

Abstract
Nicotinamide N-methyltransferase (NNMT) catalyzes the methyl transfer from cofactor S-adenosylmethionine (SAM) to nicotinamide and other pyridine-containing compounds. NNMT is an important regulator for nicotinamide metabolism and methylation potential. Aberrant expression levels of NNMT have been implicated in cancer, metabolic and neurodegenerative diseases, which makes NNMT a potential therapeutic target. Therefore, potent and selective NNMT inhibitors can serve as valuable tools to investigate the roles of NNMT in its mediated diseases. Here, we applied a rational strategy to design and synthesize the tight-binding bisubstrate inhibitor LL320 through a novel propargyl linker. LL320 demonstrates a Ki value of 1.6±0.3 nM, which is the most potent inhibitor to date. The co-crystal structure of LL320 confirms its interaction with both the substrate and cofactor binding sites on NNMT. Importantly, this is the first example of using propargyl linker to construct potent methyltransferase inhibitors, which will expand our understanding of the transition state of methyl transfer.
Graphical Abstract
INTRODUCTION
Nicotinamide N-methyltransferase (NNMT), a cytosolic enzyme that is predominantly expressed in liver and adipose tissue, catalyzes the transfer of a methyl group from cofactor S-adenosyl-L-methionine (SAM) to nicotinamide (vitamin B3) to produce S-adenosyl-L-homocysteine (SAH) and N1-methylnicotinamide.1 NNMT can also methylate pyridines, quinoline, and other structurally related heterocyclic compounds.2 Its substrate, nicotinamide, is a precursor for NAD+, an important cofactor involved in redox metabolism and energy homeostasis.3 NNMT is also implicated in methyl donor balance since its expression level is inversely proportional to the SAM/SAH ratio both in vivo and in vitro.4 Because of its dual involvement in the metabolism of both the NAD+ precursor and the methyl donor SAM, NNMT is an important regulator for nicotinamide metabolism and methylation potential. Therefore, NNMT has been linked to various diseases including cancers, neurodegenerative diseases and obesity and diabetes.3,5–8 Increased expression of NNMT enhances cell proliferation and disease progression in several cancers including 5,7,9,10 Knockdown of NNMT by siRNA or shRNA inhibits the cancer cell growth and induces apoptosis.11 In addition, NNMT is up-regulated in Parkinson’s disease, atherosclerosis, obesity and Type 2 diabetes.3,12,13 Hence, NNMT has drawn emerging attention as a potential target for the aforementioned diseases.
Potent and selective NNMT inhibitors would be valuable tools to investigate the role of its catalytic activity in its physiological and pharmacological roles. However, current reported NNMT inhibitors only have modest activity at μM levels (Figure 1). 5-amino-1-MQ is a representative example that mimics product and binds to the substrate binding site.14 5-amino-1-MQ can rejuvenate muscle stem cells to improve the regenerative capacity of aged skeletal muscle. Compound JBSNF000088 acts as a competitive but slow-turnover substrate analog.15 JBSNF000088 has been demonstrated to reduce MNA levels and drives insulin sensitization in preclinical animal models of obesity.15 SS3 is a suicide inhibitor and promotes the modification of Cys159 upon methylation.16 The inhibitor HS312, containing an alpha-chloroacetamide group, inhibits NNMT by covalently inhibiting Cys165 that is located in the SAM/SAH binding site, but loses its engagement to NNMT in cells due to its reactivity with other cysteines in situ.17
Figure 1.

Structures of reported NNMT inhibitors.
Bisubstrate analogs have the potential to offer high potency and selectivity, especially for small molecule methyltransferases like NNMT. To achieve such potential, an optimal linker is critical to orient both substrate and SAM cofactor analogs to retain their respective interactions to the target. Three previously reported bisubstrate analogs VH45, MS2756 and MS2734 demonstrated moderate activities at μM range.18,19 During the development of potent bisubstrate inhibitor for protein N-terminal methyltransferase 1 (NTMT1), a 3-C atom linker exhibited its advantage to link the substrate and SAM analog to offer potent and selective inhibition at the nM level.20 Since the distance between the sulfur atom and nucleophilic N atom is about 4 Å in both NNMT and NTMT1,21,22 we hypothesize that a 3-C atom linker may serve as an optimal linker to produce NNMT bisubstrate analogs with high potency. Here, we designed and synthesized a series of bisubstrate analogs with variable 3-C atom linkers and a 4-C atom linker to investigate the optimal linker length. Top inhibitor LL320 with a propargyl linker exhibits a Ki value at 1.6±0.3 nM, which is the most potent inhibitor for NNMT to date. In addition, we obtained co-crystal structures of both LL319 (1a) and LL320 (2a) in complex with human NNMT (hNNMT), which clearly delineate the occupancies of bisubstrate analogs at both nicotinamide and SAM binding sites. Here, we reported the first case that propargyl linker is able to construct potent and tight bisubstrate inhibitors for NNMT. The propargyl linker expands the linker pool to devise bisubstrate analogs for methyltransferases and provides a valuable approach to develop potent and selective inhibitors.
RESULTS AND DISCUSSION
Design.
There are two adjacent binding sites that are occupied by nicotinamide and SAH from the ternary complex crystal structure of the hNNMT-nicotinamide-SAH (PDB 3ROD).22 The kinetic mechanism of NNMT indicates a rapid equilibrium ordered mechanism, where NNMT binds SAM first and is followed by binding nicotinamide to form a ternary complex.23 The distance between the nicotinamide nitrogen atom and the sulfur atom of SAH ranges from 3.5 to 4.2 Å,22 which is about the linear distance of 2 to 3 methylene groups. Using a saturated 3-C-atom linker has been shown to be a feasible strategy to link both substrate and SAM analogs to generate a potent and selective inhibitor for NTMT1.20 Thus, we hypothesized that bisubstrate analogs that covalently link a SAM and a nicotinamide analog through a 3-C-atom linker would imitate the ternary complex to offer higher potency and selectivity for NNMT. In addition, there is a very narrow tunnel guiding to the SN2-type of methylation process of nicotinamide by SAM. We proposed that a more rigid unsaturated 3-C-atom like a propargyl linker may constrain the conformation to decrease the entropy loss and to yield a higher potency than a saturated 3-C-atom linker (Figure 2).
Figure 2.

Design of NNMT bisubstrate inhibitors 1a-3a.
To support our hypothesis, we docked proposed compound 1a into the SAH binding site of the crystal structure of hNNMT −nicotinamide−SAH ternary complex (3ROD) (Figure 3). Comparison of our docking model with the ternary structure depicted that the NAH moiety of 1a overlays well with SAH and the benzamide moiety inserts deeper into the substrate binding pocket to attain two extra H-bonds than nicotinamide. The oxygen atom of the benzamide forms two H-bonds with Ser201 and Ser213. Also, the nitrogen atom of the benzamide interacts with Ser213 and Asp197 residues through two additional H-bonds.
Figure 3.

Docking analysis of compound 1a in the binding sites of hNNMT. (A) Docking of compound 1a (blue) in the hNNMT (gray) structure (PDB 3ROD). H-bond interactions are shown in yellow dotted lines. (B) Overlay of the docking model with the hNNMT (gray)−nicotinamide (yellow)−SAH (pink) complex (PDB 3ROD).
Synthesis.
Syntheses of all bisubstrate analogs were accomplished in a convergent route through reductive amination of NAH and aldehydes (Figure 2).24 Protected NAH was synthesized as previously described.25 Compound S3 was synthesized according to scheme S1. To synthesize compound 1a, allyl alcohol was coupled with 3-iodobenzonitrile 4a under Heck coupling condition to yield aldehyde 5a,26 which was subject to reductive amination with protected NAH to produce 6a (Scheme 1). Oxidation by H2O2 followed by TFA deprotection provided 1a.19 Other analogs 1e-1j were prepared using the similar procedure from commercially available aldehydes 5e-5j and protected NAH or S3 (Schemes 2 and 3). Compound 7a was synthesized from commercially available 3-iodobenzonitrile through Sonogashira cross-coupling reaction (Scheme 4).27 Then 7a was subjected to Dess-Martin oxidation to yield aldehyde 8a, which was then subjected to reductive amination with protected NAH to produce 9a. Subsequent oxidation by H2O2 and TFA deprotection produced 2a. Likewise, Sonogashira reaction with 3-butynl-1-ol offered 10, which underwent hydrogenation and subsequent oxidation to provide aldehyde 11 (Scheme 5). Reductive amination of 11 with protected NAH through reductive amination afforded 12a, which was then subject to H2O2 oxidation and acid deprotection to yield 3a. Ester prodrugs 1a* and 2a* were synthesized for the cell-based study (Schemes 1 and 5). MS2756 was re-synthesized as reported before and served as a positive control for inhibition study28.
Scheme 1.

Synthesis of bisubstrate inhibitors 1a - 1d and 1a*a
Scheme 2.

Synthesis of bisubstrate inhibitors 1e - 1ia
Scheme 3.

Synthesis of bisubstrate inhibitor 1ja
Scheme 4.

Synthesis of bisubstrate inhibitor 2a - 2d and 2a*a
Scheme 5.

Synthesis of bisubstrate inhibitors 3a - 3ba
aReagents and conditions: (a) Pd(OAc)2, Bu4NCl, NaHCO3, DMF, 3 h, 45 – 84%; (b) protected NAH or S3, NaBH3CN, AcOH, MeOH, 2 h, 56 – 68%; (c) H2O2, K2CO3, DMSO; then TFA, CH2Cl2, 0 °C to rt, 6 h, 49 – 61%; (d) SOCl22, EtOH, 0 °C to 50 °C, 51–59%; (e) TFA, CH2Cl2, 0 °C to rt, 6 h, 49 – 59%; (f) Pd(PPh3)2Cl2, CuI, Et3N, CH3CN, 12 h, 87 – 95%; (g) DMP, CH2Cl2, 0 °C to rt 1 h, 55 – 61%; (h) Pd/C, H2, THF, 12 h; then DMP, CH2Cl2, 0 °C to rt 1 h, 60%.
Biochemical Characterization.
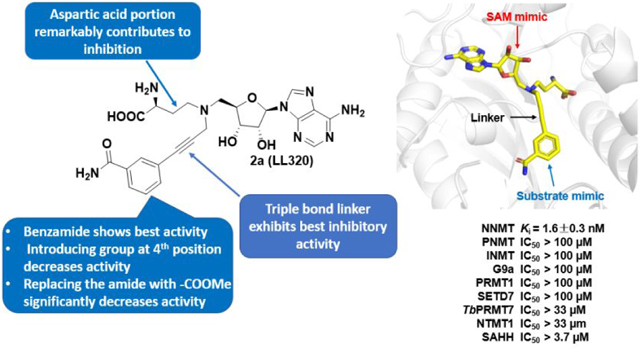
The SAH hydrolase (SAHH)-coupled fluorescence assay was employed to evaluate the inhibitory activities of all synthesized compounds continuously through monitoring the production of SAH.28,29 Under our SAHH-coupled assay condition, the Km values for nicotinamide and SAM were determined to be around 10 μM, which are comparable to reported values (Figure S1).28 Inhibition studies were performed with both SAM and nicotinamide at their respective Km values. All compounds were incubated with NNMT for 10 min before initiating the reaction through addition of nicotinamide. Inhibitory activity of each compound was first analyzed by obtaining initial velocities at increasing concentrations and followed by nonlinear regression fit. Compounds 1a-2a displayed IC50 values close to the enzyme concentration, suggesting that they are tight binding inhibitors.30 Therefore, both tight binding inhibitors were re-analyzed using Morrison’s quadratic equation to determine their apparent Ki values.30,31 Compared to the control MS2756 (Ki = 10 ± 0.35 μM), 1a (Ki = 43 ± 5.0 nM), 2a (Ki = 6.8 ± 2.3 nM), and 3a (Ki = 840 ± 50 nM) exhibit over 200-fold, 1,000-fold, and 10-fold increased inhibition, respectively. Therefore, the 3-C-atom linker used to construct 1a and 2a is superior to the 4-C-atom linker used to build 3a for development of NNMT bisubstrate inhibitors. Among them, the propargyl linker is the best linker because compound 2a showed the highest potency (Figure 4).
Figure 4.

Biochemical Characterization of compound LL320 (2a). (A) SAH hydrolase (SAHH)-coupled fluorescence assay of compound LL320 ranging in concentrations from 0 μM to 10 μM; (B) Concentration–response plots fitted to Morrison’s quadratic equation for compound LL320.
Structure-Activity Relationship (SAR) Studies.
After the 3-C-atom linker was established as the optimal length, SAR studies were carried out to understand the contribution of each portion of a potent NNMT bisubstrate inhibitor (Figure 5). Analogs of compounds 1a were initially synthesized for the SAR studies since compound 1a was designed as a standard approach to build a potent bisubstrate analog for NNMT.
Figure 5.

SARs of NNMT inhibitors. * The values of Ki for the compounds were determined with duplicate experiments (n = 2).
Nicotinamide analogs.
We began SAR analysis with the investigation on nicotinamide portion. Our synthetic scheme can accommodate the introduction of nicotinamide analogs through a late-stage reductive amination reaction. Quinoline and 4-methylnicotinamide have shown Km values comparable to nicotinamide,32 with isoquinoline showing about a 3-fold lower Km value than nicotinamide.32 Therefore, substituted benzamide and naphthalene moieties were introduced as a nicotinamide analog to produce 1a-1j. Compared with 1a, introducing a methyl group to the ortho-position of the benzamide marginally affected the inhibition as shown in compound 1c, while the activity decreased more than 3-fold when introducing methyl group at the para-position of benzamide as shown in compound 1b. When the benzamide moiety was replaced by a naphthalene ring to produce compounds 1g and 1h, the inhibitory activities dropped about 100-fold to 300-fold compared with 1a. Replacing the amide group with an ester, compound 1j lost about 700-fold activity compared with 1a, which further supported the importance of the amide group for the binding to NNMT.
Introduction of a methoxy group at the para-position of the benzamide of 2a produced 2b, resulting in a 7-fold loss of activity. When a methyl group was introduced at the ortho-position of the benzamide of 2a to generate 2c, comparable activity was observed.
Aspartic acid portion.
MS2734 showed over 10-fold higher inhibitory activity than MS 2756 by replacing the aspartic amino acid with glutamic acid in the SAM portion.28 However, similar alteration of 1a to 1d failed to display such trends, as 1d exhibited a 70-fold decreased activity. We also noticed that this alteration produced different effects on parent compounds as 1e, 1f and 3b showed either comparable or 3-fold decreased activities when compared to their respective parent compound 1g, 1h and 3a.
Deconstruction strategy.
In order to understand the contributions of each moiety of LL320, we applied a deconstruction strategy. When the aspartate portion is removed to yield 2d, there is about a 500-fold loss of activity compared to 2a. Furthermore, the Ki value of benzamide is more than 100 μM, resulting in over 10,000-fold loss of activity compared to 2a. This suggests that each portion of the bisubstrate analog is important for the potent inhibition.
Slow-binding inhibition.
Tight-binding inhibitors exhibit slow binding characteristics. Therefore, we investigated the optimal preincubation time that will ensure the equilibrium condition reached for Morrison’s analysis. Previously, all synthesized compounds were only preincubated with NNMT for 10 min. Therefore, different preincubation times (30 min, 60 min, 90 min, and 120 min) were examined to ensure the steady state condition can be achieved (Figure 6). From the results (Figure 6B), the system reaches equilibrium after a 60 min preincubation, as there is a negligible difference in Ki values at a longer preincubation time. In addition, rapid dilution study suggested the recovery of NNMT activity at 3 nM of LL320 (Figure S2), suggesting that compound LL320 is a reversible, tight binding inhibitor for NNMT.
Figure 6.

Time-dependent inhibition study of compound LL320 (n=2). (A) Concentration–response plots for different pre-incubation times fitted to Morrison’s quadratic equation for compound LL320; (B) Nonlinear regression of Morrison Ki values with pre-incubation time.
Selectivity Studies.
Bisubstrate analogs are known to display high selectivity to their target.20 To evaluate the selectivity of 2a, we investigated its inhibitory activity over a panel of methyltransferases including two closely-related methyltransferases to NNMT like phenylethanolamine N-methyltransferase (PNMT) and indolethylamine N-methyltransferase (INMT), two representative members from protein lysine methyltransferase PKMT (G9a and SETD7) and protein arginine methyltransferase PRMT (PRMT1 and TbPRMT7), respectively (Figure S3). We also included NTMT1, sharing a SAM cofactor binding pocket with NNMT. In addition, SAHH is included in the selectivity study because it has a SAH binding site and is used in the coupled fluorescence assay. As shown in Table 2 and Figure S3, compound 2a is more than 2,000-fold selective over SAM-dependent protein methyltransferases including PNMT, INMT, G9a, SETD7, and PRMT1 since it barely displayed any inhibition at 100 μM. In addition, 2a displayed about 1,000fold selectivity for TbPRMT7 and NTMT1, and 100-fold selectivity for SAHH.
Table 2.
Selectivity evaluation of compound LL320 (2a)
| Enzyme activity (%)a | [LL320] (μM) |
||||
|---|---|---|---|---|---|
| 3.7 | 11 | 33 | 100 | IC50 (μM) | |
| PNMT | 96 | 93 | 85 | 70 | >100 |
| INMT | 98 | 94 | 84 | 78 | >100 |
| G9a | 97 | 97 | 100 | 106 | >100 |
| SETD7 | 88 | 98 | 106 | 78 | >100 |
| PRMT1 | 93 | 95 | 86 | 60 | >100 |
| TbPRMT7 | 96 | 89 | 67 | 37 | >33 |
| NTMT1 | 64 | 85 | 65 | 39 | >33 |
| SAHH | 61 | 42 | 22 | 6 | >3.7 |
The values of enzyme activity are mean values of duplicate experiments (n = 2).
Co-crystal Structure of LL319 (1a) and LL320 (2a) in Complex with NNMT.
To elucidate the molecular interactions of bisubstrate analogs with NNMT, we determined the X-ray co-crystal structure of NNMT in complex with LL319 (PDB ID: 6PVE) and LL320 (PDB ID: 6PVS) (Figure 7). Both LL319 and LL320 were found to occupy both the cofactor and nicotinamide binding sties of NNMT. Specifically, the adenine part of 1a and 2a in the binary complex binds nearly identically with that of SAH. The inhibitor-superimposition of our NNMT-LL319 and NNMT-LL320 with the published NNMT-nicotinamide-SAH ternary complex (PDB ID: 3ROD) gave an RMSD value of 0.421 Å and 0.442 Å (across all residues of chain A), respectively. The interactions of inhibitors-NNMT are retained in a similar manner as previously observed with NNMT-SAH-nicotinamide in the ternary complex of nicotinamide/SAH and NNMT-MS2756 binary complex (Figure S4)22. For example, the carboxyl group of the NAH portion forms four H-bonds with Tyr 20, Tyr25, Tyr69, and Thr163. The amino group of NAH forms two H-bonds with Gly63 and Thr163 (Figure 7D and E). Meanwhile, the adenine moiety of both 1a and 2a forms two H-bonds with Asp142 and Val143, and two H-bonds with two water molecules. Hydroxyl groups of the ribose also form H-bonds with both Asp85 and Asn90. Meanwhile, the benzamide portion of 1a and 2a also binds very similarly to the nicotinamide through H-bond interactions with Ser201, Ser213, and one water molecule. Extensive examination of H-bonding interactions of 2 with NNMT indicates that additional H-bonds were formed between the amino group of NAH with Ser64 and Thr67, and between the benzamide portion with two surrounding Tyr residues 203 and 204. Superimposed structures also suggested that both 1a and 2a extended a bit further to interact with NNMT (Figure S4A).
Figure 7.

The structures of NNMT in complex with LL319 and LL320. (A) The structure of NNMT (gray cartoon) bound with LL319 (green stick) (PDB ID 6PVE). An 2Fo-Fc omit map contoured to 1.0 σ is shown for LL319 as a transparent green isosurface. (B) The structure of NNMT (gray cartoon) bound with LL320 (yellow stick) (PDB ID 6PVS). An Fo-Fc omit map contoured to 3.0 σ is shown for LL320 as a transparent yellow isosurface. (C) A structural alignment of LL319 (green stick) and LL320 (yellow stick) within the binding pocket of NNMT. A ligand interaction diagram is shown for LL319 (D) and LL320 (E) with NNMT.
Cell Permeability Evaluation.
We tested the cell permeability of compounds 1a-3a through MS detection of their cellular levels.33 However, the compounds were barely detected at 100 μM inside cells. In order to increase the cell permeability, corresponding ethyl ester prodrugs 1a* and 2a* of compounds 1a and 2a were designed and synthesized (Scheme 1 and 5). Unfortunately, prodrug 1a* barely exhibited enhanced cell permeability and 2a* only displayed limited permeability at 100 μM in comparison with TAT peptide through a cellular MS assay, respectively (Figure S5–7).
CONCLUSIONS
In summary, we designed and synthesized a series of potent bisubstrate inhibitors of NNMT. This potency supports the optimal length of the methyl transfer tunnel. To our knowledge, 2a is the most potent NNMT inhibitors reported so far with a Ki value of 1.6 ± 0.3 nM in the SAHH-coupled fluorescence assay. 2a exhibited around 1,000-fold selectivity for NNMT over other methyltransferases and almost 100-fold selectivity for SAHH. Furthermore, the co-crystal structure of NNMT in complex with compound 1a and 2a clearly showed that the bisubstrate inhibitor occupied both cofactor SAM and substrate binding sites, which is consistent with our design strategy and the docking study.
Despite their high potency and selectivity, limited cell permeability restricts them from cell-based studies. However, these tight and slow binding bisubstrate inhibitors are valuable probes for development of future cell-potent inhibitors for NNMT. More importantly, 2a is the first example to adopt a propargyl linker to conjugate SAM and a substrate analog to yield a methyltransferase bisubstrate inhibitor. In addition, the propargyl linker of 2a provides insights into the orientation of the methyl transfer of NNMT. Furthermore, the strategy described here not only applies to target NNMT, but also may have widespread impact on the development of potent inhibitors for other methyltransferases.
Note added:
During the revision of this manuscript, Shair and coworkers developed another tight binding bisubstrate inhibitor NS1 with alkynyl linker for NNMT41. The Ki value of the compound was around 500 pM and its analogs showed modest effect on cell-based study.
EXPERIMENTAL SECTION
Chemistry General Procedures.
The reagents and solvents were purchased from commercial sources (Fisher and Sigma-Aldrich) and used directly. Analytical thin-layer chromatography was performed on ready-to-use plates with silica gel 60 (Merck, F254). Flash column chromatography was performed over silica gel (grade 60, 230−400 mesh) on Teledyne Isco CombiFlash purification system. Final compounds were purified on preparative reversed phase high-pressure liquid chromatography (RP-HPLC) was performed on Agilent 1260 Series system. The Agilent 1260 Infinity II Variable Wavelength Detector (G7114A, UV = 254 nM) and a Waters BEH C18 (130Å, 5 μm, 10 mm X 250 mm) at a flow rate of 4 mL/min using a solvent system of 100% water with 0.1% TFA to 50% methanol over 30min were used to purify the final compounds. NMR spectra were acquired on a Bruker AV500 instrument (500 MHz for 1H-NMR, 126 MHz for 13C-NMR). TLC-MS were acquired using Advion CMS-L MS. Matrix-assisted laser desorption ionization mass spectra (MALDI-MS) data were acquired in positive-ion mode using a Sciex 4800 MALDI TOF/TOF MS. The Agilent 1260 Infinity II Variable Wavelength Detector (G7114A, UV = 254 nM) and an Agilent ZORBAX RR SB-C18 (80Å, 3.5 μm, 4.6 × 150 mm) at a flow rate of 1 mL/min using a solvent system of 100% water with 0.1% TFA to 40 or 60% methanol over 20min were used to assess purity of final compounds. All the purity of target compounds showed >95% in RP-HPLC.
General procedure A (Heck coupling).
Tetrabutyl ammonium chloride (4.9 mmol) and sodium bicarbonate (12.2 mmol) were taken in dry DMF (4 mL) and cooled to 0°C and 3-iodobenzonitril (4a-c) (4.9 mmol) was added. Allyl alcohol (7.3 mmol) was added to the reaction mixture, followed by a catalytic amount of Pd(OAc)2 (0.15 mmol) and the mixture was stirred at 0°C for 30 min. The reaction mixture was slowly warmed up to room temperature and further stirred for 2 h. The reaction mixture was diluted with water and the organic product was extracted with ether. The combined extracts were dried over anhydrous Na2SO4 and concentrated with under reduced pressure to get the crude product. The residue was purified with silica gel column to provide desired product.
General procedure B (Sonogashira cross coupling).
Under nitrogen condition, Pd(PPh3)2Cl2 (0.1mmol), CuI (0.2 mmol) and iodine substrates (4a-c) (5 mmol) were successively added to a 25 mL vial equipped with a stir bar. MeCN (5 mL) was added using a syringe, then propargyl alcohol (6mmol) was added to the mixture. E3N (15 mmol) was added at last. The reaction was stirred at room temperature for 8h. Solvent was removed in vacuo to leave a crude mixture, which is purified by silica gel column chromatography to afford pure product.
General procedure C (Oxidation of alcohols).
To a solution of the alcohol (7a-c) (2.5 mmol) in DCM (5 mL) were added DMP (2.5 mmol) at 0 °C. The resulting solution was stirred for 1 h at room temperature. The reaction was quenched with saturated sodium hyposulfite solution (5mL), extracted with DCM (3×25mL), washed with Na2S2O3 three times and dried over anhydrous Na2SO4. After removal of solvent, the residue was purified with silica gel column to provide the aldehydes.
General procedure D (Reductive amination).
To a solution of protected NAH, S1 or S3 (0.06 mmol) in 0.5 mL MeOH was added the aldehyde (5a-j, 8a-c and 11) (0.08 mmol) followed by 2 drops of AcOH. The resulting mixture was stirred for 30 min before NaBH3CN (0.1 mmol) was added. After being stirred for 2 h at rt, the reaction was quenched with saturated NaHCO3 and extracted with DCM (5×). The combined organic layers were dried over Na2SO4, filtered, and concentrated. The residue was purified with silica gel column to provide desired product.
General procedure E (Oxidation and deprotection of bisubstrate inhibitors).
To a solution of the cyano compound (6a-d, 9a-d, and 12a-b) (0.05 mmol) in DMSO (1 mL) was added K2CO3 (0.2 mmol). The mixture was cooled to 0 °C and treated with H2O2 (0.2 mL). The reaction mixture was warmed to rt and stirred for 3h at rt. The reaction was diluted with water and extracted with EtOAc (3×). The combined organic layers were dried over Na2SO4, filtered, and concentrated to afford the crude compound. The crude protected inhibitor was dissolved in DCM (0.5 mL). The resulting solution was cooled to 0 °C and treated with TFA (0.2 mL). The reaction mixture was warmed to rt and stirred for 6 h. The solution was concentrated under reduced pressure, dissolved in ddH2O and purified by reverse HPLC to get the corresponding bisubstrate compound.
General procedure F (Deprotection of bisubstrate inhibitors).
The protected inhibitor (6e-j) was dissolved in DCM (0.5 mL). The resulting solution was cooled to 0 °C and treated with TFA (0.2 mL). The reaction mixture was warmed to rt and stirred for 6 h. The solution was concentrated under reduced pressure, dissolved in ddH2O and purified by reverse HPLC to get the corresponding bisubstrate compound.
General procedure G (Esterification of bisubstrate analogs).
To a solution of bisubstrate analog 1a or 2a (0.02mmol) in anhydrous EtOH (3 mL), SOCl2 (0.1mmol) was added at 0 °C. The mixture was stirred at 50 °C for 2 h. The volatiles were removed under reduced pressure. The residue was dissolved in ddH2O and purified by RP-HPLC to provide the ester prodrugs of the respective bisubstrate inhibitors.
(S)-2-amino-4-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-(3-carbamoylphenyl)propyl)amino)butanoic acid (1a).
Compound 1a was prepared according to the general procedure E. 1H NMR (500 MHz, Methanol-d4) δ 8.40 (s, 1H), 8.32 (s, 1H), 7.71 – 7.61 (m, 2H), 7.39 – 7.21 (m, 2H), 6.11 – 5.95 (m, 1H), 4.71 – 4.61 (m, 1H), 4.47 – 4.32 (m, 2H), 4.04 – 3.92 (m, 1H), 3.84 – 3.70 (m, 1H), 3.67 – 3.52 (m, 2H), 3.50 – 3.39 (m, 1H), 3.29 – 3.18 (m, 2H), 2.78 – 2.65 (m, 2H), 2.44 – 2.27 (m, 1H), 2.24 – 2.12 (m, 1H), 2.12 – 1.97 (m, 2H). 13C NMR (126 MHz, Methanol-d4) δ 170.93, 170.21, 151.82, 148.27, 145.87, 142.66, 140.50, 133.91, 131.52, 128.42, 127.20, 125.26, 119.67, 90.70, 78.73, 73.19, 72.14, 54.97, 51.19, 46.92, 46.75, 31.65, 24.72, 24.55. MALDI-MS (positive) m/z: calcd for C24H33N8O6 [M + H]+ m/z 529.2523, found m/z 529.4907.
Ethyl (S)-2-amino-4-((((2R,3S,4R,5S)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-(3-carbamoylphenyl)propyl)amino)butanoate (1a*).
Compound 1a* was prepared according to the general procedure G. 1H NMR (500 MHz, D2O) δ 8.22 (s, 1H), 8.16 (s, 1H), 7.43 (d, J = 7.7 Hz, 1H), 7.34 (s, 1H), 7.21 (t, J = 7.5 Hz, 1H), 7.14 (d, J = 7.8 Hz, 1H), 5.92 (d, J = 3.6 Hz, 1H), 4.58 (t, J = 4.2 Hz, 1H), 4.39 – 4.29 (m, 2H), 4.20 (q, J = 7.1 Hz, 2H), 4.15 – 4.10 (m, 1H), 3.76 – 3.68 (m, 1H), 3.57 (d, J = 14.3 Hz, 1H), 3.52 – 3.43 (m, 1H), 3.43 – 3.34 (m, 1H), 3.24 (t, J = 8.3 Hz, 2H), 2.63 – 2.54 (m, 1H), 2.54 – 2.46 (m, 1H), 2.41 – 2.30 (m, 1H), 2.27 – 2.18 (m, 1H), 2.00 – 1.80 (m, 2H), 1.17 (t, J = 7.1 Hz, 3H). MALDI-MS (positive) m/z: calcd for C26H36N8O6Na [M + Na]+ m/z 579.2656, found m/z 579.1624.
(S)-2-amino-4-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4dihydroxytetrahydrofuran-2-yl)methyl)(3-(5-carbamoyl-2methylphenyl)propyl)amino)butanoic acid (1b).
Compound 1b was prepared according to the general procedure E. 1H NMR (500 MHz, Methanol-d4) δ 8.33 (d, J = 4.3 Hz, 1H), 8.26 (s, 1H), 7.62 (d, J = 2.0 Hz, 1H), 7.57 (dd, J = 7.8, 2.0 Hz, 1H), 7.16 (d, J = 7.8 Hz, 1H), 6.04 (t, J = 4.2 Hz, 1H), 4.70 (t, J = 4.2 Hz, 1H), 4.48 – 4.38 (m, 2H), 3.91 (dd, J = 9.6, 3.6 Hz, 1H), 3.80 – 3.70 (m, 1H), 3.62 (d, J = 13.9 Hz, 1H), 3.54 (td, J = 8.5, 8.0, 4.3 Hz, 1H), 3.50 – 3.42 (m, 1H), 3.35 (s, 3H), 2.75 – 2.60 (m, 2H), 2.39 – 2.29 (m, 1H), 2.22 (s, 2H), 2.12 (d, J = 6.5 Hz, 2H), 2.03 (p, J = 7.7 Hz, 2H). 13C NMR (126 MHz, Methanol-d4) δ 172.35, 155.10, 150.24, 149.89, 143.20, 141.70, 139.89, 132.71, 131.47, 128.90, 126.71, 121.09, 109.28, 92.11, 80.48, 74.33, 73.65, 56.09, 53.77, 49.85, 49.51, 30.41, 26.38, 24.85, 19.30.
(S)-2-amino-4-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4dihydroxytetrahydrofuran-2-yl)methyl)(3-(3-carbamoyl-4methylphenyl)propyl)amino)butanoic acid (1c).
Compound 1c was prepared according to the general procedure E. 1H NMR (500 MHz, Methanol-d4) δ 8.43 (s, 1H), 8.28 (s, 1H), 7.22 (s, 1H), 7.12 (d, J = 7.8 Hz, 1H), 7.06 (d, J = 7.9 Hz, 1H), 6.08 (d, J = 4.1 Hz, 1H), 4.73 – 4.68 (m, 1H), 4.49 – 4.35 (m, 2H), 4.08 – 3.93 (m, 1H), 3.80 – 3.71 (m, 1H), 3.63 (d, J = 13.8 Hz, 1H), 3.58 – 3.50 (m, 1H), 3.49 – 3.41 (m, 1H), 3.29 – 3.18 (m, 2H), 2.71 – 2.56 (m, 2H), 2.38 (s, 3H), 2.36 – 2.29 (m, 1H), 2.23 – 2.12 (m, 1H), 2.08 – 1.96 (m, 2H). 13C NMR (126 MHz, Methanol-d4) δ 174.24, 170.38, 151.72, 148.30, 145.69, 142.85, 137.44, 136.31, 133.35, 130.75, 129.54, 126.54, 120.04, 90.63, 78.77, 73.18, 72.14, 54.91, 51.08, 46.87, 46.29, 31.17, 24.67, 24.49, 18.03. MALDI-MS (positive) m/z: calcd for C25H35N8O6 [M + H]+ m/z 543.2680, found m/z 543.3776.
(S)-2-amino-5-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-(3-arbamoylphenyl)propyl)amino)pentanoic acid (1d).
Compound 1d was prepared according to the general procedure E. 1H NMR (500 MHz, Methanol-d4) δ 8.34 (d, J = 32.5 Hz, 2H), 7.79 – 7.58 (m, 2H), 7.41 – 7.20 (m, 2H), 6.07 (d, J = 3.8 Hz, 1H), 4.75 – 4.62 (m, 2H), 4.50 – 4.32 (m, 2H), 3.97 (d, J = 7.0 Hz, 1H), 3.78 (dd, J = 13.9, 9.8 Hz, 1H), 3.62 (d, J = 13.3 Hz, 1H), 3.36 – 3.32 (m, 1H), 3.27 (d, J = 8.2 Hz, 3H), 2.82 – 2.60 (m, 2H), 2.04 (dt, J = 13C NMR (126 MHz, 12.0, 6.0 Hz, 2H), 1.97 (d, J = 19.4 Hz, 2H), 1.93 – 1.81 (m, 2H). Methanol-d4) δ 170.92, 156.31, 148.48, 140.46, 134.01, 133.98, 133.94, 131.48, 128.45, 127.24, 126.24, 125.24, 90.80, 78.73, 73.06, 72.20, 52.59, 50.00, 46.94, 46.77, 31.67, 27.11, 24.43, 19.63.
(S)-2-amino-5-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-(naphthalen-1-yl)propyl)amino)pentanoic acid (1e).
Compound 1e was prepared according to the general procedure F. 1H NMR (500 MHz, Methanol-d4) δ 8.29 (s, 1H), 8.19 (s, 1H), 8.01 – 7.87 (m, 1H), 7.87 – 7.75 (m, 1H), 7.70 (d, J = 8.2 Hz, 1H), 7.51 – 7.38 (m, 2H), 7.32 (t, J = 7.6 Hz, 1H), 7.19 (d, J = 6.8 Hz, 1H), 5.99 (d, J = 3.4 Hz, 1H), 4.66 – 4.55 (m, 1H), 4.49 – 4.30 (m, 2H), 4.00 – 3.88 (m, 1H), 3.84 – 3.73 (m, 1H), 3.59 (d, J = 13.8 Hz, 1H), 3.49 – 3.33 (m, 3H), 3.29 – 3.25 (m, 1H), 3.20 – 3.08 (m, 1H), 3.08 – 2.96 (m, 1H), 2.23 – 2.04 (m, 2H), 2.04 13C NMR (126 MHz, Methanol-d4) δ 170.13, 152.86, 148.22, 145.75, 142.05, – 1.78 (m, 4H).135.86, 133.93, 131.38, 128.48, 126.89, 125.76, 125.64, 125.30, 125.08, 122.80, 119.70, 90.85, 78.26, 73.17, 72.12, 53.30, 52.83, 51.95, 48.45, 48.23, 28.87, 27.13, 19.76. MALDI-MS (positive) m/z: calcd for C28H36N7O5 [M + H]+ m/z 550.2778, found m/z 550.3173.
(S)-2-amino-5-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-(naphthalen-2-yl)propyl)amino)pentanoic acid (1f).
Compound 1f was prepared according to the general procedure F. 1H NMR (500 MHz, Methanol-d4) δ 8.31 (s, 1H), 8.27 (s, 1H), 7.80 – 7.75 (m, 1H), 7.71 (d, J = 8.1 Hz, 2H), 7.54 (s, 1H), 7.46 – 7.38 (m, 2H), 7.19 (d, J = 8.2 Hz, 1H), 6.00 (d, J = 3.7 Hz, 1H), 4.70 – 4.56 (m, 1H), 4.48 – 4.33 (m, 2H), 3.93 (t, J = 6.1 Hz, 1H), 3.85 – 3.70 (m, 1H), 3.70 – 3.55 (m, 1H), 3.39 – 3.32 (m, 2H), 3.30 – 3.24 (m, 2H), 2.87 – 2.67 (m, 2H), 2.19 – 2.03 (m, 2H), 2.03 – 1.74 (m, 4H). 13C NMR (126 MHz, Methanol-d4) δ 170.27, 153.68, 148.45, 141.67, 141.54, 137.33, 133.59, 132.25, 127.90, 127.22, 127.05, 126.25, 126.14, 125.79, 125.16, 119.80, 90.77, 78.44, 73.06, 72.17, 54.71, 53.04, 52.75, 48.37, 32.06, 27.16, 24.53, 19.72. MALDI-MS (positive) m/z: calcd for C28H36N7O5 [M + H]+ m/z 550.2778, found m/z 550.3164.
(S)-2-amino-4-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4dihydroxytetrahydrofuran-2-yl)methyl)(3-(naphthalen-1yl)propyl)amino)butanoic acid (1g).
Compound 1g was prepared according to the general procedure F. 1H NMR (500 MHz, Methanol-d4) δ 8.26 (s, 1H), 8.12 (s, 1H), 7.87 (d, J = 5.8 Hz, 1H), 7.73 (d, J = 6.9 Hz, 1H), 7.61 (d, J = 8.2 Hz, 1H), 7.35 (dd, J = 6.2, 3.2 Hz, 2H), 7.25 (t, J = 7.6 Hz, 1H), 7.14 (d, J = 5.4 Hz, 1H), 5.93 (s, 1H), 5.42 (s, 1H), 4.55 (s, 1H), 4.44 – 4.22 (m, 2H), 3.94 – 3.80 (m, 1H), 3.68 (s, 1H), 3.57 – 3.43 (m, 2H), 3.37 (s, 2H), 3.06 (d, J = 7.2 Hz, 2H), 3.01 – 2.91 (m, 1H), 2.28 (s, 1H), 2.08 (p, J = 8.1 Hz, 3H). 13C NMR (126 MHz, CD3OD_SPE) δ 172.62, 154.64, 151.16, 149.70, 143.46, 137.36, 135.35, 132.83, 129.89, 128.26, 127.19, 127.06, 126.70, 126.48, 124.29, 92.10, 80.38, 74.57, 73.58, 56.00, 54.80, 53.55, 30.31, 28.04, 26.37, 25.70.
(S)-2-amino-4-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4dihydroxytetrahydrofuran-2-yl)methyl)(3-(naphthalen-2-yl)propyl)amino)butanoic acid (1h).
Compound 1h was prepared according to the general procedure F. 1H NMR (500 MHz, Methanol-d4) δ 8.38 – 8.22 (m, 2H), 7.76 (d, J = 7.6 Hz, 1H), 7.74 – 7.68 (m, 2H), 7.55 (s, 1H), 7.45 – 7.36 (m, 2H), 7.25 – 7.18 (m, 1H), 6.04 5.96 (m, 1H), 4.68 – 4.59 (m, 1H), 4.48 – 4.35 (m, 2H), 4.03 – 3.90 (m, 1H), 3.83 – 3.69 (m, 1H), 3.69 – 3.59 (m, 1H), 3.59 – 3.50 (m, 1H), 3.50 – 3.42 (m, 1H), 3.39 – 3.32 (m, 1H), 3.28 3.22 (m, 1H), 2.86 – 2.73 (m, 2H), 2.42 – 2.28 (m, 1H), 2.22 – 2.05 (m, 3H). MALDI-MS (positive) m/z: calcd for C27H34N7O5 [M + H]+ m/z 536.2621, found m/z 536.3082.
(S)-2-amino-4-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(2-(naphthalen-2-yl)ethyl)amino)butanoic acid (1i).
Compound 1i was prepared according to the general procedure F. 1H NMR (500 MHz, Methanol-d4) δ 8.38 (s, 1H), 8.21 (s, 1H), 7.80 – 7.73 (m, 1H), 7.67 (dd, J = 18.8, 6.8 Hz, 2H), 7.55 (s, 1H), 7.47 – 7.40 (m, 2H), 7.23 (d, J = 8.0 Hz, 1H), 6.10 (d, J = 4.7 Hz, 1H), 4.75 (t, J = 4.9 Hz, 1H), 4.57 (d, J = 7.5 Hz, 1H), 4.44 (t, J = 5.1 Hz, 1H), 3.99 (dd, J = 9.2, 3.7 Hz, 1H), 3.85 – 3.72 (m, 2H), 3.72 – 3.63 (m, 1H), 3.63 – 3.50 (m, 3H), 3.35 (s, 1H), 3.25 – 3.12 (m, 2H), 2.51 – 2.38 (m, 1H), 2.21 (s, 1H). 13C NMR (126 MHz, Methanol-d4) δ 172.43, 154.81, 149.89, 143.31, 134.84, 134.68, 133.85, 129.55, 128.64, 128.43, 128.35, 127.66, 127.45, 127.05, 121.12, 91.93, 80.63, 74.30, 73.72, 55.99, 53.25, 40.42(2C), 30.84, 26.16.
(S)-2-amino-4-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-(3-(methoxycarbonyl)phenyl)propyl)amino)butanoic acid (1j).
Compound 1j was prepared according to the general procedure F. 1H NMR (500 MHz, Methanol-d4) δ 8.41 (s, 1H), 8.31 (s, 1H), 7.81 (d, J = 5.1 Hz, 2H), 7.35 (d, J = 6.8 Hz, 2H), 6.05 (d, J = 3.8 Hz, 1H), 4.73 – 4.64 (m, 1H), 4.48 – 4.38 (m, 2H), 4.06 – 3.94 (m, 1H), 3.89 (s, 3H), 3.84 – 3.73 (m, 1H), 3.68 – 3.40 (m, 4H), 3.35 (s, 1H), 2.37 (s, 1H), 2.24 – 2.13 (m, 1H), 2.11 – 1.93 (m, 2H). 13C NMR (126 MHz, Methanol-d4) δ 168.42, 164.26, 154.00, 149.83, 145.65, 143.60, 142.09, 134.21, 133.49, 131.58, 130.28, 129.87, 128.57, 91.89, 80.36, 74.54, 73.57, 56.29, 53.82,52.69, 40.44(2C), 33.08, 26.33, 26.15.
(S)-2-amino-4-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-(3-carbamoylphenyl)prop-2-yn-1-yl)amino)butanoic acid (2a).
Compound 2a was prepared according to the general procedure E.1H NMR (500 MHz, Methanol-d4) δ 8.46 (d, J = 11.5 Hz, 1H), 8.34 (s, 1H), 7.95 (s, 1H), 7.88 (d, J = 7.7 Hz, 1H), 7.58 (d, J = 7.6 Hz, 1H), 7.46 (t, J = 7.8 Hz, 1H), 6.20 – 6.00 (m, 1H), 4.72 (t, J = 4.6 Hz, 1H), 4.53 – 4.32 (m, 2H), 4.28 – 4.13 (m, 2H), 4.12 – 4.01 (m, 1H), 13C NMR 3.66 – 3.47 (m, 2H), 3.47 – 3.33 (m, 2H), 2.48 – 2.28 (m, 1H), 2.27 – 2.04 (m, 1H). (126 MHz, Methanol-d4) δ 170.39, 169.76, 151.36, 148.37, 145.17, 142.78, 134.44, 134.16, 130.83, 128.56, 127.86, 121.85, 119.58, 90.36, 87.60, 80.18, 79.57, 73.57, 72.17, 55.80, 51.13, + m/z 525.221, 48.30, 42.77, 25.48. MALDI-MS (positive) m/z: calcd for C24H29N8O6 [M + H] 41 found m/z 525.3198.
Ethyl (S)-2-amino-4-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-(3-carbamoylphenyl)prop-2-yn-1-yl)amino)butanoate (2a*).
Compound 2a* was prepared according to the general procedure G. 1H NMR (500 MHz, Methanol-d4) δ 8.46 (s, 1H), 8.37 (s, 1H), 7.95 (t, J = 1.8 Hz, 1H), 7.89 – 7.84 (m, 1H), 7.59 – 7.54 (m, 1H), 7.46 (t, J = 7.8 Hz, 1H), 6.11 (d, J = 3.9 Hz, 1H), 4.71 (t, J = 4.2 Hz, 1H), 4.44 – 4.38 (m, 2H), 4.31 – 4.23 (m, 2H), 4.19 (t, J = 6.3 Hz, 1H), 4.13 – 4.03 (m, 2H), 3.47 – 3.35 (m, 2H), 3.24 (t, J = 7.0 Hz, 2H), 2.35 – 2.25 (m, 1H), 2.23 – 13C NMR (126 MHz, Methanol-d4) δ 171.13, 169.85, 2.12 (m, 1H), 1.28 (t, J = 7.1 Hz, 3H). 152.78, 149.77, 146.63, 144.08, 135.73, 135.56, 132.20, 129.89, 128.95, 123.66, 121.01, 91.77, 88.05, 82.52, 82.26, 75.04, 73.57, 64.01, 57.51, 52.99, 51.52, 44.29, 27.23, 14.32. MALDI-MS (positive) m/z: calcd for C26H33N8O6 [M + H]+ m/z 553.2523, found m/z 553.2736.
(S)-2-amino-4-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-(5-carbamoyl-2-methoxyphenyl)prop-2-yn-1-yl)amino)butanoic acid (2b).
Compound 2b was prepared according to the general procedure E. 1H NMR (500 MHz, Methanol-d4) δ 8.45 (s, 1H), 8.30 (s, 1H), 7.97 – 7.87 (m, 2H), 7.09 (d, J = 8.7 Hz, 1H), 6.10 (d, J = 4.4 Hz, 1H), 4.76 (t, J = 4.8 Hz, 1H), 4.52 – 4.44 (m, 1H), 4.39 (t, J = 5.1 Hz, 1H), 4.16 (s, 2H), 4.04 (d, J = 6.5 Hz, 1H), 3.91 (s, 3H), 3.56 (t, J = 11.8 Hz, 1H), 3.50 – 3.42 (m, 1H), 3.38 (d, J = 6.6 Hz, 1H), 3.34 (s, 2H), 2.40 – 2.25 (m, 1H), 2.17 – 2.04 (m, 1H). 13C NMR (126 MHz, Methanol-d4) δ 170.43, 164.43, 154.12, 148.44, 142.32, 139.19, 134.23, 131.50, 127.10, 124.76, 120.52, 112.17, 111.72, 91.39, 85.10, 81.97, 74.89, 73.66, 61.00, 57.12, 56.76, 52.92, 49.85, 44.08, 27.01.
(S)-2-amino-4-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-(3-carbamoyl-4-methylphenyl)prop-2-yn-1-yl)amino)butanoic acid (2c).
Compound 2c was prepared according to the general procedure E. 1H NMR (500 MHz, Methanol-d4) δ 8.28 (s, 1H), 8.24 (d, J = 4.1 Hz, 2H), 7.47 (d, J = 1.8 Hz, 1H), 7.34 (dd, J = 7.7, 2.0 Hz, 2H), 7.24 (d, J = 8.2 Hz, 2H), 6.02 (d, J = 4.5 Hz, 2H), 4.77 (d, J = 4.4 Hz, 1H), 4.37 (d, J = 6.4 Hz, 6H), 3.96 (d, J = 9.3 Hz, 9H), 3.24 – 2.94 (m, 3H), 2.44 (s, 5H), 2.24 (s, 2H), 2.01 (s, 3H). MALDI-MS (positive) m/z: calcd for C25H31N8O6 [M + H]+ m/z 539.2367, found m/z 539.1651.
3-(3-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)amino)prop-1-yn-1-yl)benzamide (2d).
Compound 2d was prepared according to the general procedure E. 1H NMR (500 MHz, Methanol-d4) δ 8.47 – 8.32 (m, 2H), 8.00 – 7.94 (m, 1H), 7.94 – 7.86 (m, 1H), 7.66 – 7.58 (m, 1H), 7.53 – 7.44 (m, 1H), 6.18 – 6.05 (m, 1H), 4.82 – 4.78 (m, 1H), 4.51 – 4.46 (m, 1H), 4.45 – 4.39 (m, 1H), 4.26 (d, J = 4.5 13C NMR (126 MHz, Methanol-d4) δ 169.65, 152.17, 148.41, Hz, 2H), 3.78 – 3.59 (m, 2H). 146.27, 142.82, 134.49, 134.30, 130.86, 128.65, 128.13, 121.50, 119.97, 90.72, 87.25, 80.11, 79.10, 73.53, 71.89, 48.38, 37.05. MALDI-MS (positive) m/z: calcd for C20H21N7NaO4 [M + + m/z 446.1553, found m/z 446.1817. Na]
(S)-2-amino-4-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4dihydroxytetrahydrofuran-2-yl)methyl)(4-(3-carbamoylphenyl)butyl)amino)butanoic acid (3a).
Compound 3a was prepared according to the general procedure E. 1H NMR (500 MHz, Methanol-d4) δ 8.43 (d, J = 6.5 Hz, 1H), 8.33 (s, 1H), 7.81 – 7.57 (m, 2H), 7.45 – 7.23 (m, 2H), 6.10 (d, J = 3.8 Hz, 1H), 4.72 (t, J = 4.5 Hz, 1H), 4.51 – 4.39 (m, 2H), 4.03 – 3.91 (m, 1H), 3.79 – 3.67 (m, 1H), 3.62 (d, J = 13.8 Hz, 1H), 3.57 – 3.49 (m, 1H), 3.49 – 3.38 (m, 1H), 3.30 – 3.18 (m, 2H), 2.64 (t, J = 7.2 Hz, 2H), 2.46 – 2.28 (m, 1H), 2.25 – 2.07 (m, 1H), 1.83 – 1.51 (m, 4H).13C NMR (126 MHz, Methanol-d4) δ 171.16, 170.53, 152.16, 148.42, 146.46, 142.49, 141.83, 133.64, 131.66, 128.33, 127.32, 124.93, 119.55, 90.44, 78.76, 73.19, 72.12, 54.96, 53.20, 51.40, 48.25, 34.37, 27.57, 24.74, 22.60. MALDI-MS (positive) m/z: calcd for C25H35N8O6 [M + H]+ m/z 543.2680, found m/z 543.3305.
(S)-2-amino-5-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(4-(3-carbamoylphenyl)butyl)amino)pentanoic acid (3b).
Compound 3b was prepared according to the general procedure E. 1H NMR (500 MHz, Methanol-d4) δ 8.38 (brs, 2H), 7.82 – 7.51 (m, 2H), 7.48 – 7.16 (m, 2H), 6.11 – 6.04 (m, 1H), 4.73 (t, J = 4.4 Hz, 1H), 4.46 – 4.40 (m, 2H), 3.95 (brs, 1H), 3.79 – 3.71 (m, 1H), 3.63 – 3.56 (m, 1H), 3.28 – 3.19 (m, 4H), 2.69 – 2.56 (m, 2H), 2.02 – 1.81 (m, 4H), 1.77 – 1.59 (m, 4H). MALDI-MS (positive) m/z: calcd for C26H37N8O6 [M + H]+ m/z 557.2836, found m/z 557.4778.
3-(3-oxopropyl)benzonitrile (5a).
Compound 5a was synthesized according to general procedure A. 1H NMR (500 MHz, Chloroform-d) δ 9.81 (s, 1H), 7.37–7.50 (m, 4H), 2.98 (t, J = 5 Hz, 2H), 2.82 (t, J = 5 Hz, 2H); 13C NMR (150 MHz, Chloroform-d) δ 200.4, 141.9, 133.1, 131.9, 130.1, 129.4, 118.8, 112.6, 44.7, 27.5.
4-methyl-3-(3-oxopropyl)benzonitrile (5b).
Compound 5b was synthesized according to general procedure A. 1H NMR (500 MHz, Chloroform-d) δ 9.82 (d, J = 3.7 Hz, 1H), 7.38 (d, J = 8.0 Hz, 2H), 7.22 (d, J = 7.5 Hz, 1H), 2.93 (t, 2H), 2.77 (t, J = 7.6 Hz, 2H), 2.36 (s, 3H). 13C NMR (126 MHz, Chloroform-d) δ 200.45, 141.99, 140.03, 131.86, 131.09, 130.03, 119.03, 109.87, 43.28, 24.77, 19.70.
2-methyl-5-(3-oxopropyl)benzonitrile (5c).
Compound 5c was synthesized according to general procedure A. 1H NMR (500 MHz, Chloroform-d) δ 9.88 – 9.60 (m, 1H), 7.41 (s, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.22 (d, J = 8.0 Hz, 1H), 2.92 (d, J = 7.3 Hz, 2H), 2.79 (d, J = 7.2 Hz, 2H), 2.48 (s, 3H).
Tert-butyl -(S)-4-((((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(3-(3-cyanophenyl)propyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (6a).
Compound 6a was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.29 (d, J = 3.0 Hz, 1H), 7.91 (s, 1H), 7.52 – 7.27 (m, 4H), 6.11 – 6.04 (m, 1H), 6.04 – 5.86 (m, 2H), 5.60 – 5.35 (m, 2H), 5.12 – 4.94 (m, 1H), 4.51 – 4.26 (m, 1H), 4.23 – 4.04 (m, 1H), 2.96 – 2.26 (m, 8H), 2.07 – 1.89 (m, 1H), 1.86 – 1.65 (m, 3H), 1.61 (s, 3H), 1.53 – 1.26 (m, 21H).
Tert-butyl (S)-4-((((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(3-(5-cyano-2methylphenyl)propyl)aino)-2-((tert-butoxycarbonyl)amino)butanoate (6b).
Compound 6b was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.27 (s, 1H), 7.90 (s, 1H), 7.41 – 7.31 (m, 2H), 7.20 – 7.14 (m, 1H), 6.09 – 6.02 (m, 1H), 5.94 (s, 2H), 5.60 (d, J = 8.2 Hz, 1H), 5.54 – 5.43 (m, 1H), 5.04 – 4.95 (m, 1H), 4.34 – 4.26 (m, 1H), 4.21 – 4.14 (m, 1H), 2.80 – 2.72 (m, 1H), 2.64 – 2.40 (m, 7H), 2.29 (s, 3H), 1.97 – 1.87 (m, 1H), 1.74 – 1.61 (m, 3H), 1.59 (s, 3H), 1.51 – 1.30 (m, 21H).
Tert-butyl (S)-4-((((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(3-(3-cyano-4methylphenyl)propyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (6c).
Compound 6c was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.28 (s, 1H), 7.90 (s, 1H), 7.36 (s, 1H), 7.23 (d, J = 8.1 Hz, 1H), 7.17 (d, J = 7.8 Hz, 1H), 6.04 (s, 1H), 6.01 (s, 2H), 5.67 (d, J = 8.1 Hz, 1H), 5.49 (d, J = 6.5 Hz, 1H), 5.03 – 4.93 (m, 1H), 4.32 – 4.23 (m, 1H), 4.21 – 4.10 (m, 1H), 2.77 – 2.71 (m, 1H), 2.60 – 2.52 (m, 3H), 2.51 – 2.46 (m, 4H), 2.45 – 2.41 (m, 2H), 2.40 – 2.35 (m, 1H), 1.94 – 1.86 (m, 1H), 1.70 – 1.62 (m, 3H), 1.60 (s, 3H), 1.48 – 1.36 (m, 21H).
Tert-butyl (S)-5-((((3aR,4R,6S,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(3-(3-cyanophenyl)propyl)amino)-2-((tert-butoxycarbonyl)amino)pentanoate (6d).
Compound 6d was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.30 (s, 1H), 7.92 (s, 1H), 7.49 – 7.44 (m, 1H), 7.42 (s, 1H), 7.35 (d, J = 4.8 Hz, 2H), 6.05 (s, 1H), 5.72 (s, 2H), 5.51 (d, J = 6.3 Hz, 1H), 5.18 (d, J = 8.4 Hz, 1H), 5.00 – 4.92 (m, 1H), 4.30 (s, 1H), 4.18 – 4.09 (m, 1H), 2.70 – 2.35 (m, 8H), 1.76 – 1.53 (m, 9H), 1.46 – 1.39 (m, 21H).
Tert-butyl (S)-5-((((3aR,4R,6S,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(3-(naphthalen-1-yl)propyl)amino)-2-((tert-butoxycarbonyl)amino)pentanoate (6e).
Compound 6e was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.30 (s, 1H), 8.00 (dd, J = 8.2, 1.5 Hz, 1H), 7.92 (s, 1H), 7.84 (dd, J = 7.6, 1.7 Hz, 1H), 7.70 (d, J = 8.2 Hz, 1H), 7.52 – 7.43 (m, 2H), 7.40 – 7.35 (m, 1H), 7.27 (d, J = 6.9 Hz, 1H), 6.07 – 6.00 (m, 1H), 5.70 – 5.61 (m, 2H), 5.50 (dd, J = 6.4, 2.1 Hz, 1H), 5.20 (d, J = 8.4 Hz, 1H), 4.97 (dd, J = 6.8, 3.2 Hz, 1H), 4.34 (s, 1H), 4.15 (s, 1H), 3.10 – 2.94 (m, 2H), 2.76 – 2.67 (m, 1H), 2.63 – 2.42 (m, 5H), 1.86 – 1.70 (m, 6H), 1.60 (s, 3H), 1.42 (s, 18H), 1.38 (s, 3H).
Tert-butyl (S)-5-((((3aR,4R,6S,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(3-(naphthalen-2-yl)propyl)amino)-2-((tert-butoxycarbonyl)amino)pentanoate (6f).
Compound 6f was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.31 (s, 1H), 7.92 (s, 1H), 7.81 – 7.72 (m, 3H), 7.57 (s, 1H), 7.47 – 7.38 (m, 2H), 7.28 (d, J = 8.0 Hz, 1H), 6.03 (s, 1H), 5.78 (s, 2H), 5.49 (d, J = 6.4 Hz, 1H), 5.20 (d, J = 8.4 Hz, 1H), 4.99 – 4.90 (m, 1H), 4.37 – 4.27 (m, 1H), 4.20 – 4.11 (m, 1H), 2.78 – 2.63 (m, 3H), 2.63 – 2.40 (m, 5H), 1.84 – 1.66 (m, 3H), 1.62 – 1.55 (m, 4H), 1.45 – 1.40 (m, 20H), 1.38 (s, 3H).
Tert-butyl (S)-4-((((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(3-(naphthalen-1-yl)propyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (6g).
Compound 6g was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.30 zs, 1H), 7.99 (d, J = 8.2 Hz, 1H), 7.91 (s, 1H), 7.87 – 7.82 (m, 1H), 7.71 (d, J = 8.2 Hz, 1H), 7.53 – 7.44 (m, 2H), 7.38 (t, J = 7.6 Hz, 1H), 7.28 – 7.18 (m, 1H), 6.06 (d, J = 2.0 Hz, 1H), 5.92 (s, 2H), 5.64 (brs, 1H), 5.46 (s, 1H), 5.04 (brs, 1H), 4.41 (brs, 1H), 4.17 (s, 1H), 3.12 – 2.96 (m, 2H), 2.96 – 2.39 (m, 6H), 2.05 – 1.75 (m, 4H), 1.62 (s, 3H), 1.49 – 1.35 (m, 21H).
Tert-butyl (S)-4-((((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(3-(naphthalen-2-yl)propyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (6h).
Compound 6h was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.30 (s, 1H), 7.86 (s, 1H), 7.81 – 7.69 (m, 3H), 7.54 (s, 1H), 7.47 – 7.37 (m, 2H), 7.24 (brs, 1H), 6.08 – 5.98 (m, 1H), 5.92 – 5.75 (m, 2H), 5.43 (brs, 1H), 5.01 (s, 1H), 4.38 (brs, 1H), 4.14 (s, 1H), 2.85 – 2.41 (m, 8H), 1.99 (brs, 1H), 1.82 (brs, 3H), 1.59 (s, 3H), 1.46 – 1.36 (m, 21H).
Tert-butyl (S)-4-((((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(2-(naphthalen-2yl)ethyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (6i).
Compound 6i was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.37 (s, 1H), 7.91 (s, 1H), 7.81 – 7.70 (m, 3H), 7.53 (s, 1H), 7.46 – 7.37 (m, 2H), 7.22 (dd, J = 8.4, 1.7 Hz, 1H), 6.04 (d, J = 2.1 Hz, 1H), 5.71 (s, 2H), 5.61 – 5.44 (m, 2H), 4.95 (dd, J = 6.4, 3.4 Hz, 1H), 4.35 (s, 1H), 4.23 – 4.12 (m, 1H), 2.93 – 2.53 (m, 8H), 2.02 – 1.95 (m, 1H), 1.89 – 1.83 (m, 1H), 1.59 (s, 3H), 1.44 (s, 9H), 1.43 (s, 9H), 1.33 (s, 3H).
Methyl 3-(3-((((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)((S)-4-(tert-butoxy)-3((tert-butoxycarbonyl)amino)-4-oxobutyl)amino)propyl)benzoate (6j).
Compound 6j was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.29 (s, 1H), 7.90–7.80 (m, 3H), 7.35 – 7.27 (m, 2H), 6.03 (s, 1H), 5.74 (s, 2H), 5.72 – 5.67 (m,1H), 5.63 – 5.59 (m, 1H), 5.10 – 4.95 (m, 1H), 4.30 – 4.26 (m, 1H), 4.21 – 4.10 (m, 1H), 3.89 (s, 3H), 2.78 – 2.33 (m, 8H), 2.00 – 1.65 (m, 4H), 1.60 (s, 3H), 1.50 –1.30 (m, 21H).
3-(3-hydroxyprop-1-yn-1-yl)benzonitrile (7a).
Compound 7a was synthesized according to general procedure B. 1H NMR (500 MHz, Chloroform-d) δ 7.75 – 7.66 (m, 1H), 7.69 – 7.54 (m, 2H), 7.43 (t, J = 7.9 Hz, 1H), 4.50 (s, 2H), 1.95 (brs, 1H).
3-(3-hydroxyprop-1-yn-1-yl)-4-methoxybenzonitrile (7b).
Compound 7b was synthesized according to general procedure B.1H NMR (500 MHz, Chloroform-d) δ 7.66 – 7.47 (m, 2H), 6.90 (d, J = 8.7 Hz, 1H), 4.49 (s, 2H), 3.88 (s, 3H). 13C NMR (126 MHz, Chloroform-d) δ 163.02, 137.17, 133.93, 118.40, 113.66, 111.34, 103.97, 94.02, 79.12, 56.25, 51.32.
5-(3-hydroxyprop-1-yn-1-yl)-2-methylbenzonitrile (7c).
Compound 7c was synthesized according to general procedure B. 1H NMR (500 MHz, Chloroform-d) δ 7.60 (d, J = 1.4 Hz, 1H), 7.49 (dd, J = 8.0, 1.6 Hz, 1H), 7.24 (d, J = 8.0 Hz, 1H), 4.47 (s, 2H), 2.51 (s, 3H), 2.39 (s, 1H). 13C NMR (126 MHz, Chloroform-d) δ 142.11, 135.66, 135.36, 130.51, 37121.33, 117.34, 113.16, 89.09, 83.26, 51.40, 20.52.
3-(3-oxoprop-1-yn-1-yl)benzonitrile (8a).
Compound 8a was synthesized according to general procedure C. 1H NMR (500 MHz, Chloroform-d) δ 9.43 (s, 1H), 7.88 (s, 1H), 7.84 – 7.74 (m, 2H), 7.68 – 7.49 (m, 1H).
4-methoxy-3-(3-oxoprop-1-yn-1-yl)benzonitrile (8b).
Compound 8b was synthesized according to general procedure C. 1H NMR (500 MHz, Chloroform-d) δ 9.44 (s, 1H), 7.91 – 7.60 (m, 2H), 7.01 (d, J = 8.8 Hz, 1H), 3.98 (s, 3H). 13C NMR (126 MHz, Chloroform-d) δ 176.48, 164.37, 138.72, 136.70, 117.73, 111.93, 110.58, 104.78, 93.01, 88.52, 56.60.
2-methyl-5-(3-oxoprop-1-yn-1-yl)benzonitrile (8c).
Compound 8c was synthesized according to general procedure C. 1H NMR (500 MHz, Chloroform-d) δ 9.40 (s, 1H), 7.81 (d, J = 1.5 Hz, 1H), 7.68 (dd, J = 8.1, 1.7 Hz, 1H), 7.38 (d, J = 8.0 Hz, 1H), 2.59 (s, 3H).
Tert-butyl (S)-4-((((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(3-(3-cyanophenyl)prop-2yn-1-yl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (9a).
Compound 9a was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.32 (s, 1H), 7.95 (s, 1H), 7.62 (s, 1H), 7.58 – 7.51 (m, 2H), 7.42 – 7.35 (m, 1H), 6.07 (s, 1H), 5.75 (s, 2H), 5.50 (d, J = 7.3 Hz, 1H), 5.04 (s, 1H), 4.39 (s, 1H), 4.31 – 4.18 (m, 1H), 3.70 – 3.60 (m, 2H), 2.90 – 2.83 (m, 1H), 2.81 – 2.74 (m, 1H), 2.66 – 2.60 (m, 2H), 2.01 – 1.96 (m, 1H), 1.85 – 1.77 (m, 1H), 1.61 (s, 3H), 1.42 (s, 18H), 1.40 (s, 3H).
Tert-butyl (S)-4-((((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(3-(5-cyano-2-methoxyphenyl)prop-2-yn-1-yl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (9b).
Compound 9b was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.32 (s, 1H), 7.96 (s, 1H), 7.61 – 7.50 (m, 2H), 6.89 (d, J = 8.7 Hz, 1H), 6.07 (s, 1H), 5.94 – 5.77 (m, 2H), 5.57 – 5.44 (m, 2H), 5.06 – 4.97 (m, 1H), 4.47 – 4.33 (m, 1H), 4.25 – 4.21 (m, 1H), 3.88 (s, 3H), 3.74 – 3.63 (m, 2H), 2.91 – 2.74 (m, 2H), 2.64 (t, J = 7.0 Hz, 2H), 2.01 – 1.93 (m, 1H), 1.86 – 1.79 (m, 1H), 1.61 (s, 3H), 1.46 – 1.36 (m, 21H).
Tert-butyl (S)-4-((((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(3-(3-cyano-4methylphenyl)prop-2-yn-1-yl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (9c).
Compound 9c was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.37 – 8.28 (m, 1H), 7.94 (d, J = 12.2 Hz, 1H), 7.56 (d, J = 11.9 Hz, 1H), 7.46 – 7.36 (m, 1H), 7.24 – 7.17 (m, 1H), 6.06 (d, J = 12.6 Hz, 1H), 5.84 – 5.70 (m, 2H), 5.55 – 5.44 (m, 1H), 5.34 – 5.25 (m, 1H), 5.08 – 4.96 (m, 1H), 4.45 – 4.31 (m, 1H), 4.31 – 4.17 (m, 1H), 3.73 – 3.55 (m, 2H), 2.90 – 2.72 (m, 2H), 2.66 – 2.56 (m, 2H), 2.54 – 2.47 (m, 3H), 1.99 – 1.92 (m, 1H), 1.85 – 1.77 (m, 1H), 1.63 – 1.58 (m, 3H), 1.46 – 1.34 (m, 21H).
3-(3-((((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)amino)prop-1-yn-1yl)benzonitrile (9d).
Compound 9d was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.30 (s, 1H), 7.90 (s, 1H), 7.66 – 7.63 (m, 1H), 7.58 – 7.51 (m, 2H), 7.38 (t, J = 7.8 Hz, 1H), 6.01 (d, J = 3.2 Hz, 3H), 5.46 (dd, J = 6.4, 3.2 Hz, 1H), 5.08 (dd, J = 6.4, 3.2 Hz, 1H), 4.42 (dt, J = 6.0, 3.7 Hz, 1H), 3.71 – 3.60 (m, 2H), 3.12 – 2.97 (m, 2H), 2.33 (brs, 1H), 1.61 (s, 3H), 1.38 (s, 3H).
3-(4-hydroxybut-1-yn-1-yl)benzonitrile (10).
Compound 10 was synthesized according to general procedure B. 1H NMR (500 MHz, Chloroform-d) δ 7.63 (s, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.36 (t, J = 7.8 Hz, 1H), 3.80 (t, J = 6.3 Hz, 2H), 13C NMR (126 MHz, Chloroform-d) δ 135.87, 135.02, 131.11, 129.21, 12 2.66 (t, J = 6.4 Hz, 2H). 125.12, 118.16, 112.58, 89.76, 79.98, 60.87, 23.70.
3-(4-oxobutyl)benzonitrile (11).
To a solution of 10 (50 mg, 0.29 mmol) in THF was added 10% Pd/C (10 mg). The mixture stirred for overnight under H2 atmosphere. After filtration, the solvent was removed in vacuum. The residue was dissolved in DCM (5mL). Then DMP (175 mg, 0.4 mmol) was added to the solution at 0 °C. The resulting solution was stirred for 1 h at room temperature. The reaction was quenched with saturated sodium hyposulfite solution (5mL), extracted with DCM (3×5mL), washed with Na2S2O3 three times and dried over anhydrous Na2SO4. After removal of solvent, the residue was purified with silica gel column to get 28mg colorless oil (60%). 1H NMR (500 MHz, Chloroform-d) δ 9.77 (d, J = 1.3 Hz, 1H), 7.47 (d, J = 10.7 Hz, 2H), 7.43 – 7.35 (m, 2H), 2.76 – 2.59 (m, 2H), 2.48 (td, J = 7.2, 1.3 Hz, 2H), 2.06 – 1.81 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 201.72, 142.82, 133.12, 132.01, 130.05, 129.38, 118.98, 112.57, 42.96, 34.60, 23.31.
Tert-butyl (S)-4-((((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(4-(3cyanophenyl)butyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (12a).
Compound 12a was synthesized according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.33 (s, 1H), 7.90 (s, 1H), 7.49 – 7.41 (m, 2H), 7.40 – 7.31 (m, 2H), 6.04 (d, J = 1.6 Hz, 1H), 5.69 (s, 2H), 5.58 – 5.46 (m, 2H), 4.98 (s, 1H), 4.32 (s, 1H), 4.20 – 4.10 (m, 1H), 2.78 – 2.69 (m, 1H), 2.56 (t, J = 7.7 Hz, 4H), 2.49 – 2.33 (m, 3H), 1.91 (s, 1H), 1.82 – 1.69 (m, 3H), 1.60 (s, 3H), 1.43 (s, 9H), 1.42 (s, 9H), 1.39 (s, 3H).
Tert-butyl (S)-5-((((3aR,4R,6S,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(4-(3-cyanophenyl)butyl)amino)-2-((tert-butoxycarbonyl)amino)pentanoate (12b).
Compound 12b was prepared according to the general procedure D. 1H NMR (500 MHz, Chloroform-d) δ 8.33 (s, 1H), 7.92 (s, 1H), 7.48 – 7.41 (m, 2H), 7.39 – 7.32 (m, 2H), 6.05 (s, 1H), 5.80 (s, 2H), 5.56 – 5.47 (m, 1H), 5.20 (d, J = 8.3 Hz, 1H), 4.98 – 4.88 (m, 1H), 4.31 (s, 1H), 4.17 – 4.12 (m, 1H), 2.67 – 2.53 (m, 4H), 2.46 – 2.35 (m, 4H), 1.72 – 1.68 (m, 1H), 1.59 (s, 3H), 1.58 – 1.47 (m, 5H), 1.44 – 1.38 (m, 23H).
Protein Expression and Purification.
Expression and purification of full-length human NNMT wild type and triple mutant K100A E101A E103A (hNNMTtm) were performed as previously described.22 Briefly, full-length hNNMT (amino acids 1–270) cloned in pET28a(+) with an N-terminal TEV cleavage site was synthesized by Genscript for biochemical assays and ITC study. The full-length hNNMTtm cloned in pET28a-LIC was obtained from Addgene (#40734) was used for crystallization. Protein was expressed in E. coli BL21-CodonPlus(DE3)-RIPL competent cells and induced by 0.3 mM isopropyl-D-1-thiogalactopyranoside at 16 °C for 20 hours. Harvested cells were resuspended in 10 volumes of 50 mM KH2PO4/K2HPO4 (pH = 7.4) with 500 mM NaCl, 25 mM imidazole, 5 mM ß-mercaptoethanol, 100 μM PMSF, then lysed through sonication (Qsonica Q55 cell disruptor) on ice at 80% power using 5–7 cycles of 30s pulse, 30s rest. The cell lysate was centrifuged at 25,000 × g for 30 minutes at 4 °C and the supernatant was then applied to two 1 mL HiTrap FF Ni-NTA columns connected in series on a GE AKTA Prime purification system using 50 mM KH2PO4/K2HPO4 (pH = 7.4) with with 500 mM NaCl and 0.5 mM TCEP, washed and eluted using a step gradient of imidazole (0.025, 0.05, 0.1, 0.25 and 0.5 M). The peak fractions were verified by SDS-PAGE analysis and the purest fractions were combined. For enzymatic assays, combined His-NNMT were dialyzed in the dialysis buffer (25 mM Tris, pH = 7.5, 150 mM NaCl, 50 mM KCl) and concentrated to 1.5 mg/mL for biochemical assays. For crystallography study, the sample was then applied to an S200 Sephacryl HR size exclusion column (26/100, GE) using 50 mM Tris-HCl, pH = 8.0, 100 mM NaCl, 0.5 mM TCEP, 5% glycerol. Fractions containing purest NNMT were combined, concentrated to 0.8 mg/mL and supplemented with additional TCEP to a final concentration of 1 mM. G9a, SETD7, PRMT1, TbPRMT7, NTMT1, PNMT and INMT were expressed and purified as described before 28,34–37,41–42.
Docking Studies with Glide.
For the crystal structure 3ROD, the incomplete side-chains was reconstructed by Prime. Then the solvent was removed and the structure was minimized. SAH binding site was defined as docking site with 10-Å. The 3D structure for compounds were prepared in Maestro from the 2D structure and parameterized using the OPLS3 force-field. Rigid-receptor/flexible-ligand docking was performed using extra-precision mode in Glide. The pose with highest docking score was exported and was analyzed by PyMOL.
NNMT Biochemical Assays and Enzyme Kinetics Study.
The fluorescence-based SAHH-coupled assay was applied to study the IC50 values of compounds to monitor the production of SAH. The assay was performed under the following conditions in a final well volume of 40 μL: 25 mM Tris (pH = 7.5), 50 mM KCl, 0.01% Triton X-100, 5 μM SAHH, 0.1 μM NNMT, 10 μM AdoMet, and 10 μM ThioGlo4. After 10 min incubation with inhibitors at 37 °C , reactions were initiated by the addition of 10 μM nicotinamide (Km value). Fluorescence signal was monitored on a BMG CLARIOstar microplate reader with excitation 400 nm and emission 465 nm. Data were processed by using GraphPad Prism software 7.0.
Time-dependent inhibition study.
The fluorescence-based SAHH-coupled assay was applied to this study. The assay was performed under the following conditions in a final well volume of 40 μL: 25 mM Tris (pH = 7.5), 50 mM KCl, 0.01% Triton X-100, 5 μM SAHH, 0.1 μM NNMT, and 10 μM AdoMet. The inhibitors were added at concentrations ranging from 0.15 nM to 10 μM. After incubation for 30min, 60min, 90min and 120min, reactions were initiated by the addition of 10 μM nicotinamide and 10 μM Thioglo4. Fluorescence was monitored on a BMG CLARIOstar microplate reader with excitation 400 nm and emission 465 nm. Data were processed by using GraphPad Prism software 7.0 with orrison’s quadratic equation.
Rapid Dilution Study.
10 μM NNMT and compound LL320 were pre-incubated for 30min. Then 1 μL pre-incubation mixture was added into 99 μL reaction mixture: 25 mM Tris (pH = 7.5), 50 mM KCl, 0.01% Triton X-100, 5 μM SAHH, 10 μM ThioGlo4, 10 μM AdoMet and 10 μM nicotinamide. Fluorescence signal was monitored on a BMG CLARIOstar microplate reader with excitation 400 nm and emission 465 nm. Data were processed by using GraphPad Prism software 7.0.
Selectivity Assays.
A fluorescence-based SAHH-coupled assay was applied to study the effect of compound on methyltransferase activity of PNMT, INMT, G9a, SETD7, PRMT1, TbPRMT7 and NTMT1. For PNMT, the assay was performed in a final well volume of 40 μL: 25 mM potassium phosphate buffer (pH = 7.6), 1 mM EDTA, 2 mM MgCl2, 0.01% Triton X-100, 5 μM SAHH, 0.2 μM PNMT, 3 μM AdoMet, and 10 μM ThioGlo4. The inhibitor was added at four compound concentrations: 3.7μM, 11 μM, 33 μM and 100 μM. After 10 min incubation with inhibitor, reactions were initiated by the addition of 40 μM 51 norepinephrine. For INMT, the assay was performed in a final well volume of 40 μL: 25 mM HEPES (pH = 7.5), 5 μM SAHH, 0.2 μM INMT, 30 μM AdoMet, and 10 μM ThioGlo4. The inhibitor was added at four compound concentrations: 3.7μM, 11 μM, 33 μM and 100 μM. After 10 min incubation with inhibitor, reactions were initiated by the addition of 2.5 mM tryptamine. For G9a, the assay was performed in a final well volume of 40 μL: 25 mM potassium phosphate buffer (pH = 7.6), 1 mM EDTA, 2 mM MgCl2, 0.01% Triton X-100, 5 μM SAHH, 0.1 μM His-G9a, 10 μM AdoMet, and 10 μM ThioGlo4. The inhibitor was added at four compound concentrations: 3.7μM, 11 μM, 33 μM and 100 μM. After 10 min incubation with inhibitor, reactions were initiated by the addition of 4 μM H3–21 peptide. For NTMT1, the assay was performed in a final well volume of 40 μL: 25 mM Tris (pH = 7.5), 50 mM KCl, 0.01% Triton X-100, 5 μM SAHH, 0.1 μM NTMT1, 3 μM AdoMet, and 10 μM ThioGlo4. After 10 min incubation with inhibitor, reactions were initiated by the addition of 0.5 μM GPKRIA peptide. All experiments were determined in duplicate. Fluorescence was monitored on a BMG CLARIOstar microplate reader with excitation 400 nm and emission 465 nm.
For SETD7, the assay was performed in a final well volume of 40 μL: 25 mM potassium phosphate buffer (pH = 7.6), 0.01% Triton X-100, 5 μM SAHH, 1 μM His-SETD7, 2 μM AdoMet, and 15 μM ThioGlo1. After 10 min incubation with inhibitor, reactions were initiated by the addition of 90 μM H3–21 peptide. For PRMT1, the assay was also performed in a final well volume of 40 μL: 2.5 mM HEPES (pH = 7.0), 25 mM NaCl, 25 μM EDTA, 50 μM TCEP, 0.01% Triton X-100, 5 μM SAHH, 0.2 μM PRMT1, 10 μM AdoMet, and 15 μM ThioGlo1. After 10 min incubation with inhibitor, reactions were initiated by the addition of 5 μM H4–21 peptide. For TbPRMT7, the assay was performed in a final well volume of 40 μL: 25 mM Tris (pH = 7.5), 50 mM KCl, 0.01% Triton X-100, 5 μM SAHH, 0.2 μM TbPRMT7, 3 μM AdoMet, and 15 μM ThioGlo1. After 10 min incubation with inhibitor, reactions were initiated by the addition of 60 μM H4–21 peptide. All experiments were determined in duplicate. Fluorescence was monitored on a BMG CLARIOstar microplate reader with excitation 380 nm and emission 505 nm.
Compound was evaluated its effect on SAHH activity, the coupled enzyme. The assay was performed in a final well volume of 40 μL: 25 mM Tris (pH = 7.5), 50 mM KCl, 0.01% Triton X-100, 0.1 μM SAHH and 15 μM ThioGlo1. After 10min incubation with compound, 0.5 μM SAH was added to initiate the reactions. The experiment was determined in duplicate. Fluorescence was monitored on a BMG CLARIOstar microplate reader with excitation 380 nm and emission 505 nm.
Co-crystallization and Structure Determination.
For co-crystallization, 20 mg/mL hNNMT triple mutant was mixed with either LL319 or LL320 at a 1:4 molar ratio in 50 mM Tris-HCl, pH = 8.0, with 0.5 mM TCEP and 5% glycerol, incubated for 1 hour at 4 °C. Broad matrix crystallization screening was performed using a Mosquito-LCP high throughput crystallization robot (TTP LabTech) using hanging-drop vapor diffusion method, incubated at 20 °C, and crystals harvested directly from the 96-well crystallization plates. Crystals containing LL319 or LL320 were grown in 0.1 M bicine, pH 9.0, and 1.6 M ammonium sulfate (space group P1); all crystals were harvested directly from the drop and flash cooled into liquid nitrogen. Data were collected on single crystals at 12.0 keV at the GM/CA-CAT ID-D beamline at the Advanced Photon Source, Argonne National Laboratory. The data were processed using the on-site automated pipeline with AutoProc and/or manually with HKL2000 and molecular replacement performed with Phaser-MR (PHENIX)38,39. All model building was performed using COOT and subsequent refinement 39,40. Structure factors and model coordinates have done using phenix.refine (PHENIX) been deposited in the Protein Data Bank with ID 6PVE (LL319) and 6PVS (LL320). Data collection parameters and refinement statistics are summarized in Table S1. Structure-related figures were made with PyMOL (Schrödinger) and annotated and finalized with Adobe Photoshop and Illustrator.
Cell Permeability Evaluation by MALDI.
The colon cancer cell line HCT116 was cultured in Mccoy’s media supplemented with 10% fetal bovine serum and 1% Antibiotic-Antimycotic (Gibco). The cells were cultured in tissue culture dish (Falcon 353003). Cells were maintained in cell culture flasks until seeding into a 12 well tissue culture plate (Falcon 353047). Media was removed and the cells were washed with DPBS (1 mL) twice followed by treating with TrypLE Express (1 mL) into a 100 × 20 mm culture flask. The reaction was quenched by addition of 4 mL of media and the cells were counted. Cells were seeded into a 12 well tissue culture plate (Falcon 353047) at a density of 0.1 × 106 cells/mL and incubated overnight at 37 °C, 5% CO2, and 95% humidity, with the lid on.
They were then treated with the inhibitors at different concentrations and incubation was continued for the specified time. TAT peptide (GRKKRRQRRR-NH2) was also incubated as a control experiment. The media was removed, and the cells were washed with 1X PBS three times to remove any residual compound or peptide attached to the cell surface. Then 100 μL of 1X PBS was added and the cells were snap freeze in liquid nitrogen twice. The cell lysate was then analyzed with MALDI using DHB matrix to identify the presence of the compound inside the cell.
Supplementary Material
General Procedures for the Synthesis of compound S3; NMR spectra of compounds S3, 6a-i, 9a-d, and 12a-b; NMR, MALDI-MS and HPLC spectra of compounds 1a-j, 1a*, 2a-d, 2a* and 3a-b; Supporting Figure S1. Kinetic parameter determination for NNMT; Supporting Figure S2. Rapid dilution assay for compound LL320 (2a); Supporting Figure S3. The selectivity study of LL320 (2a); Supporting Figure S4. Superimposed co-crystal structures; Supporting Figure S5. Cell 45permeability evaluation of TAT peptide as positive control; Supporting Figure S6. Cell permeability evaluation of 1a*; Supporting Figure S7. Cell permeability evaluation of 2a*; Supporting Table S1. Crystallography data and refinement statistics (PDB ID: 6PVE and 6PVS) Molecular formula strings (CSV)
ACKNOWLEDGMENT
We appreciate Dr. Ayad A. Al-Hamashi for his initial assistance with docking studies and Guangping Dong for initial cellular concentration evaluation. We thank Dr. Darrel L. Peterson for purification of SAHH for biochemical assays. Furthermore, we thank Dr. Lawrence M. Szewczuk for valuable discussion on tight-binding analysis. The authors acknowledge the support from NIH grants R01GM117275 (RH), U01CA214649 (RH), K22 AI113078-02 (NN), 1R01GM127896-01 (NN), 1R01AI127793 (NN), and P30 CA023168 (Purdue University Center for Cancer Research).
We also thank supports from the Department of Medicinal Chemistry and Molecular Pharmacology (RH) and Department of Biological Sciences (NN) at Purdue University.
Funding Sources
NIH grants R01GM117275 (RH), U01CA214649 (RH), K22 AI113078-02 (NN), 1R01GM127896-01 (NN), 1R01AI127793 (NN), and P30 CA023168 (Purdue University Center for Cancer Research)
ABBREVIATIONS
- NNMT
nicotinamide N‑methyltransferase
- SAM
S-5’-adenosyl-L-methionine
- SAH
S-5’-adenosyl-L-homocysteines
- SAHH
SAH hydrolase
- PNMT
phenylethanolamine N methyltransferase
- INMT
indolethylamine N-methyltransferase
- NTMT1
protein N-terminal methyltransferase 1
- PKMT
protein lysine methyltransferase
- PRMT
protein arginine methyltransferase
- rt
room temperature
- TFA
trifluoroacetic acid
Footnotes
ASSOCIATED CONTENT
Supporting Information. The Supporting Information is available free of charge on the
ACS Publications website.
Accession Codes
The structures of human NNMT in complex of LL319 and LL320 have been deposited under PDB ID 6PVE and 6PVS, respectively. Authors will release the atomic coordinates and experimental data upon article publication.
The authors declare no competing financial interests.
REFERENCES
- (1).Aksoy S; Szumlanski CL; Weinshilboum RM Human Liver Nicotinamide N-Methyltransferase. cDNA Cloning, Expression, and Biochemical Characterization. J. Biol. Chem 1994, 269 (20), 14835–14840. [PubMed] [Google Scholar]
- (2).Pissios P. Nicotinamide N-Methyltransferase: More Than a Vitamin B3 Clearance Enzyme. Trends Endocrinol. Metab 2017, 28 (5), 340–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kraus D; Yang Q; Kong D; Banks AS; Zhang L; Rodgers JT; Pirinen E; Pulinilkunnil TC; Gong F; Wang YC; Cen Y; Sauve AA; Asara JM; Peroni OD; Monia BP; Bhanot S; Alhonen L; Puigserver P; Kahn BB Nicotinamide N-Methyltransferase Knockdown Protects against Diet-Induced Obesity. Nature 2014, 508 (7495), 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hong S; Zhai B; Pissios P. Nicotinamide N-Methyltransferase Interacts with Enzymes of the Methionine Cycle and Regulates Methyl Donor Metabolism. Biochemistry 2018, 57 (40), 5775–5779. [DOI] [PubMed] [Google Scholar]
- (5).Xie X; Yu H; Wang Y; Zhou Y; Li G; Ruan Z; Li F; Wang X; Liu H; Zhang J. Nicotinamide N-Methyltransferase Enhances the Capacity of Tumorigenesis Associated with the Promotion of Cell Cycle Progression in Human Colorectal Cancer Cells. Arch. Biochem. Biophys 2014, 564, 52–66. [DOI] [PubMed] [Google Scholar]
- (6).Palanichamy K; Kanji S; Gordon N; Thirumoorthy K; Jacob JR; Litzenberg KT; Patel Di.; Chakravarti A. NNMT Silencing Activates Tumor Suppressor PP2A, Inactivates Oncogenic STKs, and Inhibits Tumor Forming Ability. Clin. Cancer Res 2017, 23 (9), 2325–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Eckert MA; Coscia F; Chryplewicz A; Chang JW; Hernandez KM; Pan S; Tienda SM; Nahotko DA; Li G; Blaženović I; Lastra RR; Curtis M; Yamada SD; Perets R; McGregor SM; Andrade J; Fiehn O; Moellering RE; Mann M; Lengyel E. Proteomics Reveals NNMT as a Master Metabolic Regulator of Cancer Associated Fibroblasts. Nature 2019, 569 (7758), 723–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ulanovskaya OA; Zuhl AM; Cravatt BF NNMT Promotes Epigenetic Remodeling in Cancer by Creating a Metabolic Methylation Sink. Nat. Chem. Biol 2013, 9 (5), 300–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sartini D; Morganti S; Guidi E; Rubini C; Zizzi A; Giuliante R; Pozzi V; Emanuelli M. Nicotinamide N-Methyltransferase in Non-Small Cell Lung Cancer: Promising Results for Targeted Anti-Cancer Therapy. Cell Biochem. Biophys 2013, 67 (3), 865–873. [DOI] [PubMed] [Google Scholar]
- (10).Tomida M; Ohtake H; Yokota T; Kobayashi Y; Kurosumi M. Stat3 Up-Regulates Expression of Nicotinamide N-Methyltransferase in Human Cancer Cells. J. Cancer Res. Clin. Oncol 2008, 134 (5), 551–559. [DOI] [PubMed] [Google Scholar]
- (11).Zhang J; Wang Y; Li G; Yu H; Xie X. Down-Regulation of Nicotinamide N-Methyltransferase Induces Apoptosis in Human Breast Cancer Cells via the Mitochondria-Mediated Pathway. PLoS One 2014, 9 (2), e89202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mateuszuk Ł; Khomich TI; Słomińska E; Gajda M; Wójcik L; Łomnicka M; Gwóźdź P; Chłopicki S. Activation of Nicotinamide N-Methyltrasferase and Increased Formation of 1-Methylnicotinamide (MNA) in Atherosclerosis. Pharmacol. Reports 2009, 61 (1), 76–85. [DOI] [PubMed] [Google Scholar]
- (13).Liu KY; Mistry RJ; Aguirre CA; Fasouli ES; Thomas MG; Klamt F; Ramsden DB; Parsons RB Nicotinamide N-Methyltransferase Increases Complex i Activity in SH-SY5Y Cells via Sirtuin 3. Biochem. Biophys. Res. Commun 2015, 467 (3), 491–496. [DOI] [PubMed] [Google Scholar]
- (14).Neelakantan H; Wang HY; Vance V; Hommel JD; McHardy SF; Watowich SJ Structure-Activity Relationship for Small Molecule Inhibitors of Nicotinamide N-Methyltransferase. J. Med. Chem 2017, 60 (12), 5015–5028. [DOI] [PubMed] [Google Scholar]
- (15).Kannt A; Rajagopal S; Kadnur SV; Suresh J; Bhamidipati RK; Swaminathan S; Hallur MS; Kristam R; Elvert R; Czech J; Pfenninger A; Rudolph C; Schreuder H; Chandrasekar DV; Mane VS; Birudukota S; Shaik S; Zope BR; Burri RR; Anand NN; Thakur MK; Singh M; Parveen R; Kandan S; Mullangi R; Yura T; Gosu R; Ruf S; Dhakshinamoorthy S. A Small Molecule Inhibitor of Nicotinamide N-Methyltransferase for the Treatment of Metabolic Disorders. Sci. Rep 2018, 8 (1), 3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sen S; Mondal S; Zheng L; Salinger AJ; Fast W; Weerapana E; Thompson PR Development of a Suicide Inhibition-Based Protein Labeling Strategy for Nicotinamide N-Methyltransferase. ACS Chem. Biol 2019, 14 (4), 613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lee HY; Suciu RM; Horning BD; Vinogradova EV; Ulanovskaya OA; Cravatt BF Covalent Inhibitors of Nicotinamide N-Methyltransferase (NNMT) Provide Evidence for Target Engagement Challenges in Situ. Bioorganic Med. Chem. Lett 2018, 28 (16), 2682–2687. [DOI] [PubMed] [Google Scholar]
- (18).van Haren MJ; Taig R; Kuppens J; Sastre Toraño J.; Moret EE; Parsons RB; Sartini D; Emanuelli M; Martin NI Inhibitors of Nicotinamide N-Methyltransferase Designed to Mimic the Methylation Reaction Transition State. Org. Biomol. Chem 2017, 15 (31), 6656–6667. [DOI] [PubMed] [Google Scholar]
- (19).Babault N; Allali-Hassani A; Li F; Fan J; Yue A; Ju K; Liu F; Vedadi M; Liu J; Jin J. Discovery of Bisubstrate Inhibitors of Nicotinamide N-Methyltransferase (NNMT). J. Med. Chem 2018, 61 (4), 1541–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chen D; Dong G; Noinaj N; Huang R. Discovery of Bisubstrate Inhibitors for Protein N-Terminal Methyltransferase 1. J. Med. Chem 2019, 62 (7), 3773–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Dong C; Mao Y; Tempel W; Qin S; Li L; Loppnau P; Huang R; Min J. Structural Basis for Substrate Recognition by the Human N-Terminal Methyltransferase 1. Genes Dev. 2015, 29 (22), 2343–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Peng Y; Sartini D; Pozzi V; Wilk D; Emanuelli M; Yee VC Structural Basis of Substrate Recognition in Human Nicotinamide N-Methyltransferase. Biochemistry 2011, 50 (36), 7800–7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Loring HS; Thompson PR Kinetic Mechanism of Nicotinamide N-Methyltransferase. Biochemistry 2018, 57 (38), 5524–5532. [DOI] [PubMed] [Google Scholar]
- (24).Borch RF; Bernstein MD; Durst HD Cyanohydridoborate Anion as a Selective Reducing Agent. J. Am. Chem. Soc 1971, 93 (12), 2897–2904. [Google Scholar]
- (25).Zhang G; Richardson SL; Mao Y; Huang R. Design, Synthesis, and Kinetic Analysis of Potent Protein N-Terminal Methyltransferase 1 Inhibitors. Org. Biomol. Chem 2015, 13 (14), 4149–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Heck KF; Nolley JP Palladium-Catalyzed Vinylic Hydrogen Substitution Reactions with Aryl, Benzyl, and Styryl Halides. J. Org. Chem 1972, 37 (14), 2320–2322. [Google Scholar]
- (27).Sonogashira K. Development of Pd-Cu Catalyzed Cross-Coupling of Terminal Acetylenes with Sp2-Carbon Halides. J. Organomet. Chem 2002, 653 (1–2), 46–49. [Google Scholar]
- (28).Babault N; Allali-Hassani A; Li F; Fan J; Yue A; Ju K; Liu F; Vedadi M; Liu J; Jin J. Discovery of Bisubstrate Inhibitors of Nicotinamide N -Methyltransferase (NNMT). J. Med. Chem 2018, 61 (4), 1541–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Richardson SL; Mao Y; Zhang G; Hanjra P; Peterson DL; Huang R. Kinetic Mechanism of Protein N-Terminal Methyltransferase 1. J. Biol. Chem 2015, 290 (18), 11601–11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Copeland RA Tight Binding Inhibitors In Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd Ed., John Wiley & Sons: New Jersey, 2013; pp245–285. [PubMed] [Google Scholar]
- (31).Morrison JF The Slow-Binding and Slow, Tight-Binding Inhibition of Enzyme Catalysed Reactions. Trends Biochem. Sci 1982, 7 (3), 102–105. [Google Scholar]
- (32).Van Haren MJ; Sastre Toraño J.; Sartini D; Emanuelli M; Parsons RB; Martin NI A Rapid and Efficient Assay for the Characterization of Substrates and Inhibitors of Nicotinamide N-Methyltransferase. Biochemistry 2016, 55 (37), 5307–5315. [DOI] [PubMed] [Google Scholar]
- (33).Gordon LJ; Allen M; Artursson PER; Hann MM; Leavens BJ; Mateus A; Readshaw SIMON; Valko K; Wayne GJ; West ANDY Direct Measurement of Intracellular Compound Concentration by RapidFire Mass Spectrometry Offers Insights into Cell Permeability. J. Biomol. Screen 2016, 21 (2), 156–164. [DOI] [PubMed] [Google Scholar]
- (34).Wu H; Min J; Lunin VV; Antoshenko T; Dombrovski L; Zeng H; Allali-Hassani A; Campagna-Slater V; Vedadi M; Arrowsmith CH; Plotnikov AN; Schapira M. Structural Biology of Human H3K9 Methyltransferases. PLoS One 2010, 5 (1), e8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Feng Y; Xie N; Jin M; Stahley MR; Stivers JT; Zheng YG A Transient Kinetic Analysis of PRMT1 Catalysis. Biochemistry 2011, 50 (32), 7033–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Barsyte-Lovejoy D; Li F; Oudhoff MJ; Tatlock JH; Dong A; Zeng H; Wu H; Freeman SA; Schapira M; Senisterra GA; Kuznetsova E; Marcellus R; Allali-Hassani A; Kennedy S; Lambert JP; Couzens AL; Aman A; Gingras AC; Al-Awar R; Fish PV; Gerstenberger BS; Roberts L; Benn CL; Grimley RL; Braam MJ; Rossi FM; Sudol M; Brown PJ; Bunnage ME; Owen DR; Zaph C; Vedadi M; Arrowsmith CH (R)-PFI-2 Is a Potent and Selective Inhibitor of SETD7 Methyltransferase Activity in Cells. Proc. Natl. Acad. Sci 2014, 111 (35), 12853–12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Debler EW; Jain K; Warmack RA; Feng Y; Clarke SG; Blobel G; Stavropoulos P. A Glutamate/Aspartate Switch Controls Product Specificity in a Protein Arginine Methyltransferase. Proc. Natl. Acad. Sci 2016, 113 (8), 2068–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Winter G; Ashton A. Fast_dp. Methods Zenodo. 2014, 55, 81–93. [Google Scholar]
- (39).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. Sect. D Biol. Crystallogr 2010, 66 (2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Emsley P; Lohkamp B; Scott WG; Cowtan K. Features and Development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr 2010, 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Policarpo RL; Decultot L; May E; Kuzmic P; Carlson S; Huang D; Chu V; Wright BA; Dhakshinamoorthy S; Kannt A; Rani S; Dittakavi S; Panarese JD; Gaudet R; Shair MD High-Affinity Alkynyl Bisubstrate Inhibitors of Nicotinamide N ‑ Methyltransferase (NNMT). J. Med. Chem DOI: 10.1021/acs.jmedchem.9b01238. Published Online: October 7, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Stratton CF; Poulin MB; Du Q; Schramm VL Kinetic Isotope Effects and Transition State Structure for Human Phenylethanolamine N ‑ Methyltransferase. ACS Chem. Biol 2017, 12(2), 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General Procedures for the Synthesis of compound S3; NMR spectra of compounds S3, 6a-i, 9a-d, and 12a-b; NMR, MALDI-MS and HPLC spectra of compounds 1a-j, 1a*, 2a-d, 2a* and 3a-b; Supporting Figure S1. Kinetic parameter determination for NNMT; Supporting Figure S2. Rapid dilution assay for compound LL320 (2a); Supporting Figure S3. The selectivity study of LL320 (2a); Supporting Figure S4. Superimposed co-crystal structures; Supporting Figure S5. Cell 45permeability evaluation of TAT peptide as positive control; Supporting Figure S6. Cell permeability evaluation of 1a*; Supporting Figure S7. Cell permeability evaluation of 2a*; Supporting Table S1. Crystallography data and refinement statistics (PDB ID: 6PVE and 6PVS) Molecular formula strings (CSV)