
Figure 1.
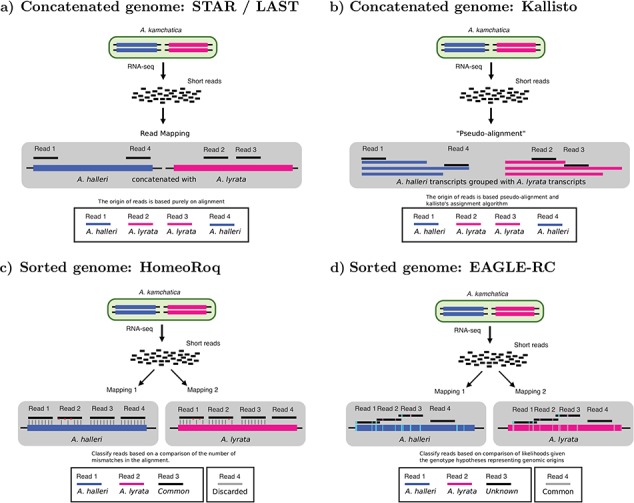
We compare ‘four different’ approaches for quantifying homeolog expression: (a) a standard genome alignment-based RNA-seq analysis on an allopolyploid (concatenated) reference genome using alignment tools STAR and LAST. (b) A pseudo-alignment workflow using Kallisto that is performed on the (concatenated) transcripts of the allopolyploid. (c) A subgenome-classification analysis ‘using HomeoRoq’ that performs alignment on each subgenome’s reference separately then performs read classification based on number of mismatches. (d) A subgenome-classification analysis ‘using EAGLE-RC’ that performs read alignment on each subgenome’s reference separately then performs read classification based on the likelihood of the read to the genotype, discarding common reads.