

Abstract
Retinal dystrophies (RDs) comprise relatively rare but devastating causes of progressive vision loss. They represent a spectrum of diseases with marked genetic and clinical heterogeneity. Mutations in the same gene may lead to different diagnoses, for example, retinitis pigmentosa or cone dystrophy. Conversely, mutations in different genes may lead to the same phenotype. The age at symptom onset, and the rate and characteristics of peripheral and central vision decline, may vary widely per disease group and even within families. For most RD cases, no effective treatment is currently available. However, preclinical studies and phase I/II/III gene therapy trials are ongoing for several RD subtypes, and recently the first retinal gene therapy has been approved by the US Food and Drug Administration for RPE65-associated RDs: voretigene neparvovec-rzyl (Luxturna). With the rapid advances in gene therapy studies, insight into the phenotypic spectrum and long-term disease course is crucial information for several RD types. The vast clinical heterogeneity presents another important challenge in the evaluation of potential efficacy in future treatment trials, and in establishing treatment candidacy criteria. This perspective describes these challenges, providing detailed clinical descriptions of several forms of RD that are caused by genes of interest for ongoing and future gene or cell-based therapy trials. Several ongoing and future treatment options will be described.
Keywords: antisense oligonucleotides, gene therapy, genetics, inherited retinal dystrophies, stem cell therapy
Retinal dystrophies (RDs) comprise a collection of degenerative diseases characterized by the usually progressive and sometimes stationary dysfunction of rods and/or cones. With a prevalence of 1:3000 individuals,1,2 RDs are not particularly rare. However, due to their genetic heterogeneity, with >200 disease genes identified to date, each genetic subtype may be exceedingly rare. These diagnoses have a profoundly distressing impact on patient's lives, progressively affecting their mobility, professional functioning, and independence. Patients are often uncertain of their prognosis, questioning whether and when they will go blind, and whether they will pass this disease onto their children. Children or adolescents diagnosed with RD need to be informed on their prognosis, to make sound decisions on life planning such as future career paths. Special lighting or magnification requirements need to be tended to at their home, school, or professional environment, or they may need to visit a special needs school altogether.
THE EVOLUTION OF GENE-BASED THERAPEUTIC TRIALS
Due to the monogenic nature of most RDs, and the relative immune privilege of the eye, its accessibility, and the ability to noninvasively monitor its function and structure, the eye is a particularly suitable target for investigational gene therapy. The blood-retinal barrier restricts the degree of vector dissemination outside the eye, and limits immune responses to the viral vector and gene product. Another advantage of the eye over other organs is the lack of cell division in most retinal cells. Thus, the viral vector DNA does not have to integrate into the host cell genome to remain available in daughter cells after cell division, and the risk of malignancy is reduced.
Autosomal recessive disorders are characterized by loss of function or even (near) absence of the protein produced encoded by the gene. Therefore, for autosomal recessive disorders, gene therapy can be based “simply” on gene augmentation or replacement through the delivery of the normal gene. However, in autosomal dominant disease, such as rhodopsin (RHO)-associated retinitis pigmentosa (RP), the phenotype is typically the result of gain-of-function mutations, where one gene copy expresses a normally functioning protein, and the other gene copy expresses a detrimental protein that needs to be suppressed. For autosomal dominant disease, therapeutic intervention generally focuses on the suppression or inactivation on the gain-of-function gene.
Important advances have been made with the turn of the millennium in the development of (gene) therapies that aim to slow or (temporarily) halt the disease progression in RDs, or even to restore some visual function. The first successful gene therapy was applied in patients with RPE65-RD.3 Several trials have found compelling results in other RD subtypes, such as choroideremia,4,5 and many other trials are ongoing (Table 1 ) or in the basic experimental or preclinical phase.6,7 However, an imbalance exists between the rapid advances in (gene) therapy development and the available literature on the clinical disease course and the phenotypic spectrum for each specific gene of interest. Until relatively recently, longitudinal studies on the detailed clinical characteristics and disease progression were scarce for several RD subtypes, which had been the focus of interest in gene therapy development. Prospective phenotyping study had been even rarer in these often relatively small patient populations. However, such information is crucial in determining the window of therapeutic opportunity, patient eligibility criteria, and clinical endpoints in ongoing and future trials to assess treatment efficacy.
TABLE 1.
An Overview of Ongoing or Recently Completed Human Gene Therapy Trials for Inherited Retinal Degenerations
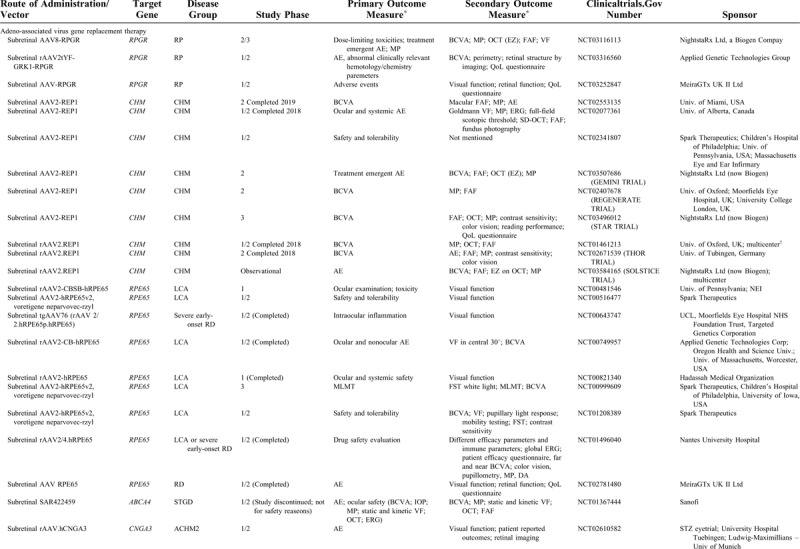
TABLE 1 (Continued).
An Overview of Ongoing or Recently Completed Human Gene Therapy Trials for Inherited Retinal Degenerations
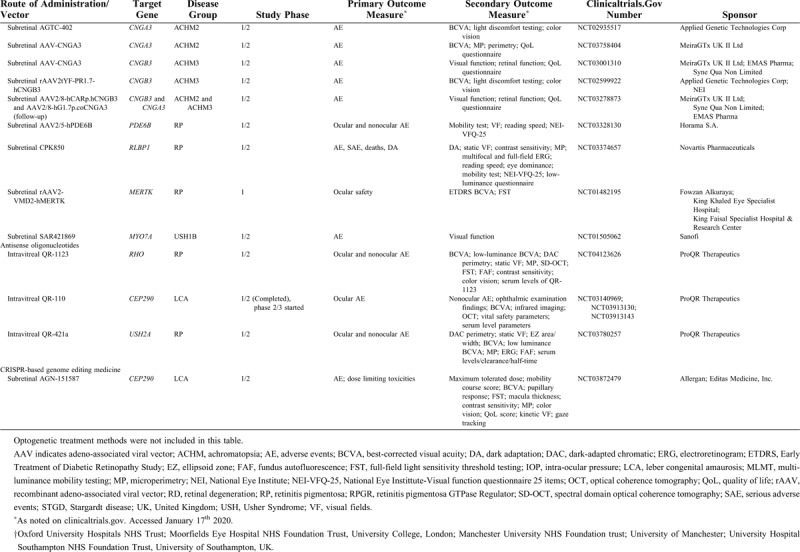
This perspective focuses on the clinical characteristics of RDs and the different ongoing and emerging treatment options, focusing on RDs caused by mutations in the CRB1 gene, the RPGR gene, the CHM gene, and the LRAT gene as an example.
Clinical Perspectives in Autosomal Recessive RDs
An autosomal recessive inheritance mode is observed in 50% to 60% of all RP cases,2,8 and in most cases of Leber congenital amaurosis.9 The most common form of macular dystrophy, Stargardt disease, is inherited in the autosomal recessive form.10 After the success of subretinal gene therapy in RPE65-associated RDs, which has led to the market approval of Luxturna by the Food and Drug Administration,11 preclinical and clinical (gene) therapy studies are ongoing for several genes responsible for autosomal recessive RD subtypes, such as ABCA4, CNGA3, CNGB3, PDE6B, RLBP1, MERTK, and MYO7A.12
Clinical Perspectives in CRB1-Associated RDs
Particularly interesting advances have been made in RDs caused by mutations in the CRB1 gene,6 which account for 3% to 9% of nonsyndromic cases of autosomal recessive RP,13 and 7% to 17% of Leber congenital amaurosis (LCA) cases.13,14 With the ongoing development of human CRB1 gene therapy,6,7,15 a detailed understanding of the phenotypic and genotypic characteristics of CRB1-associated RDs is essential. Until relatively recently, studies from literature had mostly been case reports, case series, or genetic studies with only brief descriptions of the clinical phenotype, providing limited detail.16–30
A recent Dutch retrospective cohort is the largest described to date, which allowed for statistical analysis and robust results on clinical signs and course of visual decline, which were further validated in a Belgian population, although the phenotypic and genotypic variability was higher in the Belgian population.31 Furthermore, in the Belgian population, a larger proportion of patients had a more severe diagnosis of LCA or early-onset severe retinal dystrophy (EOSRD), as compared with the Dutch population, where most patients had RP. Although the classic RP features, such as optic disc pallor, vascular attenuation, and bone-spicule-like pigmentation, were commonly found in CRB1-RP, certain characteristics outline a specific and typical CRB1-associated phenotype, such as hyperopia, nanophthalmos, a shallow anterior chamber, peri-arteriolar preservation of the retinal pigment epithelium (RPE), optic disc drusen, and Coats’-like exudative vasculopathy.24,25,29,31–42 Furthermore, these large studies highlighted the need to monitor this patient group for the risk of developing acute angle-closure glaucoma. In the Dutch cohort, optic disc drusen were found in the genetic isolate only, which prompted the suggestion of a potential genotype–phenotype correlation. However, in the Belgian cohort, optic disc drusen and hamartomas were found in patients with several different genotypes. Coats’-like exudative vasculopathy had been described before in CRB1-assocatied disease,35,39,42–45 and the large studies in the cohorts described earlier have shown cohort-wide prevalence of these vasculopathies in 10% of Dutch RP-patients, and 13% of Belgian patients.
It should be noted that although these features together form a “typical” CRB1-associated phenotype, each feature may be found in other RD subtypes as well. Optic disc drusen have been described, for example, in Usher syndrome,46 albeit to a much rarer degree, and hyperopia has been a classic feature of BEST1-associated phenotypes,47,48 where it can also be associated with angle-closure glaucoma.49 Mutations in MFRP are associated with RP along with nanophthalmos, optic disc drusen, and foveoschisis.50–53 Aside from its association with CRB1,16,31,54,55 an initial diagnosis of uveitishas has also been described in association with PRPF31,54RP1,54 Stargardt disease,56 and Usher syndrome.54,57 Although the exact mechanism of uveitis in RP remains unknown, several explanations have been suggested for the association between uveitis and RP. Circulating immune complexes have been detected in 43.5% of patients in a study, along with reduced levels of complement C3 and C4.58 A B-lymphocyte-mediated autoimmune response against retinal S-antigen, which is present in rod photoreceptors, has been shown in some RP patients, showing a low-level auto-immune responsiveness in RP.59 An as of yet unidentified genetic or auto-immune factor—or a combination thereof—may play a role.
An interesting recurrent finding is the Coats’-like exudative vasculopathy, which has a strong association with CRB1. In one case, it has been described in an RP patient from a pedigree where an RPGR ORF15 mutation segregated with disease, and where no other genes were tested.60 It has also been reported in a single case of RHO-associated RP, where no CRB1 mutations were found.61 Otherwise, it has not been associated with another RD gene, although it has regularly been described in genetically undifferentiated case reports or series,62–64 particularly in older studies where genetic analysis had not been performed.65 In some studies wherein the associated gene had not been identified, other features, such as perivascular retinal sparing,62 or nanophthalmos,66 point toward an association with CRB1. Vasoproliferative retinal lesions have been reported in association with Usher syndrome type I or II, based on the presence of RP and congenital hearing impairment, but not on genetic analysis.67,68 Few other reports, again lacking genetic analysis, have found Coats’-like vasculopathy in RP thought to be X-linked or autosomal dominant, based on pedigree analysis.69,70 The underlying mechanism of Coats’-like exudative vasculopathy includes an abnormal vascular permeability, which may be an element of CRB1-RDs. The CRB1 protein is crucial in the regulation of the number and size of Muller glia cells.71 Since Muller cells function as regulators of the tightness of the blood-retinal barrier,72 this may in some way relate to the vascular abnormalities seen in some patients with CRB1-RDs.
Another distinctive finding that we frequently observed in the Dutch and Belgian CRB1-RD patient cohorts, was thickening of the inner retina on spectral-domain optical coherence tomography (SD-OCT), which was in line with earlier reports,23,24,32,34,39,43,73–75 although some other studies have reported retinal thinning.20,37,76,77 Mouse studies have shown retinal thickening to be caused by proliferating retinal progenitor cells, resulting in an increase in the number of rod photoreceptors, Müller cells, and bipolar cells,78 or by ectopic photoreceptors.79 In both studies, the Crb2 protein has been postulated to play a role in the retinal thickening mechanism. Some other studies have suggested inner retinal thickening to be due to a remodeling process in association with loss of the outer nuclear layer,80,81 which could hinder efficacy of gene therapy. However, we found no correlation between outer retinal thinning and inner retinal thickening.
A crucial aspect of the CRB1-associated phenotype, is the (dis)organization of the normal retinal layers (lamination), and the degree of preservation of the external limiting membrane, which is assumed to include the Crumbs (CRB) complex and thus is at least in part CRB1 gene therapy's target. The CRB complex plays a role in the adhesion between photoreceptors and Müller cells, and also between photoreceptors.82,83 Reports on the laminar structure have varied, with some describing loss of lamination,19,23 and others reporting normal lamination.27,84 Reasonably well-preserved lamination was a frequent observation in our cohorts, with 91% and 41% of the Dutch and Belgian patients with available SD-OCT scans, respectively.31 This again confirmed a generally more severe phenotype in the Belgian CRB1 cohort.
Findings of CRB1-associated disease do not only involve the retina, but also other ocular structures, pointing to a role of protein CRB1 in the ocular development, as has been suggested before in BEST1-associated disease.48,85 In the retina, Crumbs proteins have a crucial role in the retinal vascular development,86 in the photoreceptor-to-photoreceptor adhesion and photoreceptor-to-Müller cell adhesion.79 Müller cells span throughout the entire neuroretina, from the inner limiting membrane to the external limiting membrane, and they are responsible for the structural stabilization of the retina.87 They are essential for the survival of photoreceptors and neurons. Furthermore, they take up neurotransmitters, such as glutamate and gamma-Aminobutyric acid (GABA), and thus are involved in regulating the synaptic activity in the inner retina. As for the role of Crumbs proteins outside of the retina, current knowledge remains limited, and this role is suggested mostly by the clinical findings. In Drosophila, crumbs proteins are involved in the development and organization of epithelial cells.88 Further research is necessary in mammalian eyes to elucidate the role of the Crumbs complex outside of the retina.
The only truly robust genotype–phenotype correlation that we were able to elucidate, is the link between the p.Ile167_Gly169del CRB1 mutation, either in homozygous or compound heterozygous form, and an isolated maculopathy. This association was observed in both Dutch and Belgian populations, and has been described in British patients as well.89 In fact, this mutation has been present in at least one allele in all patients with CRB1-associated maculopathy described so far, and also in cases of CRB1-associated foveal retinoschisis.77
A striking degree of interfamilial variability was observed in both cohorts, even in the Dutch genetic isolate, where most patients had RP with variable visual results, as some patients became blind at a relatively early age, whereas others maintained ambulatory vision well into the later decades of life, and 1 patient had a cone-rod dystrophy, with macular atrophy and barely any (mid-)peripheral retinal changes. Although interindividual variability in CRB1-associated RDs has been described,18 the particularity here is that this variability occurred despite the same homozygous mutation, and the same origin from a village.90–92 This may indicate the involvement of genetic and possibly environmental modifiers, which have been implicated before in several RD subtypes.93,94 Although RDs are monogenic, the retina is a complex tissue involving numerous proteins to survive and function normally, that may influence the phenotypic outcome of monogenic diseases considerably.95 Mouse studies may give direction on possible research on genetic modifiers in human CRB1-RDs.96
Clinical Perspectives in LRAT-Associated RDs
In light of emerging therapeutic options, few studies have also focused on an extremely rare RD subtype: LRAT-associated RDs. Having previously only been described in a small number of case reports or series,14,30,97–100LRAT-associated RDs are estimated to account for <1% of RD cases, usually exhibiting Leber congenital amaurosis, early-onset severe RD, or retinitis punctata albescens. A retrospective study in 13 patients—to our knowledge the largest series described in literature so far—has broadened the phenotypic spectrum, by describing a subset of patients with relatively preserved vision into mid- and late adulthood.101 As this study consisted largely of patients from a genetic isolate, carrying the same homozygous c.12del LRAT mutation, it was able to elucidate the intrafamilial variability, with some patients carrying an RP phenotype and other a cone-rod dystrophy (CORD) phenotype. It also provided a comparison with one patient from outside the genetic isolate, who had an overall more severe phenotype of panretinal dysfunction. The specific mutation in the genetic isolate may be the most prominent cause of the relatively slow disease course in these patients, as opposed to the early blindness described in literature. However, this remains a suggestion, as the size of the cohort did not allow for sound statistical comparisons.
LRAT encodes protein lecithin:retinol acyltransferase (LRAT), one of the retinoid cycle proteins. Protein LRAT forms a complex with RPE65 to act as the isomerol hydrolase in the regeneration of visual pigment in the retinoid cycle. Having this closely connected biological function, both LRAT-RD and RPE65-RD have been targeted in a single treatment phase I trial investigating the safety and efficacy of oral QLT091001, a synthetic chromophore 11-cis-retinal.102,103 This study enrolled patients with Leber congenital amaurosis due to mutations in RPE65 or LRAT. Although mouse studies have shown phenotypic similarities between Rpe65(-/-) and Lrat(-/-) mice, human studies comparing these phenotypes are lacking. In one study, clinical findings in a retrospective cohort of patients with LRAT-RD and earlier literature in LRAT-RD patients were compared with the available literature on RPE65-RD, and this assessment demonstrated that there is a degree of phenotypic variability, with considerable overlap in this spectrum. To further substantiate any conclusion drawn from our study, a natural history study would ideally include extensive phenotyping of patients with LRAT-RDs and RPE65-RDs in the same study.
Clinical Perspectives in X-Linked RDs
X-linked RDs comprise a clinically heterogeneous group. X-linked RP, which accounts for 10% to 20% of RP cases,2,8 has often been found to be more severe than most autosomal recessive or autosomal dominant forms of the disease. X-linked RP is caused by mutations in the RPGR gene in approximately 75% of cases, whereas most remaining cases are caused by RP2 mutations.
Cone dystrophies (COD) and CORD may be inherited in all modes of Mendelian inheritance, but the autosomal recessive form is the most common. However, the underlying genetic cause is often unknown in cases of autosomal recessive inheritance, 104–106 whereas the most common genetic association with X-linked COD/CORD is known to be the RPGR gene.107,108
Another X-linked RD subtype that has been a focus of interest in the development of gene therapy is X-linked juvenile retinoschisis caused by mutations in the RS1 gene.109,110 This entity is characterized by a “spoke-wheel” pattern of retinal fluid collections in the macula. However, in 50% of patients, abnormalities occur in the periphery, such as schisis or neovascularization.
Clinical Perspectives in RPGR-Associated RDs
As several human gene therapy trials for RPGR-associated RP emerge (NCT03116113; NCT03252847; NCT 03316560),111,112 recent studies have also focused on the clinical and genotypic characteristics of RPGR-associated RDs. A recurring finding in literature is the association between the ORF15 mutational hotspot and a COD or CORD phenotype, particularly if the mutation is located at the 3’ end of ORF15.107,113–115 Symptom onset was in the first decade of life in RP. In COD/CORD, the median age at symptom onset was 23 years, approximately 10 years later than some earlier reports of COD/CORD,104 although reports have varied, some describing a much later symptom onset.116 This study showed a particularly high variability in the age at symptom onset in COD/CORD, followed by rapid decline of visual acuity, and a probability of being blind, defined by the World Health Organization as a best-corrected visual acuity of <20/400, at the age of 40 of 55%, as opposed to 20% in RP patients. Cystoid macular edema, an otherwise relatively common finding in RP, was not observed at any point during follow-up in this cohort of RPGR-RD patients, in line with other studies of RPGR-RD.116,117
We previously described that intrafamilial variability was particularly apparent in 2 families that comprised patients of RP and CORD phenotypes within the same family.113 A pivotal factor here is time, as in later disease stages, both RP and CORD progressed to panretinal dysfunction and became indistinguishable from each other in some cases. Still, this variability in the early disease stage is striking. An earlier report has even shown variability in a pair of dizygotic twins, one with RP and the other with CORD,118 and in other siblingships.119
A prominent finding in all subtypes of RPGR-associated RD is myopia.113,120,121 Mild, moderate, or high myopia was present in 84% of male RPGR patients and 73% of female carriers. Patients became more myopic with increasing age. The Dutch study in male patients showed high myopia to be an evident risk factor for visual acuity loss in all RD subtypes, and for visual field loss in RP. RPGR mutations have been shown to coincide with the highest degree of myopia in RDs,47 and this study elucidated the quantitative effect of myopia on disease progression in RPGR-RDs. Other studies have followed, confirming the link between myopia and more severe retinal degeneration.117 Refractive errors are not uncommon in RDs,47 and in some cases, the location and function of the protein product have been postulated to explain the refractive error. Although mutations in some genes such as RPGR are associated with (high) myopia, other genes (eg, CRB1 and BEST1) are associated with hyperopia. The protein retinitis pigmentosa GTPase regulator (RPGR) is located in the connecting cilium of the photoreceptor, the transport area between the inner and outer segment. Several genes that encode connecting cilium proteins have been linked to myopia, such as RP1 and RP2, although this does not apply to all connecting cilium proteins.122,123 A study on induced refractive errors in a chick model has implicated a range of photoreceptor-related proteins involved in the development of myopia or hyperopia, and these implicated proteins were primarily linked to photoreceptor dystrophies, such as CNGB1, RS1, RPE65, and RLBP1.124 The study unfortunately did not shed light on RPGR, and further studies are needed.
An important consideration is the phenotypic spectrum in female carriers of RPGR mutations.125 Like in affected males, myopia has a deleterious effect on visual acuity. In one large study, visual symptoms were relatively common, being present in 40% of subjects, and complete expression of a disease, that is, RP or CORD was found in 23% of subjects. Likewise, some earlier studies have identified RPGR mutations in disease-affected female patients,121,126–129 some of whom were presumed to have sporadic or autosomal dominant RP.130 This sheds light on multiple essential questions: Why do some female carriers develop disease and others don’t, and can we predict either outcome? And what implications do these findings hold in the clinical and genetic counselling of female carriers? Regarding the first matter, random X-inactivation and variable mosaicism could account for the phenotypic variation observed among female individuals,131 or skewed X-inactivation,132,133 as the relatively family-based aggregation of affected female carriers makes random X-inactivation unlikely as a sole factor. In symptomatic female carriers of choroideremia, another X-linked RD, severely skewed X-inactivation has indeed been demonstrated.134 Genetic modifiers may also play a role. It is not yet possible to predict which female carrier will develop disease, although the phenotype in other heterozygotes from the same family may be predictive.125 Presence of the tapetal-like reflex does not hold predictive value, and is not associated with symptoms or pigmentary retinal changes. Regarding the counseling of female carriers of RPGR mutations, it is vital that clinicians convey the risk of developing disease, while acknowledging that a complete disease expression does not occur in most heterozygotes.
When looking at genotype–phenotype correlations, studies have found a robust correlation between RPGR-ORF15 mutations and the COD/CORD phenotype.108,113,114,116 Mutations in the ORF15 region have been associated with a higher degree of myopia.113 In Dutch male RP patients, an RPGR-ORF15 mutation signified a higher hazard (twice as high) of reaching low vision or severe visual impairment than a mutation in exon 1–14.113,135 Also, RPGR-ORF15 mutations were associated with higher myopia, a significantly thinner central retina, and a significantly faster visual field decline. Previous literature, however, has shown the opposite finding of more severe disease in patients with mutation in exon 1–14 than those with mutations in RPGR-ORF15.136,137 This discrepancy may be explained by the higher degree of clinical variability in patients with mutations in RPGR-ORF15,125 which may lead to skewed findings in one population compared with the next. Peculiarly enough, in female heterozygotes, mutations in RPGR-ORF15 were associated with a less severe phenotype.125 Previous literature on genotype–phenotype correlations in female subjects is limited, but has shown the opposite effect, with worse visual function in female subjects with RPGR-ORF15 than those with mutations in exon 1–14.138 Genetic and/or environmental modifiers may again play a role in this clinical variability. Some proteins, such as RPGRIP1L and CEP290, have been shown to biochemically interact with RPGR,94,137 but additional research on such potential modifiers is needed.
Clinical Perspectives in Choroideremia
Choroideremia is a rare X-linked RD caused by mutations in the CHM gene. It has been proposed that this dystrophy primarily affects the retinal pigment epithelium, and secondarily the photoreceptors and choroid.139 Advances in gene therapy have resulted in multiple human gene therapy trials worldwide (Table 1 ), which have recently reached phase III.4,140–144 These advances prompted several studies on the associated phenotype and long-term clinical course. Symptoms of choroideremia are usually noticed in the 1st or 2nd decade of life, and the degeneration usually starts in the midperiphery, after which it gradually extends centripetally toward the periphery and the fovea.145–148 Prolonged relative sparing of foveal structure and function accounts for the long-term preservation of visual acuity, which usually remains until the 5th decade of life. This striking feature of foveal sparing, which is also typical for eg, late-onset Stargardt disease and central areolar choroidal dystrophy,149,150 remains of unknown etiology. One study investigated the kinetics of the progression of macular atrophy in several macular diseases, and found nearly identical patterns across several RD subtypes and age-related macula degeneration, suggesting a disease-independent mechanism.150 One proposed mechanism has been the metabolic difference between different macular regions in susceptibility to atrophy of rods and cones, RPE, and choroid.151 One longitudinal clinical study in a cohort of choroideremia patients showed a stable plateau of good vision until the 5th decade of life, and generally a turning point in the visual acuity decline from the 4th decade of life onwards.145 Outer retinal tubulations, which have been associated with age-related macular degeneration and various other degenerative conditions,152 were found in the majority of choroideremia patients (69%–94%) in several studies,145,153–157 although smaller numbers have been reported as well.158 They have also been demonstrated in symptomatic carriers, where they colocalized with areas of RPE atrophy and severe hypo-autofluorescence.134 Outer retinal tubulations presumably result from the rearrangement of degenerating photoreceptors,159 and areas containing them may be prone to surgical complications, such as macular hole formation as a result of subretinal injection of gene therapy vector solution. Moreover, areas containing outer retinal tubulations in choroideremia patients reportedly lack visual sensitivity despite the concomitant presence of viable cone inner segments in that same area.158 This may guide in assessing which retinal areas of a particular patient are amenable to (gene) therapy. Choroideremia patients in general are at risk of developing a macular hole, the surgery of which seems to be effective in achieving anatomic closure.160,161 No clear genotype–phenotype correlations have been established in choroideremia.162
Clinical Perspectives in Autosomal Dominant RDs
Autosomal dominant RDs, which are caused by mutations in the rhodopsin (RHO) gene in up to 30% to 40% of cases,163 present another challenge for gene therapy development. After all, the dominant disease is usually the result of a deleterious “gain-of-function” mechanism, for example, where the altered gene product adversely affects the normal gene product from the wild-type allele. Mere gene supplementation would not suffice in slowing the disease process that is caused by a toxic gain-of-function mutant protein, and the gain-of-function effect leading to disease would have to be diminished. For RHO-associated RP, momentous advances have been made, with knockdown-and-replacement strategies,164 CRISPR/Cas9 gene editing,165,166 and antisense oligonucleotides.167 “Simple” gene augmentation could still provide some therapeutic benefit in RHO-associated RP, even when the disease is caused by a dominant-negative effect.168
Considering these advances, several studies aimed to establish a detailed clinical profile and natural history in a large cohort of patients with RHO-associated RP. One study in Dutch and Belgian patients found an appreciable difference in disease progression between patients with sectorial RP (25% of our cohort) and those with generalized RP, as visual acuity decline was relatively stationary in the sectorial form, with the first case of blindness occurring after the 8th decade of life (manuscript accepted). In previous literature, an initial sectorial RP phenotype has relatively rarely been reported to progress to the generalized form.169 In the study of Dutch and Belgian patients, no patients were found with sectorial RP whose phenotype progressed to generalized RP, although this study was restricted by the limited availability of follow-up full-field fundus photographs in its retrospective study design.
In the Dutch and Belgian RHO-RP populations (sectorial and generalized forms), best-corrected visual acuity generally remained well-preserved, with a median age of reaching mild visual impairment of 72 years (manuscript accepted). Based on visual fields, the median ages to reaching low vision and blindness were 52 and 79 years, respectively. This is in line with previous studies that have shown that RHO-associated RP is a slowly progressive disease where patients generally maintain a good central visual function,170–173 as opposed to, for example, RPGR-associated or CRB1-associated RP. This points to a particularly lengthy window of therapeutic opportunity for ongoing and future (gene) therapy trials. On the contrary, the slow disease progression may complicate ways to clearly show a potential treatment effect. Some studies have referred to the sectorial disease phenotype as a “class B” phenotype, defined by an altitudinal (hemifield) loss of photoreceptor function.170,174,175 The degree of light exposure of the retina has been suggested to play a role in the retinal degeneration, and has been hypothesized due to the altitudinal degeneration mostly affecting the inferior retinal hemisphere.169,176,177 In support of this theory, animals with RHO-RP, including the RHOP23Hmouse and rat,178,179 and the RHOT4R dog,180 that have been reared in complete darkness, have shown slower retinal degeneration. Mice that remain in red-tinted cages that filter short-wavelength light (<600 nm) have been shown to maintain a thicker photoreceptor layer and higher amplitudes of electroretinography responses than mice in nontinted cages.181 However, this effect has not been proven in humans, and would be challenging to prove in a clinical trial setting. Thus, it remains a controversial claim.
Over 150 mutations have been reported in RHO. Several studies have been performed in the RhoP23H/+ mouse, as the p.Pro23His mutation is historically the first RHO mutation discovered, and one of the most common mutations in patients in the United States of America.182 To our knowledge, this mutation has not been reported in European studies, including ours. In our Dutch cohort, 37% of patients had the p.Glu181Lys mutation. With regard to genotype-phenotype correlations, several studies found an association between the mild or sectorial RP form, and mutations that correspond to the extracellular domain (ie, the intradiscal domain),174,175 particularly in comparison with mutations in the transmembrane domain.183 However, the study in Dutch and Belgian patients still found variation in the phenotype, for example, sectorial versus generalized RP, in patients with identical genotypes, such as the p.Glu181Lys mutation. Conversely, mild and sectorial phenotypes have been reported in association with mutations in other domains.184,185 Extreme intrafamilial variability in the RHO-RP phenotype has been reported.186 In fact, both RP and congenital stationary night blindness have been reported in the same family carrying the c.337G>A (p.Glu113Lys) mutation,187 which was not present in the Dutch and Belgian patients cohorts.
Clinical Heterogeneity and Potential Modifiers in RDs
A recurring finding in nearly all RD subtypes, including the ones studied in this thesis, is clinical heterogeneity. The same gene or even the same mutation may cause different phenotypes, and a nearly identical phenotype may be caused by different genes. The presence of a genetic isolate in a Dutch cohort of CRB1-RDs,31,32,91 consisting of patients carrying the same homozygous p.Met1041Thr mutation, provided the opportunity to investigate not only genotype-phenotype correlations, but also the intrafamilial variability. Although the phenotype was generally severe, and some hallmark features of CRB1-RDs were elucidated, one 41-year old patient had a mild CORD phenotype, whereas age-matched relatives had advanced RP. Even more variability was observed in the Dutch cohort of LRAT-RDs, again consisting largely of a genetic isolate. Similarly, in 2 families of RPGR-RD, some had RP, whereas others had CORD. This last finding should be nuanced by the idea that different RDs may not be entirely different entities, but members of a continuum. Advanced stages of CORD may be indistinguishable from RP, and it may prove difficult to retrieve early medical records in a retrospective setting. Nonetheless, variable degrees of intrafamilial variability were evident in several cohorts,31,32,101 and patients at roughly similar ages may still have different phenotypes (CORD or RP).113 Environmental and genetic modifiers, such as heterozygous mutations in other RD genes or single nucleotide polymorphisms, may have a role, and may influence the degree of severity. In conclusion, although RDs are typically monogenic diseases, and rare cases of putative digenic inheritance have been reported188,189 or suggested,190 the retina and RPE are complex tissues, whose survival and function depend on the proteins encoded by >18,000 genes for each tissue.95
Current Patient Management
Before the advances made in gene therapy studies in this millennium, the management of RD patients consisted of the regular follow-up and monitoring of disease progression, genetic and prenatal counselling, low vision aids where needed, and potential enrollment in a clinical trial. For patients who are blind due to outer retinal degeneration, but have maintained the inner retinal structure and an intact optic nerve, 2 retinal prostheses, the Argus II epiretinal prosthesis system, and the Alpha IMS (first generation) and Alpha AMS (second generation) subretinal prostheses, may aid in gaining some mobility or performing specific daily tasks. However, they require careful preoperative screening and expectation management, counseling, and a comprehensive postoperative rehabilitation program at a specialized center.191,192 The 2 most studied epiretinal implants, the Argus II and alpha-IMS/AMS, have shown performance results that can overall be considered similar, despite large differences in implant design.193 While most patients with a retinal prosthesis show an improvement in mobility and orientation tasks, approximately one-third experiences measurable visual acuity improvement.194 Reading speed can be improved in a subset of patients, although single-letter recognition may still take up to several minutes.195 Preoperative counseling should comprise the advice that the output from the prosthesis is an entirely new type of functional vision rather than the recovery of previous vision.196 Due to the guarded benefit, and the frequent visits and intensive rehabilitation required to achieve it, patient selection and expectation management are key.
The recent approval of voretigene neparvovec (Luxturna), a prescription gene therapy for RPE65-RD, has marked the dawn of a new era: the availability of an RD treatment to preserve and improve retinal function. However, for other RD forms, therapeutic options, if applicable, are being investigated in a clinical trial setting, or are in an earlier preclinical investigative phase.
Associated ocular conditions, such as cystoid macular edema (CME), should be monitored for development and treated. CME has been treated with different modalities. Topical and oral carbonic anhydrase inhibitors have shown morphological improvement with reduction of the CME,197 although the effect on visual acuity has been inconsistent between studies and remains inconclusive.198–202 One study has found that CME in the outer nuclear layer showed a better response to treatment with topical or oral carbonic anhydrase inhibitors than CME in the inner nuclear layer, where CME in RD is commonly found.203 An intravitreal dexamethasone implant (Ozurdex) has shown improvement of visual acuity and edema resolution,197,204 whereas intravitreal triamcinolone acetonide showed anatomical improvement without improvement in visual acuity.205,206 When using steroids, the development of cataract, and perhaps more importantly, elevation of intraocular pressure should be closely monitored in these patients, who are at an increased risk of developing both.31 Intravitreal injection of anti-vascular endothelial growth factor (VEGF) has shown inconsistent results with resolution of CME in some studies,207,208 and no effect in other studies.209 No evident visual acuity improvement was established with the use of anti-VEGFs.208 Intravenous immunoglobulin therapy has been reported in the treatment of concomitant CME and uveitis in 1 patient, and has shown complete resolution of CME at 4 months and 1 year.210 Octreotide has been postulated to have a role in the treatment of uveitis-associated CME,211 and has been successful in reducing CME and stabilizing visual acuity in dominant cystoid macular dystrophy.212 Its effect on CME in retinitis pigmentosa has not been reported to date.
Treatment options for Coats’-like exudative vasculopathy have included laser photocoagulation or cryotherapy. This can lead to regression of the exudates and to improved or stabilized vision,60,64,69,213,214 but it has also been complicated by a vitreous hemorrhage requiring vitrectomy.64 In the case of an exudative retinal detachment, treatment with vitrectomy and endolaser has been described, with the aim of salvaging the eye and maintaining any remaining vision.70,215 More recently, the intravitreal injection of conbercept, a new anti-VEGF, has been described in RP patients with exudative retinal detachment due to Coats’-like exudative vasculopathy.216 This led to complete resolution of the subfoveal serous detachment and improvement of the visual acuity. In a patient with RHO-associated RP and Coats’-like exudation, along with treatment-resistant CME, the intravitreal injection of a dexamethasone implant (Ozurdex) led to resolution of the exudation, along with a reduction in the CME, and maintenance of a well-preserved visual acuity.61 All these case reports appear to meager to establish a clear guideline for the treatment of CME in the context of RDs.
Implications of Natural History Studies for Gene Therapy Trials
The findings in this thesis have several implications for ongoing and future gene therapy trials. Crucial factors in the design of a (gene) therapy trial, are the determination of:
-
a)
a window of therapeutic opportunity
-
b)
patient eligibility criteria
-
c)
disease symmetry between eyes and the suitability of the contralateral eye as the untreated control; and
-
d)
defining endpoints for the evaluation of clinical efficacy.
Window of Opportunity
The window of therapeutic opportunity refers to the time span within which potential treatments may still prevent disease or positively modify the natural history. As gene therapy uses viral vectors that need to infect viable retinal cells, the window of opportunity closes when no viable photoreceptors remain, and no useful vision remains to be rescued. In a trial setting, the therapy is ideally applied in an early or intermediate disease stage, when enough vision remains to be rescued, and the natural disease progression is fast enough for a therapeutic effect to be detected, that is, a change in the rate of disease progression. However, in treatment settings, intervening as early as possible in the disease course may provide the best protective effect. In the Dutch cohort of patients with CRB1-RP, the median ages for reaching visual acuity-based low vision, severe visual impairment, and blindness were 18, 32, and 44 years, respectively. Thus, the window of therapeutic opportunity spans the first 3 decades of life, and could be expanded in some patients to the fourth decade of life. In CRB1-LCA or EOSRD, intervention would ideally be much earlier, within the first decade of life, as any remaining useful vision usually degenerates in this period. In contrast, the window of opportunity is considerably broader in patients with RHO-RP. In RPGR-RDs, the window of opportunity depends on the phenotype intended to treat in the trial: patients with COD/CORD have a 55% likelihood of being blind at the age of 40, as opposed to 20% in patients with RP. Patients with mutations in the ORF15 region had a higher risk of becoming blind at an earlier age, and would thus also require earlier therapeutic intervention, according to our study. In Dutch patients with LRAT-associated RDs, the window of therapeutic opportunity may be particularly broad as well.101
In RPGR- and RHO-associated RDs, the presence of a hyperautofluorescent ring may aid in determining which retinal area is most likely to benefit from a subretinal gene therapy injection, as this ring signifies the transitional zone between degenerated retina and relatively preserved—and thus rescuable—retina. In RPGR-RP, this ring was present in 47% of patients with RPGR-RP and 71% of patients with RPGR-COD/CORD. Although the ring provides useful information on the location of the transitional zone between atrophic and relatively preserved retina, it is unknown whether it has additional value in determining the likelihood of benefit from therapeutic intervention.
Patient Eligibility Criteria
Patient eligibility criteria for inclusion in a future trial are largely dependent on the window of therapeutic opportunity, and thus the patient age and remaining visual function. The presence of CME may render the macula more susceptible to the formation of a secondary macular hole, when subretinal injection of a viral vector in gene therapy increases the retinal stretching.217 Even if such a complication would not occur, the natural fluctuation in the extent of CME and the visual acuity may confound any potential therapeutic effect. On the contrary, successful gene augmentation via gene therapy may also have a beneficial effect on the resolution of CME. Patients with CRB1-RDs should be assessed for the risk of developing acute angle-closure glaucoma, and a prophylactic peripheral iridotomy or, if appropriate, cataract extraction may be warranted to reduce this risk before enrollment in a clinical trial that requires frequent mydriasis.
An extremely important point for consideration is the a priori amenability of the retina to (gene) therapy. A point of concern, particularly in some patients with CRB1-RD, would be the retinal disorganization, which would indicate a limited availability of viable cells for the viral vector to infect and/or the inability for the gene to function due to structural disintegration. Therefore, the degree of laminar disorganization was an area of focus in our retrospective and prospective studies. In the baseline report of our prospective study, the retinal laminar organization was preserved in 24% and showed only mild coarsening without disorganization in 38% of patients, indicating an amenability of the retina for gene therapy in 64% of patients. In the other 38% of patients, the retinal laminar organization was relatively disorganized, indicating a decreased amenability.
In choroideremia, the lengthy preservation of central visual function and foveal sparing affords a broad window of therapeutic opportunity for gene therapy.4,145,148 Outer retinal tubulations, when present, may provide clues of areas retaining viable photoreceptors and remaining visual function, as they have been found to be present around areas of surviving retina.145 Full-thickness macular holes have sporadically been described in choroideremia,160,161 and although successful closure may be achieved surgically, these patients may be at a higher risk of iatrogenic damage in a gene therapeutic setting.
Gene therapy trials for male patients with RPGR-associated RP may take the additional detrimental effect of the associated high myopia into consideration when assessing patient eligibility and when interpreting safety and efficacy data, as high myopia is associated with worse visual function and a thinner retina.113 High myopia may thus be a complicating factor in the rescue of the remaining photoreceptors.
Gene therapy trials for RPGR may consider enrolling affected female heterozygotes in future trial phases, as women may express a full disease phenotype. Most female heterozygotes are mildly affected or asymptomatic, and treatment in these patients may not be necessary.
The usefulness of gene therapy is impeded in cases of extensive atrophy of the photoreceptors, RPE, and choriocapillaris. In these patients, stem cell-based therapeutic options, which are not gene-specific, may provide more benefit. Examples include the intravitreal or subretinal administration of induced pluripotent stem cells or retinal progenitor cells.218 These studies are in the early stages: one phase 1/2 clinical trial on human embryonic stem cell-derived RPE cells has been completed in age-related macular degeneration and Stargardt disease,219 and has shown an acceptable safety profile and possible improvement in visual function. Clinical trials using induced pluripotent stem cells are expected, but are yet to be initiated. In patients with advanced atrophy, stem cells may need to differentiate into multiple cell types, such as RPE and choriocapillaris, and this may require multiple injections with each treatment session. The injected cells then have to successfully convert into each mature and functional cell structure individually, and organize into a structurally and functionally intact unit. Although these challenges complicate the treatment options for these patients, in vitro and in vivo studies have shown some promising results.220,221
So far, subretinal gene augmentation therapy trials have treated the posterior pole/macular region,3,4,222,223 whereas patients with RP or choroideremia may experience visual field constriction as a major problem. Indeed, 1 study has surveyed patient-reported visual complaints and their effects on daily life, and has found that most choroideremia patients (70%) reported peripheral visual field constriction as the most debilitating symptom.145 In these patients, expectation management before enrollment in a clinical gene therapy trial is crucial, as the peripheral rods responsible for the visual field are not targeted through conventional subretinal gene therapy. Intravitreal gene therapy administration may theoretically provide a better outcome in the peripheral visual function these patients, although it currently holds a higher risk of inflammation and systemic biodistribution,224–226 and a lower degree of efficacy than subretinal administration in the eyes of primates.227 Should intravitreal gene therapy administration develop a better profile in the future, intervention would ideally happen at a much earlier stage, as rods degenerate already in the earlier disease stages, while central cone function and visual acuity may remain preserved for many years.
Interocular Symmetry
As most retinal (gene) therapy studies have treated one eye, usually the worse-seeing eye, inter-eye symmetry within the same patient is an important aspect. Interocular symmetry enables the use of the contralateral eye as an ideal untreated control. A high degree of inter-eye symmetry has been confirmed in several RD subtypes of interest for ongoing and future gene- and cell-based therapy trials.31,32,101,113,148,170,173,228–231 Interocular symmetry or lack thereof should be determined before enrollment in an interventional trial, and investigators should aim to identify a potential cause of significant asymmetry, if this can be determined.
Defining Endpoints for Evaluation of Treatment Efficacy
For many RD subtypes, it has proven to be challenging to define clinical endpoints for the evaluation of treatment efficacy. A thorough understanding and quantification of important parameters in the natural disease course is crucial, as this may help define the most appropriate efficacy endpoints. Using the appropriate endpoint may be pivotal in the process of for market-approval of gene therapy by regulatory bodies. To be an expeditious efficacy endpoint for a treatment aimed at slowing disease progression, a parameter would have to be expected to show significant decline within the clinical trial period, and a faster decline than any expected test–retest variability. Visual acuity, a measure of central cone function, usually shows significant decline over several decades of life, but may remain relatively stable over the course of a few years, whereas the duration of a clinical treatment trial is usually not much longer than 2 years. Visual acuity survival curves in CRB1-RP in the Dutch cohort have shown a relative plateau during the second decade of life. Meanwhile, the visual acuity decline rate was 0.03 logMAR per year, corresponding to 7.2% per year. Similar rates were demonstrated in the decline of the visual field area. To calculate how long a trial should last in order for a true treatment effect to be detected, test–retest variability in the visual function values should be determined in the study population. The estimated time needed to detect a significant change may be longer than the trial period in most patients, but longitudinal prospective studies must further investigate this. In patients with RPGR-RP, visual acuity did not show any significant decline before the age of 20 years in one study,113 indicating that in these young patients, visual acuity is not a sensitive marker for change. However, it would be a judicious safety marker, as any significant visual acuity decline may be an indicator of iatrogenic damage to the retina.
Several studies have indicated that the ellipsoid zone width and ellipsoid zone area on SD-OCT may be sensitive biomarkers for disease progression,229,232 even within a time span of 2 years of follow-up.233–235 In our study of RHO-RP, we found similar results for ellipsoid zone width. Several challenges accompany this particular biomarker: while this biomarker appears to be useful for instance in RHO-RP or RPGR-RDs, in CRB1-RDs, the ellipsoid zone disintegration will probably be at a too advanced stage to be able to sensitively detect a significant change in decline rate. Moreover, regulatory bodies such as the US Food and Drug Administration (FDA) and the European Medicines Agency, have not yet approved structural biomarkers as defining parameters for the approval of a therapy for retinal disease.236 For such structural biomarkers to serve as surrogate endpoints, their reliability, and their strong correlation to direct measures of the patient's visual function (eg, visual acuity), should be established. In a study on CRB1-RP (manuscript under review), the ellipsoid zone width did not maintain its significant correlation with visual acuity after correction for multiple testing. The thickness of the photoreceptor and RPE complex (ie, as measured from the external limiting membrane to the RPE at the fovea), however, did correlate with visual acuity. Its rate of decline (−0.6%/year), however, was much slower than that of the ellipsoid zone band width (−3.8%/year), which means that the expected time needed to detect a treatment effect is much longer.
Looking back at the RPE65 gene therapy trial that led to market approval of Luxturna, useful endpoints have included the full-field stimulus testing,237 which we have also employed in our prospective natural history study of CRB1-RD. Full-field stimulus testing is a psychophysical measure to determine the maximum retinal sensitivity in the full field, and chromatic stimuli can be added to determine whether this sensitivity is rod-mediated, cone-mediated, or mediated by a combination of the two.238,239 It may be employed in patients with nondetectable dark-adapted and light-adapted responses on the electroretinogram, and is therefore particularly helpful in patients who are (nearly) blind. Another useful endpoint in studies leading to marked approval of voretigene neparvovec (Luxturna) was the multi-luminance mobility test. This is a navigation course, wherein patients must maneuver past obstacles at different levels of environmental illumination, ranging from 1 lux (a moonless night) to 400 lux (a brightly lit office). It provides a reliable measure of functional vision, that is meaningful with regard to the patient's daily life. Although an impractical measure in natural history studies, it has proven useful in interventional trials, and its validity has been demonstrated in a nontrial setting.240 Other mobility courses and artificial platforms for mobility and for the simulation of daily activities have been developed, such as The StreetLab and HomeLab platforms designed by the Institut de la Vision (Paris).241
In gene therapy trials, primary outcome measures should ideally focus not only on the objective improvement in visual acuity and other visual and structural parameters, but also on the efficacy of treatments to significantly improve parameters that are important of patients’ daily lives, such as level of independence, quality of life, and other patient-reported outcomes. This is challenging, as no standardized questionnaires have been established for such quality of life and social functioning aspects for this specific population with severe visual impairment due to RDs.
Emerging Therapies and Future Perspectives
Before the emergence of gene therapeutic trials, no evidence-based treatment options existed for RDs that led to a clinically measurable improvement in visual function. The development of therapies for rare diseases has historically been challenging due to small patient populations for trials, and the challenges in post-approval marketing.
The great advances in gene therapy in the last 2 decades have led to market approval of voretigene neparvovec (Luxturna) subretinal gene therapy for RPE65-associated early-onset RD/Leber congenital amaurosis. This success, along with other advances in gene therapy development,6 has led to a spectacular expansion in the field of retinal gene therapy. Subretinal gene therapy is under development for CRB1-RDs,6 and clinical trials ongoing for RPGR-associated RP, choroideremia, achromatopsia (associated with CNGB3 and CNGA3), Stargardt disease (associated with ABCA4), X-linked retinoschisis (associated with RS1), and several other entities (Table 1 ), are in the pipeline.6,164
Gene Replacement and Gene Silencing
Gene transfer to the target cells in the retina may happen through viral vectors, mostly adenoviruses, lentiviruses, or adeno-associated viruses (AAV), the latter representing the most efficient and stable gene transfer in most RD forms.11,15 AAV vectors are currently the most used viral vectors in gene therapy, due to the extensive experience with AAV, and their excellent safety profile: in the retina, the risk for immunogenicity is low,242 and they have low inflammatory and low retinal toxicity potential.242,243 Furthermore, they do not integrate their genome into the host-cell genome,243 thus eliminating the risk of iatrogenic activation of oncogenes. Virtually all AAV serotypes are able to infect the RPE, and serotypes 2, 5, and 7 to 9 are able to infect photoreceptors.244 Drawbacks of AAV vectors include their small size, which leads to a limited transgene capacity of 4.5 to 5.0 kb. In contrast, the larger lentivirus vectors have a transgene capacity of up to 10 kb.245 However, they integrate their genome into the host-cell genome with great efficiency, although it has been shown that they do not preferentially integrate their genome in the vicinity of oncogenes.246 Although the potential of viral vectors has been demonstrated repeatedly, nonviral gene delivery systems have been investigated as well. These transfer methods, using for instance nanoparticles, liposomes, or naked plasmid DNA, are cheaper and easier to produce, and have a lower risk of inducing an immune response. However, as of yet, they have not shown promising potential for safe gene delivery, due to, for example, lack of persistent transgene expression (naked DNA and nanoparticles), or the potential for retinal toxicity (liposomes).247,248
Although gene replacement or supplementation should be sufficient in autosomal recessive RDs, in which a lack of gene expression leads to a deficit in the gene product, (additional) gene silencing is necessary in autosomal dominant RDs. In autosomal dominant RPs, the gene mutations often lead to mutant gene expression resulting in altered protein products that impair normal function of the wild-type protein, leading to a toxic effect. In such cases, gene therapy is aimed at repairing or silencing the mutated gene, and gene supplementation in the case of additional haplo-insufficiency.
Such gene silencing has been proposed through the use of allele-specific inhibitors that induce the degeneration of the mutated messenger RNA (mRNA).249 Another approach is the suppression of both the mutated and wild-type allele, and their replacement by a wild-type nonsilenced allele.250 Both strategies can be mediated for instance by small RNA inhibitors or ribozymes,251–253 each with their own set of advantages and disadvantages,254 such as a need for repeated injections.
Antisense Oligonucleotides
Antisense oligonucleotides (AONs) consist of small DNA or RNA molecules that are able to modulate splicing after binding to pre-mRNA. Preclinical studies using, for example, fibroblasts from affected patients, and animal studies have shown promising results for CEP290-LCA,255 and for RHO-RP.167 AONs can be administered “naked” through intravitreal injections, or through subretinal injections with an adenoviral-associated viral vector, and have shown minimal toxic or immunological adverse effects.256 As naked AONs are small-sized molecules, they may be able to reach their destination cells, the photoreceptors, more easily after intravitreal injections. This approach would require repeated injections throughout life, whereas a subretinal injection of an AAV-mediated AON may give a considerably more durable therapeutic benefit. However, intravitreal AONs target the entire retina, and the need for a vitrectomy and its associated complications is circumvented. A recent phase 1/2 trial investigating the effect of intravitreal AONs in the treatment of 10 patients with CEP290-associated LCA found no serious adverse events, and a clinically meaningful improvement in vision, defined in the study as 0.3 logMAR, in 5 patients.257 These encouraging results are followed up in a phase 2/3 trial, the ILLUMINATE study (NCT03913143).
Gene Editing: CRISPR/Cas9
An exciting potential alternative to gene replacement strategies is the therapeutic approach of gene editing. In gene editing, the genome can be altered by inducing double-stranded DNA breaks, single-stranded DNA breaks, or specific base changes in the DNA at target sites to correct the deleterious gene mutation. This can be achieved using several methods, such as zinc finger nucleases, meganucleases, and, more recently, clustered regularly interspaced short palindromic repeats CRISPR-Cas9-associated protein 9 (Cas9).258 CRISPR/Cas9 gene editing is a fast, cheap, and relatively efficient method to edit the genome and repair genetic mutations, typically by inducing double-stranded breaks. CRISPR is guided by RNA sequences, and multiple guide RNA sequences may be packaged into one targeted delivery system (eg, a viral vector). Thereby, CRISPR has the unique ability to target >1 genetic location.259
CRISPR/Cas9-based therapies have been used successfully in mouse models, for instance PDE6B,260CEP290,261 and RHO.165,262 In mouse models of RHO-RP, CRISPR/Cas9 has been used in a mutation-independent “ablate-and-replace” technique. Moreover, CRISPR/Cas9 has been used to generate accurate mouse models for RP and LCA.263,264
Drawbacks of the CRISPR/Cas9 gene editing system include concerns on its accuracy and the potential of off-target effects.265 Additionally, its efficiency may vary. In induced pluripotent stem cells of a patient with RPGR-RP, CRISPR-Cas9 was applied to correct the gene mutation and convert it to the wild-type allele.266 This succeeded in 13% of RPGR gene copies, which still spectacularly exceeds previous gene correction rates of 1% to 3%.267 Furthermore, it is a large-sized system that cannot be packaged into a single viral vector, and typically a dual vector system is employed.268
The challenges associated with the CRISPR/Cas9 approach have driven the exploration of alternative precision gene editing approaches. One such approach is the recently published prime editing strategy,269 which can alter DNA with single-nucleotide precision, potentially with greater safety, and with great versatility. It combines Cas9-mediated RNA-guided DNA breakage (or nicking) with reverse transcriptase-mediated DNA synthesis at the same target site. Different types of mutations, including insertions and deletions, can be corrected. It has been proposed that it can correct up to 89% of pathogenic human variants that have been described in the ClinVar archive of genetic variants in any part of the genome, which spectacularly broadens the range of mutations that can be corrected. The promising results of prime editing, as demonstrated in vitro, remain to be investigated in vivo.
Stem Cell-Based Strategies
When retinal cells have already died, genetic therapies to correct mutated genes in the affected target cells appear useless as an isolated therapeutic approach. In these cases, replacement of these dead cells by new functional cells may prove to be a viable future treatment option.220,270 Human embryonic stem cells have been investigated as a treatment for several retinal disorders, and have shown some visual improvement in RD rat models.271,272 In humans, a phase 1/2 trial transplanting human embryonic stem cells to the subretinal space in patients with Stargardt disease or atrophic age-related macular degeneration has shown some modest visual improvement in more than half of the treated eyes.219 However, the use of human embryonic stem cells as a therapy has raised ethical concerns, and concerns over immunological responses and/or the need for immunosuppression.
Fibroblast-derived induced pluripotent stem cells (iPSCs) are derived from the patient, and have been used in the treatment of several mouse and rat models of RD, where they have led to potential preservation of the visual function.273,274 Concerns regarding the use of iPSCs as a treatment modality include immunogenicity,275 and tumor formation due to incompletely differentiated iPSCs.276 A safer and particularly exciting application of iPSCs has been in the generation of retinal organoids,277 where they aid in the examination of underlying disease mechanism and in the in-vitro study of treatment options, such as in CRB1-RDs.15 In autologous iPSC-based cultured retinal cells of patients with RDs, the genetic defect may be corrected in vitro, using, for instance, AAV-based gene replacement or gene editing techniques,221 in preparation for subretinal administration. A key challenge may be not only to achieve a correct anatomical integration of such stem cells into the retina after surgical administration, but certainly also subsequent cellular function and interaction, leading to genuine functional improvement that matters to the patients.220,270,278Another important aspect when considering cell transplantation for advanced RD is the fact that such cases do not only have photoreceptor atrophy, but also atrophy of the photoreceptor's “nursing cells,” the RPE, and choriocapillaris. After all, the photoreceptor-RPE-Bruch membrane-choriocapillaris interface normally forms a closely connected and interdependent functional unit. This means that administration of such a combination of cells, possibly using cell sheets and/or a cell-carrying scaffold, may be mandatory to achieve a (close to) normal cellular interaction for a durable and functionally relevant treatment effect.
Bone-marrow-derived mesenchymal stem cells have been used in intravitreal injections in phase I clinical trials for RD patients, and in commercial “stem cell clinics” in the United States, where resulting vision-threatening complications, such as vitreous hemorrhage and rhegmatogenous retinal detachment, and blindness have been reported in patients with RD and with age-related macular degeneration.279,280
Optogenetics
In patients who are blind due to photoreceptor degeneration while still retaining a relatively intact inner retina, optogenetics may be a tool to reintroduce light perception. Optogenetics is a strategy whereby a gene encoding a photosensitive protein (an opsin) is introduced in inner retinal cells, that is, retinal ganglion cells and bipolar cells, with the aim of sensitizing these inner retinal cells to light in the absence of photoreceptors.281 It thus provides an alternative visual cycle to improve retinal activity. Opsins may have a microbial origin (type 1), such as channelrhodopsins or halorhodopsins, which function as light-gated ion channels, or an animal origin (type 2), such as melanopsin or rhodopsin. Preclinical data have suggested that blind patients with preservation of the photoreceptor nuclei, as visible on OCT, may be eligible for functional photoreceptor restoration through optogenetics.282
Portable Vision Devices
Among new low-vision aids that are aimed at improving patients’ quality of life, is the OrCam, a portable and spectacle-attached device capable of recognizing optical characters, text, currency denominations, and, as programmed, faces and objects. The device is activated when pointed, pressed, or tapped on. In small series of visually impaired patients, usage of this device led to an increase in scores on a daily function test.283
Expectation Management in Interventional Clinical Trials
Although the advances of the last 2 decades have propelled research forward toward clinical application, with exciting new treatment possibilities for RD patients, expectations should be managed and critically reconsidered. Gene supplementation therapy is notably costly to develop, to test, and to implement, and of the several gene replacement therapy trials that have been performed in the RPE65-RD population, only one has led to considerable long-term success and market approval to date: voretigene neparvovec (Luxturna), priced at approximately US $850.000. Although the bench-to-bedside success of this first commercially available retinal gene therapy has further energized patient and research communities alike, the long-term effects of the therapy on visual function in the other RPE65-gene therapy trials have been more guarded.222,223 For example, it has been found that retinal degeneration may continue, and the longevity of interventional therapy will be limited if the degree of photoreceptor degeneration has exceeded a certain limit before treatment.284 Indeed, in most patients, retinal degeneration will have progressed to intermediate or advanced stages at the time of intervention. Although any degree of visual restoration and/or preservation is a revolutionary move forward in an otherwise untreatable disease entity, FDA documents have revealed that approximately half of treated patients met the FDA criteria for minimally meaningful improvement.285 The other half did not achieve the criteria for meaningful improvement, and 2 patients had permanent vision loss, due to injection-related macular thinning in one patient, and irreversible optic nerve atrophy due to increased intraocular pressure in the other patient, who received ocular steroids for the treatment of a Staphylococcus infection.285 These results may be particularly disappointing to the patient, having undergone the surgical procedure and a period of recovery and frequent hospital visits.
In choroideremia, gene therapy has led to a median gain in visual acuity of 4.5 letters in the treatment cohort, versus a visual acuity loss of 1.5 letters in the untreated eye at the 2-year post-treatment point. In some patients, this vision improvement was sustained at up to 5 years of follow-up.141 Nonetheless, complications arose in 2 of 14 patients (14%)—surgery-related retinal thinning and incomplete vector dosing in one patient, and postoperative inflammation in the other. This has led to the prolongation of the postoperative immune suppression regimen.
The irreversibility of disease in cell populations that have already degenerated should be stressed to any potential participants in gene therapy trials. In the case of subretinal gene therapy that only targets the posterior pole, the treatment effect will be confined largely to the macula. Therefore, it should be explained to patients that peripheral visual field preservation is not expected when this is not the targeted area.
Issues regarding the cost of gene therapy remain a point of concern. As gene therapies for orphan indications, defined as diseases affecting <200,000 people in the United States, target specific genetic entities, and thus pertain to small patient populations, they remain among the most expensive drugs.
These considerations indicate that clinicians and researchers should exert caution not to oversell the capacities of investigative (gene) therapeutic strategies to patients, who are often driven by hope. In the context of informed consent, it is evident that eligible patients—who may already be small in number—are to be informed well on the risks of intervention, its investigative nature, and thus uncertain outcome, and on the lengthy post-intervention trajectory.
CONCLUDING REMARKS
New treatment opportunities emerge for RDs at an exceedingly rapid pace, offering hopeful perspectives to many RD patients worldwide. Given these developments and the need to approve effective treatments for clinical use, prospective natural history studies are of eminent importance. However, this thesis has shown that retrospective studies, despite their inherent limitations, can provide robust and useful information on important disease characteristics, variability, and course of many years. National collaborations, such as the RD5000 consortium in the Netherlands, or international collaborations, as within the European context of European Reference Network - Eye Diseases (ERN-EYE), are important to further strengthen the outcome of such studies in these relatively small patient populations. For example, access to the Delleman archive for hereditary eye diseases at the Amsterdam University Medical Centers/Academic Medical Center in Amsterdam, and access to the database for hereditary eye diseases at the Ghent University Hospital, have provided the unique opportunity to ascertain large sample sizes, and to assemble some of the largest retrospective cohorts described to date. Indeed, prospective studies will not be able to provide all the answers on disease progression and visual survival, and they will still have limitations, such as a limited capacity to include many patients, and a limited study duration. However, the limitations of retrospective research are well-described and include the lack of standardization of patient visits, interval censoring, and a limited availability of multimodal imaging. Improvement of phenotyping and genetic characterization remain of critical importance. Ongoing and future prospective studies should be geared at further assessing the rate of disease progression through different visual function parameters and biomarkers on multimodal retinal imaging, and at investigating correlations between these measures. In the end, retrospective and prospective studies have the powerful capacity to augment each other. Such studies are pivotal for well-balanced decision-making on patient eligibility for treatments, and endpoint selection to test treatment efficacy.
Footnotes
Financial support: Curing Retinal Blindness Foundation, Janivo Stichting, Stichting Blindenhulp.
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis 2006; 1:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet 2006; 368:1795–1809. [DOI] [PubMed] [Google Scholar]
- 3.Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med 2008; 358:2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacLaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet 2014; 383:1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cehajic Kapetanovic J, Barnard AR, MacLaren RE. Molecular therapies for choroideremia. Genes (Basel) 2019; 10:738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pellissier LP, Quinn PM, Alves CH, et al. Gene therapy into photoreceptors and Muller glial cells restores retinal structure and function in CRB1 retinitis pigmentosa mouse models. Hum Mol Genet 2015; 24:3104–3118. [DOI] [PubMed] [Google Scholar]
- 7.Quinn PM, Pellissier LP, Wijnholds J. The CRB1 complex: following the trail of crumbs to a feasible gene therapy strategy. Front Neurosci 2017; 11:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verbakel SK, van Huet RAC, Boon CJF, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res 2018; 66:157–186. [DOI] [PubMed] [Google Scholar]
- 9.den Hollander AI, Roepman R, Koenekoop R, PM Cremers F. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog Retin Eye Res 2008; 27:391–419. [DOI] [PubMed] [Google Scholar]
- 10.Michaelides M, Hunt DM, Moore AT. The genetics of inherited macular dystrophies. J Med Genet 2003; 40:641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziccardi L, Cordeddu V, Gaddini L, et al. Gene therapy in retinal dystrophies. Int J Mol Sci 2019; 20:5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garafalo AV, Cideciyan AV, Heon E, et al. Progress in treating inherited retinal diseases: early subretinal gene therapy clinical trials and candidates for future initiatives. Prog Retin Eye Res 2019; 100827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corton M, Tatu SD, Avila-Fernandez A, et al. High frequency of CRB1 mutations as cause of early-onset retinal dystrophies in the Spanish population. Orphanet J Rare Dis 2013; 8:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vallespin E, Cantalapiedra D, Riveiro-Alvarez R, et al. Mutation screening of 299 Spanish families with retinal dystrophies by Leber congenital amaurosis genotyping microarray. Invest Ophthalmol Vis Sci 2007; 48:5653–5661. [DOI] [PubMed] [Google Scholar]
- 15.Quinn PM, Buck TM, Mulder AA, et al. Human iPSC-derived retinas recapitulate the fetal CRB1 CRB2 complex formation and demonstrate that photoreceptors and muller glia are targets of AAV5. Stem Cell Reports 2019; 12:906–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murro V, Mucciolo DP, Sodi A, et al. Retinal capillaritis in a CRB1-associated retinal dystrophy. Ophthalmic Genet 2017; 38:555–558. [DOI] [PubMed] [Google Scholar]
- 17.Morarji J, Lenassi E, Black GC, Ashworth JL. Atypical presentation of CRB1 retinopathy. Acta Ophthalmol 2016; 94:e513–e514. [DOI] [PubMed] [Google Scholar]
- 18.Ghofrani M, Yahyaei M, Brunner HG, et al. Homozygosity mapping and targeted sanger sequencing identifies three novel CRB1 (Crumbs homologue 1) mutations in iranian retinal degeneration families. Iran Biomed J 2017; 21:294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kousal B, Dudakova L, Gaillyova R, et al. Phenotypic features of CRB1-associated early-onset severe retinal dystrophy and the different molecular approaches to identifying the disease-causing variants. Graefes Arch Clin Exp Ophthalmol 2016; 254:1833–1839. [DOI] [PubMed] [Google Scholar]
- 20.Hasan SM, Azmeh A, Mostafa O, Megarbane A. Coat's like vasculopathy in leber congenital amaurosis secondary to homozygous mutations in CRB1: a case report and discussion of the management options. BMC Res Notes 2016; 9:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vamos R, Kulm M, Szabo V, et al. Leber congenital amaurosis: first genotyped Hungarian patients and report of 2 novel mutations in the CRB1 and CEP290 genes. Eur J Ophthalmol 2016; 26:78–84. [DOI] [PubMed] [Google Scholar]
- 22.Wolfson Y, Applegate CD, Strauss RW, et al. CRB1-related maculopathy with cystoid macular edema. JAMA Ophthalmol 2015; 133:1357–1360. [DOI] [PubMed] [Google Scholar]
- 23.Kuniyoshi K, Ikeo K, Sakuramoto H, et al. Novel nonsense and splice site mutations in CRB1 gene in two Japanese patients with early-onset retinal dystrophy. Doc Ophthalmol 2015; 130:49–55. [DOI] [PubMed] [Google Scholar]
- 24.Cordovez JA, Traboulsi EI, Capasso JE, et al. Retinal dystrophy with intraretinal cystoid spaces associated with mutations in the Crumbs homologue (CRB1) gene. Ophthalmic Genet 2015; 36:257–264. [DOI] [PubMed] [Google Scholar]
- 25.Srilekha S, Arokiasamy T, Srikrupa NN, et al. Homozygosity mapping in leber congenital amaurosis and autosomal recessive retinitis pigmentosa in South Indian families. PLoS One 2015; 10:e0131679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jalkh N, Guissart C, Chouery E, et al. Report of a novel mutation in CRB1 in a Lebanese family presenting retinal dystrophy. Ophthalmic Genet 2014; 35:57–62. [DOI] [PubMed] [Google Scholar]
- 27.Tsang SH, Burke T, Oll M, et al. Whole exome sequencing identifies CRB1 defect in an unusual maculopathy phenotype. Ophthalmology 2014; 121:1773–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jonsson F, Burstedt MS, Sandgren O, et al. Novel mutations in CRB1 and ABCA4 genes cause Leber congenital amaurosis and Stargardt disease in a Swedish family. Eur J Hum Genet 2013; 21:1266–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yzer S, Leroy BP, De Baere E, et al. Microarray-based mutation detection and phenotypic characterization of patients with Leber congenital amaurosis. Invest Ophthalmol Vis Sci 2006; 47:1167–1176. [DOI] [PubMed] [Google Scholar]
- 30.den Hollander AI, Lopez I, Yzer S, et al. Identification of novel mutations in patients with Leber congenital amaurosis and juvenile RP by genome-wide homozygosity mapping with SNP microarrays. Invest Ophthalmol Vis Sci 2007; 48:5690–5698. [DOI] [PubMed] [Google Scholar]
- 31.Talib M, van Schooneveld MJ, van Genderen MM, et al. Genotypic and phenotypic characteristics of CRB1-associated retinal dystrophies: a long-term follow-up study. Ophthalmology 2017; 124:884–895. [DOI] [PubMed] [Google Scholar]
- 32.Mathijssen IB, Florijn RJ, van den Born LI, et al. Long-term follow-up of patients with retinitis pigmentosa type 12 caused by crb1 mutations: a severe phenotype with considerable interindividual variability. Retina 2017; 37:161–172. [DOI] [PubMed] [Google Scholar]
- 33.Paun CC, Pijl BJ, Siemiatkowska AM, et al. A novel crumbs homolog 1 mutation in a family with retinitis pigmentosa, nanophthalmos, and optic disc drusen. Mol Vis 2012; 18:2447–2453. [PMC free article] [PubMed] [Google Scholar]
- 34.Zenteno JC, Buentello-Volante B, Ayala-Ramirez R, Villanueva-Mendoza C. Homozygosity mapping identifies the Crumbs homologue 1 (Crb1) gene as responsible for a recessive syndrome of retinitis pigmentosa and nanophthalmos. Am J Med Genet A 2011; 155a:1001–1006. [DOI] [PubMed] [Google Scholar]
- 35.Henderson RH, Mackay DS, Li Z, et al. Phenotypic variability in patients with retinal dystrophies due to mutations in CRB1. Br J Ophthalmol 2011; 95:811–817. [DOI] [PubMed] [Google Scholar]
- 36.Riveiro-Alvarez R, Vallespin E, Wilke R, et al. Molecular analysis of ABCA4 and CRB1 genes in a Spanish family segregating both Stargardt disease and autosomal recessive retinitis pigmentosa. Mol Vis 2008; 14:262–267. [PMC free article] [PubMed] [Google Scholar]
- 37.Simonelli F, Ziviello C, Testa F, et al. Clinical and molecular genetics of Leber's congenital amaurosis: a multicenter study of Italian patients. Invest Ophthalmol Vis Sci 2007; 48:4284–4290. [DOI] [PubMed] [Google Scholar]
- 38.Bernal S, Calaf M, Garcia-Hoyos M, et al. Study of the involvement of the RGR, CRPB1, and CRB1 genes in the pathogenesis of autosomal recessive retinitis pigmentosa. J Med Genet 2003; 40:e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacobson SG, Cideciyan AV, Aleman TS, et al. Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum Mol Genet 2003; 12:1073–1078. [DOI] [PubMed] [Google Scholar]
- 40.Lotery AJ, Malik A, Shami SA, et al. CRB1 mutations may result in retinitis pigmentosa without para-arteriolar RPE preservation. Ophthalmic Genet 2001; 22:163–169. [DOI] [PubMed] [Google Scholar]
- 41.Lotery AJ, Jacobson SG, Fishman GA, et al. Mutations in the CRB1 gene cause Leber congenital amaurosis. Arch Ophthalmol 2001; 119:415–420. [DOI] [PubMed] [Google Scholar]
- 42.den Hollander AI, Heckenlively JR, van den Born LI, et al. Leber congenital amaurosis and retinitis pigmentosa with Coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) gene. Am J Hum Genet 2001; 69:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aleman TS, Cideciyan AV, Aguirre GK, et al. Human CRB1-associated retinal degeneration: comparison with the rd8 Crb1-mutant mouse model. Invest Ophthalmol Vis Sci 2011; 52:6898–6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coppieters F, Casteels I, Meire F, et al. Genetic screening of LCA in Belgium: predominance of CEP290 and identification of potential modifier alleles in AHI1 of CEP290-related phenotypes. Hum Mutat 2010; 31:E1709–E1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galvin JA, Fishman GA, Stone EM, Koenekoop RK. Evaluation of genotype-phenotype associations in leber congenital amaurosis. Retina 2005; 25:919–929. [DOI] [PubMed] [Google Scholar]
- 46.Edwards A, Grover S, Fishman GA. Frequency of photographically apparent optic disc and parapapillary nerve fiber layer drusen in Usher syndrome. Retina 1996; 16:388–392. [DOI] [PubMed] [Google Scholar]
- 47.Hendriks M, Verhoeven VJM, Buitendijk GHS, et al. Development of refractive errors-what can we learn from inherited retinal dystrophies? Am J Ophthalmol 2017; 182:81–89. [DOI] [PubMed] [Google Scholar]
- 48.Boon CJ, Klevering BJ, Leroy BP, et al. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog Retin Eye Res 2009; 28:187–205. [DOI] [PubMed] [Google Scholar]
- 49.Othman MI, Sullivan SA, Skuta GL, et al. Autosomal dominant nanophthalmos (NNO1) with high hyperopia and angle-closure glaucoma maps to chromosome 11. Am J Hum Genet 1998; 63:1411–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weng CY, Barnett D. Nanophthalmos-retinitis pigmentosa-foveoschisis-optic disc drusen syndrome (MERP). Ophthalmol Retina 2018; 2:1162. [DOI] [PubMed] [Google Scholar]
- 51.Zenteno JC, Buentello-Volante B, Quiroz-Gonzalez MA, Quiroz-Reyes MA. Compound heterozygosity for a novel and a recurrent MFRP gene mutation in a family with the nanophthalmos-retinitis pigmentosa complex. Mol Vis 2009; 15:1794–1798. [PMC free article] [PubMed] [Google Scholar]
- 52.Crespi J, Buil JA, Bassaganyas F, et al. A novel mutation confirms MFRP as the gene causing the syndrome of nanophthalmos-renititis pigmentosa-foveoschisis-optic disk drusen. Am J Ophthalmol 2008; 146:323–328. [DOI] [PubMed] [Google Scholar]
- 53.Ayala-Ramirez R, Graue-Wiechers F, Robredo V, et al. A new autosomal recessive syndrome consisting of posterior microphthalmos, retinitis pigmentosa, foveoschisis, and optic disc drusen is caused by a MFRP gene mutation. Mol Vis 2006; 12:1483–1489. [PubMed] [Google Scholar]
- 54.Hettinga YM, van Genderen MM, Wieringa W, et al. Retinal dystrophy in 6 young patients who presented with intermediate uveitis. Ophthalmology 2016; 123:2043–2046. [DOI] [PubMed] [Google Scholar]
- 55.Verhagen F, Kuiper J, Nierkens S, et al. Systemic inflammatory immune signatures in a patient with CRB1 linked retinal dystrophy. Expert Rev Clin Immunol 2016; 12:1359–1362. [DOI] [PubMed] [Google Scholar]
- 56.Bax NM, Lambertus S, Cremers FPM, et al. The absence of fundus abnormalities in Stargardt disease. Graefes Arch Clin Exp Ophthalmol 2019; 257:1147–1157. [DOI] [PubMed] [Google Scholar]
- 57.Benson MD, MacDonald IM. Bilateral uveitis and Usher syndrome: a case report. J Med Case Rep 2015; 9:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heredia CD, Huguet J, Cols N, et al. Immune complexes in retinitis pigmentosa. Br J Ophthalmol 1984; 68:811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reid DM, Campbell AM, Forrester JV. EB-virus transformed human lymphocytes from uveitis and retinitis pigmentosa patients secrete antibodies to retinal antigens. J Clin Lab Immunol 1988; 26:107–111. [PubMed] [Google Scholar]
- 60.Demirci FY, Rigatti BW, Mah TS, Gorin MB. A novel RPGR exon ORF15 mutation in a family with X-linked retinitis pigmentosa and Coats’-like exudative vasculopathy. Am J Ophthalmol 2006; 141:208–210. [DOI] [PubMed] [Google Scholar]
- 61.Patil L, Lotery AJ. Coat's-like exudation in rhodopsin retinitis pigmentosa: successful treatment with an intravitreal dexamethasone implant. Eye (Lond) 2014; 28:449–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jain S, Gupta S, Kumar V. Ultra-widefield imaging in Coats’-type retinitis pigmentosa. Indian J Ophthalmol 2018; 66:997–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang Y, Lim J, Janowicz M. Cholesterol crystals secondary to Coats-like response with retinitis pigmentosa. JAMA Ophthalmol 2017; 135:e173132. [DOI] [PubMed] [Google Scholar]
- 64.Ghassemi F, Akbari-Kamrani M. Retinitis pigmentosa associated with vasoproliferative tumors and Coats-like fundus. J Ophthalmic Vis Res 2013; 8:268–270. [PMC free article] [PubMed] [Google Scholar]
- 65.Pruett RC. Retinitis pigmentosa: clinical observations and correlations. Trans Am Ophthalmol Soc 1983; 81:693–735. [PMC free article] [PubMed] [Google Scholar]
- 66.Urgancioglu B, Ozdek S, Hasanreisoglu B. Coats’-like retinitis pigmentosa variant and nanophthalmos. Can J Ophthalmol 2007; 42:877–878. [DOI] [PubMed] [Google Scholar]
- 67.Murthy R, Honavar SG. Secondary vasoproliferative retinal tumor associated with Usher syndrome type 1. J AAPOS 2009; 13:97–98. [DOI] [PubMed] [Google Scholar]
- 68.Kiratli H, Ozturkmen C. Coats-like lesions in Usher syndrome type II. Graefes Arch Clin Exp Ophthalmol 2004; 242:265–267. [DOI] [PubMed] [Google Scholar]
- 69.De Salvo G, Gemenetzi M, Luff AJ, Lotery AJ. Cystoid macular oedema successfully treated by cryotherapy in retinitis pigmentosa with Coats’-like retinal exudation. Eye (Lond) 2011; 25:821–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bansal S, Saha N, Woon WH. The management of “coats’ response” in a patient with x-linked retinitis pigmentosa-a case report. ISRN Surg 2011; 2011:970361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van de Pavert SA, Sanz AS, Aartsen WM, et al. Crb1 is a determinant of retinal apical Muller glia cell features. Glia 2007; 55:1486–1497. [DOI] [PubMed] [Google Scholar]
- 72.Tout S, Chan-Ling T, Hollander H, Stone J. The role of Muller cells in the formation of the blood-retinal barrier. Neuroscience 1993; 55:291–301. [DOI] [PubMed] [Google Scholar]
- 73.Al Sulaiman H, Schatz P, Nowilaty SR, et al. Diffuse retinal vascular leakage and cone-rod dystrophy in a family with the homozygous missense C.1429G>A (PGLY477ARG) mutation in CRB1. Retin Cases Brief Rep 2020; 14:203–210. [DOI] [PubMed] [Google Scholar]
- 74.Shah N, Damani MR, Zhu XS, et al. Isolated maculopathy associated with biallelic CRB1 mutations. Ophthalmic Genet 2017; 38:190–193. [DOI] [PubMed] [Google Scholar]
- 75.Khan AO, Aldahmesh MA, Abu-Safieh L, Alkuraya FS. Childhood cone-rod dystrophy with macular cystic degeneration from recessive CRB1 mutation. Ophthalmic Genet 2014; 35:130–137. [DOI] [PubMed] [Google Scholar]
- 76.Bujakowska K, Audo I, Mohand-Said S, et al. CRB1 mutations in inherited retinal dystrophies. Hum Mutat 2012; 33:306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vincent A, Ng J, Gerth-Kahlert C, et al. Biallelic mutations in CRB1 underlie autosomal recessive familial foveal retinoschisis. Invest Ophthalmol Vis Sci 2016; 57:2637–2646. [DOI] [PubMed] [Google Scholar]
- 78.Pellissier LP, Alves CH, Quinn PM, et al. Targeted ablation of CRB1 and CRB2 in retinal progenitor cells mimics Leber congenital amaurosis. PLoS Genet 2013; 9:e1003976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Quinn PM, Alves CH, Klooster J, Wijnholds J. CRB2 in immature photoreceptors determines the superior-inferior symmetry of the developing retina to maintain retinal structure and function. Hum Mol Genet 2018; 27:3137–3153. [DOI] [PubMed] [Google Scholar]
- 80.Aleman TS, Cideciyan AV, Sumaroka A, et al. Inner retinal abnormalities in X-linked retinitis pigmentosa with RPGR mutations. Invest Ophthalmol Vis Sci 2007; 48:4759–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aleman TS, Cideciyan AV, Sumaroka A, et al. Retinal laminar architecture in human retinitis pigmentosa caused by Rhodopsin gene mutations. Invest Ophthalmol Vis Sci 2008; 49:1580–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alves CH, Pellissier LP, Wijnholds J. The CRB1 and adherens junction complex proteins in retinal development and maintenance. Prog Retin Eye Res 2014; 40:35–52. [DOI] [PubMed] [Google Scholar]
- 83.Spaide RF, Curcio CA. Anatomical correlates to the bands seen in the outer retina by optical coherence tomography: literature review and model. Retina 2011; 31:1609–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McKay GJ, Clarke S, Davis JA, et al. Pigmented paravenous chorioretinal atrophy is associated with a mutation within the crumbs homolog 1 (CRB1) gene. Invest Ophthalmol Vis Sci 2005; 46:322–328. [DOI] [PubMed] [Google Scholar]
- 85.Toto L, Boon CJ, Di Antonio L, et al. BESTROPHINOPATHY: a spectrum of ocular abnormalities caused by the c.614T>C mutation in the BEST1 gene. Retina 2016; 36:1586–1595. [DOI] [PubMed] [Google Scholar]
- 86.Son S, Cho M, Lee J. Crumbs proteins regulate layered retinal vascular development required for vision. Biochem Biophys Res Commun 2020; 521:939–946. [DOI] [PubMed] [Google Scholar]
- 87.Reichenbach A, Bringmann A. Glia of the human retina. Glia 2020; 68:768–796. [DOI] [PubMed] [Google Scholar]
- 88.Tepass U, Theres C. Knust E. crumbs encodes an EGF-like protein expressed on apical membranes of Drosophila epithelial cells and required for organization of epithelia. Cell 1990; 61:787–799. [DOI] [PubMed] [Google Scholar]
- 89.Khan KN, Robson A, Mahroo OAR, et al. A clinical and molecular characterisation of CRB1-associated maculopathy. Eur J Hum Genet 2018; 26:687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van Soest S, van den Born LI, Gal A, et al. Assignment of a gene for autosomal recessive retinitis pigmentosa (RP12) to chromosome 1q31-q32.1 in an inbred and genetically heterogeneous disease population. Genomics 1994; 22:499–504. [DOI] [PubMed] [Google Scholar]
- 91.van den Born LI, van Soest S, van Schooneveld MJ, et al. Autosomal recessive retinitis pigmentosa with preserved para-arteriolar retinal pigment epithelium. Am J Ophthalmol 1994; 118:430–439. [DOI] [PubMed] [Google Scholar]
- 92.Mathijssen IB, Henneman L, van Eeten-Nijman JM, et al. Targeted carrier screening for four recessive disorders: high detection rate within a founder population. Eur J Med Genet 2015; 58:123–128. [DOI] [PubMed] [Google Scholar]
- 93.Ebermann I, Phillips JB, Liebau MC, et al. PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J Clin Invest 2010; 120:1812–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Khanna H, Davis EE, Murga-Zamalloa CA, et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet 2009; 41:739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li M, Jia C, Kazmierkiewicz KL, et al. Comprehensive analysis of gene expression in human retina and supporting tissues. Hum Mol Genet 2014; 23:4001–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Markand S, Saul A, Tawfik A, et al. Mthfr as a modifier of the retinal phenotype of Crb1(rd8/rd8) mice. Exp Eye Res 2016; 145:164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thompson DA, Li Y, McHenry CL, et al. Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat Genet 2001; 28:123–124. [DOI] [PubMed] [Google Scholar]
- 98.Senechal A, Humbert G, Surget MO, et al. Screening genes of the retinoid metabolism: novel LRAT mutation in leber congenital amaurosis. Am J Ophthalmol 2006; 142:702–704. [DOI] [PubMed] [Google Scholar]
- 99.Littink KW, van Genderen MM, van Schooneveld MJ, et al. A homozygous frameshift mutation in LRAT causes retinitis punctata albescens. Ophthalmology 2012; 119:1899–1906. [DOI] [PubMed] [Google Scholar]
- 100.Dev Borman A, Ocaka LA, Mackay DS, et al. Early onset retinal dystrophy due to mutations in LRAT: molecular analysis and detailed phenotypic study. Invest Ophthalmol Vis Sci 2012; 53:3927–3938. [DOI] [PubMed] [Google Scholar]
- 101.Talib M, van Schooneveld MJ, van Duuren RJG, et al. Long-term follow-up of retinal degenerations associated with LRAT mutations and their comparability to phenotypes associated with RPE65 mutations. Transl Vis Sci Technol 2019; 8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koenekoop RK, Sui R, Sallum J, et al. Oral 9-cis retinoid for childhood blindness due to Leber congenital amaurosis caused by RPE65 or LRAT mutations: an open-label phase 1b trial. Lancet 2014; 384:1513–1520. [DOI] [PubMed] [Google Scholar]
- 103.Scholl HP, Moore AT, Koenekoop RK, et al. Safety and proof-of-concept study of oral QLT091001 in retinitis pigmentosa due to inherited deficiencies of retinal pigment epithelial 65 protein (RPE65) or lecithin:retinol acyltransferase (LRAT). PLoS One 2015; 10:e0143846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Thiadens AA, Phan TM, Zekveld-Vroon RC, et al. Clinical course, genetic etiology, and visual outcome in cone and cone-rod dystrophy. Ophthalmology 2012; 119:819–826. [DOI] [PubMed] [Google Scholar]
- 105.Wycisk KA, Zeitz C, Feil S, et al. Mutation in the auxiliary calcium-channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am J Hum Genet 2006; 79:973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Michaelides M, Aligianis IA, Ainsworth JR, et al. Progressive cone dystrophy associated with mutation in CNGB3. Invest Ophthalmol Vis Sci 2004; 45:1975–1982. [DOI] [PubMed] [Google Scholar]
- 107.Demirci FY, Rigatti BW, Wen G, et al. X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am J Hum Genet 2002; 70:1049–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang Z, Peachey NS, Moshfeghi DM, et al. Mutations in the RPGR gene cause X-linked cone dystrophy. Hum Mol Genet 2002; 11:605–611. [DOI] [PubMed] [Google Scholar]
- 109.Michaelides M, Hunt DM, Moore AT. The cone dysfunction syndromes. Br J Ophthalmol 2004; 88:291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rahman N, Georgiou M, Khan KN, Michaelides M. Macular dystrophies: clinical and imaging features, molecular genetics and therapeutic options. Br J Ophthalmol 2020; 104:451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cehajic Kapetanovic J, McClements ME, Martinez-Fernandez de la Camara C, MacLaren RE. Molecular strategies for rpgr gene therapy. Genes (Basel) 2019; 10:674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Giacalone JC, Andorf JL, Zhang Q, et al. Development of a molecularly stable gene therapy vector for the treatment of RPGR-associated X-linked retinitis pigmentosa. Hum Gene Ther 2019; 30:967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Talib M, van Schooneveld MJ, Thiadens AA, et al. Clinical and genetic characteristics of male patients with RPGR-associated retinal dystrophies: a long-term follow-up study. Retina 2019; 39:1186–1199. [DOI] [PubMed] [Google Scholar]
- 114.Ebenezer ND, Michaelides M, Jenkins SA, et al. Identification of novel RPGR ORF15 mutations in X-linked progressive cone-rod dystrophy (XLCORD) families. Invest Ophthalmol Vis Sci 2005; 46:1891–1898. [DOI] [PubMed] [Google Scholar]
- 115.Zahid S, Khan N, Branham K, et al. Phenotypic conservation in patients with X-linked retinitis pigmentosa caused by RPGR mutations. JAMA Ophthalmol 2013; 131:1016–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Thiadens AA, Soerjoesing GG, Florijn RJ, et al. Clinical course of cone dystrophy caused by mutations in the RPGR gene. Graefes Arch Clin Exp Ophthalmol 2011; 249:1527–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kurata K, Hosono K, Hayashi T, et al. X-linked retinitis pigmentosa in Japan: clinical and genetic findings in male patients and female carriers. Int J Mol Sci 2019; 20:1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Walia S, Fishman GA, Swaroop A, et al. Discordant phenotypes in fraternal twins having an identical mutation in exon ORF15 of the RPGR gene. Arch Ophthalmol 2008; 126:379–384. [DOI] [PubMed] [Google Scholar]
- 119.Ruddle JB, Ebenezer ND, Kearns LS, et al. RPGR ORF15 genotype and clinical variability of retinal degeneration in an Australian population. Br J Ophthalmol 2009; 93:1151–1154. [DOI] [PubMed] [Google Scholar]
- 120.Parmeggiani F, Barbaro V, De Nadai K, et al. Identification of novel X-linked gain-of-function RPGR-ORF15 mutation in Italian family with retinitis pigmentosa and pathologic myopia. Sci Rep 2016; 6:39179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Koenekoop RK, Loyer M, Hand CK, et al. Novel RPGR mutations with distinct retinitis pigmentosa phenotypes in French-Canadian families. Am J Ophthalmol 2003; 136:678–687. [DOI] [PubMed] [Google Scholar]
- 122.Littink KW, Pott J-WR, Collin RWJ, et al. A novel nonsense mutation in CEP290 induces exon skipping and leads to a relatively mild retinal phenotype. Invest Ophthalmol Vis Sci 2010; 51:3646–3652. [DOI] [PubMed] [Google Scholar]
- 123.Walia S, Fishman GA, Jacobson SG, et al. Visual acuity in patients with Leber's congenital amaurosis and early childhood-onset retinitis pigmentosa. Ophthalmology 2010; 117:1190–1198. [DOI] [PubMed] [Google Scholar]
- 124.Riddell N, Faou P, Murphy M, et al. The retina/RPE proteome in chick myopia and hyperopia models: commonalities with inherited and age-related ocular pathologies. Mol Vis 2017; 23:872–888. [PMC free article] [PubMed] [Google Scholar]
- 125.Talib M, van Schooneveld MJ, Van Cauwenbergh C, et al. The spectrum of structural and functional abnormalities in female carriers of pathogenic variants in the RPGR gene. Invest Ophthalmol Vis Sci 2018; 59:4123–4133. [DOI] [PubMed] [Google Scholar]
- 126.Kousal B, Skalicka P, Valesova L, et al. Severe retinal degeneration in women with a c.2543del mutation in ORF15 of the RPGR gene. Mol Vis 2014; 20:1307–1317. [PMC free article] [PubMed] [Google Scholar]
- 127.Rozet JM, Perrault I, Gigarel N, et al. Dominant X linked retinitis pigmentosa is frequently accounted for by truncating mutations in exon ORF15 of the RPGR gene. J Med Genet 2002; 39:284–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Al-Maskari A, O’Grady A, Pal B, McKibbin M. Phenotypic progression in X-linked retinitis pigmentosa secondary to a novel mutation in the RPGR gene. Eye (Lond) 2009; 23:519–521. [DOI] [PubMed] [Google Scholar]
- 129.Jacobson SG, Buraczynska M, Milam AH, et al. Disease expression in X-linked retinitis pigmentosa caused by a putative null mutation in the RPGR gene. Invest Ophthalmol Vis Sci 1997; 38:1983–1997. [PubMed] [Google Scholar]
- 130.Birtel J, Gliem M, Mangold E, et al. Next-generation sequencing identifies unexpected genotype-phenotype correlations in patients with retinitis pigmentosa. PLoS One 2018; 13:e0207958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lyon MF. X-chromosome inactivation and human genetic disease. Acta Paediatr Suppl 2002; 91:107–112. [DOI] [PubMed] [Google Scholar]
- 132.Plenge RM, Hendrich BD, Schwartz C, et al. A promoter mutation in the XIST gene in two unrelated families with skewed X-chromosome inactivation. Nat Genet 1997; 17:353–356. [DOI] [PubMed] [Google Scholar]
- 133.Plenge RM, Tranebjaerg L, Jensen PK, et al. Evidence that mutations in the X-linked DDP gene cause incompletely penetrant and variable skewed X inactivation. Am J Hum Genet 1999; 64:759–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Syed R, Sundquist SM, Ratnam K, et al. High-resolution images of retinal structure in patients with choroideremia. Invest Ophthalmol Vis Sci 2013; 54:950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Andreasson S, Breuer DK, Eksandh L, et al. Clinical studies of X-linked retinitis pigmentosa in three Swedish families with newly identified mutations in the RP2 and RPGR-ORF15 genes. Ophthalmic Genet 2003; 24:215–223. [DOI] [PubMed] [Google Scholar]
- 136.Yang L, Yin X, Feng L, et al. Novel mutations of RPGR in Chinese retinitis pigmentosa patients and the genotype-phenotype correlation. PLoS One 2014; 9:e85752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fahim AT, Bowne SJ, Sullivan LS, et al. Allelic heterogeneity and genetic modifier loci contribute to clinical variation in males with X-linked retinitis pigmentosa due to RPGR mutations. PLoS One 2011; 6:e23021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Comander J, Weigel-DiFranco C, Sandberg MA, Berson EL. Visual function in carriers of X-linked retinitis pigmentosa. Ophthalmology 2015; 122:1899–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tolmachova T, Anders R, Abrink M, et al. Independent degeneration of photoreceptors and retinal pigment epithelium in conditional knockout mouse models of choroideremia. J Clin Invest 2006; 116:386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Edwards TL, Jolly JK, Groppe M, et al. Visual acuity after retinal gene therapy for choroideremia. N Engl J Med 2016; 374:1996–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Xue K, Jolly JK, Barnard AR, et al. Beneficial effects on vision in patients undergoing retinal gene therapy for choroideremia. Nat Med 2018; 24:1507–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dimopoulos IS, Hoang SC, Radziwon A, et al. Two-year results after AAV2-mediated gene therapy for choroideremia: the Alberta experience. Am J Ophthalmol 2018; 193:130–142. [DOI] [PubMed] [Google Scholar]
- 143.Lam BL, Davis JL, Gregori NZ, et al. Choroideremia gene therapy phase 2 clinical trial: 24-month results. Am J Ophthalmol 2019; 197:65–73. [DOI] [PubMed] [Google Scholar]
- 144.Fischer MD, Ochakovski GA, Beier B, et al. Changes in retinal sensitivity after gene therapy in choroideremia. Retina 2020; 40:160–168. [DOI] [PubMed] [Google Scholar]
- 145.van Schuppen SM, Talib M, Bergen AA, et al. Long-Term follow-up of patients with choroideremia with scleral pits and tunnels as a novel observation. Retina 2018; 38:1713–1724. [DOI] [PubMed] [Google Scholar]
- 146.Hariri AH, Velaga SB, Girach A, et al. Measurement and reproducibility of preserved ellipsoid zone area and preserved retinal pigment epithelium area in eyes with choroideremia. Am J Ophthalmol 2017; 179:110–117. [DOI] [PubMed] [Google Scholar]
- 147.Hariri AH, Ip MS, Girach A, et al. Macular spatial distribution of preserved autofluorescence in patients with choroideremia. Br J Ophthalmol 2019; 103:933–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jolly JK, Xue K, Edwards TL, et al. Characterizing the natural history of visual function in choroideremia using microperimetry and multimodal retinal imaging. Invest Ophthalmol Vis Sci 2017; 58:5575–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Westeneng-van Haaften SC, Boon CJ, Cremers FP, et al. Clinical and genetic characteristics of late-onset Stargardt's disease. Ophthalmology 2012; 119:1199–1210. [DOI] [PubMed] [Google Scholar]
- 150.Bax NM, Valkenburg D, Lambertus S, et al. Foveal sparing in central retinal dystrophies. Invest Ophthalmol Vis Sci 2019; 60:3456–3467. [DOI] [PubMed] [Google Scholar]
- 151.Bird AC, Bok D. Why the macula? Eye (Lond) 2018; 32:858–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Goldberg NR, Greenberg JP, Laud K, et al. Outer retinal tubulation in degenerative retinal disorders. Retina 2013; 33:1871–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Aleman TS, Han G, Serrano LW, et al. Natural history of the central structural abnormalities in choroideremia: a prospective cross-sectional study. Ophthalmology 2017; 124:359–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sun LW, Johnson RD, Williams V, et al. Multimodal imaging of photoreceptor structure in choroideremia. PLoS One 2016; 11:e0167526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Xue K, Oldani M, Jolly JK, et al. Correlation of optical coherence tomography and autofluorescence in the outer retina and choroid of patients with choroideremia. Invest Ophthalmol Vis Sci 2016; 57:3674–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Charng J, Cideciyan AV, Jacobson SG, et al. Variegated yet non-random rod and cone photoreceptor disease patterns in RPGR-ORF15-associated retinal degeneration. Hum Mol Genet 2016; 25:5444–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jain N, Jia Y, Gao SS, et al. Optical coherence tomography angiography in choroideremia: correlating choriocapillaris loss with overlying degeneration. JAMA Ophthalmol 2016; 134:697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tuten WS, Vergilio GK, Young GJ, et al. Visual function at the atrophic border in choroideremia assessed with adaptive optics microperimetry. Ophthalmol Retina 2019; 3:888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zweifel SA, Engelbert M, Laud K, et al. Outer retinal tubulation: a novel optical coherence tomography finding. Arch Ophthalmol 2009; 127:1596–1602. [DOI] [PubMed] [Google Scholar]
- 160.Zinkernagel MS, Groppe M, MacLaren RE. Macular hole surgery in patients with end-stage choroideremia. Ophthalmology 2013; 120:1592–1596. [DOI] [PubMed] [Google Scholar]
- 161.Talib M, Koetsier LS, MacLaren RE, Boon CJF. Outcome of full-thickness macular hole surgery in choroideremia. Genes (Basel) 2017; 8:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Heon E, Alabduljalil T, Iii DB, et al. Visual function and central retinal structure in choroideremia. Invest Ophthalmol Vis Sci 2016; 57:377–387. [DOI] [PubMed] [Google Scholar]
- 163.Ferrari S, Di Iorio E, Barbaro V, et al. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics 2011; 12:238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cideciyan AV, Sudharsan R, Dufour VL, et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc Natl Acad Sci U S A 2018; 115:E8547–E8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tsai YT, Wu WH, Lee TT, et al. Clustered regularly interspaced short palindromic repeats-based genome surgery for the treatment of autosomal dominant retinitis pigmentosa. Ophthalmology 2018; 125:1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Giannelli SG, Luoni M, Castoldi V, et al. Cas9/sgRNA selective targeting of the P23H Rhodopsin mutant allele for treating retinitis pigmentosa by intravitreal AAV9.PHP.B-based delivery. Hum Mol Genet 2018; 27:761–779. [DOI] [PubMed] [Google Scholar]
- 167.Murray SF, Jazayeri A, Matthes MT, et al. Allele-specific inhibition of rhodopsin with an antisense oligonucleotide slows photoreceptor cell degeneration. Invest Ophthalmol Vis Sci 2015; 56:6362–6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mao H, James T, Jr, Schwein A, et al. AAV delivery of wild-type rhodopsin preserves retinal function in a mouse model of autosomal dominant retinitis pigmentosa. Hum Gene Ther 2011; 22:567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ramon E, Cordomi A, Aguila M, et al. Differential light-induced responses in sectorial inherited retinal degeneration. J Biol Chem 2014; 289:35918–35928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sumaroka A, Cideciyan AV, Charng J, et al. Autosomal dominant retinitis pigmentosa due to class b rhodopsin mutations: an objective outcome for future treatment trials. Int J Mol Sci 2019; 20:E5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wang J, Xu D, Zhu T, et al. Identification of two novel RHO mutations in Chinese retinitis pigmentosa patients. Exp Eye Res 2019; 188:107726. [DOI] [PubMed] [Google Scholar]
- 172.Roshandel D, Rafati M, Khorami S, et al. Rhodopsin gene mutation analysis in Iranian patients with autosomal dominant retinitis pigmentosa. Int Ophthalmol 2019; 39:2523–2531. [DOI] [PubMed] [Google Scholar]
- 173.Coussa RG, Basali D, Maeda A, et al. Sector retinitis pigmentosa: report of ten cases and a review of the literature. Mol Vis 2019; 25:869–889. [PMC free article] [PubMed] [Google Scholar]
- 174.Jacobson SG, McGuigan DB, 3rd, Sumaroka A, et al. Complexity of the class B phenotype in autosomal dominant retinitis pigmentosa due to rhodopsin mutations. Invest Ophthalmol Vis Sci 2016; 57:4847–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cideciyan AV, Hood DC, Huang Y, et al. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci U S A 1998; 95:7103–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Paskowitz DM, LaVail MM, Duncan JL. Light and inherited retinal degeneration. Br J Ophthalmol 2006; 90:1060–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Athanasiou D, Aguila M, Bellingham J, et al. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog Retin Eye Res 2018; 62:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Naash ML, Peachey NS, Li ZY, et al. Light-induced acceleration of photoreceptor degeneration in transgenic mice expressing mutant rhodopsin. Invest Ophthalmol Vis Sci 1996; 37:775–782. [PubMed] [Google Scholar]
- 179.Organisciak DT, Darrow RM, Barsalou L, et al. Susceptibility to retinal light damage in transgenic rats with rhodopsin mutations. Invest Ophthalmol Vis Sci 2003; 44:486–492. [DOI] [PubMed] [Google Scholar]
- 180.Iwabe S, Ying GS, Aguirre GD, Beltran WA. Assessment of visual function and retinal structure following acute light exposure in the light sensitive T4R rhodopsin mutant dog. Exp Eye Res 2016; 146:341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Orlans HO, Merrill J, Barnard AR, et al. Filtration of short-wavelength light provides therapeutic benefit in retinitis pigmentosa caused by a common rhodopsin mutation. Invest Ophthalmol Vis Sci 2019; 60:2733–2742. [DOI] [PubMed] [Google Scholar]
- 182.Dryja TP, McGee TL, Reichel E, et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 1990; 343:364–366. [DOI] [PubMed] [Google Scholar]
- 183.Oh KT, Oh DM, Weleber RG, et al. Genotype-phenotype correlation in a family with Arg135Leu rhodopsin retinitis pigmentosa. Br J Ophthalmol 2004; 88:1533–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Shah SP, Wong F, Sharp DM, Vincent AL. A novel rhodopsin point mutation, proline-170-histidine, associated with sectoral retinitis pigmentosa. Ophthalmic Genet 2014; 35:241–247. [DOI] [PubMed] [Google Scholar]
- 185.Napier ML, Durga D, Wolsley CJ, et al. Mutational analysis of the rhodopsin gene in sector retinitis pigmentosa. Ophthalmic Genet 2015; 36:239–243. [DOI] [PubMed] [Google Scholar]
- 186.Abdulridha-Aboud W, Kjellstrom U, Andreasson S, Ponjavic V. Characterization of macular structure and function in two Swedish families with genetically identified autosomal dominant retinitis pigmentosa. Mol Vis 2016; 22:362–373. [PMC free article] [PubMed] [Google Scholar]
- 187.Reiff C, Owczarek-Lipska M, Spital G, et al. The mutation p.E113K in the Schiff base counterion of rhodopsin is associated with two distinct retinal phenotypes within the same family. Sci Rep 2016; 6:36208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Liu YP, Bosch DG, Siemiatkowska AM, et al. Putative digenic inheritance of heterozygous RP1L1 and C2orf71 null mutations in syndromic retinal dystrophy. Ophthalmic Genet 2017; 38:127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Dryja TP, Hahn LB, Kajiwara K, Berson EL. Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Invest Ophthalmol Vis Sci 1997; 38:1972–1982. [PubMed] [Google Scholar]
- 190.Kariminejad A, Bozorgmehr B, Najafi A, et al. Retinitis pigmentosa, cutis laxa, and pseudoxanthoma elasticum-like skin manifestations associated with GGCX mutations. J Invest Dermatol 2014; 134:2331–2338. [DOI] [PubMed] [Google Scholar]
- 191.Ho AC, Humayun MS, Dorn JD, et al. Long-term results from an epiretinal prosthesis to restore sight to the blind. Ophthalmology 2015; 122:1547–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Edwards TL, Cottriall CL, Xue K, et al. Assessment of the electronic retinal implant alpha AMS in restoring vision to blind patients with end-stage retinitis pigmentosa. Ophthalmology 2018; 125:432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Stronks HC, Dagnelie G. The functional performance of the Argus II retinal prosthesis. Expert Rev Med Devices 2014; 11:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ahuja AK, Behrend MR. The Argus II retinal prosthesis: factors affecting patient selection for implantation. Prog Retin Eye Res 2013; 36:1–23. [DOI] [PubMed] [Google Scholar]
- 195.da Cruz L, Coley BF, Dorn J, et al. The Argus II epiretinal prosthesis system allows letter and word reading and long-term function in patients with profound vision loss. Br J Ophthalmol 2013; 97:632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Farvardin M, Afarid M, Attarzadeh A, et al. The Argus-II retinal prosthesis implantation; from the global to local successful experience. Front Neurosci 2018; 12:584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Veritti D, Sarao V, De Nadai K, et al. Dexamethasone implant produces better outcomes than oral acetazolamide in patients with cystoid macular edema secondary to retinitis pigmentosa. J Ocul Pharmacol Ther 2020; 36:190–197. [DOI] [PubMed] [Google Scholar]
- 198.Huang Q, Chen R, Lin X, Xiang Z. Efficacy of carbonic anhydrase inhibitors in management of cystoid macular edema in retinitis pigmentosa: a meta-analysis. PLoS One 2017; 12:e0186180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Cox SN, Hay E, Bird AC. Treatment of chronic macular edema with acetazolamide. Arch Ophthalmol 1988; 106:1190–1195. [DOI] [PubMed] [Google Scholar]
- 200.Genead MA, Fishman GA. Efficacy of sustained topical dorzolamide therapy for cystic macular lesions in patients with retinitis pigmentosa and usher syndrome. Arch Ophthalmol 2010; 128:1146–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ikeda Y, Hisatomi T, Yoshida N, et al. The clinical efficacy of a topical dorzolamide in the management of cystoid macular edema in patients with retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol 2012; 250:809–814. [DOI] [PubMed] [Google Scholar]
- 202.Liew G, Moore AT, Webster AR, Michaelides M. Efficacy and prognostic factors of response to carbonic anhydrase inhibitors in management of cystoid macular edema in retinitis pigmentosa. Invest Ophthalmol Vis Sci 2015; 56:1531–1536. [DOI] [PubMed] [Google Scholar]
- 203.Strong SA, Hirji N, Quartilho A, et al. Retrospective cohort study exploring whether an association exists between spatial distribution of cystoid spaces in cystoid macular oedema secondary to retinitis pigmentosa and response to treatment with carbonic anhydrase inhibitors. Br J Ophthalmol 2019; 103:233–237. [DOI] [PubMed] [Google Scholar]
- 204.Srour M, Querques G, Leveziel N, et al. Intravitreal dexamethasone implant (Ozurdex) for macular edema secondary to retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol 2013; 251:1501–1506. [DOI] [PubMed] [Google Scholar]
- 205.Ozdemir H, Karacorlu M, Karacorlu S. Intravitreal triamcinolone acetonide for treatment of cystoid macular oedema in patients with retinitis pigmentosa. Acta Ophthalmol Scand 2005; 83:248–251. [DOI] [PubMed] [Google Scholar]
- 206.Scorolli L, Morara M, Meduri A, et al. Treatment of cystoid macular edema in retinitis pigmentosa with intravitreal triamcinolone. Arch Ophthalmol 2007; 125:759–764. [DOI] [PubMed] [Google Scholar]
- 207.Yuzbasioglu E, Artunay O, Rasier R, et al. Intravitreal bevacizumab (Avastin) injection in retinitis pigmentosa. Curr Eye Res 2009; 34:231–237. [DOI] [PubMed] [Google Scholar]
- 208.Artunay O, Yuzbasioglu E, Rasier R, et al. Intravitreal ranibizumab in the treatment of cystoid macular edema associated with retinitis pigmentosa. J Ocul Pharmacol Ther 2009; 25:545–550. [DOI] [PubMed] [Google Scholar]
- 209.Melo GB, Farah ME, Aggio FB. Intravitreal injection of bevacizumab for cystoid macular edema in retinitis pigmentosa. Acta Ophthalmol Scand 2007; 85:461–463. [DOI] [PubMed] [Google Scholar]
- 210.Ediriwickrema LS, Chhadva P, Rodger DC, et al. Intravenous immunoglobulin in the treatment of juvenile retinitis pigmentosa-associated cystoid macular edema and uveitis. Retin Cases Brief Rep 2018; 12:242–246. [DOI] [PubMed] [Google Scholar]
- 211.Missotten T, van Laar JA, van der Loos TL, et al. Octreotide long-acting repeatable for the treatment of chronic macular edema in uveitis. Am J Ophthalmol 2007; 144:838–843. [DOI] [PubMed] [Google Scholar]
- 212.Hogewind BF, Pieters G, Hoyng CB. Octreotide acetate in dominant cystoid macular dystrophy. Eur J Ophthalmol 2008; 18:99–103. [DOI] [PubMed] [Google Scholar]
- 213.Sarao V, Veritti D, Prosperi R, et al. A case of CRB1-negative Coats-like retinitis pigmentosa. J AAPOS 2013; 17:414–416. [DOI] [PubMed] [Google Scholar]
- 214.Kan E, Yilmaz T, Aydemir O, et al. Coats-like retinitis pigmentosa: reports of three cases. Clin Ophthalmol 2007; 1:193–198. [PMC free article] [PubMed] [Google Scholar]
- 215.Lee SY, Yoon YH. Pars plana vitrectomy for exuduative retinal detachment in coats-type retinitis pigmentosa. Retina 2004; 24:450–452. [DOI] [PubMed] [Google Scholar]
- 216.Chu X, Du W, Xu M, et al. Intravitreal conbercept combined with laser photocoagulation for exudative retinal detachment in a patient with Coats-like retinitis pigmentosa. Ophthalmic Genet 2019; 40:581–583. [DOI] [PubMed] [Google Scholar]
- 217.Xue K, Groppe M, Salvetti AP, MacLaren RE. Technique of retinal gene therapy: delivery of viral vector into the subretinal space. Eye (Lond) 2017; 31:1308–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Tang Z, Zhang Y, Wang Y, et al. Progress of stem/progenitor cell-based therapy for retinal degeneration. J Transl Med 2017; 15:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Schwartz SD, Regillo CD, Lam BL, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt's macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet 2015; 385:509–516. [DOI] [PubMed] [Google Scholar]
- 220.Singh MS, Park SS, Albini TA, et al. Retinal stem cell transplantation: balancing safety and potential. Prog Retin Eye Res 2019; 75:100779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Burnight ER, Gupta M, Wiley LA, et al. Using CRISPR-Cas9 to generate gene-corrected autologous iPSCs for the treatment of inherited retinal degeneration. Mol Ther 2017; 25:1999–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Bainbridge J, Mehat M, Sundaram V, et al. Long-term effect of gene therapy on Leber's congenital amaurosis. N Engl J Med 2015; 372:1887–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Jacobson SG, Cideciyan AV, Roman AJ, et al. Improvement and decline in vision with gene therapy in childhood blindness. N Engl J Med 2015; 372:1920–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Dalkara D, Byrne LC, Klimczak RR, et al. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med 2013; 5:189ra76. [DOI] [PubMed] [Google Scholar]
- 225.Reichel FF, Peters T, Wilhelm B, et al. Humoral immune response after intravitreal but not after subretinal AAV8 in primates and patients. Invest Ophthalmol Vis Sci 2018; 59:1910–1915. [DOI] [PubMed] [Google Scholar]
- 226.Seitz IP, Michalakis S, Wilhelm B, et al. Superior retinal gene transfer and biodistribution profile of subretinal versus intravitreal delivery of AAV8 in nonhuman primates. Invest Ophthalmol Vis Sci 2017; 58:5792–5801. [DOI] [PubMed] [Google Scholar]
- 227.Dias MS, Araujo VG, Vasconcelos T, et al. Retina transduction by rAAV2 after intravitreal injection: comparison between mouse and rat. Gene Ther 2019; 26:479–490. [DOI] [PubMed] [Google Scholar]
- 228.Bellingrath JS, Ochakovski GA, Seitz IP, et al. High symmetry of visual acuity and visual fields in RPGR-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci 2017; 58:4457–4466. [DOI] [PubMed] [Google Scholar]
- 229.Tee JJL, Yang Y, Kalitzeos A, et al. Natural history study of retinal structure, progression, and symmetry using ellipzoid zone metrics in RPGR-associated retinopathy. Am J Ophthalmol 2019; 198:111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Tee JJL, Yang Y, Kalitzeos A, et al. Characterization of visual function, interocular variability and progression using static perimetry-derived metrics in RPGR-associated retinopathy. Invest Ophthalmol Vis Sci 2018; 59:2422–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Seitz IP, Zhour A, Kohl S, et al. Multimodal assessment of choroideremia patients defines pre-treatment characteristics. Graefes Arch Clin Exp Ophthalmol 2015; 253:2143–2150. [DOI] [PubMed] [Google Scholar]
- 232.Tee JJL, Carroll J, Webster AR, Michaelides M. Quantitative analysis of retinal structure using spectral-domain optical coherence tomography in RPGR-associated retinopathy. Am J Ophthalmol 2017; 178:18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Birch DG, Locke KG, Wen Y, et al. Spectral-domain optical coherence tomography measures of outer segment layer progression in patients with X-linked retinitis pigmentosa. JAMA Ophthalmol 2013; 131:1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Sujirakul T, Lin MK, Duong J, et al. Multimodal imaging of central retinal disease progression in a 2-year mean follow-up of retinitis pigmentosa. Am J Ophthalmol 2015; 160:786–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Takahashi VKL, Takiuti JT, Carvalho-Jr JRL, et al. Fundus autofluorescence and ellipsoid zone (EZ) line width can be an outcome measurement in RHO-associated autosomal dominant retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol 2019; 257:725–731. [DOI] [PubMed] [Google Scholar]
- 236.Csaky K, Ferris F, 3rd, Chew EY, et al. Report From the NEI/FDA endpoints workshop on age-related macular degeneration and inherited retinal diseases. Invest Ophthalmol Vis Sci 2017; 58:3456–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Maguire AM, Russell S, Wellman JA, et al. Efficacy, safety, and durability of voretigene neparvovec-rzyl in RPE65 mutation-associated inherited retinal dystrophy: results of Phase 1 and 3 trials. Ophthalmology 2019; 126:1273–1285. [DOI] [PubMed] [Google Scholar]
- 238.Roman AJ, Cideciyan AV, Aleman TS, Jacobson SG. Full-field stimulus testing (FST) to quantify visual perception in severely blind candidates for treatment trials. Physiol Meas 2007; 28:N51–N56. [DOI] [PubMed] [Google Scholar]
- 239.Collison FT, Fishman GA, McAnany JJ, et al. Psychophysical measurement of rod and cone thresholds in stargardt disease with full-field stimuli. Retina 2014; 34:1888–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Chung DC, McCague S, Yu ZF, et al. Novel mobility test to assess functional vision in patients with inherited retinal dystrophies. Clin Exp Ophthalmol 2018; 46:247–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Lombardi M, Zenouda A, Azoulay-Sebban L, et al. Correlation between visual function and performance of simulated daily living activities in glaucomatous patients. J Glaucoma 2018; 27:1017–1024. [DOI] [PubMed] [Google Scholar]
- 242.Hareendran S, Balakrishnan B, Sen D, et al. Adeno-associated virus (AAV) vectors in gene therapy: immune challenges and strategies to circumvent them. Rev Med Virol 2013; 23:399–413. [DOI] [PubMed] [Google Scholar]
- 243.Daya S, Berns KI. Gene therapy using adeno-associated virus vectors. Clin Microbiol Rev 2008; 21:583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Day TP, Byrne LC, Schaffer DV, Flannery JG. Advances in AAV vector development for gene therapy in the retina. Adv Exp Med Biol 2014; 801:687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Kumar M, Keller B, Makalou N, Sutton RE. Systematic determination of the packaging limit of lentiviral vectors. Hum Gene Ther 2001; 12:1893–1905. [DOI] [PubMed] [Google Scholar]
- 246.Sakuma T, Barry MA, Ikeda Y. Lentiviral vectors: basic to translational. Biochem J 2012; 443:603–618. [DOI] [PubMed] [Google Scholar]
- 247.Conley SM, Cai X, Naash MI. Nonviral ocular gene therapy: assessment and future directions. Curr Opin Mol Ther 2008; 10:456–463. [PMC free article] [PubMed] [Google Scholar]
- 248.Peeters L, Sanders NN, Braeckmans K, et al. Vitreous: a barrier to nonviral ocular gene therapy. Invest Ophthalmol Vis Sci 2005; 46:3553–3561. [DOI] [PubMed] [Google Scholar]
- 249.Liang Y, Fotiadis D, Maeda T, et al. Rhodopsin signaling and organization in heterozygote rhodopsin knockout mice. J Biol Chem 2004; 279:48189–48196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Farrar GJ, Kenna PF, Humphries P. On the genetics of retinitis pigmentosa and on mutation-independent approaches to therapeutic intervention. EMBO J 2002; 21:857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.O’Neill B, Millington-Ward S, O’Reilly M, et al. Ribozyme-based therapeutic approaches for autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 2000; 41:2863–2869. [PubMed] [Google Scholar]
- 252.Gorbatyuk M, Justilien V, Liu J, et al. Preservation of photoreceptor morphology and function in P23H rats using an allele independent ribozyme. Exp Eye Res 2007; 84:44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Kiang AS, Palfi A, Ader M, et al. Toward a gene therapy for dominant disease: validation of an RNA interference-based mutation-independent approach. Mol Ther 2005; 12:555–561. [DOI] [PubMed] [Google Scholar]
- 254.Kurz D, Ciulla TA. Novel approaches for retinal drug delivery. Ophthalmol Clin North Am 2002; 15:405–410. [DOI] [PubMed] [Google Scholar]
- 255.Garanto A, Chung DC, Duijkers L, et al. In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum Mol Genet 2016; 25:2552–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Rowe-Rendleman CL, Durazo SA, Kompella UB, et al. Drug and gene delivery to the back of the eye: from bench to bedside. Invest Ophthalmol Vis Sci 2014; 55:2714–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Cideciyan AV, Jacobson SG, Drack AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat Med 2019; 25:225–228. [DOI] [PubMed] [Google Scholar]
- 258.Suzuki K, Tsunekawa Y, Hernandez-Benitez R, et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016; 540:144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Vagni P, Perlini LE, Chenais NAL, et al. Gene Editing Preserves Visual Functions in a Mouse Model of Retinal Degeneration. Front Neurosci 2019; 13:945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Maeder ML, Stefanidakis M, Wilson CJ, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med 2019; 25:229–233. [DOI] [PubMed] [Google Scholar]
- 262.Latella MC, Di Salvo MT, Cocchiarella F, et al. In vivo editing of the human mutant rhodopsin gene by electroporation of plasmid-based CRISPR/Cas9 in the mouse retina. Mol Ther Nucleic Acids 2016; 5:e389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Zhong H, Chen Y, Li Y, et al. CRISPR-engineered mosaicism rapidly reveals that loss of Kcnj13 function in mice mimics human disease phenotypes. Sci Rep 2015; 5:8366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Arno G, Agrawal SA, Eblimit A, et al. Mutations in REEP6 cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet 2016; 99:1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Fu Y, Foden JA, Khayter C, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 2013; 31:822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Bassuk AG, Zheng A, Li Y, et al. Precision medicine: genetic repair of retinitis pigmentosa in patient-derived stem cells. Sci Rep 2016; 6:19969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Yang L, Guell M, Byrne S, et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res 2013; 41:9049–9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Yu W, Wu Z. In vivo applications of CRISPR-based genome editing in the retina. Front Cell Dev Biol 2018; 6:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019; 576:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.MacLaren RE, Bennett J, Schwartz SD. Gene therapy and stem cell transplantation in retinal disease: the new frontier. Ophthalmology 2016; 123:S98–S106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Lund RD, Wang S, Klimanskaya I, et al. Human embryonic stem cell-derived cells rescue visual function in dystrophic RCS rats. Cloning Stem Cells 2006; 8:189–199. [DOI] [PubMed] [Google Scholar]
- 272.Lu B, Malcuit C, Wang S, et al. Long-term safety and function of RPE from human embryonic stem cells in preclinical models of macular degeneration. Stem Cells 2009; 27:2126–2135. [DOI] [PubMed] [Google Scholar]
- 273.Carr AJ, Vugler AA, Hikita ST, et al. Protective effects of human iPS-derived retinal pigment epithelium cell transplantation in the retinal dystrophic rat. PLoS One 2009; 4:e8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Li Y, Tsai YT, Hsu CW, et al. Long-term safety and efficacy of human-induced pluripotent stem cell (iPS) grafts in a preclinical model of retinitis pigmentosa. Mol Med 2012; 18:1312–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Zhao T, Zhang ZN, Rong Z, Xu Y. Immunogenicity of induced pluripotent stem cells. Nature 2011; 474:212–215. [DOI] [PubMed] [Google Scholar]
- 276.Pera MF. Stem cells: the dark side of induced pluripotency. Nature 2011; 471:46–47. [DOI] [PubMed] [Google Scholar]
- 277.Quinn PM, Buck TM, Ohonin C, et al. Production of iPS-derived human retinal organoids for use in transgene expression assays. Methods Mol Biol 2018; 1715:261–273. [DOI] [PubMed] [Google Scholar]
- 278.Scruggs BA, Jiao C, Cranston CM, et al. Optimizing donor cellular dissociation and subretinal injection parameters for stem cell-based treatments. Stem Cells Transl Med 2019; 8:797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Kuriyan AE, Albini TA, Townsend JH, et al. Vision loss after intravitreal injection of autologous “stem cells” for AMD. N Engl J Med 2017; 376:1047–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Satarian L, Nourinia R, Safi S, et al. Intravitreal injection of bone marrow mesenchymal stem cells in patients with advanced retinitis pigmentosa; a safety study. J Ophthalmic Vis Res 2017; 12:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Simunovic MP, Shen W, Lin JY, et al. Optogenetic approaches to vision restoration. Exp Eye Res 2019; 178:15–26. [DOI] [PubMed] [Google Scholar]
- 282.Busskamp V, Picaud S, Sahel JA, Roska B. Optogenetic therapy for retinitis pigmentosa. Gene Ther 2012; 19:169–175. [DOI] [PubMed] [Google Scholar]
- 283.Moisseiev E, Mannis MJ. Evaluation of a portable artificial vision device among patients with low vision. JAMA Ophthalmol 2016; 134:748–752. [DOI] [PubMed] [Google Scholar]
- 284.Gardiner KL, Cideciyan AV, Swider M, et al. Long-term structural outcomes of late-stage RPE65 gene therapy. Mol Ther 2020; 28:266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Darrow JJ. Luxturna: FDA documents reveal the value of a costly gene therapy. Drug Discov Today 2019; 24:949–954. [DOI] [PubMed] [Google Scholar]