential function (τfast = 1.1 ± 0.1 ms, τslow = 24.5 ± 3.8 ms; n = 33; Figure 5A, blue), while the rise time was similar to that of pure AMPAR-mediated eEPSCs (Figure S4A). The application of the AMPAR blocker GYKI 53655 revealed the presence of a KAR-mediated eEPSC component with decay kinetics similar to the τslow quantified in the mixed AMPAR-KAR eEPSCs (τ = 36.3 ± 4.4 ms; n = 11; Figure 5B, blue) that was fully blocked by CNQX (25 μM). The overexpression of LiGluK2Δ16 instead of LiGluK2 did not affect the properties of KAR-mediated currents. Indeed, mixed AMPAR-KAR-mediated eEPSCs involving the activation of LiGluK2Δ16 exhibited kinetics comparable to that observed in the presence of LiGluK2 WT (Figure 5A). Moreover, in the presence of GYKI, eEPCS in LiGluK2- and LiGluK2Δ16-expressing neurons had similar decay kinetics (τ = 36.3 ± 4.4, τ = 33.9 ± 4.1 ms, for LiGluK2 and LiGluK2Δ16, respectively; ns, Mann-Whitney U test; Figure 5B), rise time (2.8 ± 0.4 ms, 3 ± 0.3 ms; n = 11 and n = 8, respectively; ns, Mann-Whitney U test; Figure S4B), and amplitude (22 ± 2 pA, 20.5 ± 3 pA, for LiGluK2 and LiGluK2Δ16, respectively; ns, Mann-Whitney U test; Figure S4B).
Figure 5.

The Mobility of Desensitized KARs Affects the Kinetics of Mixed AMPAR/KAR-Mediated Currents
(A) Left: scatter dot plot of decay time constants (τ) of eEPSCs mediated by AMPA and mixed AMPA/LiGluK2 receptors in neurons expressing only endogenous AMPARs (untransfected, black) or expressing LiGluK2 (blue) or LiGluK2Δ16 (green). Right: representative averaged traces of eEPSCs mediated by AMPARs, AMPA/LiGluK2 receptors, and AMPA/LiGluK2Δ16 receptors. Note that for visualization purposes, the stimulation artifacts of black and green traces have been omitted.
(B) Scatter dot plot of decay time constants (τ) of eEPSC mediated by LiGluK2 (blue) or LiGluK2Δ16 receptors (green) recorded in the presence of GYKI 53655 (left) and corresponding normalized representative traces (right).
(C) Top: representative AMPA/LiGluK2 receptors-mediated mixed eEPSCs recorded before and after the delivery of the desensitizing train. Bottom: magnification of the framed areas showing AMPA/LiGluK2 receptors-mediated mixed eEPSC before, during, and after the train. Inset: superimposed normalized slow component of mixed eEPSC decay before (black) and after (blue) the train, showing acceleration in the “after” current.
(D) Left: weighted time constant (τweighted) of mixed eEPSC before and after the train. Right: relative weight of the AMPAR and LiGluK2 components in AMPA/LiGluK2 receptors-mediated mixed eEPSCs before and after the train. Please note that after the desensitizing train, current decay kinetics was accelerated, the AMPAR-mediated component increased, and the KAR-mediated component decreased.
(E) Top: representative traces of AMPA/LiGluK2Δ16 receptors-mediated mixed eEPSC before and after the desensitizing train. Bottom: magnification as in (C). Inset: superimposed normalized slow component of mixed AMPA/LiGluK2Δ16 eEPSC before (black) and after (green) the train.
(F) Left: weighted time constant (τweighted) of AMPA/LiGluK2Δ16 mixed eEPSC before and after the train, indicating unchanged current decay kinetics. Right: unaffected relative weight of AMPAR and KAR components in AMPA/LiGluK2Δ16 mixed eEPSCs before and after the train.
Data are presented as mean ± SEM, ∗∗∗p < 0.001; ns, non-significant. See also Figure S4.
It has been shown that the protein Neto-2 (neuropilin tolloid-like 2) confers slow decay kinetics to KAR currents (Straub et al., 2011), including those mediated by recombinant homomeric GluK2 receptors (Zhang et al., 2009). Since Neto2 has been reported to be expressed at negligible levels in cultured neurons (Mahadevan et al., 2015, Palacios-Filardo et al., 2016, Vernon and Swanson, 2017), we co-transfected with LiGluK2 along with Neto2. However, the presence of Neto2 failed to further slow the decay kinetics of evoked synaptic currents mediated by LiGluK2 receptors (Figure S5A). Thus, we decided to study the expression of Neto2 in our hippocampal cultures by using a biochemistry approach. Interestingly, we found that the expression of Neto2 is developmentally regulated: high at early stages (day in vitro [DIV] 7) and progressively downregulated (from DIV 14 to DIV 28; Figure S5B). Such a temporal profile of Neto2 expression in cultured neurons can account for the slow kinetics of KAR-mediated synaptic currents observed in our experiments at DIV 14 and 15 and can provide an explanation for the lack of effect of Neto2 overexpression on the GluK2-mediated currents’ decay kinetics. We then studied the kinetics of mixed AMPAR-KAR eEPSCs before and 50 ms after the application of a depolarization train (1 s at the frequency of 100 or 50 Hz; see STAR Methods) aimed at inducing massive desensitization of both synaptic AMPARs and KARs (Figure 5C). Interestingly, in neurons transfected with LiGuK2, the desensitizing train induced a significant acceleration of the mixed AMPA-KAR EPSCs’ decay kinetics (τweighted before train: 2.4 ± 0.3 ms; τweighted after train: 1.7 ± 0.2 ms; n = 21, p < 0.001, paired Wilcoxon test; Figure 5D, left), indicating that the KAR-mediated component preferentially desensitized with respect to that mediated by AMPAR. Moreover, we computed that after the train, the relative contribution of the KAR component was decreased in favor of the AMPAR component (KAR before = 7.3% ± 1.1%, after = 3.7% ± 0.7%; n = 21, p < 0.001, paired Wilcoxon test; Figure 5D, right). Interestingly, LiGluK2Δ16 transfection prevented the acceleration of EPSCs’ decay induced by the desensitizing train, as quantified by comparable time constants before and after the protocol (τweighted before train = 2.2 ± 0.3 ms; τweighted after train: 2.6 ± 0.4 ms; n = 21, paired Wilcoxon test, p > 0.05; Figure 5E), as well as the unaffected relative contribution of the KAR component (KAR before = 5.4% ± 1.0%, after = 7.2% ± 1.4%; paired Wilcoxon test, p > 0.05; Figure 5F). In a control experiment, we applied the same protocol to pure AMPA-mediated eEPSCs (in untransfected neurons), and we observed no differences in the decay kinetics before and after the train (τ before: 1.3 ± 0.1 ms; τ after: 1.3 ± 0.1 ms; n = 9, ns, paired Wilcoxon test; Figures S4C and S4D). Along the same line, we found that the amplitude of KAR-EPSCs pharmacologically isolated by using GYKI 10 μM was dramatically reduced 50 ms after the desensitizing train (before: 26.5 ± 2.5 pA; after: 6.2 ± 0.8 pA; n = 6, p < 0.005, paired Wilcoxon test; Figures S4E and S4F), thus confirming the LiGluK2-mediated currents undergo profound desensitization after such stimulation. In contrast in the same conditions, the amplitude of KAR-EPSCs upon transfection with LiGluK2Δ16 was slightly (but not significantly) reduced (before: 27.8 ± 5.0 pA; after: 20.4 ± 5.6 pA; n = 6, ns, paired Wilcoxon test; Figures S4G and S4H). These data indicate that during repetitive synaptic activation, the regulation of KARs’ lateral mobility by glutamate binding can shape the extent of the KAR-mediated component, thus modulating the kinetics of mixed AMPA-KAR EPSCs.
To provide a quantitative analysis of the relation between the desensitization of KAR-mediated currents and KARs’ lateral mobility, we performed computer modeling. This approach was used to estimate (1) the probability of KARs to exchange between the synaptic and the extrasynaptic compartments, depending on their diffusion coefficient in a realistic synaptic environment, and (2) the impact of such receptor exchange rate in the accumulation of desensitization of KAR-mediated EPSCs (see STAR Methods). Receptor diffusion coefficients imposed in the simulations were those quantified in SPT experiments when LiGluK2 receptors were either in the closed (D = 0.016 μm2s−1, “high mobility”) or in the open/desensitized states (D = 0.003 μm2s−1, “low mobility”) (Figure S6A; STAR Methods). Interestingly, “synaptic” receptors exchanged with the “extra-synaptic” naive receptors with probability p = 0.41 or p = 0.09, when considering the “high mobility” (closed receptor) or the “low mobility” (open/desensitized receptor) conditions, respectively (Figure S6A; STAR Methods). When the KAR gating was simulated according to a kinetic model (Figure S6B), the reduced “exchange probability” observed for “low mobility” receptors resulted in a significantly higher desensitization of simulated KAR-EPSCs 50 ms after a 100-Hz train (Figures S6C and S6D). These results indicate that the differences of KAR synaptic diffusion coefficients in the different KAR conformational states with SPT technique play an important role in the EPSCs’ short-term plasticity.
Discussion
In the present study, we provide a causal link between the gating and diffusion properties of synaptic KARs. In particular, we show that the entry of the LiGluK2 receptor in the open/desensitized state caused the rapid immobilization of LiGluK2 receptors at synapses. While the use of light allows the specific and fast control of LiGluK2 receptors, it is possible that the conformational change induced by light would differ from that induced by conventional glutamate binding. However, in addition to a previous study showing that LiGluK2 recapitulates the main features of GluK2 receptor gating (Reiner and Isacoff, 2014), we showed that a similar receptor immobilization at synapses was observed either with LiGluK2 activated by light or GluK2 activated by glutamate. Moreover, we observed comparable effects when both LiGluK2 and GluK2 were activated by glutamate. Thus, the potential differences between LiGluK2 (light activated) and GluK2 (glutamate activated) or between LiGluK2 and GluK2 activated by glutamate do not seem to interfere in their open/desensitized-induced trapping at glutamatergic synapses.
Our data show that the desensitization-dependent immobilization of LiGluK2 at synapses is unaffected by the deletion of the GluK2 C-terminal PDZ-interacting domain (ETMA), indicating that this phenomenon is unlikely to occur through GluK2 interactions with PSD-95, GRIP, and PICK1 proteins (Garcia et al., 1998, Hirbec et al., 2003). In contrast, such immobilization was abolished by the deletion of the GluK2 C terminus last 16 aa, a protein portion previously shown to interact with the N-cadherin/β-catenin complex (Coussen et al., 2002, Fièvre et al., 2016) that localizes at glutamatergic synapses (Uchida et al., 1996, Bamji, 2005, Mendez et al., 2010, Okuda et al., 2007). In line with this, the overexpression of N-cadherin dominant-negative mutants also abolishes the glutamate-dependent GluK2 synaptic trapping. These data, together with the observation that the interaction between GluK2 and β-catenin is increased in the presence of glutamate, allowed us to propose a model whereby glutamate binding to GluK2 induces the rearrangement of the GluK2 CTD, thus increasing its affinity for the N-cadherin/β-catenin complex and consequently promoting the immobilization of LiGluK2 receptors at synapses. While agonist-induced changes of the CTD conformation has been previously demonstrated at NMDARs and AMPARs (Dore et al., 2015, Zachariassen et al., 2016, Ferreira et al., 2017), at KARs, several studies have only focused on the interaction of the GluK2 CTD with intracellular proteins without considering the role of possible agonist-induced alterations of the CTD structure and interactions (Coussen et al., 2002, Maraschi et al., 2014). Other X-ray crystallography and cryo-EM studies have shown major GluK2 structural rearrangements following desensitization at the level of the ligand binding domain (LBD), leaving the potential changes of the GluK2 CTD during receptor opening/desensitization unaddressed (Meyerson et al., 2016, Møllerud et al., 2017). Therefore, at this stage, it cannot be excluded that in addition to the CTD, the functional interaction between the activated GluK2 and N-cadherin/β-catenin complex may be mediated by the interaction of the GluK2 LBD (known to undergo heavy rearrangements during receptor activation and desensitization) with the extracellular domain of N-cadherin. Likewise, the present data are compatible with the additional hypothesis that other KAR scaffold/accessory proteins could participate in the GluK2 binding with N-cadherin/β-catenin.
In our uncaging experiments, we observed that the activation-induced interaction of GluK2 with N-cadherin/β-catenin (which immobilizes KARs at synapses) plays a role in tuning the extent of current desensitization. Importantly, in mutants with impaired GluK2-N-cadherin/β-catenin interaction (thus lacking activation-induced KAR immobilization and showing a reduced current desensitization), the receptor immobilization by X-link restored the extent of current desensitization to the control values observed with WT LiGluK2. Surprisingly, the X-link of WT LiGluK2 did not affect the extent of desensitization of KAR-mediated uncaging currents. This result might appear in contrast with the general paradigm that the regulated mobility of desensitized receptors can modulate the accumulation of current desensitization, as demonstrated here in physiological experiments studying KAR-mediated synaptic currents and in other studies with different receptors (AMPAR and GABAAR) (Heine et al., 2008, de Luca et al., 2017). However, it has to be noted that in the experimental conditions with impaired GluK2-N-cadherin/β-catenin interaction (LiGluK2-Δ16 and LiGluK2/N-cadΔE), KARs exhibit an overall higher mobility as compared to WT LiGluK2 receptors, particularly in the open/desensitized state. This implies that the effect of the X-link protocol is clearly sizable in the mutants whereby the open/desensitized KAR diffusion coefficient is high; conversely, it can be reasoned that the lower mobility of open/desensitized WT LiGluK2 makes the additional immobilization induced by the X-link moderate, supporting the possibility that changes of WT current desensitization are poorly detectable. Additionally, the effect of the X-link protocol on the extent of desensitization of WT-LiGluK2 uncaging currents might be hindered by several factors, including (1) the relatively low spatial resolution of the uncaging technique and (2) the possible lower segregation of overexpressed KARs at synapses as compared to AMPARs and GABAARs.
A previous study has identified the regulated diffusion of KARs at synapses as a determinant for long-term plasticity (Carta et al., 2013). Here, we demonstrate that the immobilization of desensitized LiGluK2 at synapses modulates the kinetics of KAR-mediated EPSCs, therefore implicating KARs’ synaptic mobility also in glutamatergic short-term synaptic plasticity. In particular, we demonstrate that the immobilization of open/desensitized KARs occurs through increased GluK2 interaction with the N-cadherin/β-catenin complexes. In a similar although opposite paradigm, desensitized AMPARs increase their diffusion at synapses through reduced AMPAR interactions with the stargazin-PSD95 complex (Constals et al., 2015). The different players involved in the synaptic trapping of KARs and AMPARs may provide the molecular basis for the distinct action of desensitization on the lateral mobility of these two ionotropic receptors at glutamatergic synapses. We demonstrate that during sustained synaptic activity, this opposite regulation of AMPARs’ mobility can tune the relative weight of AMPAR- and KAR-mediated components in mixed AMPAR-KAR EPSCs by reducing the KAR-mediated slow component in favor of the AMPAR-mediated fast component. In these conditions, the time window for synaptic input integration is regulated by the degree of synaptic KAR desensitization that, in turn, is modulated by the frequency of synaptic activity. This emphasizes that the interplay between the desensitization and lateral mobility of AMPARs and KARs represents a form of integrated short-term plasticity at glutamatergic synapses expressing AMPAR-KAR.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-HA | Roche | Cat# 11867423001; RRID: AB_390918 |
| Rabbit anti β-catenin | Sigma | Cat# C2206; RRID: AB_476831 |
| Mouse anti N-Cadherin | BD Bioscience | Cat# 610920; RRID: AB_2077527 |
| Mouse Anti-Myc-Tag | Cell signaling | Cat# 2276; RRID:AB_331783 |
| Guinea pig Anti-Vesicular Glutamate Transporter 1 (VGLUT1) | Millipore | Cat# AB5905; RRID:AB_2301751 |
| Rabbit anti Neto2 | Abcam | Cat# ab109288; RRID:AB_10863520 |
| Pierce Anti-HA Magnetic Beads | Thermo Fisher Scientific | Cat# 88836; RRID:AB_2749815 |
| GAPDH Loading Control | Thermo Fisher Scientific | Cat# MA5-15738-HRP; RRID:AB_2537659 |
| Goat anti-Rat IgG (H+L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat# 31470; RRID:AB_228356 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat# 31460; RRID:AB_228341 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat# 32430; RRID:AB_1185566 |
| QDot 655 goat F(ab’)2 anti mouse IgG | Thermo Fisher Scientific | Cat# Q11022MP; RRID:AB_10373262 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2-APB | Tocris Bioscience | 1224 |
| D-AP5 | Tocris Bioscience | 0106/50 |
| Ryanodine | Tocris Bioscience | 1329 |
| ω-conotoxin MVIIC | Tocris Bioscience | 1084 |
| CNQX disodium salt | Tocris Bioscience | 1045/10 |
| MNI-caged-L-glutamate | Tocris Bioscience | 1490/10 |
| (S)-5-Iodowillardiine | Tocris Bioscience | 0307 |
| 1(S),9(R)-(−)-Bicuculline methiodide | Sigma | 14343 |
| GYKI 53655 | Sigma | G-7798 |
| Nifedipine | Sigma | N-7634 |
| L-Glutamic acid | Sigma | 49449 |
| DAKO fluorescent mounting medium | DAKO | S302380-2 |
| Concanavalin-A | Sigma | L7647 |
| MAG | Gift from Trauner D. and Gorostiza P. | N/A |
| Casein | VectorLab | SP 5020 |
| Critical Commercial Assays | ||
| Effectene Transfection Reagent | QIAGEN | 301427 |
| QuickChange II Site-Directed Mutagenesis Kit | Agilent Technologies | 200524 |
| Pierce BCA Protein Assay Kit | Thermo Scientific | 23225 |
| Deposited Data | ||
| Original images of western blot data | This paper | https://doi.org/10.17632/9p5bgckkyh.1 |
| Experimental Models: Cell Lines | ||
| Human Embrionic Kidney 293T | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Wild-type (C57BL/6J) | Envigo | C57BL/6JRccHsd (Cat#043) |
| Recombinant DNA | ||
| pcDNA3-Homer1c-DsRed | Petrini et al., 2009 | N/A |
| pTR-hSyn Grik2-L439C-GFP (LiGluK2-GFP) | Volgraf et al., 2006 | N/A |
| HA-LiGluK2-GFP | This paper | N/A |
| HA-LiGluK2Δ4-GFP | This paper | N/A |
| HA-LiGluK2Δ16-GFP | This paper | N/A |
| HA-GluK2-GFP | This paper | N/A |
| pcDNA3-Neto2 | Zhang et al., 2009 | N/A |
| pCS2-NcadΔE-myc | Chazeau et al., 2015 | N/A |
| Software and Algorithms | ||
| Metamorph 7.8 | Molecular Devices | https://www.moleculardevices.com/Products/Software/Meta-Imaging-Series/MetaMorph.html; RRID: SCR_002368 |
| Clampex 10.6 | Molecular Devices | https://www.moleculardevices.com/products/software/pclamp.html; RRID:SCR_011323 |
| Clampfit 10.7 | Molecular Devices | https://www.moleculardevices.com/products/software/pclamp.html; RRID:SCR_011323 |
| MATLAB | Mathworks | https://www.mathworks.com/products/matlab/; RRID:SCR_001622 |
| GraphPad Prism 6 | GraphPad | https://www.graphpad.com/; RRID: SCR_002798 |
| KyPlot 5.0 | KyensLab | https://kyenslab-inc.software.informer.com/ |
| Python Language Reference 2. 7 | Python Software Foundation | https://www.python.org; RRID: SCR_008394 |
| Custom program written for MATLAB to reconnect QD trajectories | Petrini et al., 2014; from D Choquet and L Cognet | N/A |
| Custom program for SPT quantifications | Petrini et al., 2014; from D Choquet and A Serge | N/A |
| Custom Python script to simulate the diffusion of the receptors on the synaptic disk. | This paper | N/A |
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrea Barberis (andrea.barberis@iit.it).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data And Code Avalability
The code generated during this study is available at github: https://github.com/thierrynieus/Kainate-receptor-activation-shapes-short-term-synaptic-plasticity-by-controlling-receptor-lateral-mo.
Original full-length images of western blot experiments have been deposited to Mendeley Data, https://doi.org/10.17632/9p5bgckkyh.1
Experimental Model and Subject Details
Primary neuronal cultures
All experiments have been approved by the Italian Ministry of Health (authorization 079 2015) and by the IIT animal welfare body. The experiments have been carried out in accordance with the guidelines established by the European Directive (2010/63/EU of 22 September 2010), and by the national legislation (D.Lgs.26/2014). Primary cultures of hippocampal neurons were prepared from P0-P1 C57BL/6J mice of either sex as previously published (de Luca et al., 2017). The animal were housed in 12:12 hr light-dark circles in individually ventilated cages. Neurons were plated at a density of 60 × 103 cells/cm2 on poly-D-lysine pre-coated coverslips and kept in Neurobasal-A medium (Thermo Fisher, Italy) supplemented with B-27 (Thermo Fisher, Italy) 2%, Glutamax 1% (Thermo Fisher, Italy) and gentamycin 5 mg/ml (Sigma) at 37°C in 7.4% CO2. All the experiments were performed at Days In Vitro (DIV) 14-17.
Cell lines
Human Embrionic Kidney 293T cells (ATCC) were cultured at 37°C in 5% CO2 in DMEM (Thermo Fisher, Italy) medium containing 10% fetal bovine serum (Thermo Fisher, Italy) and penicillin/streptomycin (EuroClone).
Method Details
Plasmid constructs
The pTR-hSyn Grik2-L439C-GFP plasmid (LiGluK2 plasmid) was kindly provided by Prof. E. Isacoff (Berkeley University of California). The HA-LiGluK2 was obtained by inserting the Hemagglutinin (HA) sequence at the fourth amino acid of the pTR-hSyn Grik2-L439C-GFP. Please note that throughout the text, the resulting HA-LiGluK2-GFP plasmid was referred to as HA-LiGluK2. HA-GluK2 was obtained by retromutation of the cysteine 439 of the HA-LiGluK2 to a lysine, as found in the wild-type GluK2. HA-LiGluK2Δ4 and HA-LiGluK2Δ16 were obtained by deleting the last 4 residues (ETMA) or the last 16 residues (MHTFNDRRLPGKETMA) from the HA-LiGluK2 plasmid, respectively. All the aforementioned plasmid editings were performed with the QuickChange II Site-Directed Mutagenesis Kit (Agilent). The N-cadherinΔE-myc construct was a gift from Prof. O. Thoumine (University of Bordeaux-2). Neto2 plasmid was kindly provided by Prof. S. Tomita (Yale University). Homer1c-DsRed plasmid (kindly provided by Prof. D. Choquet) encodes for DsRed at the N terminus of Homer1c (Petrini et al., 2009). All constructs were verified by DNA sequencing.
Transfection
Hippocampal neurons were transfected at DIV 7 using Effectene (QIAGEN, Germany) following the manufacturer’s protocol. HEK293T were transfected with the plasmids of interest using Lipofectamine 2000 (Thermo Fisher, Italy) according to manufacturer’s protocol.
Light-gated glutamate receptors (LiGluK2)
Light-Gated Glutamate receptors (LiGluK2), developed by the Isacoff Lab (Volgraf et al., 2006) consist of an engineered kainate receptor to bind the photoswitchable tethered ligand (PTLs) maleimide-azobenzene-glutamate (MAG). In particular, the ligand glutamate is linked to azobenzene that can reversibly photoisomerize between a trans and cis configuration in response to illumination with light at different wavelengths (380 nm and > 460 nm, respectively) (Gorostiza and Isacoff, 2008). Azobenzene, in turn, is anchored to a mutated cysteine introduced into the ligand-binding domain (LBD) of GluK2 receptor through the cysteine-reactive group maleimide. Photoswitching is operated by the reversible binding of the glutamate moiety of MAG, which is presented to the ligand-binding site in the cis configuration and withdrawn in trans. The MAG molecule was kindly provided by Dr Dirk Trauner (The Ludwig Maximilians University of Munich) and Pau Gorostiza (Institute of Bioengineering of Catalonia, Barcelona). After dilution in DMSO, MAG conjugation with LiGluK2 was performed by exploiting the photo affinity labeling procedure. In brief, MAG was diluted in the extracellular solution to 40 μM (from a 10 mM stock solution) and illuminated with 380 nm light to promote its accumulation of the cis-form, thus favoring the binding between the GluK2 glutamate binding site and the engineered cysteine in the GluK2 ligand-binding domain (LBD). Hippocampal neurons were then incubated with MAG (in cis configuration) at 37°C for 30 min, rinsed with extracellular solution to remove any unreacted MAG, and subsequently used for recordings. The LiGluK2 photoconversion was performed by a diode-based illumination device (Lumencor, SpectraX Light Engine, Optoprim, Italy) controlled by either the Clampex 10.6 software (for electrophysiology experiments) or the Metamorph software (ver. 7.7.8, Molecular Devices, USA) for the single particle tracking experiments. Samples were illuminated through a 60X oil-immersion/1.4NA Plan Apochromat immersion objective mounted on an inverted microscope (Eclipse Ti, Nikon, Japan). The light power measured at the exit of the objective was 3 mW for the 380 nm and 7 mW for the 488 nm illumination, respectively.
Electrophysiological recordings and Glutamate uncaging
Currents mediated by photoactivation of LiGluK2 receptors were recorded in the whole-cell configuration of the patch clamp technique at room temperature at holding potential of −65mV. External solution contained (in mM): 145 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 10 glucose and 10 HEPES, pH 7.4. Patch pipettes (pulled from borosilicate glass capillaries, Hilgenberg, Malsfeld, Germany) had a 3.5–4 MΩ resistance when filled with the intracellular recording solution containing (in mM): 130 KGluconate, 5 KCl, 5 sucrose, 1 EGTA, 10 HEPES and 4 MgATP (300 mOsm and pH 7.2 with KOH). Currents were acquired using Clampex 10.6 software (Molecular Devices, Sunnyvale, CA). Currents were sampled at 50 kHz, digitally filtered at 3 kHz using the 700B Axopatch amplifier (Molecular Devices) and subsequently analyzed by using the Clampfit software. The stability of the patch was checked by repetitively monitoring the input and series resistance during the experiments. Cells exhibiting 10%–15% changes were discarded from analysis. Concanavalin-A (Sigma-Aldrich, Italy) was used at a concentration of 0.1 mg/ml in the extracellular solution to suppress desensitization of LiGluK2. The maximal Concanavalin-A effect was typically achieved within 8 min after perfusion. (S)-5-Iodowillardiine (5-IW) was purchased from Tocris Bioscience (Italy).
Uncaging experiments were performed by exploiting MNI-L-Glutamate (Tocris Bioscience, Italy): MNI-Glutamate (5 mM) was dissolved in the extracellular solution and locally perfused through a patch pipette (2-4 μM tip diameter) by means of a pressure-based application system (20 psi) (Picospritzer, Parker, USA) and placed at 10 and 20 μM (x- and z axis, respectively) from the region of interest. Experiments were performed in the presence of 50 μM D-APV (Tocris) to block NMDA receptor; EPSCs mediated by kainate receptor (KAR) were pharmacologically isolated in the presence of 10 μM GYKI 53655 (Sigma). A 378 nm diode laser (Cube 378, 16 mW, Coherent Italia, Italy) was directly coupled to the microscope objective (Olympus UPlanSApo 100X oil-1.40 NA). In order to obtain the smallest laser spot size on the sample we backfilled the objective by using a beam expander placed in the optical pathway between the laser source and the objective. The measured point spread function (PSF) of the 378 nm illumination had lateral dimension of 487 ± 55 nm (FWHM, n = 6). The laser beam was steered in the field of view by means of a galvanometric mirrors-based pointing system allowing the illumination of specific regions of interest tailored around glutamatergic synapses (UGA32, Rapp OptoElectronics, Hamburg, Germany). Synchronization of optical stimulations and electrophysiological recordings was controlled with the UGA32 software interfaced with the Clampex 10.6 software (Molecular Devices, Sunnyvale, CA, USA). Currents were elicited by 500 μs light pulses with a power intensity of 80-100 μW at the exit of the objective.
For experiments with synaptic stimulation, mixed AMPAR/KARs EPSCs were recorded in the presence of 50 μM D-APV (Tocris) and 10 μM bicuculline (Sigma) whereas pure KAR EPSCs in the presence of 10 μM GYKI 53655 (Sigma). Synaptic responses were evoked by minimal stimulation (0.8 ms pulse length) through an electrode filled with extracellular solution placed nearby the cell body of the postsynaptic neuron. The duration and the amplitude of the extracellular stimulus were controlled by Model DS2A Constant Voltage Isolated Stimulator (Digitimer Ltd.) synchronized with Clampex 10.6 software (Molecular Devices). The desensitizing depolarization trains were elicited at either 100 or 50 Hz. Since the effect of trains at these two frequencies were indistinguishable, data were pooled together.
AMPAR decay kinetics were obtained using a mono exponential function and mixed AMPAR/KAR decay kinetics using a bi-exponential function: I = A1∗e (−t/τfast) + A2∗exp(−t/τslow), where A1 and A2 are the current amplitudes of the fast and slow components, respectively. τweighted values were calculated using the equation: τweighted = (A1∗τfast + A2∗τslow)/(A1+A2).
Single particle tracking
Quantum Dot (QD) staining of surface HA-LiGluK2 or HA-GluK2 receptors was performed as described previously (Petrini et al., 2014). Briefly, anti-HA antibodies (Roche) were premixed with anti-mouse QD 655 (Invitrogen, Italy) for 30 min in the presence of caseine (Vectorlab, Italy) to prevent nonspecific binding. Neurons were then incubated with the diluted antibody-QD premix for 3 minutes at room temperature to obtain a final QD concentration of 0.1 nM. For SPT experiments with VGCC blockers, we sequentially added to the extracellular solution 10 μM nifedipine, 2 μM ω-conotoxin MVIIC, 50 μM D-APV, 10 μM GYKI 53655, 100 μM 2-APB and 10 μM ryanodine and we recorded the mobility of kainate receptors in the different conformational states in the continuous presence of these drugs. 2-APB, D-APV, Ryanodine and ω-conotoxin MVIIC were purchased from Tocris Bioscience (Italy). GYKY-53655, L-Glutamate and nifedipine were purchased from Sigma-Aldrich (Italy). The specificity of QD labeling was demonstrated by the exclusive binding of anti-HA-coupled QDs to HA-tagged overexpressed proteins and by the absence of QD labeling when the primary antibody was omitted from QD-antibody premix (data not shown). QD recording were performed on an inverted microscope (Eclipse Ti, Nikon, Japan) equipped with a 60X oil-immersion/1.4NA Plan Apochromat immersion objective by acquiring 100 consecutive frames at 20 Hz with a 512x512 pixel EM-CCD camera (9100, Hamamatsu, Japan) using Metamorph software (ver. 7.7.8, Molecular Devices, USA). The highly diluted QD labeling resulted in < 20 QDs per field of view, so that individual QD receptor complexes did not overlap the trajectories of neighboring complexes. When, occasionally, two QDs were too close to unambiguously reconstruct their individual trajectories, both QDs were discarded from the analysis. Samples were illuminated in epifluorescence with a diode-based illumination device (Lumencor, SpectraX Light Engine, Optoprim, Italy) controlled by the Metamorph software providing the appropriate excitation for Homer1c and QDs. By taking advantage of their wide excitation spectrum, QDs were imaged at either 380 or 488 nm, thus allowing the opening or closing of LiGluK2 and simultaneous QD imaging. Emission wavelengths were selected by means of optical filter (Semrock, Italy) mounted on a filter wheels controlled by the Metamorph software. Experiments were performed on an inverted microscope (Eclipse Ti, Nikon, Japan) equipped with a 60X oil-immersion/1.4NA Plan Apochromat immersion objective. During the experiments, neurons were kept at 32°C in an open chamber and continuously superfused with the recording solution at 12ml/h (see below). Synapses were identified by transfection of Homer1c-DsRed. QDs, recognized by their diffraction limited fluorescence spot shape and characteristic blinking, were tracked with 50 ms time resolution. QD spatial coordinates were identified in each frame as sets of > 4 connected pixels using two-dimensional object wavelet-based localization at sub-diffraction limited resolution (∼40 nm) with MIA software based on simulated annealing algorithm. Continuous tracking between blinks was performed with an implemented version of custom software originally written in MATLAB (The Mathworks Inc., Italy) in Dr Choquet’s lab. The method is based on a QD maximal allowable displacement (4 pixels) during a maximal allowable duration of the dark period (25 frames, corresponding to 1.25 s acquisition). This stringent reconnection of trajectories across QD blinking combined with the highly diluted QD labeling have been set to avoid erroneous reconnection of neighboring QD in the same trajectory and to provide unambiguous observations of individual receptor QD complex trajectories. Receptor trajectories were defined as ‘synaptic’ or ‘extrasynaptic’ when their spatial coordinates coincided or not with those of the localization of the postsynaptic compartment, respectively.
Although the definition of the compartments was diffraction limited, the sub-wavelength resolution of the single particle detection (∼40 nm) allowed accurate description of receptor mobility within synaptic regions. Instantaneous diffusion coefficients, D, were calculated as previously described (Tardin et al., 2003) from linear fits of the n = 1–4 values of the MSD versus time plot, according to the equation: MSD(t) = < r2 > = 4Dt for two-dimensional diffusion.
MSD(t) was calculated according to the formula:
for reconstructed trajectories of 100 frames using a custom-made software developed by Dr Choquet (Bordeaux, France). To better characterize receptor mobility, receptor QDs have been distinguished into mobile and immobile populations by using as a threshold the local minimum of the bimodal distribution of synaptic diffusion coefficients (0.0075 μm2 s-1). Next, the diffusive properties of the mobile receptor population were described as their median ± IQR, defined as the interval between 25%–75% percentiles. The immobile receptor population was described by the immobile fraction defined as the relative duration of the residency of a receptor QD in a given compartment with coefficient < 0.0075 μm2 s-1.
Cross-link (X-link) protocol
The X-linking of HA-tagged kainate receptors (HA-LiGluK2, HA-LiGluK2Δ16 and HA-GluK2) was achieved by first incubating hippocampal neurons for 10 min with an excess of the anti-HA primary antibody (Roche) and subsequently with an appropriate secondary antibody for 10 min (Gerrow and Triller, 2014, Heine et al., 2008). After washing, neurons were used for the recordings of uncaging synaptic currents (uEPSCs).
Immunocytochemistry
Neurons were rinsed in extracellular solution, fixed with 4% paraformaldehyde (PFA) for 10 min and subsequently incubated with BSA (1%) for 30 min to prevent nonspecific binding. Anti-HA antibody (1:50; Roche, Italy) was incubated for 60 minutes at room temperature, followed by incubation with Alexa-647 secondary antibody for 45 min at room temperature. Immunostaining of intracellular protein Homer1c (1:500, SySy) or VGLUT1 (1:400, Millipore) was performed by permeabilizing neurons with 0.2% Triton X-100 for 10 min after fixation, and sequential incubation with primary and secondary antibodies. Control experiments without the primary antibody were performed to test fluorescence signal arising from nonspecific binding of the secondary antibody. Coverslips were mounted in DAKO fluorescent mounting medium. Images were acquired using an inverted microscope (Eclipse Ti, Nikon, Japan) equipped with a 60X oil-immersion/1.4NA Plan Apochromat immersion objective and a 512x512 pixel EM-CCD camera (9100, Hamamatsu, Japan) with pixel size 16 μm. Quantification of immunocytochemistry experiments was carried out using Metamorph 7.8 (Molecular Device, USA). For each neuron, a dendritic portion was outlined to define the region of interest (ROI) included in the analysis. The same ROI was transferred to all channels to quantify the total average fluorescence of each probe (Homer1c, LiGluK2, N-cadherin). For the cluster analysis, in order to outline cluster regions, a user-defined threshold was applied to each FFT-processed image and regions were generated around the thresholded area (within the boundaries of the dendritic ROI). Thresholds were set individually for each channel and kept constant across each experiment. Cluster quantification was performed on LiGluK2 (or N-cadherin) and Homer1c images after subtraction of background fluorescence and without thresholding. Cluster immunoreactivity was quantified as the integrated fluorescence intensity measured in the aforementioned cluster regions. Synaptic analysis was performed by considering LiGluK2 immunoreactivity colocalizing with the postsynaptic marker Homer1c or juxtaposed (within 2 pixels) to the presynaptic marker vGlut. Data from clusters were averaged to give a value per neuron. Clusters smaller than 0.07 μm2 were excluded from the analysis. For each neuron, the cluster density was computed as the total number of cluster regions divided by the area of the dendritic ROI.
Coimmunoprecipitation and Western Blot
HEK293T were transfected with HA-LiGluK2 or HA-GluK2 plasmids alone, or along with N-cadΔE-myc or with HA-LiGluK2Δ16 using Lipofectamine 2000 and after 48h cells were lysed in a buffer containing 150 mM NaCl, Tris-HCl pH 7.4 25mM, 1 mM EDTA, 1% Np-40, supplemented with a cocktail of protease (Sigma, P8340) and phosphatase inhibitors (Sigma, P5726). The lysates were incubated on ice for 10 min and then centrifuged for 10 min at 13000 g 4°C. The supernatant was incubated with 25 μL of pre-washed anti-HA magnetic beads for 1 hour in a rotating wheel at 4°C. Beads were then collected on a magnetic stand and washed three times with ice-cold washing buffer (150 mM NaCl, Tris-HCl pH 7.4 25mM, 1 mM EDTA, 1% Np-40). Beads were re-suspended in sample buffer, heated at 95°C and magnetically separated. The eluted proteins were resolved by SDS–PAGE using 4%–12% Tris-glycine gels (Novex NuPAGE SDS–PAGE gels, Invitrogen) along with the “supernatant” (i.e., the remaining solution after IP) and electroblotted onto nitrocellulose membranes. Membranes were then incubated with anti-HA antibodies (1:1000, Roche), anti β-catenin (1:2000, Sigma) or anti-myc (1:1000, Cell Signaling) overnight at 4°C, washed, and incubated for 2 hours with horseradish peroxidase (HRP)-conjugated goat anti-rat, anti-rabbit immunoglobulin or anti-mouse (IgG) antibodies, respectively (1:5000, Invitrogen).
For coimmunoprecipitation experiments of KARs in the desensitized state, cells were incubated in an extracellular solution supplemented with glutamate (100 μM) with or without calcium for 2 minutes and then lysed by following the aforementioned protocol. 10% of total extract was loaded as control input.
For Neto2 Western Blot experiments, hippocampal neurons plated on 6-well Petri dishes coated with poly-D-lysine (0.1mg/ml) at a density of 550.000/well were washed with cold PBS and then lysed in RIPA buffer (which contained 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% NaDOC) supplemented with the aforementioned cocktail of protease and phosphatase inhibitors for 30 minutes at 4°C. Lysate were cleared by centrifugation at 20000 g for 20 minutes at 4°C and the total protein level was quantified by BCA (Pierce BCA Protein Assay Kit, ThermoScientific). Equivalent amounts of total protein (10 μg) were separated on NuPAGE 4%–12% (Invitrogen, Carlsbad, CA) in reducing conditions, transferred to nitrocellulose membranes, and immunoblotted overnight at 4°C with either monoclonal anti-NETO2 (1:1000, Abcam) or anti-GAPDH-HRP (1:1000, Invitrogen) antibodies. After washing, membranes incubated with the anti-Neto2 antibody were incubated for 2 hours with HRP-conjugated anti-mouse IgG antibodies (1:5000, Invitrogen). Blots were visualized by chemiluminescence detection, scanned and quantified using ImageJ software.
Model simulations
We performed computer simulations to recapitulate our main findings concerning the role of kainate receptor (KAR) diffusion in the short-term modulation of KAR-mediated EPSCs. The modeling of kainate receptor (KAR) gating (Figure S6B) was performed by using a kinetic scheme optimized from Barberis et al., (2008) to match the KAR-receptor mediated currents obtained experimentally in the present work (Figures 5C and S4). In line with the experimental stimulation protocol represented in Figures 5C and S4, the simulated stimulation protocol consisted of a control pulse to elicit a simulated KAR-mediated EPSC followed by a train of KAR-mediated EPSCs (1 s @100Hz) to induce massive receptor desensitization, and a “test” KARs-EPSC elicited 50 ms after the train to monitor the degree of receptor desensitization. Such simulated KAR-EPSCs were elicited by 0.3 ms pulses of glutamate 1mM. The fraction of receptors exchanged during the stimulation train (1 s) with the external naive ones was quantified by using stochastic simulations. After 1 s, the exchange rate reached a steady state (i.e., simulations performed with longer time periods yielded the same result). We next considered a simplified reaction-diffusion process to assess how changes in receptor lateral mobility (modulated by receptor gating) tunes synaptic responses. At the beginning of each simulation, the receptors were uniformly distributed on the synaptic disk (of radius 0.1 μm) at the density of 1100 receptors/μm2 (i.e., 27 receptors). In order to maintain the synapse populated by the same number of receptors we imposed that any receptor leaving the synaptic disk is instantaneously replaced with a naive one randomly positioned close to the border of the synaptic disk (Figure S6A). In the model, we assumed that: the receptors exchanged at discrete time instants in coincidence with the stimulation pulses (i.e., every 10 ms).
We simulated 10 spatial random arrangements of the receptors on the synaptic disk. Each one was simulated 10 times starting with a different, randomly generated receptor seed. During the simulations, we prevented receptors overlap by introducing an elastic repulsive force.
Each receptor (n) diffused according to a standard Brownian motion:
| (1) |
where , D is the diffusion coefficient, dt the time step of the simulation and are random numbers with a normal distribution. Equations (1) were integrated with a time step of 0.01 ms.
We performed simulations by considering different diffusion coefficients to mimic the control (i.e., closed receptor, D = 0.016 μm2/s) and the bound condition (open/desensitized receptor, 0.003 μm2/s). We found that synaptic receptors exchanged with the extrasynaptic ones with probability p = 0.41 in the closed naive state, and p = 0.09 in the open/desensitized states. These probabilities were used as weighting factors given to the naive receptors and the complementary weights to the immobile receptors (i.e., always present/active in the synapse). Such difference in “exchange probability” observed in the “lower receptor mobility” configuration resulted in a significantly higher extent of desensitization, hence reduced amplitude of simulated KAR-EPSC after the 100 Hz train, thus nicely reproducing our experimental data.
Quantification and Statistical Analysis
Values are given as means ± sem or, in the case of non-normally distributed QD data, as medians ± IQR. Prism 6.04 Software (GraphPad, USA) was used to test statistical significance. Non-normally distributed datasets were tested by two-tailed non-parametric Mann-Whitney U-test. Normally distributed datasets were compared using the unpaired two-tailed Student’s t test, while for paired non-parametric datasets, Wilcoxon paired test was used. Indications of significance correspond to P values < 0.05 (∗), p < 0.01 (∗∗), p < 0.001 (∗∗∗) and non-significant (ns). For QD tracking, n represents the total number of trajectories reconstructed after different experiments performed on multiple neuronal preparations. For immunocytochemistry and electrophysiological recordings, n represents the number of neurons analyzed.
Acknowledgments
This work has been supported by Telethon-Italy (GGP11043) and Compagnia di San Paolo (ROL-4318) to A.B. and by CERCA Programme (2017-SGR-1442), Human Brain Project (SGA3 Grant 945539), MINECO (DeepRed), “la Caixa” Foundation (ID 100010434, LCF/PR/HR19/52160010) to P.G. We are grateful to Dirk Trauner for providing us the MAG compound, Ehud Y. Isacoff for sharing the LiGluK2 plasmid, Susumu Tomita for providing the NETO2 plasmids, Olivier Thoumine for providing the N-cadherinΔE-myc construct, and Daniel Choquet for providing the Homer1c-DsRed plasmid. We thank Miquel Bosch and Aida Garrido-Charles for critical reading of the manuscript. We also thank Andrea Contestabile for technical help and Nelson Rebola, Mario Carta, Christophe Mulle, and Daniel Choquet for helpful discussion.
Author Contributions
Conceptualization, A.B.; Methodology, A.P., P.G., E.M.P., and A.B.; Validation, A.P., E.M.P., and A.B.; Formal Analysis, A.P., T.N., E.M.P., and A.B.; Investigation, A.P. and T.N.; Resources, S.G; Writing – Original Draft, A.B., P.G., and A.P.; Writing – Review & Editing, A.B., A.P., and E.M.P.; Visualization, A.P., E.M.P., T.N., and A.B.; Supervision, E.M.P. and A.B.; Project Administration, A.B.; Funding Acquisition, A.B.
Declaration of Interests
The authors declare no competing interests.
Published: June 9, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107735.
Supplemental Information
Representative Reconstructed Trajectory (Yellow) of a Single HA-LiGluK2 Receptor Diffusing at an Individual Glutamatergic Synapse (Red) in a Cultured Hippocampal Neuron, Related to Figure 1 Synapses were identified by the DsRed fluorescence of overexpressed Homer1c-DsRed. The LiGluK2 receptor was illuminated with 488 nm light in order to lock the receptor in the closed unbound state. Quantum dot has been tethered to the receptor by means of a primary antibody directed against the HA tag of HA-LiGluK2 receptor. The movie has been recorded for 3 s at 20 Hz frame rate by using a 512x512 pixel EM-CCD camera (9100, Hamamatsu, Japan). The spatial accuracy of the quantum dot location is 40 nm. Scale bar 1 μm.
Please note that the transition between the closed unbound to the open/desensitized states leads to the reduction of the receptor diffusion and to increased receptor confinement. Scale bar 1 μm.
References
- Ashby M.C., Maier S.R., Nishimune A., Henley J.M. Lateral diffusion drives constitutive exchange of AMPA receptors at dendritic spines and is regulated by spine morphology. J. Neurosci. 2006;26:7046–7055. doi: 10.1523/JNEUROSCI.1235-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamji S.X. Cadherins: actin with the cytoskeleton to form synapses. Neuron. 2005;47:175–178. doi: 10.1016/j.neuron.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Bannai H., Lévi S., Schweizer C., Inoue T., Launey T., Racine V., Sibarita J.B., Mikoshiba K., Triller A. Activity-dependent tuning of inhibitory neurotransmission based on GABAAR diffusion dynamics. Neuron. 2009;62:670–682. doi: 10.1016/j.neuron.2009.04.023. [DOI] [PubMed] [Google Scholar]
- Barberis A., Sachidhanandam S., Mulle C. GluR6/KA2 kainate receptors mediate slow-deactivating currents. J. Neurosci. 2008;28:6402–6406. doi: 10.1523/JNEUROSCI.1204-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bats C., Groc L., Choquet D. The interaction between Stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron. 2007;53:719–734. doi: 10.1016/j.neuron.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Borgdorff A.J., Choquet D. Regulation of AMPA receptor lateral movements. Nature. 2002;417:649–653. doi: 10.1038/nature00780. [DOI] [PubMed] [Google Scholar]
- Bowie D., Mayer M.L. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 1995;15:453–462. doi: 10.1016/0896-6273(95)90049-7. [DOI] [PubMed] [Google Scholar]
- Bowie D., Garcia E.P., Marshall J., Traynelis S.F., Lange G.D. Allosteric regulation and spatial distribution of kainate receptors bound to ancillary proteins. J. Physiol. 2003;547:373–385. doi: 10.1113/jphysiol.2002.033076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau I., Dieudonne S., Coussen F., Mulle C. Kainate receptor-mediated synaptic currents in cerebellar Golgi cells are not shaped by diffusion of glutamate. Proc. Natl. Acad. Sci. USA. 2000;97:6838–6843. doi: 10.1073/pnas.97.12.6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M., Opazo P., Veran J., Athané A., Choquet D., Coussen F., Mulle C. CaMKII-dependent phosphorylation of GluK5 mediates plasticity of kainate receptors. EMBO J. 2013;32:496–510. doi: 10.1038/emboj.2012.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo P.E., Malenka R.C., Nicoll R.A. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature. 1997;388:182–186. doi: 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- Chazeau A., Garcia M., Czöndör K., Perrais D., Tessier B., Giannone G., Thoumine O. Mechanical coupling between transsynaptic N-cadherin adhesions and actin flow stabilizes dendritic spines. Mol. Biol. Cell. 2015;26:859–873. doi: 10.1091/mbc.E14-06-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquet D., Triller A. The dynamic synapse. Neuron. 2013;80:691–703. doi: 10.1016/j.neuron.2013.10.013. [DOI] [PubMed] [Google Scholar]
- Constals A., Penn A.C., Compans B., Toulmé E., Phillipat A., Marais S., Retailleau N., Hafner A.S., Coussen F., Hosy E., Choquet D. Glutamate-induced AMPA receptor desensitization increases their mobility and modulates short-term plasticity through unbinding from Stargazin. Neuron. 2015;85:787–803. doi: 10.1016/j.neuron.2015.01.012. [DOI] [PubMed] [Google Scholar]
- Contractor A., Mulle C., Swanson G.T. Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci. 2011;34:154–163. doi: 10.1016/j.tins.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R., Epsztein J., Tyzio R., Becq H., Hirsch J., Ben-Ari Y., Crépel V. Quantal release of glutamate generates pure kainate and mixed AMPA/kainate EPSCs in hippocampal neurons. Neuron. 2002;35:147–159. doi: 10.1016/s0896-6273(02)00753-5. [DOI] [PubMed] [Google Scholar]
- Coussen F., Normand E., Marchal C., Costet P., Choquet D., Lambert M., Mège R.-M., Mulle C. Recruitment of the kainate receptor subunit glutamate receptor 6 by cadherin/catenin complexes. J. Neurosci. 2002;22:6426–6436. doi: 10.1523/JNEUROSCI.22-15-06426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G., Hajnóczky G. SR/ER-mitochondrial local communication: calcium and ROS. Biochim. Biophys. Acta. 2009;1787:1352–1362. doi: 10.1016/j.bbabio.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan M., Lévi S., Luccardini C., Rostaing P., Riveau B., Triller A. Diffusion dynamics of glycine receptors revealed by single-quantum dot tracking. Science. 2003;302:442–445. doi: 10.1126/science.1088525. [DOI] [PubMed] [Google Scholar]
- de Luca E., Ravasenga T., Petrini E.M., Polenghi A., Nieus T., Guazzi S., Barberis A. Inter-Synaptic Lateral Diffusion of GABAA Receptors Shapes Inhibitory Synaptic Currents. Neuron. 2017;95:63–69.e5. doi: 10.1016/j.neuron.2017.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore K., Aow J., Malinow R. Agonist binding to the NMDA receptor drives movement of its cytoplasmic domain without ion flow. Proc. Natl. Acad. Sci. USA. 2015;112:14705–14710. doi: 10.1073/pnas.1520023112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira J.S., Papouin T., Ladépêche L., Yao A., Langlais V.C., Bouchet D., Dulong J., Mothet J.-P., Sacchi S., Pollegioni L. Co-agonists differentially tune GluN2B-NMDA receptor trafficking at hippocampal synapses. eLife. 2017;6:1–22. doi: 10.7554/eLife.25492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fièvre S., Carta M., Chamma I., Labrousse V., Thoumine O., Mulle C. Molecular determinants for the strictly compartmentalized expression of kainate receptors in CA3 pyramidal cells. Nat. Commun. 2016;7:12738. doi: 10.1038/ncomms12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M., Ohliger-Frerking P. AMPA receptors and kainate receptors encode different features of afferent activity. J. Neurosci. 2002;22:7434–7443. doi: 10.1523/JNEUROSCI.22-17-07434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M., Malenka R.C., Nicoll R.A. Synaptic activation of kainate receptors on hippocampal interneurons. Nat. Neurosci. 1998;1:479–486. doi: 10.1038/2194. [DOI] [PubMed] [Google Scholar]
- Garcia E.P., Mehta S., Blair L.A., Wells D.G., Shang J., Fukushima T., Fallon J.R., Garner C.C., Marshall J. SAP90 binds and clusters kainate receptors causing incomplete desensitization. Neuron. 1998;21:727–739. doi: 10.1016/s0896-6273(00)80590-5. [DOI] [PubMed] [Google Scholar]
- Garcia M., Leduc C., Lagardère M., Argento A., Sibarita J.-B., Thoumine O. Two-tiered coupling between flowing actin and immobilized N-cadherin/catenin complexes in neuronal growth cones. Proc. Natl. Acad. Sci. USA. 2015;112:6997–7002. doi: 10.1073/pnas.1423455112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrow K., Triller A. GABAA receptor subunit composition and competition at synapses are tuned by GABAB receptor activity. Mol. Cell. Neurosci. 2014;60:97–107. doi: 10.1016/j.mcn.2014.04.001. [DOI] [PubMed] [Google Scholar]
- Goldin M., Epsztein J., Jorquera I., Represa A., Ben-Ari Y., Crépel V., Cossart R. Synaptic kainate receptors tune oriens-lacunosum moleculare interneurons to operate at theta frequency. J. Neurosci. 2007;27:9560–9572. doi: 10.1523/JNEUROSCI.1237-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorostiza P., Isacoff E.Y. Optical switches for remote and noninvasive control of cell signaling. Science. 2008;322:395–399. doi: 10.1126/science.1166022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L., Heine M., Cousins S.L., Stephenson F.A., Lounis B., Cognet L., Choquet D. NMDA receptor surface mobility depends on NR2A-2B subunits. Proc. Natl. Acad. Sci. USA. 2006;103:18769–18774. doi: 10.1073/pnas.0605238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M.K., Tang C., Verpelli C., Narayanan R., Stearns M.H., Xu R.-M., Li H., Sala C., Hayashi Y. The postsynaptic density proteins Homer and Shank form a polymeric network structure. Cell. 2009;137:159–171. doi: 10.1016/j.cell.2009.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine M., Groc L., Frischknecht R., Béïque J.-C., Lounis B., Rumbaugh G., Huganir R.L., Cognet L., Choquet D. Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science. 2008;320:201–205. doi: 10.1126/science.1152089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirbec H., Francis J.C., Lauri S.E., Braithwaite S.P., Coussen F., Mulle C., Dev K.K., Coutinho V., Meyer G., Isaac J.T.R. Rapid and differential regulation of AMPA and kainate receptors at hippocampal mossy fibre synapses by PICK1 and GRIP. Neuron. 2003;37:625–638. doi: 10.1016/s0896-6273(02)01191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob T.C., Bogdanov Y.D., Magnus C., Saliba R.S., Kittler J.T., Haydon P.G., Moss S.J. Gephyrin regulates the cell surface dynamics of synaptic GABAA receptors. J. Neurosci. 2005;25:10469–10478. doi: 10.1523/JNEUROSCI.2267-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou T.S., Stewart D.B., Stappert J., Nelson W.J., Marrs J.A. Genetic and biochemical dissection of protein linkages in the cadherin-catenin complex. Proc. Natl. Acad. Sci. USA. 1995;92:5067–5071. doi: 10.1073/pnas.92.11.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd F.L., Isaac J.T. Developmental and activity-dependent regulation of kainate receptors at thalamocortical synapses. Nature. 1999;400:569–573. doi: 10.1038/23040. [DOI] [PubMed] [Google Scholar]
- Lerma J., Marques J.M. Kainate receptors in health and disease. Neuron. 2013;80:292–311. doi: 10.1016/j.neuron.2013.09.045. [DOI] [PubMed] [Google Scholar]
- Mahadevan V., Dargaei Z., Ivakine E.A., Hartmann A.-M., Ng D., Chevrier J., Ormond J., Nothwang H.G., McInnes R.R., Woodin M.A. Neto2-null mice have impaired GABAergic inhibition and are susceptible to seizures. Front. Cell. Neurosci. 2015;9:368. doi: 10.3389/fncel.2015.00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraschi A., Ciammola A., Folci A., Sassone F., Ronzitti G., Cappelletti G., Silani V., Sato S., Hattori N., Mazzanti M. Parkin regulates kainate receptors by interacting with the GluK2 subunit. Nat. Commun. 2014;5:5182. doi: 10.1038/ncomms6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S., Bouschet T., Jenkins E.L., Nishimune A., Henley J.M. Bidirectional regulation of kainate receptor surface expression in hippocampal neurons. J. Biol. Chem. 2008;283:36435–36440. doi: 10.1074/jbc.M806447200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M.L., Vyklicky L., Jr. Concanavalin A selectively reduces desensitization of mammalian neuronal quisqualate receptors. Proc. Natl. Acad. Sci. USA. 1989;86:1411–1415. doi: 10.1073/pnas.86.4.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez P., De Roo M., Poglia L., Klauser P., Muller D. N-cadherin mediates plasticity-induced long-term spine stabilization. J. Cell Biol. 2010;189:589–600. doi: 10.1083/jcb.201003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson J.R., Chittori S., Merk A., Rao P., Han T.H., Serpe M., Mayer M.L., Subramaniam S. Structural basis of kainate subtype glutamate receptor desensitization. Nature. 2016;537:567–571. doi: 10.1038/nature19352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møllerud S., Frydenvang K., Pickering D.S., Kastrup J.S. Lessons from crystal structures of kainate receptors. Neuropharmacology. 2017;112:16–28. doi: 10.1016/j.neuropharm.2016.05.014. [DOI] [PubMed] [Google Scholar]
- Okuda T., Yu L.M.Y., Cingolani L.A., Kemler R., Goda Y. beta-Catenin regulates excitatory postsynaptic strength at hippocampal synapses. Proc. Natl. Acad. Sci. USA. 2007;104:13479–13484. doi: 10.1073/pnas.0702334104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios-Filardo J., Aller M.I., Lerma J. Synaptic Targeting of Kainate Receptors. Cereb. Cortex. 2016;26:1464–1472. doi: 10.1093/cercor/bhu244. [DOI] [PubMed] [Google Scholar]
- Paternain A.V., Rodríguez-Moreno A., Villarroel A., Lerma J. Activation and desensitization properties of native and recombinant kainate receptors. Neuropharmacology. 1998;37:1249–1259. doi: 10.1016/s0028-3908(98)00098-7. [DOI] [PubMed] [Google Scholar]
- Petrini E.M., Lu J., Cognet L., Lounis B., Ehlers M.D., Choquet D. Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron. 2009;63:92–105. doi: 10.1016/j.neuron.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrini E.M., Ravasenga T., Hausrat T.J., Iurilli G., Olcese U., Racine V., Sibarita J.-B., Jacob T.C., Moss S.J., Benfenati F. Synaptic recruitment of gephyrin regulates surface GABAA receptor dynamics for the expression of inhibitory LTP. Nat. Commun. 2014;5:3921. doi: 10.1038/ncomms4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A., Isacoff E.Y. Tethered ligands reveal glutamate receptor desensitization depends on subunit occupancy. Nat. Chem. Biol. 2014;10:273–280. doi: 10.1038/nchembio.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruano D., Lambolez B., Rossier J., Paternain A.V., Lerma J. Kainate receptor subunits expressed in single cultured hippocampal neurons: molecular and functional variants by RNA editing. Neuron. 1995;14:1009–1017. doi: 10.1016/0896-6273(95)90339-9. [DOI] [PubMed] [Google Scholar]
- Sachidhanandam S., Blanchet C., Jeantet Y., Cho Y.H., Mulle C. Kainate receptors act as conditional amplifiers of spike transmission at hippocampal mossy fiber synapses. J. Neurosci. 2009;29:5000–5008. doi: 10.1523/JNEUROSCI.5807-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas G.D., Blair L.A., Needleman L.A., Gonzales J.D., Chen Y., Li M., Singer J.D., Marshall J. Actinfilin is a Cul3 substrate adaptor, linking GluR6 kainate receptor subunits to the ubiquitin-proteasome pathway. J. Biol. Chem. 2006;281:40164–40173. doi: 10.1074/jbc.M608194200. [DOI] [PubMed] [Google Scholar]
- Straub C., Zhang W., Howe J.R. Neto2 modulation of kainate receptors with different subunit compositions. J. Neurosci. 2011;31:8078–8082. doi: 10.1523/JNEUROSCI.0024-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardin C., Cognet L., Bats C., Lounis B., Choquet D. Direct imaging of lateral movements of AMPA receptors inside synapses. EMBO J. 2003;22:4656–4665. doi: 10.1093/emboj/cdg463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi H., Abe K., Mizoguchi A., Takaoka K., Chisaka O., Takeichi M. Cadherin regulates dendritic spine morphogenesis. Neuron. 2002;35:77–89. doi: 10.1016/s0896-6273(02)00748-1. [DOI] [PubMed] [Google Scholar]
- Uchida N., Honjo Y., Johnson K.R., Wheelock M.J., Takeichi M. The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. J. Cell Biol. 1996;135:767–779. doi: 10.1083/jcb.135.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon C.G., Swanson G.T. Neto2 Assembles with Kainate Receptors in DRG Neurons during Development and Modulates Neurite Outgrowth in Adult Sensory Neurons. J. Neurosci. 2017;37:3352–3363. doi: 10.1523/JNEUROSCI.2978-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignes M., Collingridge G.L. The synaptic activation of kainate receptors. Nature. 1997;388:179–182. doi: 10.1038/40639. [DOI] [PubMed] [Google Scholar]
- Volgraf M., Gorostiza P., Numano R., Kramer R.H., Isacoff E.Y., Trauner D. Allosteric control of an ionotropic glutamate receptor with an optical switch. Nat. Chem. Biol. 2006;2:47–52. doi: 10.1038/nchembio756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariassen L.G., Katchan L., Jensen A.G., Pickering D.S., Plested A.J.R., Kristensen A.S. Structural rearrangement of the intracellular domains during AMPA receptor activation. Proc. Natl. Acad. Sci. USA. 2016;113:E3950–E3959. doi: 10.1073/pnas.1601747113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., St-Gelais F., Grabner C.P., Trinidad J.C., Sumioka A., Morimoto-Tomita M., Kim K.S., Straub C., Burlingame A.L., Howe J.R., Tomita S. A transmembrane accessory subunit that modulates kainate-type glutamate receptors. Neuron. 2009;61:385–396. doi: 10.1016/j.neuron.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative Reconstructed Trajectory (Yellow) of a Single HA-LiGluK2 Receptor Diffusing at an Individual Glutamatergic Synapse (Red) in a Cultured Hippocampal Neuron, Related to Figure 1 Synapses were identified by the DsRed fluorescence of overexpressed Homer1c-DsRed. The LiGluK2 receptor was illuminated with 488 nm light in order to lock the receptor in the closed unbound state. Quantum dot has been tethered to the receptor by means of a primary antibody directed against the HA tag of HA-LiGluK2 receptor. The movie has been recorded for 3 s at 20 Hz frame rate by using a 512x512 pixel EM-CCD camera (9100, Hamamatsu, Japan). The spatial accuracy of the quantum dot location is 40 nm. Scale bar 1 μm.
Please note that the transition between the closed unbound to the open/desensitized states leads to the reduction of the receptor diffusion and to increased receptor confinement. Scale bar 1 μm.