

Abstract
Epidermal growth factor receptor (EGFR) is a major driver of head and neck cancer, a devastating malignancy with a major sub-site in the oral cavity manifesting as oral squamous cell carcinoma (OSCC). EGFR is a glycoprotein receptor tyrosine kinase (RTK) whose activity is upregulated in >80 % OSCC. Current anti-EGFR therapy relies on the use of cetuximab, a monoclonal antibody against EGFR, although it has had only a limited response in patients. Here, we uncover a novel mechanism regulating EGFR activity, identifying a role of the nuclear branch of the Wnt/β-catenin signaling pathway, the β-catenin/CBP axis, in control of EGFR posttranslational modification with N-glycans. Genomic and structural analyses reveal that β-catenin/CBP signaling represses fucosylation on the antennae of N-linked glycans on EGFR. By employing nUPLC-MS/MS, we determined that malignant human OSCC cells harbor EGFR with a paucity of N-glycan antennary fucosylation, while indolent cells display higher levels of fucosylation at sites N420 and N579. Additionally, treatment with either ICG-001 or E7386, which are both small molecule inhibitors of β-catenin/CBP signaling, leads to increased transcriptional expression of fucosyltransferases FUT2 and FUT3, with a concomitant increase in EGFR N-glycan antennary fucosylation. In order to discover which fucosylated glycan epitopes are involved in the observed effect, we performed in-depth characterization of multiply-fucosylated N-glycans via glycopeptide tandem mass spectrometry of the EGFR tryptic glycopeptides. Data are available via ProteomeXchange with identifier PXD017060. We propose that β-catenin/CBP signaling promotes EGFR oncogenic activity in OSCC by inhibiting its N-glycan antennary fucosylation through transcriptional repression of FUT2 and FUT3.
Introduction:
Head and neck squamous cell carcinoma (HNSCC) is a debilitating disease and the sixth most common malignancy in the world, with an estimated incidence of greater than 650,000 cases and an annual toll of 350,000 deaths worldwide (1,2). Major risk factors associated with head and neck cancer include alcohol and tobacco use and human papillomavirus (HPV) infection; men are more likely than women to be diagnosed with oral cavity cancer (3). Within the head and neck region, squamous cell carcinomas can develop in multiple subsites, with the majority of cases arising in the oral cavity and oropharynx (4). Oral squamous cell carcinoma (OSCC) accounts for the majority of HNSCC cases diagnosed on a yearly basis. In the United States, a large disparity exists in the survival of black and white individuals diagnosed with cancers of the oral cavity and pharynx; overall survival is 18% lower for black patients compared to white patients, and this difference is even more pronounced among black men (4). Understanding dysregulated signaling in oral cancer at the molecular, genetic, and epigenetic levels is critical to develop effective therapies to treat patients with OSCC. Major advances in the understanding of the molecular basis of oral cancer include the roles of Wnt/β-catenin signaling (5–7), NOTCH1 (8,9), PI3K (10), and IL-6/JAK/STAT3 signaling (11,12), as well as epidermal growth factor receptor (EGFR) (13,14). Cetuximab and pembrolizumab/nivolumab, monoclonal antibodies targeting EGFR and PD-1, respectively, are the only FDA-approved targeted therapies available for the treatment of this malignancy, and even they have had only limited success (< 20%) in the clinic (2).
A major driver of OSCC is EGFR, a receptor tyrosine kinase (RTK) whose activity is aberrantly upregulated in >80% of tumors (14). EGFR is highly modified with N-linked glycans, a structurally diverse class of carbohydrates that share a tri-mannosyl chitobiose (Man3GlcNAc2) core and vary in monosaccharide composition and degree of branching. Changes in cell surface protein glycosylation have been shown to alter receptor signaling and frequently accompany malignant transformation and metastasis (15,16). For instance, RTK N-glycosylation has been shown to regulate cell proliferation and differentiation, with RTK N-linked glycans being implicated in presenting binding sites for glycan-binding proteins that oppose receptor endocytosis, leading to prolonged signaling (17–23). Accordingly, treatment of tumor cells with tunicamycin (to inhibit N-linked glycosylation) reduces RTK signaling (24–28).Both the monosaccharide types and linkages within the N-linked glycans regulate receptor dimerization. In particular, fucosylation and sialylation of antennary N-glycans at specific sites on the extracellular domain of the receptor have been demonstrated to interfere with EGFR dimerization and activation in lung adenocarcinoma (21). In contrast, core fucosylation on the glycans favors EGFR dimerization (20,21,29). Collectively, studies to date suggest that post-transcriptional modification of the N-glycans has a large impact on EGFR activity.
In OSCC, EGFR signaling converges on Wnt/β-catenin activity, known to play pivotal roles in the pathobiology of this malignancy through the interaction of nuclear β-catenin with the histone acetyltransferase CREB-binding protein (CBP) (30). The protein N-glycosylation pathway interacts with Wnt/β-catenin signaling to promote OSCC pathogenesis (31–33). N-glycosylation is regulated at multiple steps in this pathway that impact stem cell maintenance, proliferation and survival. We have shown that a small molecule inhibitor of the β-catenin-CBP interaction, ICG-001 (Fig. 1D), interferes with OSCC proliferation and aggressive features in cellular, zebrafish and murine models (6). Given that EGFR is a major driver of OSCC, we examined whether inhibition of β-catenin/CBP signaling also impacted EGFR function. We now show that OSCC-cell line induced mouse tumor xenografts exhibit reduced EGFR abundance, and genomic analyses reveal a positive correlation between ICG-001 and EGFR inhibition. Interestingly, whereas EGFR transcript levels were not affected by ICG-001 treatment, EGFR protein abundance decreased significantly, suggesting that EGFR levels were regulated on a post-translational level. Gene set expression analyses (GSEA) revealed significant increases in the expression of multiple glycosyltransferases, including fucosyltransferases FUT2 and FUT3, following inhibition of β-catenin/CBP signaling. Given that modification of EGFR N-glycans with antennary fucose residues has been reported to inhibit receptor dimerization and activity, we examined whether changes in EGFR fucosylation were associated with different aggressive characteristics of OSCC cells. Our studies suggest that β-catenin/CBP signaling in OSCC suppresses FUT2 and FUT3 expression coincident with reduced EGFR outer chain fucosylation, and that inhibition of β-catenin-CBP interaction relieves FUT2 and FUT3 suppression and promotes fucosylation of EGFR and attenuation of its oncogenic signaling. To our knowledge, this is the first report that aligns changes in EGFR fucosylation with aggressive OSCC phenotypes.
Figure 1: β-catenin/CREB-Binding Protein (CBP) Inhibition Alters EGFR Abundance and Fucosyltransferase Expression in a Mouse Tongue Squamous Cell Carcinoma Orthotopic Xenograft Model.
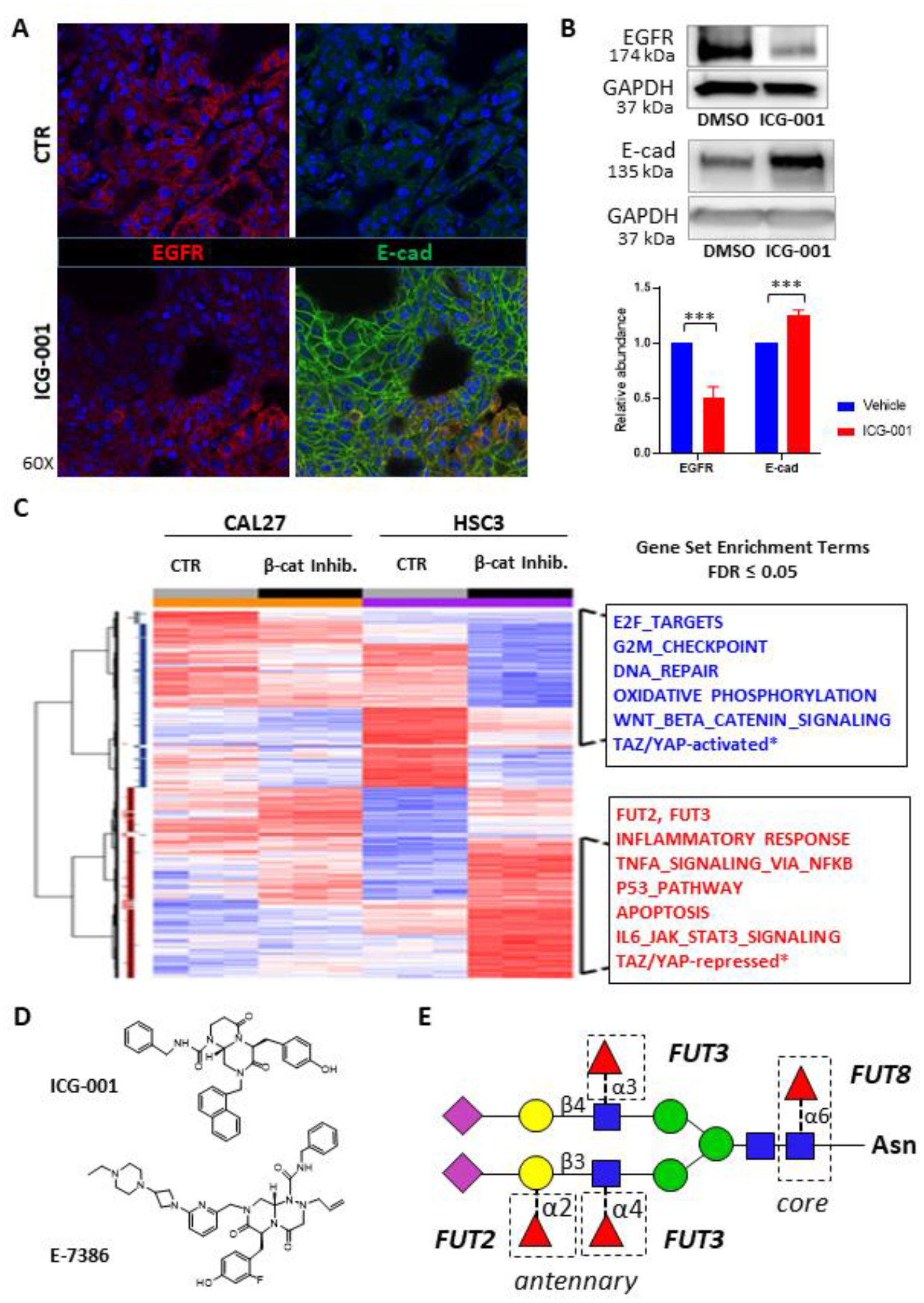
A. We used a previously established mouse tongue squamous cell carcinoma orthotopic xenograft model (36). Inhibition of β-catenin/CBP with the small molecule inhibitor ICG-001 in HSC-3-induced mouse orthotopic tongue tumor xenografts led to decreased EGFR levels (left, lower panel) compared to the vehicle control (DMSO; left, upper panel). In contrast, β-catenin/CBP inhibition resulted in an increase in E-cadherin at cell-cell interfaces (right, lower panel) compared to the control (DMSO; right, upper panel). B. Western blots of EGFR and E-cadherin from total tissue lysates (TTLs) harvested from mouse HSC-3-derived orthotopic tongue tumors treated with vehicle (DMSO) or ICG-001. GAPDH shown as a loading control. Quantification of EGFR and E-cadherin abundances are also shown, ***p <0.0001. Results of triplicate analyses are shown, +/− one standard deviation. C. Heat map comparing gene expression in CAL27 cells and HSC-3 cells treated either with vehicle (DMSO) and ICG-001. Red indicates an increase in expression, while blue indicates a decrease in expression. D. The chemical structures of ICG-001 and E7386 are shown (34,35). E. Schematic of N-linked glycan attached to an asparagine (Asn) residue, with potential linkages catalyzed by FUT2, FUT3 and FUT8 indicated. N-acetylglucosamine (blue squares), mannose (green circles), galactose (yellow circles); N-acetylneuraminic acid (pink diamonds), and fucose (red triangles) are indicated.
Results
Inhibition of Nuclear β-catenin/CBP Signaling Leads to Decreased EGFR Protein Levels and Higher Expression of Fucosyltransferases FUT2 and FUT3 in Human HSC-3-Induced Mouse Xenografts.
Our recent findings provided evidence that inhibition of β-catenin-CBP interaction with the small molecule ICG-001 abrogates OSCC cell proliferation and reduces mesenchymal traits by promoting E-cadherin adhesion (6). As EGFR is a major driver of OSCC, we sought to explore the impact of nuclear β-catenin/CBP inhibition on EGFR oncogenic activity in vivo. Thus, we evaluated human HSC-3 cell-derived orthotopic oral tongue tumor xenografts from nude mice treated with either the small molecule inhibitor ICG-001 or vehicle (DMSO) control, since we have reported that these cells form metastatic tumors (6,30,34–36). Immunofluorescence analyses revealed lower levels of membranous EGFR in tumors from ICG-001-treated mice compared to tumors from control mice (Fig. 1A, left). This was in striking contrast to increased levels of junctional E-cadherin in tumors from ICG-001-treated mice (Fig. 1A, right) (6). Furthermore, immunoblot analyses of total tissue lysates demonstrated a statistically significant (p ˂ 0.0001) decrease in EGFR abundance in tumors from mice treated with ICG-001, compared to tumors from control mice (Fig. 1B). At the same time, total tissue levels of E-cadherin increased in response to ICG-001 treatment (Fig. 1B). In this context, increased nuclear β-catenin/CBP signaling correlates with higher EGFR abundance and lower E-cadherin adhesion. This conclusion was supported by computational analyses comparing the ICG-001 treatment signatures and EGFR inhibition signatures in TCGA OSCCs which revealed a very strong positive correlation between ICG-001 and EGFR inhibition (Fig. 1C, Supplementary Figs. 1A–B), further suggesting that the ICG-001 treatment inhibits genes involved in EGFR signaling in primary oral cancer tumor tissues.
Upon disruption of nuclear β-catenin/CBP signaling with E7386, a novel β-catenin/CBP modulator that displays an activity profile that closely overlaps with that of ICG-001 but exhibits ~50 – 100-fold lower EC50 in OSCC cell lines than ICG-001, the ICG-001 inhibition-associated transcriptional signatures tracked with tumor grade and poor human OSCC patient overall survival (6). Likewise, the E7386 inhibition signature was associated with advanced TCGA OSCC tumor grade that is associated with poor overall patient survival (Supplementary Fig. 3A). Lastly, similar to ICG-001, the E7386 inhibition signature was highly correlated with EGFR inhibition in TCGA OSCC (Supplementary Fig. 3B).
The observed downregulation of EGFR abundance in response to the inhibition of β-catenin/CBP activity was not a consequence of transcriptional attenuation, as treatment with either ICG-001 or E7386 did not affect EGFR transcript levels. Since EGFR exhibits high glycoform diversity at multiple N-glycosylation sites, and since modifications of the N-glycan structures affect protein folding, targeting and activity, we postulated that ICG-001 and E7386 had impacted EGFR abundance on a post-translational level. Therefore, we assessed the impact of ICG-001 and E7386 inhibition on the expression of genes that function in the protein N-glycosylation pathway. Indeed, treatment of CAL27 and HSC-3 cells with either ICG-001 or E7386 increased expression of two antennary fucosyltransferases, FUT2 and FUT3 (Table 1, Fig. 1D), but did not alter the level of fucosyltransferase FUT8, which catalyzes core fucosylation of N-glycan structures (data not shown). While α1,6-linked (core) fucosylation of EGFR catalyzed by FUT8 has been shown to increase EGFR signaling (29), α1,3-linked antennary fucosylation has been reported to suppress EGFR signaling in lung adenocarcinoma (21). FUT2 catalyzes α1,2-fucosylation of galactose in Type-1 chains (Galβ1–3GlcNAc), while FUT3 catalyzes α1,4-fucosylation of GlcNAc in Type-1 chains (Galβ1–3GlcNAc) and α1,3-fucosylation of GlcNAc in Type-2 chains (Galβ1–4GlcNAc) (Fig. 1E). These results suggested that fucosyltransferases FUT2 and/or FUT3 affected EGFR protein levels in OSCC.
Table 1. Altered Fucosyltransferase Expression in Response to β-catenin/CBP Inhibition.
Expression change of fucosyltransferases FUT2, FUT3, FUT8, the epidermal growth factor receptor (EGFR), and E-cadherin (CDH1) after treatment with ICG-001 (top) or E7386 (bottom) compared to vehicle control (DMSO) in indolent CAL27 and metastatic HSC-3 cells. The fold change of the treatment compared to the DMSO control (Fold Δ) and q-value (q, FDR adjusted) are shown for each condition tested.
| HSC-3 | CAL27 | |||
|---|---|---|---|---|
| Fold Δ | q | Fold Δ | q | |
| FUT2 | +2.1 | 2.0 × 10−5 | +1.1 | 5.4 × 10−1 |
| FUT3 | +3.0 | 5.7 × 10−8 | +1.4 | 1.5 × 10−1 |
| FUT8 | +1.2 | 9.0 × 10−3 | +1.2 | 1.2 × 10−1 |
| EGFR | +1.1 | 3.9 × 10−1 | +1.3 | 2.1 × 10−2 |
| CDH1 | +1.2 | 5.1 × 10−2 | −1.1 | 3.6 × 10−1 |
| HSC-3 | CAL27 | |||
| Fold Δ | q | Fold Δ | q | |
| FUT2 | +2.4 | 1.4 × 10−3 | NC | - |
| FUT3 | +2.3 | 5.5 × 10−4 | +1.6 | 1.8 × 10−2 |
| FUT8 | +1.4 | 5.0 × 10−3 | +1.1 | 2.6 × 10−1 |
| EGFR | +1.5 | 3.3 × 10−3 | +1.5 | 1.3 × 10−2 |
| CDH1 | +1.2 | 8.1 × 10−2 | −1.1 | 6.3 × 10−1 |
MS/MS Analyses of EGFR from OSCC Cell Lines Identify Multiple Sites with Fucosylated N-glycans.
Next, we sought to determine the extent to which EGFR derived from untreated OSCC cell lines displayed fucosylated N-glycans. EGFR contains 12 N-glycosylation sequons in its extracellular domain, divided among the receptor’s four subdomains, I-IV (Fig. 2A). To determine the site-specific patterns of EGFR N-glycosylation, we immunoprecipitated EGFR from indolent CAL27 and metastatic HSC-3 human tongue oral squamous carcinoma cell lines. Following proteolysis of EGFR, glycopeptides were enriched on an Amide-80 trapping column, separated via C18 reversed-phase chromatography, and analyzed via Q-TOF MS/MS. Additional analyses were performed without glycopeptide enrichment, using data-dependent MS/MS methods on an Orbitrap Tribrid MS, including glycopeptide oxonium ion-triggered Higher-energy Collisional Dissociation (HCD) and Electron-Transfer Dissociation (ETD) combined with gentler HCD (EThcD) fragmentation (Fig 2B) (37–39). In total, we detected glycopeptides representing 11 of the 12 reported EGFR N-glycosylation sites (Supplementary Table 1, and Supplementary Figs. 4–14). To determine which sites were modified with high mannose, hybrid, or complex N-glycans, we surveyed the glycan compositions associated with these 11 sites. Sites N172, N328, N337, and N599 displayed high mannose N-linked glycans in both CAL27 and HSC-3 cells, indicating that these sites could not be modified by fucosyltransferases FUT2 and FUT3. In contrast, sites N32, N104, N151, N389, N420, N544, and N579 displayed complex glycans or a combination of complex and high mannose glycans in both cell lines (Fig. 2C, Supplementary Tables 1–6). Glycopeptides corresponding to site N504 were not detected. Furthermore, treatment with ICG-001 did not significantly alter N-glycosylation site occupancy (Supplementary Table 7). Of the sites bearing complex N-glycans, all seven contained at least one fucosylated glycoform. At sites N32, N104, and N544, only narrow distributions of glycoforms were detected. In contrast, extensive N-glycan heterogeneity was observed on EGFR domain I site N151, EGFR domain III sites N389 and N420, and EGFR domain IV site N579.
Figure 2: N-glycosylation of the Epidermal Growth Factor Receptor (EGFR).
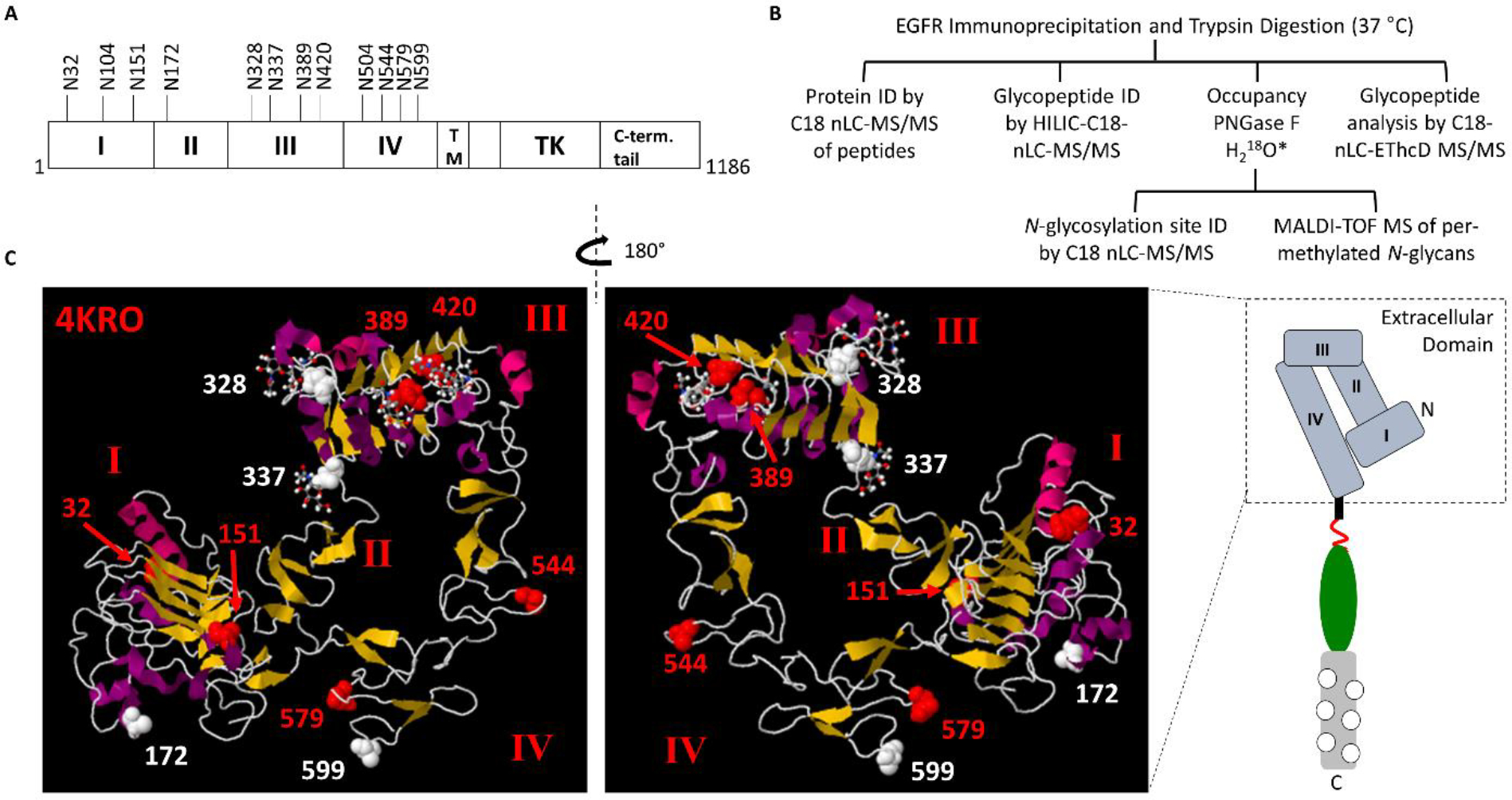
A. A schematic of EGFR is shown, with N-glycosylation sites labeled. EGFR contains four sub-domains (I-IV) within the extracellular domain, a transmembrane (TM) region, and the cytoplasmic tyrosine kinase (TK) and C-terminal tail. B. Schematic of the analytical workflow for the analysis of EGFR derived from OSCC cell lines. *PNGase F treatment was performed in the presence of H218O for peptide analyses and site-occupancy determination. For glycan analyses, H2O was used. C. Crystal structure of the EGFR extracellular domain (Protein Data Bank ID: 4KRO) (64). Based on our survey of EGFR N-glycopeptides derived from OSCC cell lines, N-glycosylated asparagine residues bearing complex glycans are shown in red, and those bearing high-mannose type glycans are shown in white. N104 is not shown because it is not resolved in this structure determination – this is likely due to the higher flexibility of this region.
EGFR from Indolent CAL27 Cells Displays a Higher Proportion of N-Glycans with Multiple Fucose Residues While EGFR from Metastatic HSC-3 Cells Displays Lower Levels of Fucosylated Glycoforms.
Next, we performed a quantitative comparison of the fucosylation status of N-glycans from indolent CAL27 cells and metastatic HSC-3 cells. To achieve this, EGFR glycopeptides were analyzed in triplicate. Extracted ion chromatograms were generated for each EGFR glycopeptide/glycoform observed, and the area of each was calculated. At EGFR site N389, all glycopeptides detected in both cell lines displayed at least one fucose on complex glycans (Fig. 3A, Supplementary Tables 2–6 and 9). Singly-fucosylated glycoforms at site N389 accounted for 85.0 +/− 2.5% of the area in CAL27 cells and 91.1 +/− 1.2% of the area in HSC3 cells, a small but statistically significant difference (p = 3.4 × 10−2). The doubly-fucosylated and sialylated N-glycan composition HexNAc4Hex5Fuc2NeuAc1 (N4H5F2S1) had higher relative abundance in CAL27 cells compared to HSC-3 cells (p = 1.4 × 10−2). Triply-fucosylated glycoforms were 4.5-fold higher in CAL27 cells compared to HSC-3 cells (p = 1.1 × 10−2) (Fig. 3D). At site N420, 40.3% +/−1.8% of glycoforms (by area) were fucosylated in CAL27 cells, whereas fucosylated glycans accounted for roughly half as much of the total (20.9 +/− 3.8%) in EGFR derived from HSC-3 cells, and this difference was highly statistically significant (p = 1.2 × 10−3) (Fig. 3B, Supplementary Tables 10A–B). Singly-, doubly-, and triply-fucosylated glycoforms were higher in CAL27 cells compared to HSC3 cells; singly-fucosylated glycans were 1.5-fold higher (p = 7.1 × 10−3) and doubly-fucosylated glycans were 3.2-fold higher (p = 2.3 × 10−3) (Fig. 3E). While triply-fucosylated glycopeptides accounted for 1.4% +/− 0.2% of the area of N420 glycopeptides in CAL27 cells, no triply-fucosylated glycopeptides were detected in HSC-3 cells. At site N579, singly-fucosylated glycoforms had lower relative abundances in CAL27 cells compared to HSC-3 cells at 66.5 +/− 4.6% and 88.3 +/− 0.2% of total N579 glycopeptide area, respectively (Fig. 3C, Supplementary Table 11). However, the relative levels of doubly-fucosylated glycans were 2.3-fold higher in CAL27 cells compared to HSC-3 cells (p = 2.3 × 10−2), accounting for 20.2 +/− 4.5% and 8.6 +/− 0.2% of N579 glycopeptide area, respectively (Fig. 3F). At sites N328, N337, and N599 displaying high mannose glycans, and sites N32, N104, and N544, where a limited distribution of glycoforms were detected, we did not observe any changes in N-glycan fucosylation. In summary, multiple (but not all) EGFR N-glycosylation sites displayed higher proportions of multiply-fucosylated N-linked glycans in indolent CAL27 cells compared to metastatic HSC-3 cells.
Figure 3: EGFR N-Glycan Fucosylation Differs Between Indolent CAL27 Cells and Metastatic HSC-3 Cells.
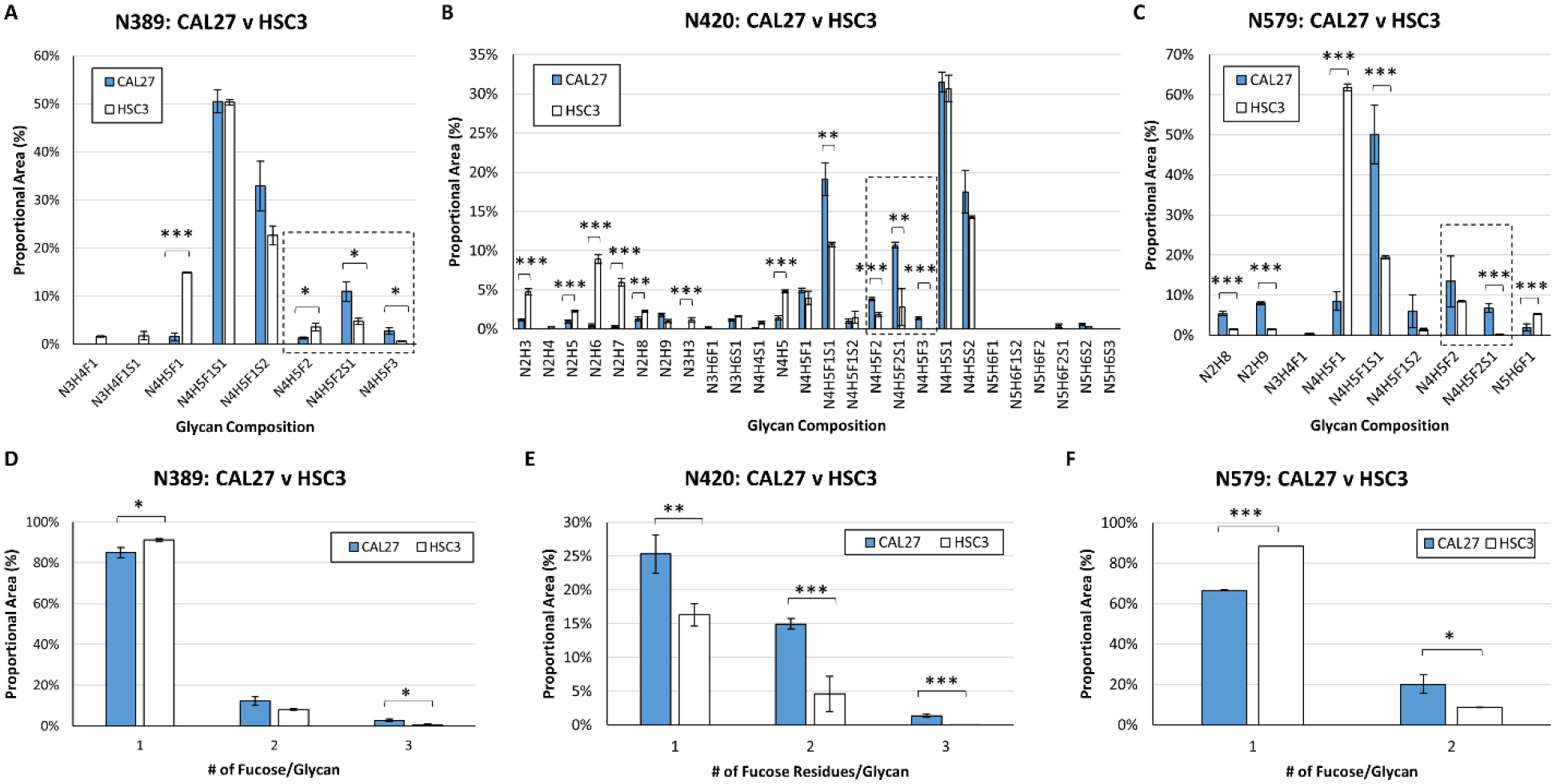
A. CAL27 (blue bars) and HSC-3 (white bars) cells display high levels of complex, singly-fucosylated glycans at site N389; proportional areas of glycan compositions are shown. B. Proportional areas of glycan compositions at site N420; glycan compositions consistent with high mannose and complex glycans were detected at this site. Singly- and multiply-fucosylated glycan compositions are more abundant in CAL27 compared to HSC-3 cells. C. Proportional areas of glycan compositions at site N579. D. Comparison of the proportional area of singly, multiply-fucosylated glycans at site N389 in CAL27 and HSC-3 cells, and for sites N420 (E) and N579 (F). For all sites, nUPLC-MS analyses were performed in triplicate (technical replicates), and the area of each glycopeptide precursor was extracted (based on m/z). Plots show the proportional area of each glycoform as a percentage of the total glycoform area for each indicated N-glycosylation site. N: N-acetylhexosamine, H: hexose (mannose or galactose), F: fucose, S: N-acetylneuraminic acid (sialic acid). Multiply-fucosylated glycan compositions are outlined (box with dashed line). * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.005.
Inhibition of β-catenin-CBP Signaling in Metastatic HSC-3 Cells Results in Increased Fucosylation of EGFR N-Glycans.
Given that expression of FUT2 and FUT3 increased in HSC-3 cells in response to β-catenin/CBP inhibition, we next investigated whether or not EGFR derived from HSC-3 cells treated with ICG-001 displayed higher levels of fucosylated N-glycans. At site N151, all detected glycoforms contained at least one fucose residue under control conditions. Inhibition of β-catenin/CBP signaling in HSC-3 cells resulted in a statistically significant increase in the relative abundance of glycans with the doubly-fucosylated glycan composition HexNAc4Hex5Fuc2 (N4H5F2). The proportional abundance of this glycan composition increased from 6.4 +/− 2.1% in the control to 18.5 +/− 0.5% after treatment with ICG-001 (Fig. 4A, Supplementary Table 8) (p=1.3 × 10−3). At site N389, the relative abundances of glycans with doubly-and triply-fucosylated glycan compositions increased dramatically in cells treated with ICG-001 compared to controls (Fig. 4B, Supplementary Table 9). At EGFR site N420, the proportional abundance of singly-, doubly-, and triply-fucosylated N-glycans increased in response to β-catenin/CBP inhibition (Fig. 4C–D, Supplementary Tables 10A–B). In contrast, EGFR from CAL27 cells had few changes in N-glycan fucosylation at site N420 in response to β-catenin/CBP inhibition (Fig. 4E, Supplementary Tables 10A–B). Indeed, EGFR from HSC-3 cells treated with ICG-001 exhibited similar proportional levels of fucosylated N-glycans as EGFR from untreated CAL27 cells at site N420 (p=6.4 × 10−3) and at additional sites (Fig. 4E, Supplementary Tables 8–11). Multiply fucosylated glycan compositions also increased at EGFR site N579 in HSC-3 cells treated with ICG-001 compared to controls (Fig. 4F, Supplementary Table 11). In contrast to the site-specific changes in EGFR N-glycosylation observed during nUPLC-MS/MS analyses of immunoprecipitated EGFR, MALDI-TOF MS analyses of released permethylated N-glycans derived from the whole lysates of HSC-3 and CAL27 cells treated with either DMSO or ICG-001 showed no significant changes (Supplementary Figs. 15A–C), though the dynamic ranges of these analyses were limited, with the result that we did not detect multiply-fucosylated species.
Figure 4: Inhibition of β-catenin/CBP in Metastatic HSC-3 Cells Leads to Higher Levels of EGFR N-Glycan Fucosylation.
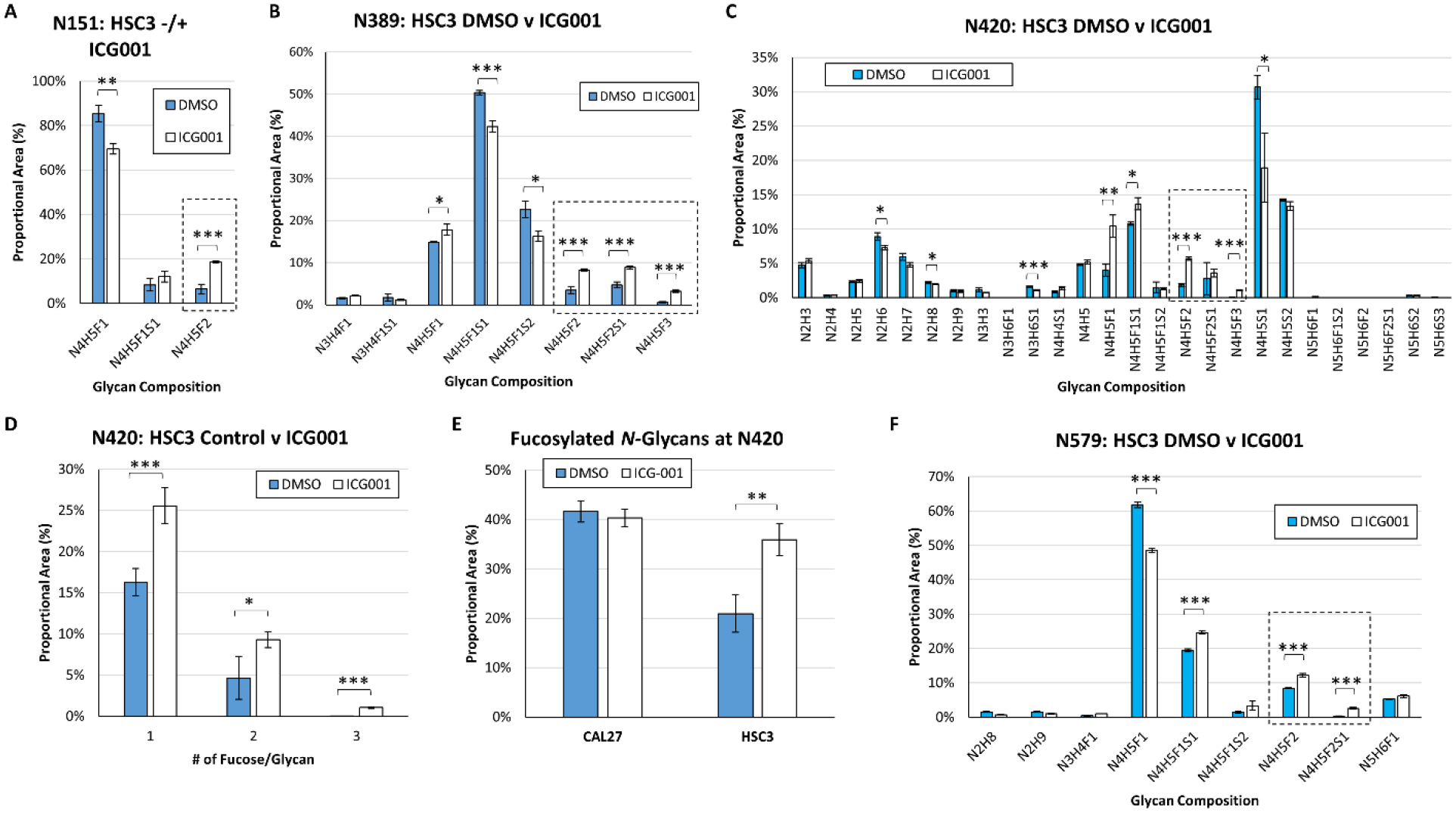
EGFR N-glycan heterogeneity at sites (A) N151, (B) N389, (C) N420, and (F) N579, in EGFR derived from HSC-3 cells treated with either a vehicle control (DMSO) or ICG-001, a small molecule inhibitor of nuclear β-catenin/CBP. Comparison of the proportional area of fucosylated glycans to total glycans at site N420 in CAL27 and HSC-3 cells in response to β-catenin/CBP inhibition (D). Proportional area of singly-, doubly-, and triply-fucosylated N-glycans at site N420 in HSC-3 cells in response to β-catenin/CBP inhibition. (E) At site N420, the proportion of fucosylated N-glycans increased in response to β-catenin/CBP inhibition in HSC-3 cells, and almost matched the levels of fucosylation observed in CAL27 cells. For all sites, nUPLC-MS analyses were performed in triplicate (technical replicates), and the area of each glycopeptide precursor was extracted (based on m/z). N: N-acetylhexosamine, H: hexose (mannose or galactose), F: fucose, S: N-acetylneuraminic acid (sialic acid). Multiply-fucosylated glycan compositions are outlined (box with dashed line). * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.005.
Inhibition of β-catenin/CBP Signaling Shifts the Balance of Fucosylated EGFR N-Glycans Toward Antennary-Fucosylation.
Having observed an increase in fucosylation in response to β-catenin/CBP inhibition in HSC-3 cells, we next made determinations of the location of the fucose residue(s) on EGFR N-linked glycans. We observed multiple values for the retention times of fucosylated glycopeptides sharing the same precursor m/z values in HSC-3 cells after treatment with ICG-001 (Fig. 5A), and hypothesized that this diversity might be due to the presence of variations in the linkage positions of the fucose residues. Because fucose residues are frequently cleaved off during fragmentation with higher-energy collisional dissociation (HCD), we performed additional analyses with (a) low-energy HCD and (b) electron transfer dissociation with supplemental activation (EThcD) to favor the generation of glycopeptide fragment ions while minimizing glycosidic bond fragmentations. This enabled us to distinguish between antennary- and core-fucosylated N-glycopeptides. An HCD MS/MS spectrum of the early-eluting glycopeptide corresponding to EGFR site N420, 406TKQHGQFSLAVVSLNITSLGLR427 + HexNAc4Hex5dHex1 ([M+4H]4+ m/z 1035.2483), provided evidence for a core-fucosylated isomer. This assignment was supported by the presence of the product ion consisting of the intact triply protonated peptide + HexNAc1dHex1 (pNF) ion at m/z 906.8231 (3+), as well as low levels of the HexNAc1Hex1dHex1 (NHF+) oxonium ion at m/z 512.1954 (Fig. 5B). Such a fragment could be generated with low efficiency by rearrangement of fucose during fragmentation (40–43). In contrast, the product ion scan of the late-eluting isomer showed very high levels of the HexNAc1Hex1dHex1 (NHF+) oxonium ion observed at m/z 512.1953, consistent with the presence of outer-arm fucosylation. As expected on the basis of this assignment, we detected high relative levels of the peptide + HexNAc1 peak without fucose at m/z 858.1374, and higher intensities of product ions associated with loss of components that would include antennary fucose, including [M-NHF]3+ observed at m/z 1209.5911 and [M-NH2F]3+ at m/z 1155.5740 (Fig. 5C). We have documented additional examples of such spectral differences in core and antennary fucosylation (Supplementary Figs. 16–18). In addition, we examined multiply-fucosylated species. EThcD fragmentation of the dominant doubly-fucosylated EGFR site N420 glycopeptide species in HSC-3 cells after treatment with ICG-001, 406TKQHGQFSLAVVSLNITSLGLR427 + HexNAc4Hex5dHex2 ([M+4H]4+ m/z 1071.7526), indicated that both fucose residues were located on the N-glycan antennae, based on the presence of product ions in the MS2 spectrum, including the triply protonated fragment ions that lack fucose: peptide + HexNAc2Hex3 (pN2H3) and peptide + HexNAc3Hex3 (pN3H3), observed at m/z 1088.2133 (3+) and m/z 1155.5729 (3+) respectively (Fig. 6, Supplementary Fig. 17A–B). These findings were consistent with fucosyl transferase expression level results.
Figure 5: β-catenin/CBP Inhibition Shifts EGFR N-Glycan Fucosylation from “Core” Fucosylation to a Signal Suppressing “Outer-Arm” Fucosylation.
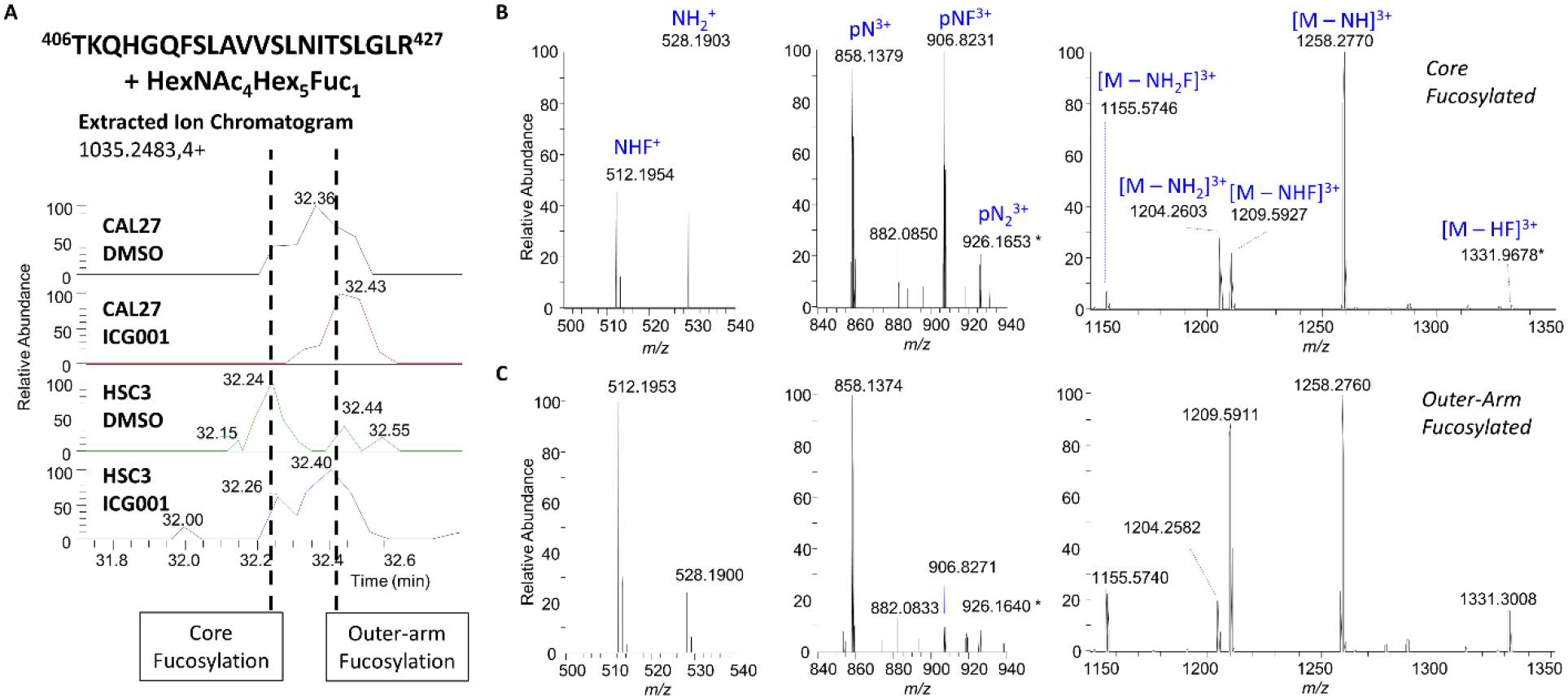
A. Extracted ion chromatograms (EICs) of the EGFR site N420 tryptic glycopeptide 406TKQHGQFSLAVVSLNITSLGLR427 + HexNAc4Hex5Fuc1 (m/z 1035.2483, z=4+) in CAL27 and HSC-3 cells under control conditions (DMSO) or after treatment with ICG-001. Note two partially-resolved peaks in the chromatograms centered at approximately 32.2 min and 32.4 min with the same m/z. Selected regions of the low-energy (10% NCE) HCD product ion spectra of the ‘early’ eluting form with annotated peaks demonstrating evidence of core fucosylation (B), and the ‘late’ eluting form with annotated peaks demonstrating evidence of antennary fucosylation (C). p = peptide, N = HexNAc, H = hexose, F = dHex.
Figure 6: EGFR Site N420 Fucosylated Glycoform, EThcD MS/MS Spectrum of EGFR Glycopeptide 406TKQHGQFSLAVVSLNITSLGLR427 + HexNAc4Hex5dHex2 from HSC-3 Cells After Treatment with ICG-001.
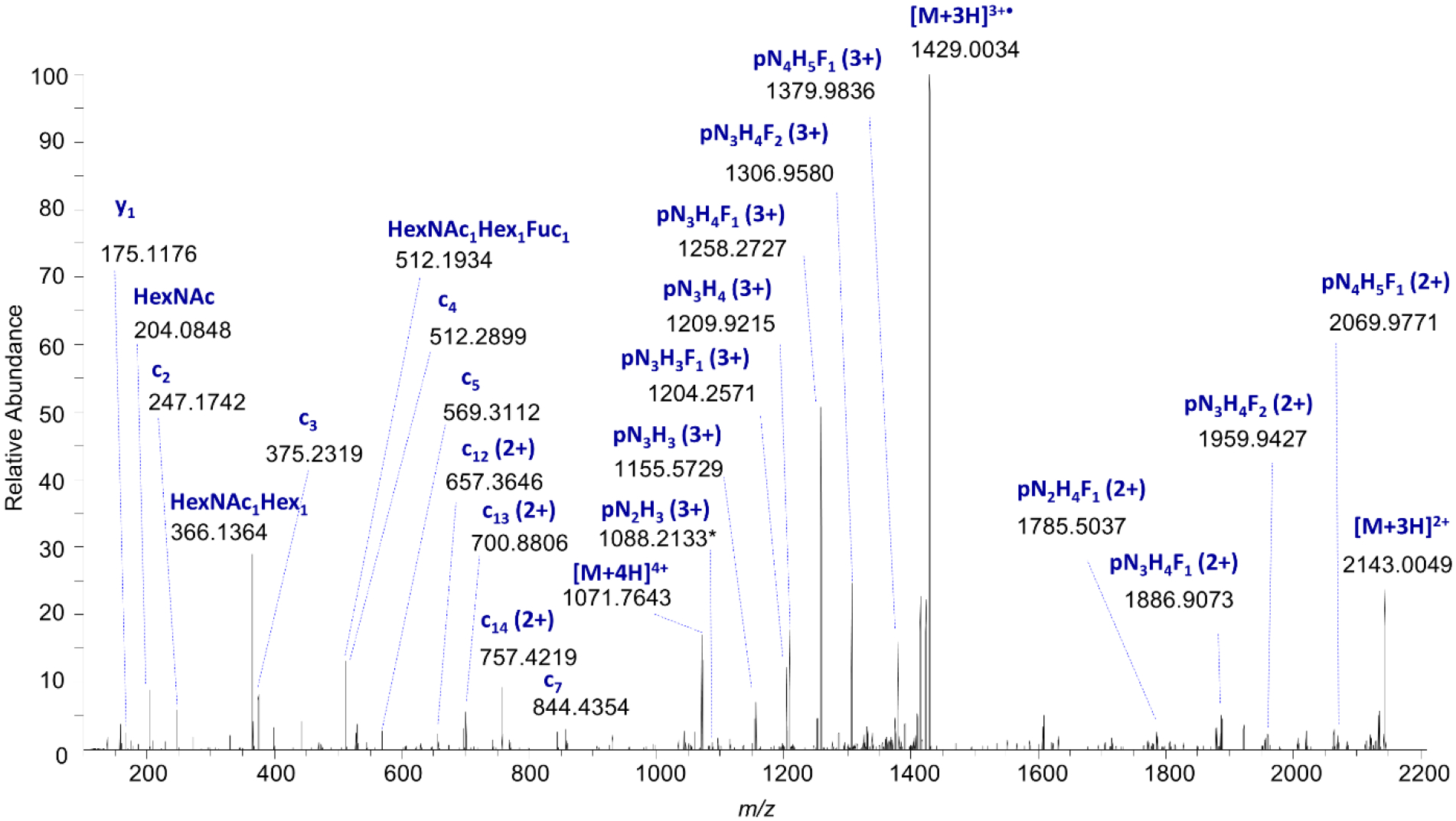
Electron-transfer/higher-energy collision dissociation (EThcD) mass spectrum of EGFR Site N420 glycopeptide, [M+4H]4+ m/z 1071.7526, demonstrating evidence of N-glycan “outer arm” fucosylation; the presence of the HexNAc+Hex+dHex oxonium ion at m/z 512.1934, and the lack of dHex shift associated with fragments that contain only the trimannosyl chitobiose core (pN2H3), suggest that both fucose residues are located on the outer arms, consistent with expression level data. p = peptide, N = HexNAc, H = hexose, F = dHex.
Discussion
The Wnt/β-catenin signaling pathway and EGFR signaling are known to cross talk at multiple levels and to enhance each other’s activities although the details of these interactions have not been well defined in OSCC (7,44,45). Our studies show for the first time that the nuclear branch of the Wnt/β-catenin pathway, the β-catenin/CBP axis, impacts EGFR protein abundance by modulating its N-glycosylation status via transcriptional repression of two fucosyltransferases, FUT2 and FUT3, shown to fucosylate outer chain N-glycans on EGFR and inhibit its activity. Our detailed structural analyses provide evidence that antennary fucosylation of EGFR N-glycans is associated with indolent OSCC phenotypes and that suppression of EGFR fucosylation aligns with malignant features. We have shown previously that HSC-3 cells displayed high nuclear β-catenin/CBP activity and that disruption of the β-catenin-CBP interaction reverted the mesenchymal phenotype of HSC-3 cells to an epithelial morphology similar to indolent CAL27 cells (6). Accordingly, disruption of the β-catenin-CBP interaction effectively relieved β-catenin/CBP-driven suppression of FUT2 and FUT3. These effects of nuclear β-catenin/CBP signaling on EGFR N-glycosylation reveal a novel level of EGFR regulation that includes transcriptional suppression of specific post-translational N-glycosylation enzyme activities.
Results showed that inhibition of β-catenin/CBP activity with ICG-001 in HSC-3-induced mouse tumor xenografts caused a decrease in total tissue levels of EGFR but not in EGFR transcript levels. Instead, there was an increase in the expression of FUT2 and FUT3 (Fig. 1A–C). Both of these fucosyltransferases transfer fucose to N-glycan antennae; FUT2 catalyzes α1,2-linkage of fucose residues to N-linked glycans, while FUT3 can catalyze the attachment of fucose via α1,3- and α1,4-linkages. The expression of an inactive form of FUT2 is associated with the development of oral cancer (46,47), suggesting that the active form of the enzyme has a protective role in OSCC. In contrast, FUT3 has the potential to contribute to the expression of sialyl-Lewis X (SLex) glycan epitopes and has been shown to play a role in establishing the invasive and metastatic properties of oral cancer stem cells (48). Thus, different fucosyltransferases may have opposing roles in oral cancer. On the other hand, the Lewis y (Ley) antigen (with both α1,2- and α1,3-linked fucose) is expressed in OSCC cell lines and tissues, but disappears in invasive regions, suggesting that fucosylated glycans may be involved in the suppression of aggressive features (49). Moreover, EGFR antennary-fucosylated glycans are associated with patient survival, and suppress receptor dimerization and signaling in lung adenocarcinoma (21). Further studies are required to elucidate specific roles of FUT2 and FUT3 in modulating EGFR and potentially other oncogenic glycoproteins in OSCC.
We identified multiple EGFR sites bearing fucosylated N-glycans (Fig. 2C). Several EGFR N-glycosylation sites appear to play key roles in receptor dimerization and signaling, including site N420 which alters EGFR dimerization (50), and N579 which plays a role in EGFR signaling by preventing the receptor from becoming activated (auto-inhibition) in the absence of ligand stimulation (51). Using nUPLC-MS/MS, we tracked the glycosylation of EGFR across multiple sites, including sites N420 and N579. While EGFR fucosylation from CAL27 and HSC3 cells differs modestly at certain sites, we observed dramatic changes in EGFR fucosylation at sites N420 and N579. EGFR from metastatic HSC-3 cells displayed low levels of fucosylated N-glycans, while EGFR from non-metastatic CAL27 cells displayed higher levels of fucosylation at multiple sites, including sites N420 and N579.
Post-translational regulation of RTKs has been implicated in cell proliferation and differentiation (19), and inhibition of N-glycosylation has been shown to disrupt RTK oncogenic signaling in tumor cells (26). Moreover, other post-translational mechanisms, such as reduced proteolytic shedding, have been implicated in RTK-mediated cancer treatment resistance (52). Our present studies suggest that the β-catenin/CBP axis modulates EGFR signaling through downregulation of FUT2 and FUT3 expression and activity. Given that decrease in fucosylation has been reported in the invasive regions of OSCC tumors, it is possible that inhibition of this modification by FUT2 and FUT3 relieves EGFR from suppression and drives aggressive OSCC phenotypes. Likewise, fucosylated epitopes reported in OSCCs may serve to suppress EGFR signaling. Thus, given that ICG-001 and E7386 have overlapping activities, inhibition of β-catenin/CBP signaling with the more soluble E7386 may serve as a therapeutic approach not only to inhibit downstream targets of β-catenin/CBP that drive disease progression, but also to downregulate EGFR pro-tumorigenic activity in OSCC.
Experimental Procedures
Reagents and Cell Lines.
PNGase F was purchased from New England Biolabs (Ipswich, MA). MS grade trypsin (tosyl phenylalanine chloromethyl ketone treated) was purchased from ThermoFisher Scientific (Waltham, MA). Pierce C18 Tips (100 uL), dithiothreitol (DTT), ammonium bicarbonate (NH4HCO3), and iodoacetamide were purchased from Sigma Aldrich (St. Louis, MO, USA). Small (10 μL) C18 ZipTips were purchased from Millipore (Billerica, MA). Heavy water (H218O, 99%) was purchased from Cambridge Isotope Laboratories (Andover, MA). CAL27 cells were purchased from ATCC while HSC-3 cells were obtained from XenoTech (Tokai, Japan). CAL27 and HSC-3 cell lines were routinely authenticated by short tandem repeat profiling every 6 months, either by ATCC or Genetica. ICG-001 was purchased from Selleckchem (Houston, TX) and E7386 was provided by Eisai Inc (Woodcliff Lake, NJ). For immunofluorescence and Western blot analyses, rabbit monoclonal antibody to EGFR [EP39Y; ab52894] and mouse monoclonal antibody to E-cadherin [4A2; ab231303] were purchased from Abcam (Cambridge, MA). DAPI and goat anti-rabbit and anti-mouse IgG derivatized with either Texas red or FITC secondary antibodies were obtained from Molecular Probes (Eugene, OR). For Western blot analyses, anti-GAPDH antibody (mouse monoclonal, G8795) was purchased from Sigma-Aldrich (St. Louis, MO).
Immunofluorescence and Western Blot Analyses.
HSC-3 cell-derived mouse tumor xenografts were generated and processed for immunofluorescence and immunoblot analyses as previously described (6). Tumor sections (5 μm) were immunostained with antibodies for EGFR and E-cadherin, counter-stained for nuclei with DAPI, followed by incubation with secondary Texas Red- and FITC-conjugated antibodies. Negative controls lacked primary antibodies. The slides were mounted in ProLong Gold Antifade and optical sections (0.5 μm intervals) were analyzed by confocal microscopy using a Zeiss LSM710-Live Due Scan confocal microscope. For comparison of fluorescence intensities between samples, settings were fixed to the most highly stained sample with all other images acquired at those settings. Images were processed with ZEN 2 and ImageJ imaging software.
For Western blot analyses, total tumor tissue lysates were prepared by grinding tumors to a fine powder in liquid nitrogen, followed by extraction of total tissue proteins with Triton/β-octylglucoside buffer. Protein concentrations were determined using the BCA assay (Pierce). Tissue lysates (20–30 μg of total protein) were fractionated via 4–12% SDS-PAGE, transferred onto polyvinylidene difluoride membranes, blocked with 5% nonfat dry milk, and incubated sequentially with primary antibodies to EGFR, E-cadherin and GAPDH. Protein-specific detection was carried out with horseradish peroxidase-labeled secondary antibodies conjugated to horseradish peroxidase (Bio-Rad), and Enhanced Chemiluminescence Plus (Amersham Biosciences). Signal intensities were normalized to GAPDH.
β-Catenin/CBP Inhibition and EGFR Immunoprecipitation.
CAL27 and HSC-3 cells were treated with 10 μM ICG-001/DMSO for 50–55 h. Cells were cultured to approximately 60% confluence, then lysed using Triton-X-100/β-octylglucoside buffer (10 mM imidazole, 100 mM NaC1, 1 mM MgC12, 5 mM Na2EDTA, 1% Triton-X-100, 0.87 mg/mL β-octylglucoside) and MS-SAFE (Sigma) protease and phosphatase inhibitor cocktail. Protein levels of total cell lysates from CAL27 and HSC3 cells were quantified by absorbance (280 nm), and 700 μg of lysate was used per immunoprecipitation. Immunoprecipitation was performed according to the manufacturer guidelines using a Pierce Crosslink Immunoprecipitation Kit from Thermo Scientific (#26147) with 3 μg of a mouse monoclonal antibody to EGFR (111.6, ThermoFisher MA5–13269). The binding of the antibody to protein A/G plus agarose, as well as the crosslinking of the bound antibody, were performed individually for each sample, followed by the addition of the pre-cleared lysate. EGFR was eluted using the proprietary elution buffer included with the kit, presumed to be a low pH and high molar solution of glycine. After elution, immunoprecipitated proteins were neutralized by addition of 1 M Tris buffer, pH 8.0, and stored at −20 °C until proteolysis.
Proteolysis.
Each EGFR sample was suspended in 50 mM NH4HCO3, pH 7.8, reduced with 5 mM DTT, and alkylated with 15 mM iodoacetamide. Excess iodoacetamide was quenched by addition of 20 mM DTT. Following reduction and alkylation, samples were incubated with trypsin at a ratio of 1:50 to total protein at 37 °C for 18 h. Samples were dried to remove volatile buffers, and peptides were purified using C18 tips (100 μL), according to the manufacturer’s protocol. The presence of EGFR in immunoprecipitated samples was confirmed by analyzing a small quantity (≤ 1/10th) of the resulting peptides (Supplementary Tables 12–15).
Gene Set Enrichment Analysis (GSEA).
GSEA v2.2.1 software was used to perform enrichment analysis on the derived ICG-001 and E7386 treatment profiles, as previously reported (6,53,54). Briefly, pre-ranked GSEA was run, using the t-statistic of microarray differential expression results comparing DMSO (vehicle control) to either ICG-001 or E7386. All default parameters of the GSEA software were used (6).
PNGase F/H218O.
Each of the EGFR peptide mixtures obtained by treatment with proteases was split into two equal amounts and dried in a centrifugal evaporator. GlycoBuffer 2 (NEB, 50 mM sodium phosphate, pH 7.5) was aliquoted and dried under vacuum; next, 20 μL of H218O (99%, Cambridge Isotopes, Andover, MA) was added. After mixing, each of the buffer solutions was transferred to a tube containing one of the dried aliquots of peptides. Next, 1 μL of PNGase F was added to one tube of peptides in each pair of aliquots; the second tube was not treated with the glycosidase and served as the control. The contents were gently mixed by vortexing, and the tubes were placed on the Thermomixer at 37 °C for 16 h. Samples were dried under vacuum and cleaned via C18 ZipTip (Millipore, Billerica, MA) according to the manufacturer’s protocol.
NanoLC-MS/MS of Peptides.
After PNGase F/H218O treatment, peptide samples were analyzed on a Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Scientific) equipped with a nanoAcquity UPLC system (Waters) and a Triversa Nanomate (Advion, Ithaca, NY). For chromatographic separation, a nanoACQUITY UPLC Symmetry C18 Trap Column (100 Å, 5 μm; 180 μm × 20 mm, Waters) column was used for trapping, and an ACQUITY UPLC Peptide BEH C18 nanoACQUITY Column (130 Å, 1.7 μm; 150 μm × 100 mm, Waters) column was used for separation. The peptide trapping step was performed at 4 μL/min for 4 min with 1% acetonitrile and 0.1% formic acid (Solvent A). Following the trapping step, peptides were separated on the analytical column according to the following conditions: 0–1 min: 2% B, 1–3 min: 2–5% B, 3–43 min: 5–40% B (Solvent B: 99% acetonitrile and 0.1% formic acid). MS scans were acquired with the following settings: 70 000 resolution @ m/z 400, scan range m/z 370–1880, 1 μscan/MS, AGC target 1e6, and a maximum injection time of 100 ms. MS2 scans were acquired with the following settings: 17 500 resolution at m/z 400, AGC target of 5e5, maximum injection time of 60 ms, isolation window of 2.0 m/z, isolation offset of 0.4 m/z, normalized collision energy (NCE) of 27%, exclusion of charge states 1 and >8, underfill ratio of 1.2%, and dynamic exclusion of 8 s. Profile data were recorded for MS and MS2 scans.
Glycopeptide nanoLC-MS/MS.
For the initial characterization of glycosylation, EGFR glycopeptides were enriched, separated, and analyzed using a 6550 Q-TOF MS with a 1200 series nanoflow HPLC-Chip-ESI source fitted with a custom HPLC-Chip with a 360 nL TSK Gel Amide-80 5 μm trapping and enrichment column and a 150 mM × 75 μm Polaris C18-A 3-μm analytical column (all from Agilent Corp., Santa Clara, CA). EGFR digests were initially dissolved in 50% acetonitrile with 0.1% TFA (55). Immediately prior to injection, the concentration of acetonitrile was adjusted to 80% acetonitrile and 0.1% TFA. After each injection of the sample onto the Amide-80 enrichment column, the column was washed at a flow rate of 1.5 μL/min for 4 min using 80% acetonitrile and 0.1% TFA, followed by elution of the sample onto the C18 analytical column with 1% acetonitrile and 0.1% formic acid. Finally, glycopeptides were separated on the analytical column using a gradient from 1% to 40% acetonitrile with 0.1% formic acid at a flow rate of 0.2 μL/min. The 6550 Q-TOF mass spectrometer was operated in positive mode using the high-resolution, extended dynamic range (2 GHz) setting. MS spectra were collected over the range m/z 150–2000, and MS2 spectra were collected from m/z 50–3000. The ion source gas temperature was set to 225 °C, and the flow was set at 11 L/min, with a capillary voltage of 1900 V. Precursors ≥1200 counts and charge states ≥2 were selected for fragmentation, and the collision energy was set according to the equation y = mx + b, with y being the collision energy, slope m = 5, x representing the charge state, and the offset b = −4.8. Spectra were collected in profile mode. The error for all peaks with signal-to-noise (S-to-N) ratio of >10 was within 5 ppm.
Glycopeptide Analysis for MS (only), HCD Oxonium Ion-Triggered Low-Energy HCD, and EThcD.
For MS-only, low-energy HCD, and EThcD analyses, EGFR tryptic peptides were analyzed on an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Scientific) equipped with an ACQUITY UPLC M-Class system (Waters) and a TriVersa NanoMate (Advion). For chromatographic separation, a nanoEase Symmetry C18 UPLC Trap Column (100 Å, 5 μm, 180 μm × 20 mm, Waters) was used for trapping, and a nanoEase MZ HSS C18 T3 UPLC Column (100 Å, 1.8 μm, 75 μm × 100 mm, Waters) was used for separation. The peptide trapping and separation were performed as described above for peptide/glycopeptide analysis. All analyses were performed in positive mode, with the RF lens set to 30%. For MS-only analyses, initial MS scans were acquired with the following settings: isolation window m/z 1.6, 30% collision energy, 120,000 resolution @ m/z 400, scan range m/z 500–2000, 100 ms maximum injection time, AGC target 4 × 105, and 2 μscans/MS. For low-energy HCD analyses, initial MS scans (30% collision energy) were acquired with the following settings: 30,000 resolution @ m/z 400, scan range m/z 100–2000, 1 μscan/MS, AGC target 5 × 105, and a maximum injection time of 60 ms. Based on the initial HCD (30%) MS2 scans, a second low-energy fragmentation step was triggered if at least two glycopeptide oxonium ions were detected (at m/z 204.0867, HexNAc; m/z 138.0545, HexNAc – CH6O3; and m/z 366.1396, HexNAc1Hex1) within a 15-ppm mass tolerance. For each sample, three independent analyses were performed, each with a different collision energy setting for the second (triggered) MS2: 10%, 15%, or 20% collision energy, based loosely on parameters published by L. Turiak, et al. (37) and M. Sanda, et al. (38). Triggered MS2 were acquired with the following parameters: 1/6 m/z isolation window, 30,000 resolution @ m/z 400, 150 ms injection time, AGC target 1 × 106, and 2 μscans/MS.
For EThcD analyses, MS scans were acquired with the following settings: positive mode, 120,000 resolution @ m/z 400, scan range m/z 350–2000, 1 μscan/MS, AGC target 4 × 105, and a maximum injection time of 50 ms. Data-dependent MS2 scans with fixed-energy HCD (30% NCE) fragmentation were performed following isolation in the quadrupole using an isolation window of 1.6 m/z. Fragment ions were detected in the Orbitrap in positive mode with 30,000 resolution @ m/z 400, 60 ms injection time, AGG target 5 × 105, 1 μscan/MS, scan range m/z 100–2000, and data were recorded as profile spectra. Based on the initial HCD MS2 scans, EThcD fragmentation was triggered if at least two glycopeptide oxonium ions were detected (at m/z 204.0867, HexNAc; m/z 138.0545, HexNAc – CH6O3; and m/z 366.1396, HexNAc1Hex1) within a 15-ppm mass tolerance. EThcD MS2 spectra were acquired after isolation in the quadrupole with the following settings: isolation window 1.6 m/z, a first mass of m/z 100, AGC target 1 × 106, 30,000 resolution @ m/z 400, 2 μscans/MS, and 150 ms maximum injection time.
Analysis of Peptide and Glycopeptide nUPLC-MS/MS Data.
To confirm protein identity, nano LC-MS/MS data were processed using Mascot search algorithm with the UniProtKB/Swiss-Prot Homo sapiens protein sequence database (modified September 29, 2019). For all data sets, cleavage rules were applied for each specific protease (trypsin: K, R, P1′ ≠ P) and peptides with up to two missed cleavages were considered. The following peptide modifications were considered: methionine oxidation (variable), deamidation (variable), deamidation with 18O (variable), and carbamidomethylation (fixed). To assign glycopeptide tandem mass spectra, nanoLC-MS/MS data were processed using Byonic (Protein Metrics), with a custom protein sequence database consisting of the forward and reverse protein sequence of human EGFR. Cleavage rules were consistent with those used in Mascot searches (trypsin). The following peptide modifications were considered: methionine oxidation (variable), carbamidomethylation (fixed), and an N-glycan database consisting of 300+ mammalian N-glycans, including multiply-fucosylated glycans.
N-glycan Release, Permethylation, and MALDI-TOF MS Analysis.
Three-hundred μg of whole cell lysates from CAL27 and HSC-3 cells treated either with DMSO (vehicle control) or 10 μM ICG-001, were subjected to N-glycan release via filter-aided N-glycan separation (56) with 500 units of PNGase F (NEB) in 50 mM ammonium bicarbonate buffer overnight at 37 °C. Samples were passed through SepPak C18 (1 cc) cartridges (Waters, Milford, MA, USA), and the flow-through was dried. Samples were resuspended in DMSO, then subjected to permethylation with periodic addition of methyl iodide (Sigma-Aldrich, St. Louis, MO, USA) according to an established protocol (57–60). Following permethylation, glycans were dried, then dissolved in 10% acetonitrile/90% water with 0.1% formic acid, then desalted using a C18 ZipTip™ (Millipore, Billerica, MA). Glycans were eluted into 10 μL of 60% acetonitrile/40% water containing 0.1% formic acid, and 1/10th of each sample was spotted onto a steel target plate with 0.5 uL of 1 mM sodium acetate, 0.5 μL 2,5-dihydroxybenzoic acid (DHB)(20 mg/mL), and dried under vacuum. MALDI-TOF MS analysis was performed on an UltrafleXtreme TOF/TOF mass spectrometer (Bruker Daltonics, Billerica, MA, USA) using 10% laser power and summing 500 shots/spectrum.
Supplementary Material
Figure 7: Model: Antennary-Fucosylated N-glycans Stabilize the Auto-Inhibited EGFR Conformation and Suppress EGFR Signaling.
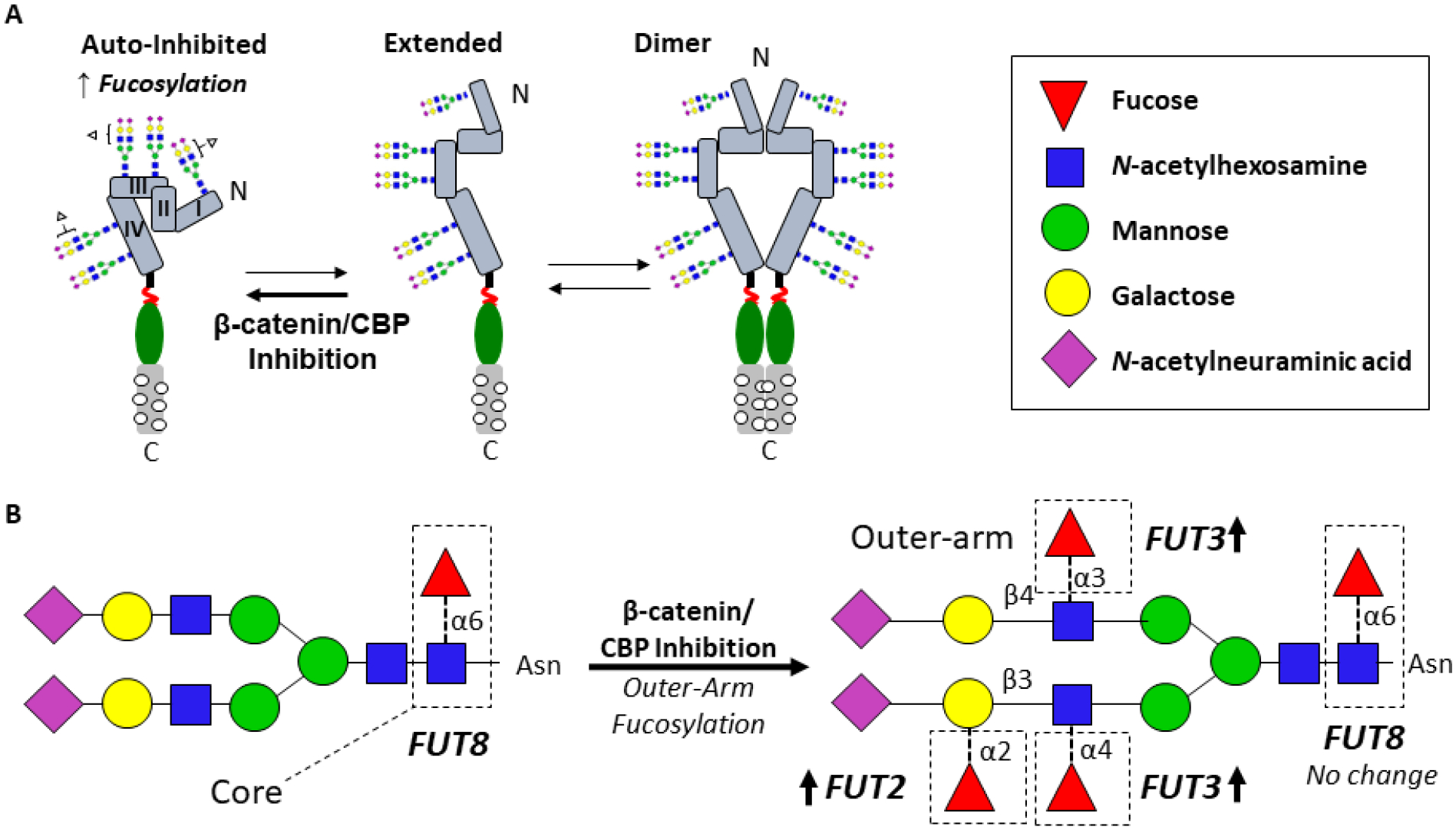
A. Proposed model, whereby EGFR antennary-fucosylated N-glycans stabilize the auto-inhibited conformation of the receptor, preventing dimerization and activation. Relief from suppression may drive aggressive OSCC phenotypes. B. β-catenin/CBP inhibition leads to higher expression of FUT2 and FUT3, involved in the synthesis of antennary-fucosylated glycans, such as the structure shown, thereby suppressing EGFR antennary fucosylation and promoting the auto-inhibited conformation.
Acknowledgements
Supported by NIH grants P41 GM104603, R24 GM134210, S10 OD010724, and S10 OD021728 (to C. E. C.), F32 CA196157 (to K. B. C.), R21 DE026892 (M.V.B), the Undergraduate Research Opportunities Program (UROP) of Boston University (to V. L. S.), and by the Evans Center for Interdisciplinary Biomedical Research ARC #9950000118 (to M. A. K.).
Footnotes
Data Availability
Peptide, glycopeptide and formerly-N-glycosylated peptide mass spectrometry data have been deposited to the ProteomeXchange Consortium (61) via the PRIDE (62) partner repository with the dataset identifier PXD017060, in compliance with MIRAGE standards (63).
Disclosure: Trustees of Boston University (“Boston University”) and Eisai, Inc. (“Eisai”) have executed a Material Transfer Agreement (“MTA”) in which Dr Kukuruzinska is a named investigator and a Sponsored Research Agreement (“SRA”) in which Dr Kukuruzinska is the named principal investigator. All other authors declare that they have no conflicts of interest.
Animal Studies
All experiments involving mice were approved by the Boston University Medical Center Institutional Animal Care and Use Committee. All institutional and national guidelines for care and use of laboratory animals were followed.
References:
- 1.Siegel RL, Miller KD, and Jemal A (2019) Cancer statistics, 2019. CA: A Cancer Journal for Clinicians 69, 7–34 [DOI] [PubMed] [Google Scholar]
- 2.Vigneswaran N, and Williams MD (2014) Epidemiologic trends in head and neck cancer and aids in diagnosis. Oral Maxillofacial Surg Clinics of North America 26, 123–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, and Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians 68, 394–424 [DOI] [PubMed] [Google Scholar]
- 4.Marur S, and Forastiere AA (2016) Head and Neck Squamous Cell Carcinoma: Update on Epidemiology, Diagnosis, and Treatment. Mayo Clinic Proceedings 91, 386–396 [DOI] [PubMed] [Google Scholar]
- 5.Varelas X, and Kukuruzinska MA (2014) Head and neck cancer: from research to therapy and cure. Ann. NY Acad Sci 1333, 1–32 [DOI] [PubMed] [Google Scholar]
- 6.Kartha VK, Alamoud KA, Sadykov K, Nguyen BC, Laroche F, Feng H, Lee J, Pai SI, Varelas X, Egloff AM, Snyder-Cappione JE, Belkina AC, Bais MV, Monti S, and Kukuruzinska MA (2018) Functional and genomic analyses reveal therapeutic potential of targeting beta-catenin/CBP activity in head and neck cancer. Genome Medicine 10, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, and Gutkind JS (2009) Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncology 45, 324–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, Carter SL, Voet D, Cortes ML, Auclair D, Berger MF, Saksena G, Guiducci C, Onofrio RC, Parkin M, Romkes M, Weissfeld JL, Seethala RR, Wang L, Rangel-Escareno C, Fernandez-Lopez JC, Hidalgo-Miranda A, Melendez-Zajgla J, Winckler W, Ardlie K, Gabriel SB, Meyerson M, Lander ES, Getz G, Golub TR, Garraway LA, and Grandis JR (2011) The mutational landscape of head and neck squamous cell carcinoma. Science 333, 1157–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, Zhang N, El-Naggar AK, Jasser SA, Weinstein JN, Trevino L, Drummond JA, Muzny DM, Wu Y, Wood LD, Hruban RH, Westra WH, Koch WM, Califano JA, Gibbs RA, Sidransky D, Vogelstein B, Velculescu VE, Papadopoulos N, Wheeler DA, Kinzler KW, and Myers JN (2011) Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 333, 1154–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lui VW, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y, Lu Y, Zhang Q, Du Y, Gilbert BR, Freilino M, Sauerwein S, Peyser ND, Xiao D, Diergaarde B, Wang L, Chiosea S, Seethala R, Johnson JT, Kim S, Duvvuri U, Ferris RL, Romkes M, Nukui T, Kwok-Shing Ng P, Garraway LA, Hammerman PS, Mills GB, and Grandis JR (2013) Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discovery 3, 761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson DE, O’Keefe RA, and Grandis JR (2018) Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nature reviews. Clinical Oncology 15, 234–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tian Y, Lin J, Tian Y, Zhang G, Zeng X, Zheng R, Zhang W, and Yuan Y (2018) Efficacy and safety of anti-EGFR agents administered concurrently with standard therapies for patients with head and neck squamous cell carcinoma: a systematic review and meta-analysis of randomized controlled trials. Int J Cancer 142, 2198–2206 [DOI] [PubMed] [Google Scholar]
- 13.Ribeiro FA, Noguti J, Oshima CT, and Ribeiro DA (2014) Effective targeting of the epidermal growth factor receptor (EGFR) for treating oral cancer: a promising approach. Anticancer Res 34, 1547–1552 [PubMed] [Google Scholar]
- 14.Xu MJ, Johnson DE, and Grandis JR (2017) EGFR-targeted therapies in the post-genomic era. Cancer Metastasis Rev 36, 463–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christiansen MN, Chik J, Lee L, Anugraham M, Abrahams JL, and Packer NH (2014) Cell surface protein glycosylation in cancer. Proteomics 14, 525–546 [DOI] [PubMed] [Google Scholar]
- 16.Chandler KB, Leon DR, Kuang J, Meyer RD, Rahimi N, and Costello CE (2019) N-Glycosylation regulates ligand-dependent activation and signaling of vascular endothelial growth factor receptor 2 (VEGFR2). J Biol Chem 294, 13117–13130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, Nabi IR, Wrana JL, and Dennis JW (2004) Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science 306, 120–124 [DOI] [PubMed] [Google Scholar]
- 18.Dennis JW, Lau KS, Demetriou M, and Nabi IR (2009) Adaptive regulation at the cell surface by N-glycosylation. Traffic 10, 1569–1578 [DOI] [PubMed] [Google Scholar]
- 19.Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, and Dennis JW (2007) Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 129, 123–134 [DOI] [PubMed] [Google Scholar]
- 20.Fernandes H, Cohen S, and Bishayee S (2001) Glycosylation-induced conformational modification positively regulates receptor-receptor association: a study with an aberrant epidermal growth factor receptor (EGFRvIII/DeltaEGFR) expressed in cancer cells. J Biol Chem 276, 5375–5383 [DOI] [PubMed] [Google Scholar]
- 21.Liu YC, Yen HY, Chen CY, Chen CH, Cheng PF, Juan YH, Chen CH, Khoo KH, Yu CJ, Yang PC, Hsu TL, and Wong CH (2011) Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc Natl Acad Sci U S A 108, 11332–11337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yen HY, Liu YC, Chen NY, Tsai CF, Wang YT, Chen YJ, Hsu TL, Yang PC, and Wong CH (2015) Effect of sialylation on EGFR phosphorylation and resistance to tyrosine kinase inhibition. Proc Natl Acad Sci U S A 112, 6955–6960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandler KB, and Costello CE (2016) Glycomics and glycoproteomics of membrane proteins and cell-surface receptors: Present trends and future opportunities. Electrophoresis 37, 1407–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nacev BA, Grassi P, Dell A, Haslam SM, and Liu JO (2011) The antifungal drug itraconazole inhibits vascular endothelial growth factor receptor 2 (VEGFR2) glycosylation, trafficking, and signaling in endothelial cells. J Biol Chem 286, 44045–44056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banerjee A, Lang JY, Hung MC, Sengupta K, Banerjee SK, Baksi K, and Banerjee DK (2011) Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. J Biol Chem 286, 29127–29138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Contessa JN, Bhojani MS, Freeze HH, Rehemtulla A, and Lawrence TS (2008) Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res 68, 3803–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez-Sambrooks C, Shrimal S, Khodier C, Flaherty DP, Rinis N, Charest JC, Gao N, Zhao P, Wells L, Lewis TA, Lehrman MA, Gilmore R, Golden JE, and Contessa JN (2016) Oligosaccharyltransferase inhibition induces senescence in RTK-driven tumor cells. Nat Chem Biol 12, 1023–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopez Sambrooks C, Baro M, Quijano A, Narayan A, Cui W, Greninger P, Egan R, Patel A, Benes CH, Saltzman WM, and Contessa JN (2018) Oligosaccharyltransferase Inhibition Overcomes Therapeutic Resistance to EGFR Tyrosine Kinase Inhibitors. Cancer Res 78, 5094–5106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Gu J, Ihara H, Miyoshi E, Honke K, and Taniguchi N (2006) Core fucosylation regulates epidermal growth factor receptor-mediated intracellular signaling. J Biol Chem 281, 2572–2577 [DOI] [PubMed] [Google Scholar]
- 30.Teo JL, and Kahn M (2010) The Wnt signaling pathway in cellular proliferation and differentiation: A tale of two coactivators. Advanced Drug Deliv Rev 62, 1149–1155 [DOI] [PubMed] [Google Scholar]
- 31.Nita-Lazar M, Noonan V, Rebustini I, Walker J, Menko AS, and Kukuruzinska MA (2009) Overexpression of DPAGT1 leads to aberrant N-glycosylation of E-cadherin and cellular discohesion in oral cancer. Cancer Res 69, 5673–5680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sengupta PK, Bouchie MP, and Kukuruzinska MA (2010) N-glycosylation gene DPAGT1 is a target of the Wnt/beta-catenin signaling pathway. J Biol Chem 285, 31164–31173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jamal B, Sengupta PK, Gao ZN, Nita-Lazar M, Amin B, Jalisi S, Bouchie MP, and Kukuruzinska MA (2012) Aberrant amplification of the crosstalk between canonical Wnt signaling and N-glycosylation gene DPAGT1 promotes oral cancer. Oral Oncology 48, 523–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, Moon RT, Teo JL, Kim HY, Moon SH, Ha JR, and Kahn M (2004) A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc Natl Acad Sci U S A 101, 12682–12687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamada K, Hori Y, Yamaguchi A, Matsuki M, Tsukamoto S, Yokoi A, Semba T, Ozawa Y, Inoue S, Yamamoto Y, Iso K, Nakamoto K, Harada H, Yoneda N, Takemura A, Matsukura M, Kubara K, Odagami T, Iwata M, Tsuruoka A, Uenaka T, Matsui J, Matsushima T, Nomoto K, Kouji H, and Owa T (2017) Abstract 5177: E7386 : First-in-class orally active CBP/beta-catenin modulator as an anticancer agent. Cancer Research 77, 5177–5177 [Google Scholar]
- 36.Bais MV, Kukuruzinska M, and Trackman PC (2015) Orthotopic non-metastatic and metastatic oral cancer mouse models. Oral Oncology 51, 476–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Acs A, Ozohanics O, Vekey K, Drahos L, and Turiak L (2018) Distinguishing Core and Antenna Fucosylated Glycopeptides Based on Low-Energy Tandem Mass Spectra. Anal Chem 90, 12776–12782 [DOI] [PubMed] [Google Scholar]
- 38.Yuan W, Wei R, Goldman R, and Sanda M (2019) Optimized Fragmentation for Quantitative Analysis of Fucosylated N-Glycoproteins by LC-MS-MRM. Anal Chem 91, 9206–9212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hogan JM, Pitteri SJ, Chrisman PA, and McLuckey SA (2005) Complementary structural information from a tryptic N-linked glycopeptide via electron transfer ion/ion reactions and collision-induced dissociation. J Proteome Res 4, 628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harvey DJ, Mattu TS, Wormald MR, Royle L, Dwek RA, and Rudd PM (2002) “Internal residue loss”: rearrangements occurring during the fragmentation of carbohydrates derivatized at the reducing terminus. Anal Chem 74, 734–740 [DOI] [PubMed] [Google Scholar]
- 41.Brüll LP, Heerma W, Thomas-Oates J, Haverkamp J, Kovácik V, and Kovác P (1997) Loss of internal 1 → 6 substituted monosaccharide residues from underivatized and per-O-methylated trisaccharides. J Amer Soc Mass Spectrom 8, 43–49 [Google Scholar]
- 42.Wuhrer M, Deelder AM, and van der Burgt YE (2011) Mass spectrometric glycan rearrangements. Mass Spectrom Rev 30, 664–680 [DOI] [PubMed] [Google Scholar]
- 43.Ernst B, Müller DR, and Richter WJ (1997) False sugar sequence ions in electrospray tandem mass spectrometry of underivatized sialyl-Lewis-type oligosaccharides. International Mass Spectrom Ion Process 160, 283–290 [Google Scholar]
- 44.Liu L, Yang Y, Liu S, Tao T, Cai J, Wu J, Guan H, Zhu X, He Z, Li J, Song E, Zeng M, and Li M (2019) EGF-induced nuclear localization of SHCBP1 activates beta-catenin signaling and promotes cancer progression. Oncogene 38, 747–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu T, and Li C (2010) Convergence between Wnt-beta-catenin and EGFR signaling in cancer. Molecular Cancer 9, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campi C, Escovich L, Moreno A, Racca L, Racca A, Cotorruelo C, and Biondi C (2012) Expression of the gene encoding secretor type galactoside 2 alpha fucosyltransferase (FUT2) and ABH antigens in patients with oral lesions. Medicina oral, patologia oral y cirugia bucal 17, e63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su KJ, Ho CC, Lin CW, Chen MK, Su SC, Yu YL, and Yang SF (2016) Combinations of FUT2 gene polymorphisms and environmental factors are associated with oral cancer risk. Tumour biology: J Intl Soc Oncodevelopmental Biol and Med 37, 6647–6652 [DOI] [PubMed] [Google Scholar]
- 48.Desiderio V, Papagerakis P, Tirino V, Zheng L, Matossian M, Prince ME, Paino F, Mele L, Papaccio F, Montella R, Papaccio G, and Papagerakis S (2015) Increased fucosylation has a pivotal role in invasive and metastatic properties of head and neck cancer stem cells. Oncotarget 6, 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hotta H, Hamamura K, Yamashita K, Shibuya H, Tokuda N, Hashimoto N, Furukawa K, Yamamoto N, Hattori H, Toyokuni S, Ueda M, and Furukawa K (2013) Lewis y antigen is expressed in oral squamous cell carcinoma cell lines and tissues, but disappears in the invasive regions leading to the enhanced malignant properties irrespective of sialyl-Lewis x. Glycoconj J 30, 585–597 [DOI] [PubMed] [Google Scholar]
- 50.Tsuda T, Ikeda Y, and Taniguchi N (2000) The Asn-420-linked sugar chain in human epidermal growth factor receptor suppresses ligand-independent spontaneous oligomerization. Possible role of a specific sugar chain in controllable receptor activation. J Biol Chem 275, 21988–21994 [DOI] [PubMed] [Google Scholar]
- 51.Whitson KB, Whitson SR, Red-Brewer ML, McCoy AJ, Vitali AA, Walker F, Johns TG, Beth AH, and Staros JV (2005) Functional effects of glycosylation at Asn-579 of the epidermal growth factor receptor. Biochemistry 44, 14920–14931 [DOI] [PubMed] [Google Scholar]
- 52.Miller MA, Oudin MJ, Sullivan RJ, Wang SJ, Meyer AS, Im H, Frederick DT, Tadros J, Griffith LG, Lee H, Weissleder R, Flaherty KT, Gertler FB, and Lauffenburger DA (2016) Reduced Proteolytic Shedding of Receptor Tyrosine Kinases Is a Post-Translational Mechanism of Kinase Inhibitor Resistance. Cancer Discovery 6, 382–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shen Y, Rahman M, Piccolo SR, Gusenleitner D, El-Chaar NN, Cheng L, Monti S, Bild AH, and Johnson WE (2015) ASSIGN: context-specific genomic profiling of multiple heterogeneous biological pathways. Bioinformatics (Oxford, England) 31, 1745–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khatri K, Staples GO, Leymarie N, Leon DR, Turiak L, Huang Y, Yip S, Hu H, Heckendorf CF, and Zaia J (2014) Confident assignment of site-specific glycosylation in complex glycoproteins in a single step. J Proteome Res 13, 4347–4355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abdul Rahman S, Bergstrom E, Watson CJ, Wilson KM, Ashford DA, Thomas JR, Ungar D, and Thomas-Oates JE (2014) Filter-aided N-glycan separation (FANGS): a convenient sample preparation method for mass spectrometric N-glycan profiling. J Proteome Res 13, 1167–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang P, Mechref Y, Klouckova I, and Novotny MV (2005) Solid-phase permethylation of glycans for mass spectrometric analysis. Rapid Commun Mass Spectrom 19, 3421–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kang P, Mechref Y, and Novotny MV (2008) High-throughput solid-phase permethylation of glycans prior to mass spectrometry. Rapid Commun Mass Spectrom 22, 721–734 [DOI] [PubMed] [Google Scholar]
- 59.Ciucanu I, F. K (1984) A Simple and Rapid Method for the Permethylation of Carbohydrates. Carbohydr Res 131, 209–217 [Google Scholar]
- 60.Ciucanu I, and Costello CE (2003) Elimination of oxidative degradation during the per-O-methylation of carbohydrates. J Am Chem Soc 125, 16213–16219 [DOI] [PubMed] [Google Scholar]
- 61.Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T, Campbell DS, Bernal-Llinares M, Okuda S, Kawano S, Moritz RL, Carver JJ, Wang M, Ishihama Y, Bandeira N, Hermjakob H, and Vizcaino JA (2017) The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res 45, D1100–d1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vizcaino JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, Xu QW, Wang R, and Hermjakob H (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44, D447–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Struwe WB, Agravat S, Aoki-Kinoshita KF, Campbell MP, Costello CE, Dell A, Ten F, Haslam SM, Karlsson NG, Khoo KH, Kolarich D, Liu Y, McBride R, Novotny MV, Packer NH, Paulson JC, Rapp E, Ranzinger R, Rudd PM, Smith DF, Tiemeyer M, Wells L, York WS, Zaia J, and Kettner C (2016) The minimum information required for a glycomics experiment (MIRAGE) project: sample preparation guidelines for reliable reporting of glycomics datasets. Glycobiology 26, 907–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmitz KR, Bagchi A, Roovers RC, van Bergen en Henegouwen PM, and Ferguson KM (2013) Structural evaluation of EGFR inhibition mechanisms for nanobodies/VHH domains. Structure (London, England : 1993) 21, 1214–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.