

Abstract
Ovarian cancer is the most lethal gynecological malignancy. Abnormal homologous recombination repair, high level of reactive oxygen species (ROS) and upregulation of antioxidant genes are characteristic features of ovarian cancer. However, the molecular mechanisms governing the redox homeostasis in ovarian cancer cells remain to be fully elucidated. We here demonstrated a critical role of RAD51, a protein essential for homologous recombination, in the maintenance of redox homeostasis. We found that RAD51 is overexpressed in high grade serous ovarian cancer and is associated with poor prognosis. Depletion or inhibition of RAD51 results in G2/M arrest, increased production of reactive oxygen species and accumulation of oxidative DNA damage. Importantly, antioxidant N-acetylcysteine (NAC) significantly attenuated the induction of DNA damage and the perturbation of proliferation caused by RAD51 depletion. We further demonstrated that RAD51 inhibition or depletion led to elevated production of mitochondrial superoxide and increased accumulation of mitochondria. Moreover, CHK1 activation is required for the G2/M arrest and the generation of mitochondrial stress in response to RAD51 depletion. Together, our results indicate that nuclear DNA damage caused by RAD51 depletion may trigger mitochondria-originated redox dysregulation. Our findings suggest that a vicious cycle of nuclear DNA damage, mitochondrial accumulation and oxidative stress may contribute to the tumor-suppressive effects of RAD51 depletion or inhibition.
Keywords: RAD51, Redox homeostasis, G2/M arrest, CHK1, Mitochondria stress, Ovarian cancer
Abbreviations: HGSOC, High grade serous ovarian cancer; FT, fallopian tube epithelia; HRR, homologous recombination repair; DSBs, double strand breaks; CHK1, checkpoint kinase 1; ROS, reactive oxygen species; NAC, N-acetylcysteine; OCR, oxygen consumption rate; ECAR, extracellular acidification rate; MMP, mitochondrial membrane potential; TMA, tissue microarray; CCCP, Carbonyl cyanide 3-chlorophenylhydrazone; MitoQ, mitoquinone; mtDNA, mitochondrial DNA
Graphical abstract
Highlights
-
•
RAD51 is overexpressed in ovarian cancer and is associated with poor prognosis.
-
•
Depletion of RAD51 leads to increased mitochondrial superoxide production and oxidative DNA damage.
-
•
Increased production of mitochondrial ROS requires CHK1-mediated G2/M arrest.
-
•
mROS increase is independent of mtDNA.
1. Introduction
Ovarian cancer is the most lethal gynecological malignancy. Because of lack of biomarkers and symptoms at early stage, most patients are diagnosed at advanced stage. Debulking surgery followed by platinum-taxane based chemotherapy is the standard of care for patients with advanced stage ovarian cancer [1,2]. However, most patients relapse with chemoresistance [2] and the overall 5-year survival is only about 30% [2,3]. The high response rate of ovarian cancer to platinum analogues is believed to be due to a high prevalence of defective homologous recombination repair (HRR) [3]. HRR plays a critical role for error-free repair of DNA double-strand breaks. The recombinase activity of the RAD51 protein forms the catalytic core of the HRR pathway for the repair of DSBs [4]. In addition, RAD51-mediated HR plays a critical role in facilitating efficient DNA replication in mammalian cells, particularly when under genotoxic stress [[5], [6], [7], [8]]. Many studies have shown that RAD51 is overexpressed in a variety of tumors [[9], [10], [11], [12], [13], [14], [15], [16], [17], [18]] and the high-expression of RAD51 is associated with poor prognosis [16,19,20]. RAD51 overexpression contributes to chemotherapy resistance in human soft tissue sarcoma cells [15] while RAD51 suppression leads to increased sensitivity to chemotherapeutics like cisplatin and PARP inhibitors, and microRNAs that target RAD51 can also sensitize cancer cells to chemotherapeutic agents [[21], [22], [23], [24], [25]]. It is also documented that RAD51 overexpression results in an increased resistance to radiation [13,14,26]. Because RAD51 is the key catalytic protein in HRR, it is reasonable to assume that the chemo- or radioresistance of cancer cells conferred by RAD51 overexpression may be related to an increased DNA repair capacity. However, BRCA1, another key player in HRR, was recently shown to promote antioxidant defense by upregulating NRF2 [27]. In addition, the cellular responses to defective HRR remain to be fully elucidated. Recent studies indicate that nuclear DNA damage can trigger mitochondrial response, and increased production of mitochondrial reactive oxygen species may exacerbate nuclear DNA damage [28,29].
Abnormal HRR, high level of reactive oxygen species (ROS) and upregulation of antioxidant genes are characteristic features of ovarian cancer [3,[30], [31], [32], [33], [34]]. While it is easy to understand that an increased antioxidant capacity may be necessary for the ovarian cancer cells to cope with the increased oxidative stress, whether the HRR proteins may indirectly contribute to the maintenance of redox homeostasis remains to be determined. Would defective HRR lead to mitochondrial response? In the present study, we investigated the role of RAD51 in the redox regulation in ovarian cancer cells. We observed that RAD51 is overexpressed in high-grade serous ovarian carcinoma compared to fallopian tubes and RAD51 depletion significantly impaired the proliferation of ovarian cancer cells. Importantly, we found that the tumor-suppressive effect of RAD51 inhibition or depletion is partly attributable to increased oxidative stress. RAD51 depletion results in increase of mitochondrial superoxide, reduced mitochondrial membrane potential (MMP) and accumulation of mitochondria. However, the mitochondrial accumulation caused by RAD51 depletion appeared to be a response to nuclear DNA damage, but not related to mtDNA. Our findings reveal a novel role of RAD51 in sustaining the malignancy of ovarian cancer and have implications in the development of new therapeutic strategies for ovarian cancer.
2. Materials and methods
2.1. Tissue samples for real-time PCR
All evaluated HGSOC and fallopian tube tissues which were collected in Qilu Hospital of Shandong University from April 2009 to July 2015. The HGSOC specimens were obtained from primary ovarian cancer patients receiving no previous surgery or chemotherapy. Fallopian tube tissues were from patients who received a total hysterectomy and bilateral salpingo-oophorectomy for uterine diseases or for benign neoplastic adnexal pathologic changes. The archived tissue sections were collected and prepared for tissue microarray (TMA) as previously described [35]. Ethics Committee of Shandong University approved the study and all participants gave written informed consent.
2.2. Tissue microarray for immunohistochemistry
Formalin-fixed and paraffin-embedded tissues were sectioned at 4 μm. Tissue slides were deparaffinized in xylene and rehydrated in a graded series of ethanol. Antigen retrieval was performed by microwave irradiation in citrate retrieval solution (pH 6.0). Nonspecific binding was blocked with donkey serum and sections were then incubated with anti-RAD51 antibody (1:200, Abcam, Cambridge, UK) overnight at 4 °C. The signal was developed by DAB detection system and counterstained with hematoxylin. The staining results were independently evaluated by two investigators blinded to the clinical data.
2.3. Cell culture
FTE-187 (immortalized normal human fallopian tube epithelial cell line) cells were as previously described [36]. A2780, HO8910 and HEK293T cell lines were purchased from China Type Culture Collection (Shanghai, China). Human osteosarcoma cell line U2OS and human breast cancer cells MDA-MB-231 were obtained from the American Type Culture Collection (Manassas, VA). HEY, SKOV3 cell lines were purchased from American Type Culture Collection. 143B.TK ρ0 cells were generously provided by Dr. Chuanzhu Yan at Shandong University. Ovarian cancer cell line SKOV3 cells were cultured in McCoy's 5A modified medium (Gibco, Invitrogen, Carlsbad, CA). A2780, HEY, U2OS, MDA-MB-231 and HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco, Invitrogen). HO8910 cells were cultured in RPMI 1640 medium (Gibco, Invitrogen). FTE187 cells were maintained in cell culture medium consisting of 1:1 Medium199 and MCDB105 medium (Sigma-Aldrich) as previously described [36]. 143B.TK ρ0 cells were cultured in DMEM medium supplied with 50 μg/mL uridine. All media contained 10% fetal bovine serum (Gibco, Invitrogen), 100 units/mL penicillin, and 100 μg/mL streptomycin. All cells were cultured in a humidified atmosphere of 5% CO2 at 37 °C.
2.4. Reagents and antibodies
Antioxidant N-acetylcysteine (A7250), Uridine (U3750), B02 (RAD51 inhibitor, 553525) and UCN-01 (CHK1 inhibitor, U6508) were purchased from Sigma-Aldrich. Mitoquinone mesylate (HY-100116A) was purchased from Medchem Express. Mounting Medium with DAPI (ab104139) was purchased from Abcam. MitoSOX™ Red Mitochondrial Superoxide Indicator (M36008) and MitoTracker™ Deep Red FM (M22426) were from Invitrogen Life Technologies. Mitochondria isolation kit were purchased from Beyotime (C3601). Mitochondria staining kit (JC-1) was purchased from MultiSciences Biotech (MJ101, China). CCCP (Carbonyl cyanide 3-chlorophenylhydrazone) was purchased from MultiSciences Biotech. CCK-8 was from Vazyme (Vazyme Biotech Co.,Ltd). Antibody against RAD51 (ab133534) was from Abcam. Antibodies against phospho-CHK1 (2348), Ki-67 (9129) and γ-H2AX (9718) were from Cell Signaling Technology. Anti-8-OHdG (sc-393871), anti-CHK1 (sc-8408) were acquired from Santa cruz Biotechnology. anti-GAPDH (60004-1-Ig), anti-Alpha-Tubulin (66031-1-Ig) and anti-COX IV (11242-1-AP) were from Proteintech.
2.5. RNA isolation and real-time quantitative PCR
Total RNA was extracted from fresh tissues with Trizol reagents (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions. mRNAs were reverse transcribed to generate complementary DNA (cDNA) using oligo (dT) primers. GAPDH was considered as the endogenous control. Quantitative reverse transcriptase-polymerase chain reaction was performed using LightCycler Fast Start DNA Master SYBR Green I on a LightCycler (Roche Diagnostics GmbH, Mannheim, Germany) system. The comparative cycle threshold method (ΔΔCt) was used to quantify the relative expression of each gene. The samples were loaded in triplicate, and the results of each sample were normalized to GAPDH. The primers used in the present study were listed in Supplementary Table S1.
2.6. Western blot analysis
Protein samples were separated by SDS-PAGE (6–15%) and electro-transferred onto PVDF membrane, which was then blocked with 5% no fat milk for 60 min. The membrane was then incubated with specific primary antibodies overnight at 4 °C. 30–60 μg proteins of interest were detected with appropriate horseradish peroxidase-conjugated secondary antibodies for 60 min at room temperature and developed using the ECL-enhanced luminol reagent (Thermo). The protein levels were normalized by α-Tubulin or GAPDH.
2.7. RNA interference
All small interfering RNAs (siRNAs) were purchased from Sigma-Aldrich. The siRNA targeting human RAD51 was applied as a mixture at a total final concentration of 50 nM. Cells were transfected with siRNAs using Liofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. siRNA sequences were listed in Supplementary Table S2.
2.8. Cell proliferation and clonogenic assay
CCK-8 assay was used to examine cell viability. In brief, cells were seeded in 96-well culture plates at 3 × 103/well the day before NAC or CHK1 inhibitor treatment. Then cells were cultured for 48 h in a humidified atmosphere of 5% CO2 at 37 °C. After treatment, 10 μL of CCK-8 reagent was added to each well and incubated for 3 h at 37 °C. The absorbance of each well was measured at 450 nm. All experiments were repeated at least three times. Clonogenic assay was performed as described previously [23]. Briefly, single-cell suspensions were prepared for each cell line and specific numbers of cells were seeded in 6-well tissue culture plates. Then cells were cultured in a humidified atmosphere of 5% CO2 at 37 °C for two weeks. In the end, the media were carefully removed by aspiration. Then, colonies were stained by crystal violet, photographed, and scored. Only colonies of more than 50 cells were counted.
2.9. Cell cycle analysis
Cells were transfected with scrambled siRNA (siScr) or siRNA targeting RAD51 (siRAD51) for 48 h. Then cells were washed three times with PBS, trypsinized, and collected by centrifugation at 800 rpm for 5 min. Then cells were fixed in 4 mL of cold 75% ethanol at −20 °C overnight. After centrifugation at 1600 rpm for 10 min, cells were washed by 4 mL cold PBS and centrifugated again, cell pellets were resuspended and incubated with 0.5 mL of PBS containing 100 μg/mL RNase (Sangon Biotech) and 5 μg/mL propidium iodide (Beyotime) at room temperature for 30 min. Cell cycle distribution was analyzed by flow cytometry (BD Biosciences, San Jose, CA).
2.10. Measurement of ROS, MitoSOX and mitochondrial mass
ROS generation was measured using oxidation sensitive fluorescent probe (DCFH-DA) according to the manufacturer's protocols (Beyotime, China). Cells were transfected scrambled siRNA or siRNA targeting RAD51 for 48 h. Then cells were harvested and stained with 10 μM DCFH-DA probe at 37 °C for 20 min. Cells were washed three times with PBS and the induction of ROS was examined by flow cytometry (BD Biosciences, San Jose, CA). Mitochondrial ROS were measured using MitoSOX following the manufacturer's instructions. For measurement of mitochondrial mass, cells were collected and resuspended in preheated medium with 100 nM Mitotracker Red incubated in a humidified 5% CO2 atmosphere at 37 °C for 30 min, then cells were rinsed and suspended in preheated medium and measured by flow cytometry. In all experiments, 20, 000 viable cells were analyzed.
2.11. EdU proliferation assay
Cells transfected scrambled siRNA or siRNA targeting RAD51 were replated in 96-well plate and incubated for 48 h, cell proliferation was detected using the incorporation of 5-ethynyl-2′-deoxyuridine (EdU) with the EdU cell proliferation assay kit (Guangzhou RiboBio Co., Ltd. Guangzhou, China). Briefly, the cells were incubated with 50 μM EdU for 2 h before fixation, permeabilization and EdU staining according to the manufacturer's protocol. The nuclei were stained with DAPI. The proportion of EdU positive cells was determined by fluorescence microscopy. EdU proliferation assay kit for flow cytometry was purchased from Beyotime (China) and measured following the manufacturer's instructions.
2.12. Lentivirus production and infection
RAD51 short hairpin RNA (shRNA) in pZIP-TRE3GS was purchased from BioSune (Shanghai, China). Lentivirus-expressed shRAD51 was produced in HEK293T cells packaged by pMD2G and psPAX2. For stable infection, 6 × 104 cells were plated in six-well plates in 2 mL of medium without antibiotics. After overnight incubation, the medium was removed and replaced with 1 mL per well of medium containing 8 μg/mL polybreen. Then, 50 μL of concentrated retroviral particles was added into each well. 48 h later, fresh medium containing 2 μg/mL puromycin was added into each well. Fresh medium containing puromycin was replaced every 3–4 days. Single colonies were obtained after 2 weeks of puromycin selection.
2.13. Xenografts in nude mice
Female BALB/c nude mice (4-6-week old) were purchased from Beijing Experimental Animal Center and kept in pathogen-free conditions and handled in accordance with the requirements of the Guideline for Animal Experiments. For xenograft experiments, equal number (5 × 106) of HO8910 shRAD51 cells were harvested and resuspended in 0.1 mL of PBS for subcutaneous injection. Animals were randomly divided into two groups (n = 4–6 for each group) and treated with either control (H2O) or doxycycline (1.2 mg/mL) given daily. Tumor growth was monitored with a caliper. Tumor volume was calculated with the use of the following formula: tumor volume (in mm3) = a × b2 × π/6, where ‘a’ is the longest diameter, ‘b’ is the shortest diameter. After the last treatment on day 24, tumors were removed and weighed. All tumors for each group were excised, fixed in 10% formalin, and subjected to routine histologic examination and immunostaining of 8-OHdG and Ki-67.
2.14. Immunofluorescence staining of DNA damage markers
Immunofluorescence staining was carried out as described previously [37]. Briefly, cells seeded on coverslips were fixed in 4% paraformaldehyde for 10 min. The cells were then permeabilized in 0.2% Triton X-100 for 10 min, and blocked in 10% normal goat serum overnight at 4 °C. The coverslips were incubated with 8-OHdG antibody overnight at 4 °C, washed with PBS, and incubated with TRITC-conjugated Goat anti-mouse secondary antibody (Jackson Immuno Research Laboratories, West Grove, PA) for 1 h at room temperature. Cells were washed in PBS three times and counterstained with DAPI. Fluorescence images were captured under a fluorescence microscope.
2.15. Immunohistochemistry of tumor xenografts
Formalin-fixed and paraffin-embedded tumor xenografts were sectioned at 4 μm. After deparaffinizayion and rehydration, the sections were boiled in citrate sodium buffer (pH 6.0) for 15 min for antigen recovery, and immersed in 3% H2O2 for 10 min to quench endogenous peroxidase. Nonspecific binding was blocked by 10% donkey serum at room temperature for 1 h. Then sections were incubated with the primary antibodies (Ki-67, CST, 1:200 dilution; 8-OHdG, Santa Cruz, 1:50 dilution) overnight at 4 °C. The signal was developed by DAB detection system and counterstained with hematoxylin.
2.16. Determination of mitochondrial membrane potential (MMP)
The mitochondrial membrane potential (MMP) was determined by Mitochondria Staining Kit (JC-1, MultiSciences Biotech, China). Briefly, ovarian cancer cells were harvested and 105 cells were loaded with JC-1 (2 μM) at 37 °C, 5% CO2 for 20 min, then analyzed by flow cytometry (BD Biosciences, San Jose, CA) excited at 488 nm and the emitted fluorescence was collected at 530 nm and 590 nm. MMP was expressed as the emitted fluorescence ratio (590/530 nm) in percentage to the initial level, whereas 0% level was recorded after complete mitochondrial depolarization with CCCP.
2.17. Measurement of oxygen consumption rate and glycolytic capacity
The real-time measurement of oxygen consumption rate (OCR) in live cultured ovarian cancer cells was performed using the Seahorse XF24 Analyzer (Seahorse Bioscience, North Billerica, MA, USA) according to the manufacturer's instructions. ATP synthase inhibitor oligmycin (1 μM), uncoupler FCCP (4 μM) and complex I inhibitors rotenone (1 μM) and antimycin A (1 μM) were injected to the wells, followed by measurement cycles for OCR. The oxygen consumption rates were calculated from the continuous average slope of the O2 decrease using a compartmentalization model. Basal OCR is (OCR with substrates) - (OCR with rotenone and antimycin A). Maximal OCR is (OCR with FCCP) - (OCR with rotenone and antimycin A). Assay for glycolytic capacity in live cultured ovarian cancer cells was performed using the Seahorse XF24 Analyzer. On the day of the assay, the medium was changed to DMEM (without serum, glucose, or bicarbonate, but with 2 mM glutamine) and incubated for 1 h before the assay in a non-CO2 incubator at 37 °C. Glucose (10 mM), oligomycin (1 mM), and 2-deoxy-d-glucose (2-DG 100 mM) were diluted in the assay medium and loaded onto ports A, B, and C, respectively. The analyzer was calibrated and the assay was performed using the glycolysis stress test assay protocol as suggested by the manufacturer. The rate of glycolysis is the extracellular acidification rate (ECAR) after the addition of glucose. The glycolytic capacity is the rate of increase in ECAR after the injection of oligomycin following glucose.
2.18. Real-time quantitative PCR for mtDNA copy number
Total genomic DNA was extracted from A2780 cells with a genomic DNA extraction kit (Vazyme Biotech Co.,Ltd). For the determination of mtDNA copy number relative to nuclear DNA in cells, the ND1 gene was used as a marker of mtDNA and the B2M gene for nuclear DNA [38]. Quantitative PCR was performed on a real-time PCR detection system using SYBR green, as described above. The primer sequences for mtDNA copy number were listed in Supplementary Table S3.
2.19. Statistical analysis
All statistical data were calculated and graphed using GraphPad Prism 6 and data were presented as mean ± standard deviation (S.D.). Statistical significance was tested using two-sided Student's t-test. p < 0.05 was considered statistically significant. Statistical significance was also taken as *p < 0.05, **p < 0.01 and ***p < 0.001, ns stands for no significant.
3. Results
3.1. RAD51 is upregulated in ovarian cancer and high RAD51 expression level is associated with poor prognosis
To investigate the expression of RAD51 in ovarian cancer, we firstly analyzed RAD51 expression in ovarian cancers using data from the TCGA and Oncomine™ databank. We found that RAD51 was overexpressed in ovarian cancer (Fig. 1A and B). Next we measured the mRNA levels of RAD51 in cancer specimens collected at Qilu Hospital by qPCR. As shown in Fig. 1C, RAD51 mRNA levels are generally higher in high-grade serous ovarian carcinoma (HGSOC, n = 46) than in fallopian tube epithelia (FT, n = 20). We previously also found that the protein levels of RAD51 were higher in ovarian cancer cells than in the immortalized normal human fallopian tube epithelial cell line FTE-187 [23]. We then examined RAD51 expression in HGSOC (n = 228) and FT (n = 41) by immunohistochemistry. The immunostaining intensity of RAD51 was significantly higher in HGSOC than in fallopian tubes (Fig. 1D). Furthermore, Kaplan-Meier plotter analysis (www.kmplot.com) showed that high RAD51 expression is associated with poor prognosis in ovarian cancer patients (Fig. 1E). These results indicate that RAD51 is generally overexpressed in ovarian cancer and is associated with poor prognosis.
Fig. 1.

RAD51 is upregulated in HGSOC and high RAD51 expression level is associated with poor prognosis. (A) Boxplot representing RAD51 expression values in ovarian cancer of the TCGA database (logarithmic values). The expression of RAD51 is higher in tumors (n = 586) than healthy tissues (n = 8). (B) Boxplot representing RAD51 expression values in ovarian cancer of the Yoshihara database (logarithmic values). The expression of RAD51 is higher in tumors (n = 43) than healthy tissues (n = 10). (C) Real-time quantitative PCR analysis of RAD51 in HGSOC tissue samples (n = 46) compared with fallopian tube tissues (FT, n = 20). The expression of RAD51 is higher in HGSOC than FT. (D) Representative images of immunohistochemistry staining of RAD51 in tissue microarray (left) and the expression level distribution of RAD51 (quantified by immunohistochemical score) in HGSOC (n = 228) compared with FT (n = 41, right). (E) Kaplan-Meier plots showing that high RAD51 expression is indicative of poor prognosis in ovarian cancer patients. Data presented as mean ± S.D. The statistical differences between the two groups were analyzed by two-sided unpaired Student's t-test(*p < 0.05, ***p < 0.001).
3.2. RAD51 knockdown reduces proliferation of ovarian cancer cells in vitro and impedes tumor growth in vivo
To determine whether RAD51 contributes to the proliferation of ovarian cancer cells, we knocked down RAD51 by transfecting ovarian cancer cells with RAD51 specific siRNA and evaluated cell cycle distribution and apoptosis by flow cytometry. RNAi efficiency of A2780, HEY and HO8910 cells were measured by Western blot analysis (Fig. 2A). Colony formation assay showed that RAD51 inhibition led to decreased proliferation (Fig. 2A). We also established a cell line with inducible RAD51 knockdown in HO8910 cells using a doxycycline (Dox)-inducible and GFP-labelled lentiviral shRNA. The knockdown efficiency of RAD51 shRNA was evaluated by Western blot analysis and GFP examination under a fluorescence microscope after doxycycline treatment for 48 h (Fig. 2B) (Fig. S1A). The colony formation ability was also significantly decreased in HO8910 shRAD51 cells (Fig. 2B). Next, we examined the proliferation-inhibitory effect of RAD51 knockdown by EdU incorporation and found a reduced rate of EdU incorporation in Dox-treated cells (low RAD51 expression) when compared with untreated cells (Fig. 2C). The cell cycle distribution in A2780, HO8910 and HEY cells transfected with RAD51 siRNA showed an increased accumulation of cells at G2/M phase (Fig. 2D) (Fig. S2A). However, no increase in apoptosis was detected after RAD51 siRNA transfection for 48 h (Figs. S2B and C). To investigate the role of RAD51 in tumor growth in vivo, HO8910 cells stably transfected with inducible RAD51 shRNA were subcutaneously inoculated into flanks of BALB/c nude mice, the tumor-bearing mice were randomly divided into two groups (4–6 mice in each group). RAD51 depletion in tumor xenografts was achieved by feeding the mice with 1.2 mg/mL doxycycline. One week later, tumor volumes were measured every 3 days. On the 24th day, the mice were euthanized and tumor weights were measured (Fig. S1B). As shown in Fig. 2E–G, the average tumor size in RAD51 knockdown group was much smaller than in the control groups, which was consistent with the in vitro studies (Fig. 2A–C). In keeping with the anti-proliferative effect of RAD51 knockdown on ovarian cancer cells in vitro, there was a significant decrease in the abundance of Ki-67 positive cells in the tumor xenografts (Fig. 2H).
Fig. 2.

RAD51 depletion reduces proliferation of ovarian cancer cells in vitro and impedes tumor growth in vivo. (A) RNAi efficiency of A2780, HEY and HO8910 cells were measured by Western blot analysis. α-Tubulin was used as a loading control (left). Band intensities were quantified by ImageJ. Colony-forming efficiency of A2780 and HO8910 cells after transfected scrambled siRNA (siScr) or siRNA targeting RAD51 (siRAD51) for two weeks (right). (B) Knockdown efficiency of RAD51 shRNA were evaluated after doxycycline treatment for 48 h by Western blot analysis (left). α-Tubulin was used as a loading control. Band intensities were quantified by ImageJ. Colony-forming efficiency of HO8910 cells stably transfected RAD51 shRNA after doxycycline treatment for two weeks (right). (C) EdU incorporation was performed after doxycycline treatment for 48 h in HO8910 cells. (D) Cell cycle distribution of A2780, HO8910 and HEY cells after transfected siScr or siRAD51 for 48 h. The cell cycle distribution was measured by flow cytometry. (E) Images of HO8910 shControl and HO8910 shRAD51 tumors of each treatment group. (F) Tumor weights at day 24. (G) Growth curves of tumors from transplanted HO8910 shControl and HO8910 shRAD51 cells in nude mice. Tumor volumes were measured every 3 days. (H) Representative images of immunohistochemistry staining using Ki-67 antibody in HO8910 tumors for each group. Scale bar: 50 μm. Data presented as mean ± S.D. are representative of three independent experiments. The statistical differences between the two groups were analyzed by two-sided unpaired Student's t-test (**p < 0.01, ***p < 0.001).
3.3. RAD51 knockdown leads to increased oxidative stress and oxidative DNA damage in ovarian cancer cells
Ovarian cancer cells rely on high level of reactive oxygen species (ROS) for their proliferation [30,31]. To determine the role of RAD51 in defending oxidative stress, we measured the ROS levels in ovarian cancer cells in which RAD51 was depleted or inhibited. We observed that either RAD51 knockdown or RAD51 inhibitor (B02) treatment caused a significant increase in ROS in all four ovarian cancer cell lines tested (Fig. 3A and B). We also found a significant elevation in ROS in immortalized fallopian tube epithelial cell line FTE-187, human osteosarcoma cell line U2OS and human breast cancer cells MDA-MB-231 when RAD51 was knocked down (Fig. S3). Next we examined the level of oxidative base damage in RAD51 knockdown cells. As shown in Fig. 3C, there was a significant increase in the level of 8-OHdG, a marker of oxidative base damage. Moreover, there was a significant increase in 8-OHdG staining intensity in the tumor xenografts formed by RAD51 knockdown HO8910 cells (Fig. 3D). These results indicate that RAD51 contributes to maintaining redox homeostasis and its depletion may lead to increased oxidative DNA damage.
Fig. 3.

RAD51 knockdown increases the ROS levels and oxidative DNA damage of ovarian cancer cells. (A) ROS distribution measured by flow cytometry in A2780, HO8910, HEY and SKOV3 cells after transfected siScr or siRAD51 for 48 h. RFI, relative fluorescence intensity. (B) ROS distribution measured by flow cytometry in A2780, HO8910, HEY and SKOV3 cells after 10 μM B02 (RAD51 inhibitor) treatment for 48 h. (C) Immunofluorescence staining of 8-OHdG in A2780 and HO8910 cells transfected siScr or siRAD51 for 48 h. Scale bar: 20 μm. (D) Representative images of immunohistochemistry staining using 8-OHdG antibody in HO8910 tumors for each group. Scale bar: 50 μm. Data presented as mean ± S.D. are representative of three independent experiments. The statistical differences between the two groups were analyzed by two-sided unpaired Student's t-test (**p < 0.01, ***p < 0.001).
3.4. Antioxidant rescues proliferation and reduces oxidative DNA damage of RAD51 knockdown cells
Because RAD51 is central to HRR [4,16], it can be assumed that the anti-proliferative effect exerted by RAD51 inhibition is due to impaired DNA repair. To test whether the increased oxidative stress as a consequence of RAD51 depletion may also mediate the anti-proliferative effect, we examined whether the anti-proliferative effect of RAD51 knockdown could be rescued by antioxidant. As shown in Fig. 4A–C and Fig. S4, the proliferation abilities of RAD51-depleted ovarian cancer cells, as measured by colony formation, cell viability and EdU incorporation, were significantly rescued by antioxidant N-acetylcysteine (NAC). NAC also rescued the colony formation of cancer cells that were treated with RAD51 inhibitor (Fig. 4D). Importantly, the levels of DSBs caused by RAD51 knockdown were significantly reduced by NAC (Fig. 4E and F). These results suggest that oxidative stress also mediates the anti-proliferative effect of RAD51 knockdown.
Fig. 4.

Antioxidant rescues proliferation ability and reduces oxidative DNA damage of RAD51 knockdown cells. (A) Clonogenic assay of A2780 cells transfected siScr or siRAD51 treated with or without 10 mM NAC. (B) Cell viability was measured by CCK-8 assay. A2780 cells transfected siScr or siRAD51 treated with or without 10 mM NAC. (C) EdU incorporation of A2780 cells transfected siScr or siRAD51 treated with or without 10 mM NAC. (D) Clonogenic assay of A2780 cells treated with or without 5 μM B02 and 10 mM NAC. (E) Western blot analysis of γ-H2AX protein levels in A2780 and HEY cells transfected siScr or siRAD51 treated with or without 10 mM NAC. (F) Western blot analysis of γ-H2AX protein levels in HO8910 cells stably transfected RAD51 shRNA treated with or without 10 mM NAC. α-Tubulin was used as a loading control. Band intensities were quantified by ImageJ. Data presented as mean ± S.D. are representative of three independent experiments. The statistical differences between the two groups were analyzed by two-sided unpaired Student's t-test (**p < 0.01, ***p < 0.001, ns stand for no significant).
3.5. RAD51 knockdown results in mitochondrial oxidative stress
We next examined whether mitochondria are the source of ROS in RAD51 knockdown cells. As shown in Fig. 5A, the level of mitochondrial superoxides, as measured by MitoSOX, was significantly increased in A2780, HO8910 and HEY cells in which RAD51 was depleted. RAD51 inhibitor (B02) treatment also caused a significant increase in mitochondrial superoxides in all four ovarian cancer cell lines tested (Fig. S5A). Of note, mitoquinone (MitoQ, a mitochondria-targeted antioxidant) treatment significantly attenuated the elevation of ROS in RAD51 knockdown cells (Fig. 5B), suggesting that increased ROS are mainly derived from mitochondria. As expected, mitoquinone treatment significantly attenuated the elevation of MitoSOX in RAD51 depleted HEY cells (Fig. S5B). Furthermore, the mitochondrial membrane potential (MMP) was decreased in RAD51 knockdown cells (Fig. 5C). The maximal oxygen consumption rate (OCR) was significantly increased in RAD51-depleted A2780 cells (Fig. 5D). Both the basal and maximal OCR were increased in RAD51-depleted HEY and HO8910 cells (Figs. S5C and D), demonstrating that RAD51 knockdown enhances mitochondial reserve capacity [39]. RAD51 knockdown also increased the glycolytic capacity in A2780 and HEY cells (Fig. 5E and Fig. S5C). Consistent with the increased OCR and glycolysis, the intracellular ATP levels were increased in RAD51 knockdown cells (Fig. S5E). These results indicate that RAD51 inhibition could cause mitochondrial oxidative stress in ovarian cancer cells.
Fig. 5.

RAD51 knockdown results in mitochondrial dysfunction. (A) MitoSOX distribution measured by flow cytometry in A2780, HO8910 and HEY cells transfected siScr or siRAD51 for 48 h. (B) ROS distribution measured by flow cytometry in A2780, HO8910 and HEY cells transfected siScr or siRAD51 treated with or without 500 nM MitoQ. (C) Measurements of MMP (by JC-1) in A2780, HO8910 and HEY cells transfected siScr or siRAD51 for 48 h. (D) Oxygen consumption rate (OCR) of A2780 cells transfected siScr or siRAD51 for 48 h. The OCR was measured under baseline conditions and after oligomycin, FCCP and rotenone/antimycin injection, as indicated by the arrows. (E) Extracellular acidification rate (ECAR) of A2780 cells transfected siScr or siRAD51 for 48 h. The ECAR was measured under baseline conditions and after glucose, FCCP and 2-deoxy-d-glucose (2-DG) injection, as indicated by the arrows. Data presented as mean ± S.D. are representative of three independent experiments. The statistical differences between the two groups were analyzed by two-sided unpaired Student's t-test (*p < 0.05, **p < 0.01, ***p < 0.001, ns stand for no significant).
3.6. CHK1 activation mediates the increased mitochondrial oxidative stress caused by RAD51 depletion
We next determined whether the increased OCR in RAD51-depleted cancer cells was associated with increased accumulation of mitochondria. Depletion of RAD51 in A2780 cells displayed a significant increase in mitochondrial content, as quantified by flow cytometry of cells stained with Mitotracker (Fig. 6A, middle). As a control, cancer cells treated with Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) exhibited a decreased Mitotracker staining (Fig. 6A, left). RAD51 inhibition similarly induced an increased buildup of mitochondria (Fig. 6A right and Fig. S5F). Cells arrested at G2/M phase usually harbor more mitochondria and exhibit a higher level of ROS than at other phases of the cell cycle [40]. Because CHK1 is known to mediate G2/M arrest [41,42] and the ovarian cancer cells are arrested at G2/M when RAD51 are depleted, we speculated that CHK1 might mediate the G2/M arrest and the generation of mitochondrial oxidative stress in response to RAD51 depletion. Indeed, the protein level of phosphorylated CHK1, which marks activated CHK1, was greatly increased when RAD51 was depleted (Fig. 6B). Importantly, inhibition of CHK1 kinase activity by UCN-01 or CHK1 siRNA abolished G2/M arrest caused by RAD51 depletion (Fig. 6C and Fig. S6C). Moreover, the mitochondrial accumulation caused by RAD51 depletion was abrogated by the CHK1 inhibition (Fig. 6D and Fig. S6D). Consistently, increase in the level of mitochondrial superoxides was attenuated by the inhibition of CHK1 (Fig. 6E and Fig. S6E). We next determined the proliferation of A2780 cells in response to RAD51 depletion, CHK1 inhibition or the combined treatment of the two. While RAD51 depletion or UCN-01 alone each resulted in a reduction in the EdU-positive cells, the two in combination led to no further reduction in the proliferation of cancer cells (Fig. 6F and Fig. S7), suggesting that the alleviation of mitochondrial oxidative stress by CHK1 inhibition may have prevented an additive inhibitory effect on proliferation. In contrast to the lack of additive inhibitory effect on ovarian cancer cells by RAD51 and CHK1 inhibitors when used in combination, it was reported that the two inhibitors acted synergistically to inhibite the proliferation of HeLa cells [43]. We therefore tested whether the lack of additive effect by the two inhibitors was cell-type specific. Indeed, as shown in Fig. S8, the two in combination led to further reduction in the proliferation of HeLa cells. Thus the rescuing effect of CHK1 inhibition may not apply to other types of cancer cells. Nevertheless, at least in some categories of cancer cells, depletion of RAD51 can lead to CHK1-dependent G2/M arrest and the subsequent mitochondrial oxidative stress, which may cause further DNA damage and sustain a feedforward loop.
Fig. 6.

CHK1 activation-mediates the increased mitochondrial oxidative stress caused by RAD51 depletion. (A) Mitochondrial content decreased significantly after 12.5 μM CCCP treatment for 48 h (left). Mitochondrial content was elevated in A2780 cells after RAD51 knockdown (middle) or 10 μM B02 treatment (right), cells were stained with Mitotracker and quantified by flow cytometry. (B) Western blot analysis of phosphorylation of CHK1 in A2780, HO8910 and HEY cells after transfected siScr or siRAD51 for 48 h. CHK1 serverd as loading control. Band intensities were quantified by ImageJ. (C) Cell cycle distribution of A2780 and HEY cells transfected siScr or siRAD51 treated with or without 300 nM CHK1 inhibitor (UCN-01). The cell cycle distribution was measured by flow cytometry. (D) Mitochondrial content was quantified by flow cytometry in A2780 cells transfected siScr or siRAD51 treated with or without 300 nM CHK1 inhibitor (UCN-01). (E) MitoSOX distribution measured by flow cytometry in A2780 cells transfected siScr or siRAD51 treated with or without 300 nM CHK1 inhibitor (UCN-01). (F) EdU incorporation measured by flow cytometry in A2780 cells transfected siScr or siRAD51 treated with or without 300 nM CHK1 inhibitor (UCN-01). Data presented as mean ± S.D. are representative of three independent experiments. The statistical differences between the two groups were analyzed by two-sided unpaired Student's t-test (*p < 0.05, **p < 0.01, ***p < 0.001, ns stand for no significant).
3.7. Mitochondrial oxidative stress caused by RAD51 knockdown was independent of mtDNA
Recent studies showed that RAD51 can localize in human mitochondria and play an important role in the maintenance of the mitochondrial genome integrity [44]. We isolated mitochondria from several human ovarian cancer cells and found that RAD51 could indeed be detected in mitochondria of ovarian cancer cells (Fig. S5G). It has been shown that depletion of RAD51 results in a significant decrease in mtDNA copy number following oxidative stress, supporting that RAD51-mediated recombination contributes to the maintenance of mtDNA genome stability [44]. We therefore examined whether mitochondrial dysregulation induced by RAD51 knockdown was related to the maintenance of mtDNA genome integrity. As shown in Fig. 7A, mtDNA copy number decreased significantly in RAD51 knockdown cells. Rho0 cells, which are depleted of mtDNA, were used as a control to show the absence of mtDNA. Surprisingly, the levels of ROS, MitoSOX and Mitotracker were all increased in rho0 cells by RAD51 depletion (Fig. 7B–D). These results revealed that mitochondrial dysregulation caused by RAD51 knockdown can occur independent of mtDNA.
Fig. 7.

RAD51 depletion-induced mitochondrial dysregulation is independent of mtDNA. (A) mtDNA copy number quantified in rho0 cells (left). mtDNA copy number decreased in A2780 cells knockdown RAD51 (right). (B) ROS distribution measured by flow cytometry in rho0 cells after transfected siScr or siRAD51 for 48 h. (C) MitoSOX distribution measured by flow cytometry in rho0 cells after transfected siScr or siRAD51 for 48 h. (D) Mitochondrial content was quantified by flow cytometry in rho0 cells after transfected siScr or siRAD51 for 48 h. Data presented as mean ± S.D. are representative of three independent experiments. The statistical differences between the two groups were analyzed by two-sided unpaired Student's t-tes t(**p < 0.01, ***p < 0.001).
4. Discussion
RAD51 overexpression is common in human cancers and is believed to contribute to the malignant features of cancer cells, such as metastasis and chemoresistance. However, the consequences of RAD51 depletion or inhibition in cancer cells and the underlying mechanisms remain to be fully determined. Does RAD51 simply function to maintain genomic stability and thereby promote cell survival and proliferation? Does RAD51 depletion have consequences beyond genomic instability? Here, we showed that RAD51 was significantly upregulated in HGSOC and high expression of RAD51 is associated with poor prognosis, which are consistent with previous studies. Importantly, we found that RAD51 contributes to the maintenance of redox homeostasis in ovarian cancer cells. RAD51 depletion or inhibition led to increased ROS level and oxidative DNA damage in cancer cells. Furthermore, the accumulation of DSBs and impaired proliferation caused by RAD51 depletion or inhibition can be significantly reduced by antioxidant NAC. Therefore, besides a direct impairment of DNA repair, the cytotoxic effect of RAD51 depletion or inhibition can be further amplified by the associated increase in oxidative stress.
We showed that the oxidative stress caused by RAD51 inhibition or depletion is primarily derived from mitochondria. We observed an increased level of mitochondrial superoxide, along with increased accumulation of mitochondria, when RAD51 is depleted or inhibited. We further showed that the increased mitochondrial oxidative stress is dependent on CHK1 activation. While RAD51 depletion led to CHK1-mediated G2/M arrest, mitochondrial buildup and increased generation of superoxide, inhibition of CHK1 can greatly attenuate or abrogate those changes. CHK1 thus acts as a critical mediator of the deleterious effect of RAD51 depletion on cell proliferation (Fig. 8). These results are consistent with a recent report that CHK1 overactivation due to defective chaperone-mediated autophagy may drive the accumulation of more DNA damage [45]. DNA damage response (DDR) is generally believed to be a consequence of oxidative stress [46], our results indicate that DDR, such as the activation of CHK1, can be the cause of oxidative stress. A recent study also showed that oxidative stress peaks in mitosis and is exacerbated by mitotic arrest [47]. Together, these findings indicate that DNA damage signaling does not only respond to oxidative stress, but may also contribute to it.
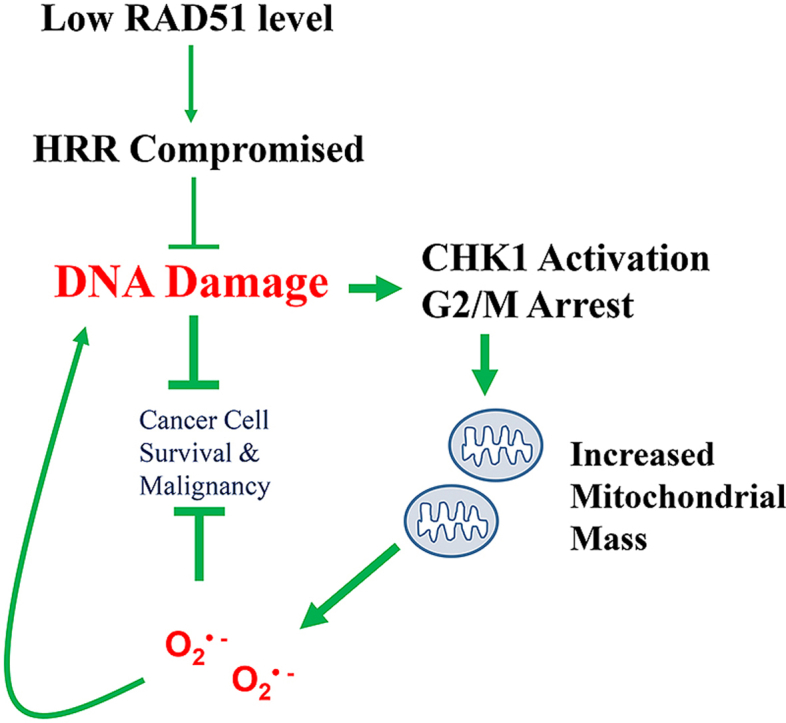
Fig. 8.

A working model for RAD51 depletion induced mitochondrial oxidative stress.
High RAD51 level in cancer cell promotes HRR, reduces the level of DNA damage and the associated CHK1 activation, ultimately leading to cancer cell survival and malignancy. Low RAD51 level compromises HRR, causing more DNA damage, activating CHK1 and arresting cells at G2/M, and consequently leading to mitochondrial oxidative stress, which may aggravate nuclear DNA damage and reinforce a vicious feedforward loop. The increased oxidative stress inhibits cancer cell survival and malignancy.
In addition to its function in nucleus, RAD51 has been reported to be localized in human mitochondria and to play an important role in the maintenance of the mitochondrial genome integrity [44]. It is therefore possible that the mitochondrial stress in response to RAD51 depletion may be attributed to an impaired maintenance of mtDNA. However, while we observed a decreased copy number of the mtDNA in RAD51-depleted ovarian cancer cells, the levels of overall ROS and mitochondrial superoxide and the mitochondrial mass can still be increased in mtDNA-devoid rho0 cells, indicating that the mitochondrial stress caused by RAD51 depletion is probably not related to mtDNA.
We found that the ovarian cancer cells were arrested at G2/M phase in response to RAD51 depletion, which is consistent with the reported G2/M arrest in Rad51 knockout chicken DT40 lymphocytes [48]. We further showed that this G2/M arrest and the associated mitochondrial stress were dependent on CHK1. Thus, CHK1 activation exerts a tumor suppressive effect in this context. CHK1 inhibition has been explored as a cancer therapeutic strategy in several studies [[49], [50], [51]], our findings indicate that caution should be taken when prescribing CHK1 inhibitor in cancer therapy, especially in scenarios where the cancer-killing effect relies on the induction of oxidative stress. Instead of compromising cancer cell survival, such treatment may relieve the cancer cells of oxidative stress and promote survival. In addition, CHK1 inhibitors have also been shown to activate other pro-survival mechanisms, such as the compensatory activation of ATM and ERK1/2 [52], which may account for the less than expected anti-cancer efficacy of CHK1 inhibitors in many pre-clinical studies.
Taken together, our work indicates that in addition to participating in HRR, RAD51 indirectly contributes to the maintenance of mitochondrial redox homeostasis and the increased mitochondrial oxidative stress also mediates the anti-proliferative effect of RAD51 depletion in ovarian cancer cells. Moreover, RAD51 depletion-induced mitochondria oxidative stress is not due to loss of mtDNA integrity, but is instead caused by a CHK1-dependent G2/M arrest and mitochondrial accumulation. Our findings suggest that a vicious cycle of nuclear DNA damage, mitochondrial oxidative stress may contribute to the tumor-suppressive effects of RAD51 depletion or inhibition.
Declaration of competing interest
Designed, supervised the experiments, finalized the manuscript and provided financial support: Changshun Shao.
Conducted most of the experiments, data analysis and manuscript preparation: Limei Xu.
Give assistance to some experiments: Tingting Wu, Shihua Lu, Xiaohe Hao, Junchao Qin, Jing Wang.
Critical revision of manuscript for important intellectual content: Zhaojian Liu, Yaoqin Gong, Beihua Kong, Xiyu Zhang, Qiao Liu.
All authors approved the final manuscript.
The authors declare that they have no competing interests.
Acknowledgements
We thank Dr. Jinsong Liu and Chuanzhu Yan for providing FTE-187 and 143B.TK ρ0 cell lines, respectively. This study was supported by National Natural Science Foundation of China (81372241, 81572785) and the State Key Laboratory of Radiation Medicine and Protection, Soochow University (GZN1201804).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101604.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Cannistra Stephen A. Cancer of the ovary. N. Engl. J. Med. 2004;351:2519–2529. doi: 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- 2.Vaughan S., Coward J.I., Bast R.C., Jr., Berchuck A., Berek J.S., Brenton J.D. Rethinking ovarian cancer: recommendations for improving outcomes. Nat. Rev. Canc. 2011;11:719–725. doi: 10.1038/nrc3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konstantinopoulos P.A., Ceccaldi R., Shapiro G.I., D'Andrea A.D. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Canc. Discov. 2015;5:1137–1154. doi: 10.1158/2159-8290.CD-15-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baumann P., West S.C. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci. 1998;23:247–251. doi: 10.1016/s0968-0004(98)01232-8. [DOI] [PubMed] [Google Scholar]
- 5.Groth P., Orta M.L., Elvers I., Majumder M.M., Lagerqvist A., Helleday T. Homologous recombination repairs secondary replication induced DNA double-strand breaks after ionizing radiation. Nucleic Acids Res. 2012;40:6585–6594. doi: 10.1093/nar/gks315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lundin C., Schultz N., Arnaudeau C., Mohindra A., Hansen L.T., Helleday T. RAD51 is involved in repair of damage associated with DNA replication in mammalian cells. J. Mol. Biol. 2003;328:521–535. doi: 10.1016/s0022-2836(03)00313-9. [DOI] [PubMed] [Google Scholar]
- 7.Saintigny Y., Delacôte F., Varès G., Petitot F., Lambert S., Averbeck D. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001;20:3861–3870. doi: 10.1093/emboj/20.14.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dungrawala H., Bhat K.P., Le Meur R., Chazin W.J., Ding X., Sharan S.K. RADX promotes genome stability and modulates chemosensitivity by regulating RAD51 at replication forks. Mol. Cell. 2017;67:374–386. doi: 10.1016/j.molcel.2017.06.023. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maacke H., Opitz S., Jost K., Hamdorf W., Henning W., Krüger Stefan. Over-expression of wild-type Rad51 correlates with histological grading of invasive ductal breast cancer. Int. J. Canc. 2000;88:907–913. doi: 10.1002/1097-0215(20001215)88:6<907::aid-ijc11>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 10.Chen Q., Cai D., Li M., Wu X. The homologous recombination protein RAD51 is a promising therapeutic target for cervical carcinoma. Oncol. Rep. 2017;38:767–774. doi: 10.3892/or.2017.5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maacke H., Jost K., Opitz S., Miska S., Yuan Y., Hasselbach L. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene. 2000;19:2791–2795. doi: 10.1038/sj.onc.1203578. [DOI] [PubMed] [Google Scholar]
- 12.Mitra A., Jameson C., Barbachano Y., Sanchez L., Kote-Jarai Z., Peock S. Overexpression of RAD51 occurs in aggressive prostatic cancer. Histopathology. 2009;55:696–704. doi: 10.1111/j.1365-2559.2009.03448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Q., Jiang H., Liu Z., Wang Y., Zhao M., Hao C. Berberine radiosensitizes human esophageal cancer cells by downregulating homologous recombination repair protein RAD51. PLoS One. 2011;6 doi: 10.1371/journal.pone.0023427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.King H.O., Brend T., Payne H.L., Wright A., Ward T.A., Patel K. RAD51 is a selective DNA repair target to radiosensitize glioma stem cells. Stem Cell Rep. 2017;8:125–139. doi: 10.1016/j.stemcr.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hannay J.A., Liu J., Zhu Q.S., Bolshakov S.V., Li L., Pisters P.W. Rad51 overexpression contributes to chemoresistance in human soft tissue sarcoma cells: a role for p53/activator protein 2 transcriptional regulation. Mol. Canc. Therapeut. 2007;6:1650–1660. doi: 10.1158/1535-7163.MCT-06-0636. [DOI] [PubMed] [Google Scholar]
- 16.Gachechiladze M., Skarda J., Soltermann A., Joerger M. RAD51 as a potential surrogate marker for DNA repair capacity in solid malignancies. Int. J. Canc. 2017;141:1286–1294. doi: 10.1002/ijc.30764. [DOI] [PubMed] [Google Scholar]
- 17.Woditschka S., Evans L., Duchnowska R., Reed L.T., Palmieri D., Qian Y. DNA double-strand break repair genes and oxidative damage in brain metastasis of breast cancer. J. Natl. Cancer Inst. 2014;106 doi: 10.1093/jnci/dju145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raderschall E., Stout K., Freier S., Suckow V., Schweiger S., Haaf T. Elevated levels of Rad51 recombination protein in tumor cells. Canc. Res. 2002;62:219–225. [PubMed] [Google Scholar]
- 19.Tennstedt P., Fresow R., Simon R., Marx A., Terracciano L., Petersen C. RAD51 overexpression is a negative prognostic marker for colorectal adenocarcinoma. Int. J. Canc. 2013;132:2118–2126. doi: 10.1002/ijc.27907. [DOI] [PubMed] [Google Scholar]
- 20.Qiao G.B., Wu Y.L., Yang X.N., Zhong W.Z., Xie D., Guan X.Y. High-level expression of Rad51 is an independent prognostic marker of survival in non-small-cell lung cancer patients. Br. J. Canc. 2005;93:137–143. doi: 10.1038/sj.bjc.6602665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai M.S., Kuo Y.H., Chiu Y.F., Su Y.C., Lin Y.W. Down-regulation of Rad51 expression overcomes drug resistance to gemcitabine in human non-small-cell lung cancer cells. J. Pharmacol. Exp. Therapeut. 2010;335:830–840. doi: 10.1124/jpet.110.173146. [DOI] [PubMed] [Google Scholar]
- 22.Wang B., Hou D., Liu Q., Wu T., Guo H., Zhang X. Artesunate sensitizes ovarian cancer cells to cisplatin by downregulating RAD51. Canc. Biol. Ther. 2015;16:1548–1556. doi: 10.1080/15384047.2015.1071738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hou D., Xu G., Zhang C., Li B., Qin J., Hao X. Berberine induces oxidative DNA damage and impairs homologous recombination repair in ovarian cancer cells to confer increased sensitivity to PARP inhibition. Cell Death Dis. 2017;8 doi: 10.1038/cddis.2017.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu G., Yang D., Rupaimoole R., Pecot C.V., Sun Y., Mangala L.S. Augmentation of response to chemotherapy by microRNA-506 through regulation of RAD51 in serous ovarian cancers. J. Natl. Cancer Inst. 2015;107 doi: 10.1093/jnci/djv108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang J.W., Wang Y., Dhillon K.K., Calses P., Villegas E., Mitchell P.S. Systematic screen identifies miRNAs that target RAD51 and RAD51D to enhance chemosensitivity. Mol. Canc. Res. 2013;11:1564–1573. doi: 10.1158/1541-7786.MCR-13-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choudhury A., Zhao H., Jalali F., Al Rashid S., Ran J., Supiot S. Targeting homologous recombination using imatinib results in enhanced tumor cell chemosensitivity and radiosensitivity. Mol. Canc. Therapeut. 2009;8:203–213. doi: 10.1158/1535-7163.MCT-08-0959. [DOI] [PubMed] [Google Scholar]
- 27.Gorrini C., Baniasadi P.S., Harris I.S., Silvester J., Inoue S., Snow B. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013;210:1529–1544. doi: 10.1084/jem.20121337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang E.F., Scheibye-Knudsen M., Chua K.F., Mattson M.P., Croteau D.L., Bohr V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016;17:308–321. doi: 10.1038/nrm.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saki M., Prakash A. DNA damage related crosstalk between the nucleus and mitochondria. Free Radic. Biol. Med. 2017;107:216–227. doi: 10.1016/j.freeradbiomed.2016.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu Y., Rosen D.G., Zhou Y., Feng L., Yang G., Liu J. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. J. Biol. Chem. 2005;280:39485–39492. doi: 10.1074/jbc.M503296200. [DOI] [PubMed] [Google Scholar]
- 31.Xia C., Meng Q., Liu L.Z., Rojanasakul Y., Wang X.R., Jiang B.H. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Canc. Res. 2007;67:10823–10830. doi: 10.1158/0008-5472.CAN-07-0783. [DOI] [PubMed] [Google Scholar]
- 32.Graham K.A., Kulawiec M., Owens K.M., Li X., Desouki M.M., Chandra D. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Canc. Biol. Ther. 2010;10:223–231. doi: 10.4161/cbt.10.3.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konstantinopoulos P.A., Spentzos D., Fountzilas E., Francoeur N., Sanisetty S., Grammatikos A.P. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Canc. Res. 2011;71:5081–5089. doi: 10.1158/0008-5472.CAN-10-4668. [DOI] [PubMed] [Google Scholar]
- 34.Van der Wijst M.G., Brown R., Rots M.G. Nrf2, the master redox switch: the Achilles' heel of ovarian cancer? Biochim. Biophys. Acta. 2014;1846:494–509. doi: 10.1016/j.bbcan.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Wei J.J., Wu J., Luan C., Yeldandi A., Lee P., Keh P. HMGA2: a potential biomarker complement to p53 for detection of early-stage high-grade papillary serous carcinoma in fallopian tubes. Am. J. Surg. Pathol. 2010;34:18–26. doi: 10.1097/PAS.0b013e3181be5d72. [DOI] [PubMed] [Google Scholar]
- 36.Shan W., Mercado-Uribe I., Zhang J., Rosen D., Zhang S., Wei J. Mucinous adenocarcinoma developed from human fallopian tube epithelial cells through defined genetic modifications. Cell Cycle. 2012;11:2107–2113. doi: 10.4161/cc.20544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Z., Liu Q., Xu B., Wu J., Guo C., Zhu F. Berberine induces p53-dependent cell cycle arrest and apoptosis of human osteosarcoma cells by inflicting DNA damage. Mutat. Res. 2009;662:75–83. doi: 10.1016/j.mrfmmm.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 38.Correia-Melo C., Marques F.D., Anderson R., Hewitt G., Hewitt R., Cole J. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016;35:724–742. doi: 10.15252/embj.201592862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dranka B.P., Hill B.G., Darley-Usmar V.M. Mitochondrial reserve capacity in endothelial cells: the impact of nitric oxide and reactive oxygen species. Free Radic. Biol. Med. 2010;48:905–914. doi: 10.1016/j.freeradbiomed.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamamori T., Yasui H., Yamazumi M., Wada Y., Nakamura Y., Nakamura H. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 2012;53:260–270. doi: 10.1016/j.freeradbiomed.2012.04.033. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y., Liu Q., Liu Z., Li B., Sun Z., Zhou H. Berberine, a genotoxic alkaloid, induces ATM-Chk1 mediated G2 arrest in prostate cancer cells. Mutat. Res. 2012;734:20–29. doi: 10.1016/j.mrfmmm.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 42.Liu Q., Guntuku S., Cui X.S., Matsuoka S., Elledge S.J. Chk1 is an essential kinase that is regulated by ATR and required for the G2/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 43.Krajewska M., Fehrmann R.S.N., Schoonen P.M. ATR inhibition preferentially targets homologous recombination-deficient tumor cells. Oncogene. 2015;34:3474–3481. doi: 10.1038/onc.2014.276. [DOI] [PubMed] [Google Scholar]
- 44.Sage J.M., Gildemeister O.S., Knight K.L. Discovery of a novel function for human Rad51: maintenance of the mitochondrial genome. J. Biol. Chem. 2010;285:18984–18990. doi: 10.1074/jbc.M109.099846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park C., Suh Y., Cuervo A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015;6:6823. doi: 10.1038/ncomms7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo Z., Kozlov S., Lavin M.F., Person M.D., Paull T.T. ATM activation by oxidative stress. Science. 2010;330:517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 47.Patterson J.C., Joughin B.A., van de Kooij B., Lim D.C., Lauffenburger D.A., Yaffe M.B. ROS and oxidative stress are elevated in mitosis during asynchronous cell cycle progression and are exacerbated by mitotic arrest. Cell Syst. 2019;8:163–167 e2. doi: 10.1016/j.cels.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takata M., Sasaki M.S., Sonoda E., Morrison C., Hashimoto M., Utsumi H. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17:5497–5508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma C.X., Janetka J.W., Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol. Med. 2011;17:88–96. doi: 10.1016/j.molmed.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Daud A.I., Ashworth M.T., Strosberg J., Goldman J.W., Mendelson D., Springett G. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J. Clin. Oncol. 2015;33:1060–1066. doi: 10.1200/JCO.2014.57.5027. [DOI] [PubMed] [Google Scholar]
- 51.Parmar K., Kochupurakkal B.S., Lazaro J.B., Wang Z.C., Palakurthi S., Kirschmeier P.T. The CHK1 inhibitor prexasertib exhibits monotherapy activity in high-grade serous ovarian cancer models and sensitizes to PARP inhibition. Clin. Canc. Res. 2019;25:6127–6140. doi: 10.1158/1078-0432.CCR-19-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dent P., Tang Y., Yacoub A. CHK1 inhibitors in combination chemotherapy: thinking beyond the cell cycle. Mol. Interv. 2011;11:133–140. doi: 10.1124/mi.11.2.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.