

Abstract
Nuclear quantum effects have significant contributions to thermodynamic quantities and structural properties; furthermore, very expensive methods are necessary for their accurate computation. In most calculations, these effects, for instance, zero-point energies, are simply neglected or only taken into account within the quantum harmonic oscillator approximation. Herein, we present a new method, Generalized Smoothed Trajectory Analysis, to determine nuclear quantum effects from molecular dynamics simulations. The broad applicability is demonstrated with the examples of a harmonic oscillator and different states of water. Ab initio molecular dynamics simulations have been performed for ideal gas up to the temperature of 5000 K. Classical molecular dynamics have been carried out for hexagonal ice, liquid water, and vapor at atmospheric pressure. With respect to the experimental heat capacity, our method outperforms previous calculations in the literature in a wide temperature range at lower computational cost than other alternatives. Dynamic and structural nuclear quantum effects of water are also discussed.
1. Introduction
Calculations of reaction free energy profiles and activation barriers are routinely performed within the rigid-rotor and harmonic-oscillator approximation;1 meanwhile, the more accurate computation of thermodynamic quantities or vibrational spectra is still a great challenge.2−10 The inclusion of nuclear quantum effects (NQEs), such as zero-point energy (ZPE) or tunneling, is even more difficult.11−13 Path-integral molecular dynamics (PIMD) and path integral Monte Carlo (PIMC) simulations are accurate, yet highly expensive methods to incorporate NQEs.14,15 The computational cost of PIMD simulations can be significantly reduced by advanced techniques.16−18 Recently developed algorithms, such as a colored noise thermostat and quantum thermal bath, are more effective to add quantum effects to classical simulations,19−21 but settings need to be chosen carefully to prevent zero-point energy leakage.22,23
When empirical water models were used in PIMD simulations, several properties deviated more from the experiments than in the classical simulations.24−27 In these quantum simulations, the liquid water becomes less structured and less viscous. This has been explained by double counting of quantum effects: once in the parameter optimization using experimental data, second in the quantum simulations. This is why several water models were reparametrized for accurate PIMD simulations resulting in q-SPC/Fw,28 q-TIP4P/f,24 and TIP4PQ/2005 models.29 Another solution to avoid double counting is the application of PIMD with ab initio methods9 or force fields trained on ab initio data.25,27,30 Numerous methodological developments have also been made to calculate quantum free energy values from PIMD simulations.31−35
In routine DFT calculations, with the optimized geometry in hand, the free energies are summed for all the different motions such as translation, rotation, and vibration, using the partition functions of the particle in the box, rigid rotor, and harmonic oscillator (RRHO) models.36 This approach works satisfactorily for small molecules at ambient temperatures, where the normal modes of vibrations can be considered as decoupled harmonic oscillators. For systems, where anharmonicity is significant, and/or the conformational space is extended, the RRHO fails to reproduce the exact thermodynamic quantities. Recognizing the need to address this issue, more sophisticated approaches use slightly modified partition functions on optimized geometries.37,38 There are a few methods which can estimate quantum corrections from classical MD trajectories, for example, one- and two-phase thermodynamics methods (1PT, 2PT).39−41 In those cases the vibrational density of states (VDOS) is determined from molecular dynamics by the Fourier transformation of the velocity autocorrelation function. Quantum corrections are computed by the multiplication of VDOS with weight functions derived from the partition functions of motions. In the 1PT model, only vibrational modes are considered as harmonic oscillators; in the 2PT model, gas phase motions such as rotational and translational modes are also taken into account. The 2PT model is an improved method based on the original work of Berens et al. which corresponds to the 1PT method with anharmonic correction (1PT+AC).39
The 2PT and 1PT+AC methods were successfully applied for the calculation of thermodynamic properties of several systems such as Lennard-Jones fluids,40 water,39,41−47 organic liquids,48,49 carbon dioxide,50 adsorbed urea to cellulose,51 ionic liquids,52,53 carbohydrates,54 mixtures,55 and interfaces.56 Heat capacity is generally used as a reference property for the benchmark of force fields.42,48,49,53 The 1PT/2PT methods are still in continuous development in respect to accuracy and applicability.57−63
Here, we propose the Generalized Smoothed Trajectory Analysis (GSTA) method, which is numerically beneficial to the 1PT/2PT methods and, moreover, addresses their limitations arising from the used approximations. Our theory is demonstrated on the exact reproduction of heat capacity and internal energy of a harmonic oscillator. We have chosen different states of water as real-world examples as the heat capacity varies widely between phases, and it is still one of the most investigated materials in computations.64 Beyond thermodynamic properties, structural and dynamic NQEs are also investigated.
2. Theory
In a molecular dynamics simulations, the velocity autocorrelation function (VACF) can be defined as follows:
| 1 |
where v is the velocity as a function of the time (t). Here, we refer to mass (m)-weighted VACF, but we assume identical masses in the derivations for simplicity. The vibrational density of states (VDOS) is the representation of the autocorrelation function (VACF) in the Fourier domain:
| 2 |
where ν is the frequency. Since in our calculations real numbers are used, the Fourier cosine transform is applied. Consequently, the VACF function is the inverse Fourier transform of the VDOS function:
| 3 |
Using t = 0, we get the norm of the VDOS function:
| 4 |
Applying ν = 0 in eq 2, we get the norm of the VACF function:
| 5 |
Originally, Berens et al. proposed that quantum corrected density of states can be determined by the multiplication of VDOS with an appropriate weight function w:39
| 6 |
2.1. Corrections of Vibrational Density of States (VDOS)
2.1.1. One-Phase Thermodynamics (1PT)
In the 1PT method, only vibrational motions are considered. The internal energy is
| 7 |
where U0 is the reference energy and β = (kBT)−1, kB is the Boltzmann constant, and T is the temperature.
The vibrational weight function wU for the energy originates from the quantum harmonic oscillator model:36
| 8 |
where h is the Planck constant and coth denotes the hyperbolic cotangent function.
The heat capacity is the temperature derivative of the internal energy:
| 9 |
The last term in the integral is the weight function for the heat capacity:36
| 10 |
The weight functions are shown in Figure 1. Using this weight function wcV(ν), the heat capacity can be obtained directly from the classical VDOS by integrating over the frequency domain:
| 11 |
In the classical limit (h → 0), the 1PT method always gives kB for the heat capacity, which corresponds to the classical harmonic value, so it cannot model anharmonicity.
Figure 1.
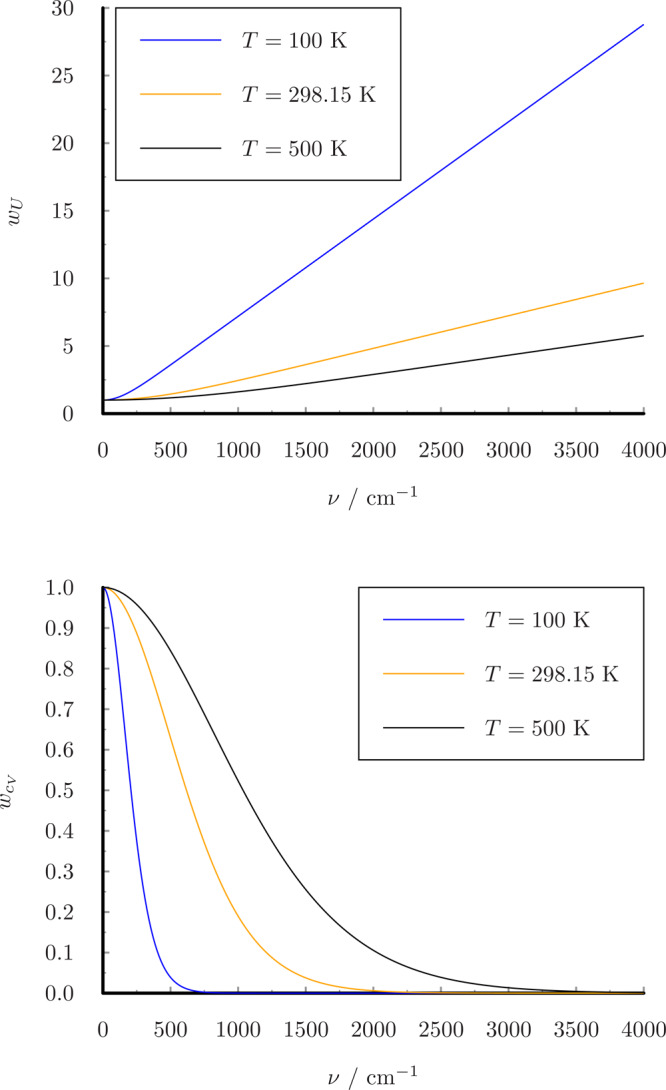
Weight functions for VDOS.
2.1.2. Two-Phase Thermodynamics (2PT)
Gas-like motions such as translations and rotations are separated from vibrations in the 2PT method. The total VDOS is decomposed into vibrations, translations, and rotations:
| 12 |
Translation and rotation are determined for the center of mass and principal axes of the molecules, respectively. Different weight functions are used for the different motions in the calculation of the thermodynamic properties:
| 13 |
The weight function of translation and rotation is 1/2 for the heat capacity within the 2PT model.41
In the classical limit (h → 0), the 2PT heat capacity can vary between kB/2 and kB per atom, not incorporating any anharmonicity, so the 2PT method cannot describe cases where the classical heat capacity is above kB. Another limitation is that the molecular topology needs to remain the same during the simulation, so bond breaking/formation, thus chemical reactions, cannot be modeled. Moreover, intramolecular rotations cannot be described properly with 2PT.
2.1.3. 1PT with Anharmonic Correction (1PT+AC)
Berens et al. proposed a quantum correction added to the classical heat capacities:39
| 14 |
The quantum correction can be determined from the quantum harmonic weight function cVΔ given by
| 15 |
If the integral terms are partitioned differently then we get:
| 16 |
where the second term is the anharmonic correction. The 1PT+AC internal energy can be gained analogously as the heat capacity:
| 17 |
where the anharmonic correction is the deviation of the classical internal energy (Ucl) from the ideal harmonic case:
| 18 |
The 1PT+AC model always satisfies the correspondence principle in contrast with the 1PT or 2PT methods:
| 19 |
This also implies that the technique is able to describe anharmonic motions to the extent of the method used to obtain the original classical trajectory.
2.2. Generalized Smoothed Trajectory Analysis (GSTA)
In the previous sections, we briefly introduced the relevant methods from the literature about the correction of the VDOS function to get quantized thermodynamic properties. In this section, we present a derivation to show that a similar correction can be performed in the time domain on the VACF function and on the coordinates.
2.2.1. Correction of Velocity Autocorrelation Function
Convolution of two functions f and g is defined as
| 20 |
is frequently used in digital processing for the smoothing of signals or the filtering of high frequency noises.65 On the basis of the convolution theorem, the multiplication of the VDOS function with a weight function w is equivalent to a convolution of the Fourier transforms of the two functions. The quantum corrected VACF function is the inverse Fourier transform of the quantum corrected VDOS function:
| 21 |
The Fourier transform of the weight function can be used for the quantum correction of the VACF function:
| 22 |
From the corrected VACF function, the thermodynamic properties can also be calculated. For instance, the 1PT+AC heat capacity:
| 23 |
where γcV is the Fourier transform of the weight function in eq 10 according to eq 22.
| 24 |
where csch is the hyperbolic cosecant function. The 1PT+AC internal energy takes the form
| 25 |
and the weight function for VACF with the internal energy is given by
| 26 |
The integral in eq 26 fails to converge as the wU weight function increases monotonously. Fortunately, in real systems, the maximum frequency in the VDOS is always finite, so the weight function can be cut at a high finite frequency with the application of an exponentially decreasing function:
| 27 |
where variable b controls the exponential decay. The decay is faster as b increases. This function becomes unity as b approaches infinity:
| 28 |
The γU function can be determined as a limit of an integral:
| 29 |
The integral can be evaluated in eq 29, and finally, the γU function can be expressed as a limit of a piecewise function:
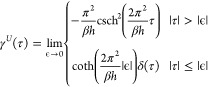 |
30 |
where δ denotes the dirac delta function. The norm of the function in eq 30 is 1. The coth function is the primitive integral of the csch2 function. Since in the simulations the data are represented at discrete time intervals Δt, it is useful to write the γU function at discrete time steps in such a way that it is directly applicable for integration, i.e.,
| 31 |
where n is the index of the time step. When n = 0, the γU function takes the simple form:
| 32 |
The weight functions of VACF are shown in Figure 2.
Figure 2.
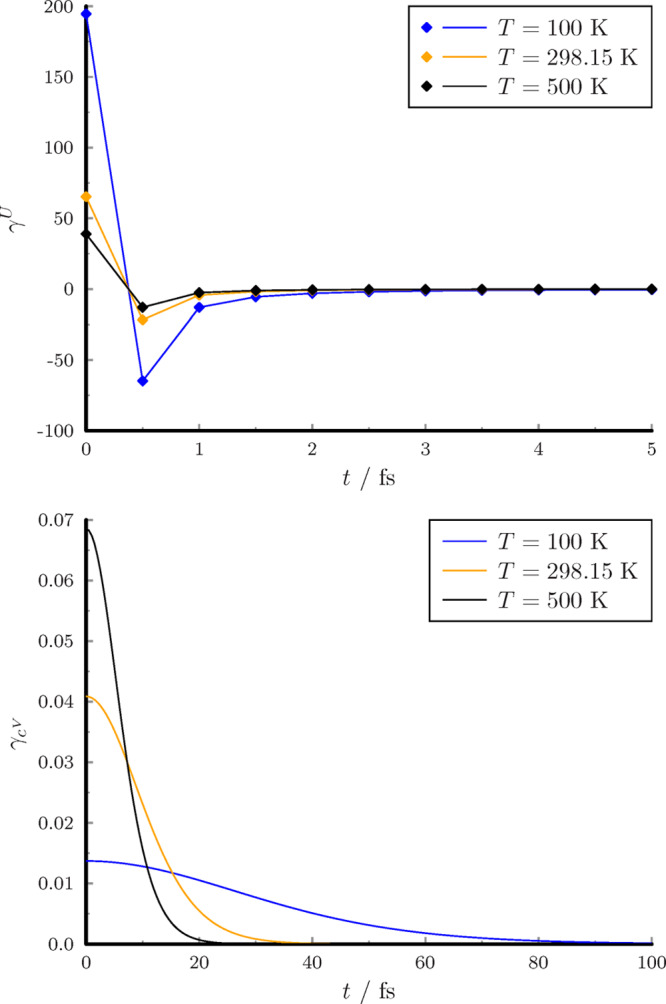
Weight functions for VACF.
2.2.2. Correction of Trajectory
The velocity autocorrelation function is actually a convolution function of the velocity with itself, i.e., f = g = v in eq 20:
| 33 |
According to eqs 1 and 2, the VDOS can be written in the form of:
| 34 |
Similarly, the quantum corrected counterpart can be written as
| 35 |
where ṽ is a modified velocity which satisfies eq 35. In the following steps, we determine ṽ.
Substituting eqs 34 and 35 into eq 6:
| 36 |
Using the convolution theorem:
| 37 |
Assuming that w(ν) is a non-negative real-valued function we get
| 38 |
In the time domain, the multiplication by w(ν) is replaced by convolution.
| 39 |
| 40 |
Thus, we arrived to a g(t) function whereby convoluting the velocities one can directly obtain ṽ and the quantum corrected vibrational density of states in eq 35. If one wants to use a general function, which can be applied any atomic velocity function, then the vibrational weight function (eq 10) can be chosen:
| 41 |
where sech means the hyperbolic secant function.
In the determination of a kernel function, the weight function of the internal energy can also be used:
| 42 |
Similarly to the determination of the γU function in eq 26, the integral does not converge here either. We could not derive an analytic form for the Fourier transform in eq 42. In order to perform this Fourier transform numerically in a practical way, the weight function of the internal energy can be split into two parts:
| 43 |
The Fourier transform of the second term in eq 43 can be readily evaluated numerically, while the Fourier transform of the first part can be determined analytically in a similar way as we did with the γU function:
| 44 |
The result can be given as a limit of a piecewise function:
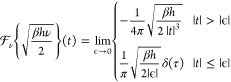 |
45 |
So the gU function can be calculated:
 |
46 |
When the convolution is performed at discrete time step, Δt, the value of the kernel function in the nth step is
| 47 |
At zero time
| 48 |
The kernels of gU and gcV are shown in Figure 3. gU is represented in discrete points with Δt = 0.5 fs.
Figure 3.

Kernels for trajectory filtration
Utilizing a kernel function defined in eq 40, one can obtain filtered time-dependent variables such as coordinates, velocities, and forces (x̃, ṽ, F̃):
| 49 |
| 50 |
| 51 |
Having these variables smoothed, we are able to derive smoothed energy components. The smoothed kinetic energy (Ẽkin) can be obtained straightforwardly from smoothed velocities.
| 52 |
2.2.3. Correction of Potential Energy
For the definition of the smoothed potential energy (Ẽpot), it can be expected to fulfill the following condition:
| 53 |
The smoothed total energy is simply the sum of the smoothed kinetic and potential energies:
| 54 |
The kernel function can depend on several parameters like the temperature or the Planck constant h. In order to connect the classical systems with the quantum systems continuously, a fictitious η variable is introduced. η = 0 corresponds to the classical picture, and η = h is the quantum one. With η, we can perform an integration from the classical state to the quantum state.
It can be shown that the total energy remains conservative upon smoothing:
| 55 |
The total differential of the smoothed total energy is
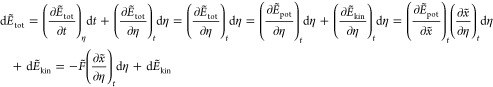 |
56 |
The negative of the first term can be called as work of smoothing:
| 57 |
The smoothed potential energy is defined at a specific time (t0) as a correction on the original potential energy (Epot) with the work of smoothing:
| 58 |
Integrating this equation according to time yields the expectation value of smoothed potential energy:
| 59 |
| 60 |
The average change of kinetic energy is equal to the average change of the potential energy (more details in Appendix A):
| 61 |
So the mean smoothed total energy is
| 62 |
Note that eq 62 corresponds to U1PT+AC in eq 17. So we arrived to the same result as the 1PT+AC method, which is true for the heat capacity as well:
| 63 |
Quantum-corrected state functions can be determined from the presented smoothed quantities, as shown by the example of heat capacity below.
3. Computational Details
3.1. Calculation of Heat Capacity
According to the Theory section, several estimators can be designed to determine the heat capacity depending on what is corrected: VDOS, VACF, or the trajectory. The quantum correction can be introduced with different functions which correspond to the heat capacity or the internal energy, but the resulting thermodynamic functions can be transformed into each other by integration/differentiation. We used the kernel function of the heat capacity (eq 41) for the filtration since it is more convenient; i.e., its analytical form is known.
We performed several (50–120) independent NVE simulations around the T target temperature. The classical temperature (Tcl) was determined simply from the average classical kinetic energy for a particular trajectory:
| 64 |
The isochoric heat capacity can be determined from the slope of the mean total energies vs Tcl functions:
| 65 |
| 66 |
The classic temperature originates from the classical normalization factor in eq 35. The isobaric heat capacity can be determined from the slope of the H vs Tcl (or H̃ vs Tcl) function:
| 67 |
| 68 |
For condensed phases, p = 1 atm was applied instead of the calculated average pressure because its fluctuation was larger than several hundred atm. We used linear regression to determine the heat capacities and their errors. The uncertainties of our calculations are given at the 95% confidence level. Representative fittings are shown in the Supporting Information.
3.2. Born–Oppenheimer Molecular Dynamics Simulations
We carried out classical microcanonical normal mode sampling66 with Gaussian 09 Rev. E for the vibrations of one water molecule using B3PW91/6-311G(d,p) level of theory.67,68 This functional was chosen since it reproduced the exact frequencies of a water molecule the most accurately with an average error of 6.1 cm–1.69 Initial energies were distributed equally between modes according to the equipartition theorem. The equations of motion for the nuclear evolution were integrated employing the velocity Verlet algorithm with a 0.1 fs time step.70 Then, 50 independent 1 ps long trajectories were generated around the desired temperature, and the total energies represented a χ2 distribution.
3.3. Classical Molecular Dynamics Simulations
Classical molecular dynamics simulations were performed with the Tinker molecular modeling software.71 The simulation box always contained 432 water molecules. For the liquid and vapor phases, a cubic box was applied. For Ih ice, water molecules were arranged according to the Bernal–Fowler ice rules72 having a vanishing net dipole moment.
First, we performed 10 ps long NpT simulations starting from previously equilibrated structures followed by 11 ps long NVE simulations. The last 1 ps data were used for the trajectory analysis. Here, 120 independent trajectories were generated to determine the heat capacity at a given temperature. In order to make the linear fitting more effective, the temperature of the thermostat was varied around the target temperature according to a χ2 distribution. The equations of motion were integrated employing the Bussi–Parrinello algorithm73 for the NpT ensemble and a modified Beeman algorithm74 for the NVE ensemble with a 0.5 fs time step. In condensed phase simulations, the particle mesh Ewald (PME) method was applied for the long-range electrostatic interaction with 9.0 Å cutoff distance.75 For the vapor phase, a larger cutoff of 113.0 Å was used with a 30 × 30 × 30 grid size for the Ewald summation.
We performed simulations for an NVT ensemble to determine the structural properties of the SPC/Fw water model at 298.15 K, with a simulation box size of 23.439 Å. Then, 500 configurations were collected, and after 11 ps long NVE simulations, the last 1 ps trajectory was filtered with the gU function with Δt = 0.1 fs to get one filtered structure from each trajectory. These 500 independent structures were used to calculate the distributions of the intramolecular distances as well as the radial distribution functions.
For the calculation of the IR absorption spectrum of the AMOEBA14 water model, 120 independent NVE trajectories were used. All simulations were 20 ps long, and they were equilibrated at 298.15 K before. We used a four term Blackman–Harris window before the Fourier transform of the dipole autocorrelation function.76
3.4. Path Integral Molecular Dynamics Calculations
PIMD simulations were carried out with AMBER1277 in a canonical ensemble for 216 water molecules using the SPC/Fw model. The settings were taken from ref (28), but 32 beads were used instead of 24. The length of the cubic simulation box was 18.68 Å, according to the equilibrium density. After 1 ns long equilibration, 1000 structures were collected for each bead in an additional 1 ns long simulation. For the calculation of the isochoric heat capacity, we performed canonical simulations at 288.15 and 308.15 K as well.
In order to determine the isobaric heat capacity for the liquid phase, NpT simulations were also performed at atmospheric pressure in the temperature range from 260.65 to 385.65 K.
4. Results and Discussion
4.1. Harmonic Oscillator Model
Here, we show the effect of two filters on the sum of two noncoupled oscillators at 298.15 K. One oscillator has high frequency (3000 cm–1), while the other has low frequency (100 cm–1). The analytic curves are shown in Figure 4. Here, gcV smooths the vibration with high frequency, while gU enhances the high frequency motion. The filtered function with gU corresponds to the quantum fluctuation.
Figure 4.
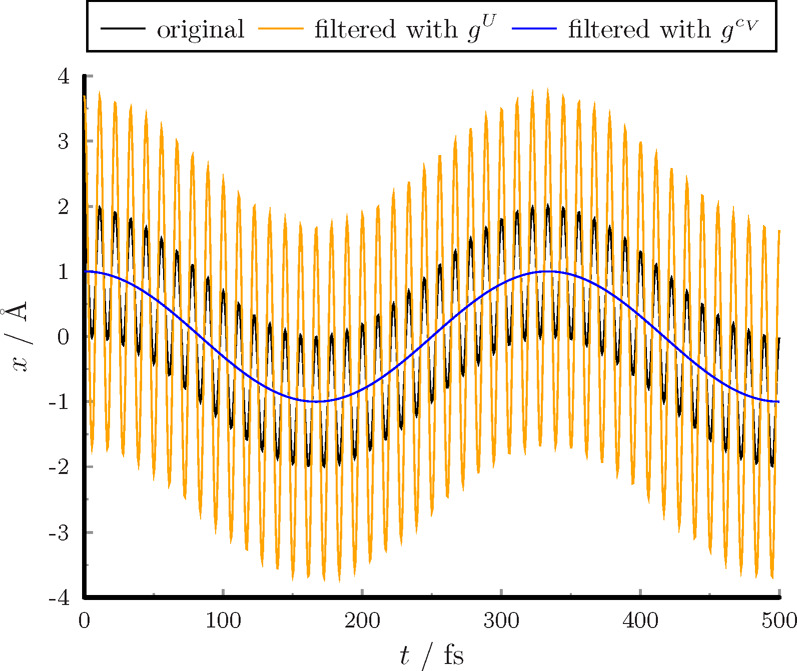
Filtration of the sum of two harmonic functions.
In the following section, we determine the fluctuation of the coordinates for a single harmonic oscillator. The time evolution of the position:
| 69 |
where X is the amplitude in a particular trajectory.
The probability distribution of the position for the harmonic oscillator in canonical ensemble:
| 70 |
where κ is the force constant of the harmonic potential. The filtered position is
| 71 |
The probability distribution of the filtered position is given by
 |
72 |
This corresponds to the exact quantum fluctuation of the position for the harmonic oscillator. So the GSTA method is exact for the harmonic model for not only in the thermodynamic properties but for the coordinate distribution as well. This is a clear advance of GSTA compared to the 2PT method, since structural quantum effects cannot be investigated with 2PT.
4.2. BOMD Simulations of One Water Molecule
Apart from cases where analytical solution exists, like a harmonic oscillator, we have chosen to test the proposed method on a single water molecule as a more realistic system. Born–Oppenheimer molecular dynamics (BOMD) simulations were carried out without rotation in the temperature range from 25 to 5000 K using the B3PW91 functional.67 The vibrational part of the isochoric heat capacity (cVvib) was obtained as the slope of the internal energy vs temperature function. For the sake of comparison, we estimated the isobaric heat capacity (including rotation and translation) based on these data as cp = cV + 1.5R + 1.5R + R = cVvib + 4R.
We compare GSTA results with heat capacities determined using several different methods in Figure 5. The graph also includes the ideal classical value, 7R = 58.2 J/K/mol (blue line) as well as the values determined with the application of the quantum harmonic oscillator model to the optimized geometry (green circles). At low temperature, all methods give very similar results, while at the higher temperature only the trajectory smoothing (red crosses) is close to the experimental values (black triangles).78 This illustrates that anharmonicity can be described properly by GSTA in contrast with the quantum harmonic oscillator model or the 1PT method (purple squares).
Figure 5.
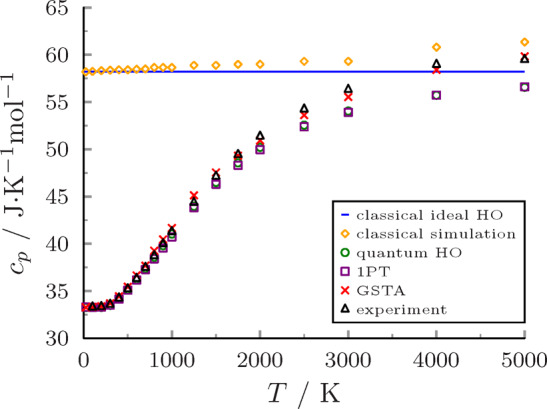
Isobaric heat capacities of a single water molecule from BOMD simulations.
4.3. Properties of Bulk Water
In order to demonstrate that our approach can work in very different circumstances, bulk water was considered as a test case in a wide temperature range. The quantum effects are decreasing while anharmonicity is increasing with temperature, and the heat capacity of water changes significantly from zero to 76.0 J/mol/K between different phases at atmospheric pressure.79,80 The intramolecular vibrations remain in the ground state at ambient temperature; therefore, many rigid water models are successful in reproducing the experimental values.64,81 For the sake of generality, a flexible model is desired with which similar approaches can be examined even at high temperatures where excited vibrations can be populated as well. Three-site models are beneficial because analytical Hessian can be calculated for the optimized structures, which is not possible for polarizable water models. Considering these requirements, we have chosen the recently developed SPC/Fw82 water model which performs well on several physical properties.81 The parameters of the SPC/Fw water model were developed for classical molecular dynamics simulations to reproduce experimental data such as self-diffusion coefficient, dielectric constant, vibrational frequencies, oxygen–oxygen radial pair distribution function, and heat of evaporation.82
4.3.1. Static Properties
Classical simulations cannot distinguish between the static properties of light and heavy water. In principle, exactly the same thermodynamic and structural properties should be determined for H2O and D2O since classical statistical thermodynamics predicts that equilibrium properties (heat capacity, molar density, surface tension, etc.) are independent of the masses of atoms. To reveal the different NQEs of classical trajectories, we need post processing methods like 1PT, 2PT, 1PT+AC, or GSTA. An important issue is to decide the reference which the computed quantities are compared to. Converged PIMD simulations provide exact static properties within the limit of the particular water model. However, empirical potentials are developed to reproduce various experimental data with classical simulations. Thus, our strategy is to compare the different approximate values to both PIMD results and experiments as well.
Heat Capacity
At a given temperature, we determined the isobaric heat capacity as the derivative of the enthalpy vs temperature function from 120 independent 1 ps microcanonical trajectories. Three states such as Ih ice, liquid water, and vapor were simulated at temperatures from 25 to 1000 K at atmospheric pressure (Figure 6). The classical ideal values are also shown with blue lines for the condensed and vapor phases in the figure (9R = 74.8 J/K/mol, 7R = 58.2 J/K/mol, respectively).
Figure 6.
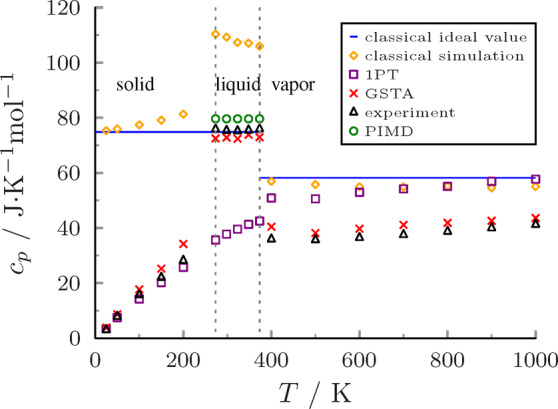
Isobaric heat capacities of bulk water in different phases.
The nuclear motions can be accurately described by independent harmonic oscillators at low temperature; therefore, the heat capacity can be estimated quite well with all methods in the case of hexagonal ice (black triangles in Figure 6). Our method (denoted with red crosses) also gives values close to the experimental ones.79 The maximum deviation is 6.0 J/mol/K at T = 200 K, probably due to the fact that the SPC/Fw water model was developed for liquid water and not for ice. A typical indication of this is that the melting point of the SPC/Fw water model is similar to that of the SPC/E water model (215 K), well below the experimental value (273.15 K).83
For liquid water, the harmonic oscillator model is qualitatively wrong as it fails to describe anharmonicity captured already by classical simulations (yellow diamonds in Figure 6). The smoothing correction successfully takes the coupled motions of the molecules into account, thus performing outstandingly when compared to the experimental values. The isobaric heat capacity calculated by PIMD is somewhat higher (green circles in Figure 6). Most importantly, GSTA performs much better than the classical model or the 1PT method no matter what reference we use, i.e., experimental value or PIMD results. Intramolecular vibrations are filtered out in the gcV-smoothed trajectories. In this aspect, water molecules behave rigidly in these liquid simulations (see the animation in the Supporting Information). This is in line with the experience that the overall performance of rigid water models is comparable to that of the flexible ones at room temperature.64,81
For the vapor phase, we used the same water model and trajectory analysis as in condensed phases (Figure 6). GSTA gives reasonable results but overestimates the experimental heat capacities by about 3.2 J/mol/K. This is probably due to the fact that the dipole moment of the SPC/Fw water model is adjusted to liquid properties and considering that it is high compared to the dipole moment of a single water molecule in the gas phase (2.39 vs 1.85 D, respectively). The 1PT method overestimates the experimental values with 16 J/mol/K as a consequence that it considers only harmonic vibrations.
Overall, the smoothed SPC/Fw simulations are able to reproduce the heat capacity with an average error of 3.1 J/mol/K in an extended temperature range at atmospheric pressure. We have found two computational studies in the literature which investigated the heat capacities of the three most important phases of water. Yeh et al. used the 2PT method with different rigid water models, and all calculated heat capacities deviated more than 6.7 J/mol/K from the experiments.45 Shinoda and Shiga performed PIMD simulations for the three phases using the flexible SPC/F water model.84 They reproduced the experimental values excellently for ice and gas phase, but the heat capacity of liquid water was significantly underestimated by 16.6 J/mol/K.
We have investigated other water models to examine their performance for the calculated heat capacities of liquid water (at T = 298.15 K, p = 1 atm). Besides the discussed SPC/Fw model, other potentials were also tested including rigid, polarizable, and four-site models. Table 1 includes results from previous works as well for comparison. Regarding the classical heat capacities (cpcl), we have reproduced the literature data with small differences, which validates our simulations. GSTA-corrected heat capacities vary between 72.8 and 80.9 J/mol/K with the different water models around the experimental value. The 1PT and 2PT methods give values from 52.2 to 86.6 J/mol/K, and generally, rigid models perform better. The PIMD heat capacities range from 58.2 to 92.8 J/mol/K. The PIMD values can be considered as the exact heat capacity for a particular water model. Previously, the PIMD heat capacity of the SPC/Fw water model was found to be 88.7 J/mol/K, significantly higher than that of the experimental (75.4 J/mol/K) or the GSTA values (72.8 J/mol/K). In our PIMD simulations on an NpT ensemble, the isobaric heat capacity is 79.6 J/mol/K. In order to resolve this contradiction, we performed PIMD simulations on a canonical ensemble with the same settings as in the literature.28 Using the same number of beads, 24, we obtain 79.8 J/mol/K for the heat capacity, but with 32 beads it decreases to 76.6 J/mol/K. This indicates that there is a discrepancy between the value obtained here and those from earlier simulations. Still, we note that our PIMD heat capacity is in reasonable agreement with the experimental and the GSTA values. Our results indicate that the SPC/Fw water model reproduces the experimental heat capacity sufficiently. On the basis of the comparisons discussed here, we conclude that GSTA is an accurate and robust method in the sense that the calculated heat capacities are less sensitive to the chosen potential.
Table 1. Calculated Isobaric Heat Capacities of Liquid Watera.
| potential | flexibility | cp | cpcl |
|---|---|---|---|
| experiment | 75.4 | – | |
| 1PT or 1PT+AC | |||
| AMOEBA14 | flexible | 85.7b | 110.6b |
| F3C | flexible | 84.5c,r | 109.6c,r |
| SPC/E | flexible | 86.6d,r | 123.7d,r |
| WATTS | flexible | 72.5e,r | 107.3e,r |
| SPC/E | rigid | 73.0d,r | 87.4d,r |
| 2PT | |||
| AIMD–PBE | flexible | 52.2f,r | 90.1f,r |
| F3C | flexible | 67.4f,r | 104.6f,r |
| ReaxFF | flexible | 73.0f,r | 108.4f,r |
| SPC-Fw | flexible | 69.3f,r | 109.5f,r |
| SPC/E | rigid | 76.7f,r; 68g | 87.3f,r; 87g |
| TIP3P | rigid | 58.3f,r; 60g | 67.5f,r; 86g |
| TIP4P/2005 | rigid | 77.5f,r; 68g | 85.5f,r; 89g |
| TIP4P-Ew | rigid | 69.0f,r; 69g | 80.8f,r; 86g |
| PIMD | |||
| SPC/F | flexible | 59.0h,r | 117.2h,r |
| SPC/F2 | flexible | 58.2i,r | 117.2i,r |
| SPC-Fw | flexible | 88.7j,r; 79.6 ± 0.5 | 113.8j,r |
| q-SPC/Fw | flexible | 92.8j,r | – |
| q-TIP4P/F | flexible | 66.7k,r | – |
| TIP4PQ/2005 | rigid | 75.3l | – |
| GSTA | |||
| AMOEBA14b | flexible | 80.9 ± 0.7 | 113.5 ± 0.6 |
| F3Cc | flexible | 76.3 ± 0.7 | 108.4 ± 0.5 |
| SPC-Fwm | flexible | 72.8 ± 0.6 | 109.3 ± 0.4 |
| TIP3Fn | flexible | 72.8 ± 0.7 | 106.9 ± 0.6 |
| TIP4P/2005Fo | flexible | 80.1 ± 0.9 | 113.4 ± 0.7 |
| TIP3Pp | rigid | 75.0 ± 1.0 | 81.2 ± 0.9 |
| TIP4P/2005q | rigid | 79.0 ± 0.7 | 87.1 ± 0.6 |
T = 298.15 K, p = 1 atm; all values are given in J/mol/K.
ref (42).
ref (43).
ref (44).
ref (39); 2PT.
ref (46).
ref (45); PIMD.
ref (84).
ref (85).
ref (28).
ref (26).
ref (86); GSTA: this work.
ref (82).
ref (87).
ref (88).
ref (89).
ref (90).
cp was estimated from the given cV as cp = cV + 0.8 J/mol/K.
Structure of Liquid Water
A quantum-corrected structure can be obtained after the filtration of the coordinates with the kernel function of the energy (gU). The largest NQE in the structural properties of water is the quantum fluctuation of the hydrogen atoms. This effect is illustrated in the distribution of the intramolecular distances in Figure 7 and also in the animation (Supporting Information). The distribution determined from PIMD simulations can be considered as the exact reference. The distributions of the intramolecular distances become broader compared to the classic results according to the Heisenberg uncertainty principle. The classical distributions are too narrow, but the average distances are almost identical (Table 2). The GSTA method reproduces almost perfectly the exact distributions with a slight shift of 0.01 Å for the maxima.
Figure 7.
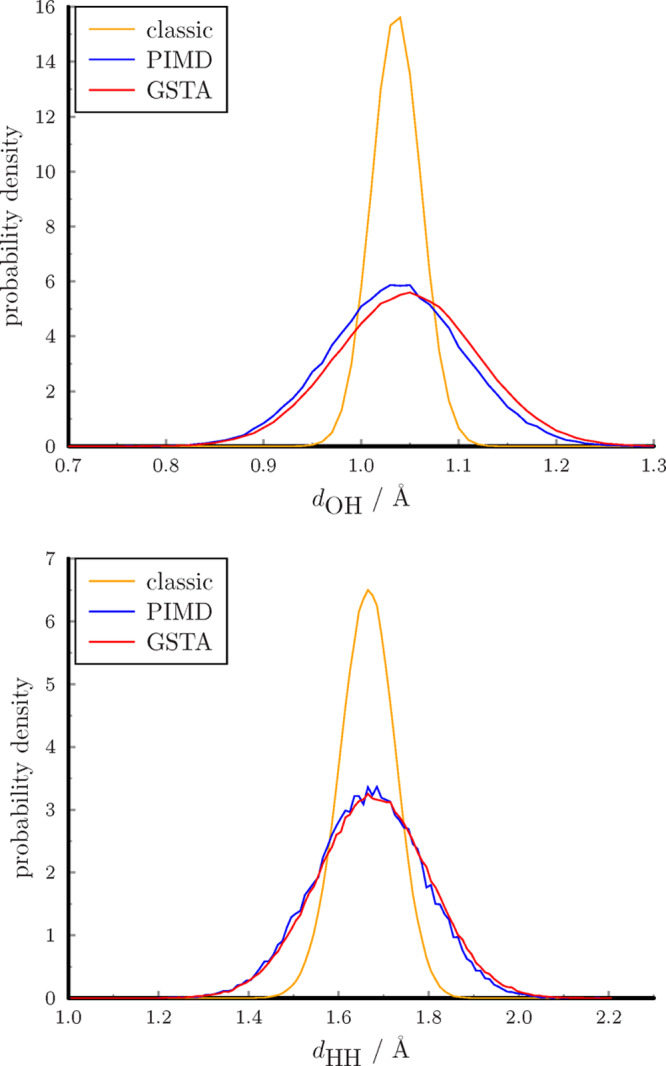
Probability distribution functions of intramolecular distances.
Table 2. Intramolecular Distances of Bulk Water.
| OH distance/Å | |||
|---|---|---|---|
| classic | GSTA | PIMD | |
| mean | 1.03 | 1.04 | 1.03 |
| standard deviation | 0.03 | 0.07 | 0.07 |
| HH distance/Å | |||
|---|---|---|---|
| classic | GSTA | PIMD | |
| mean | 1.66 | 1.68 | 1.66 |
| standard deviation | 0.06 | 0.12 | 0.12 |
The classical and the filtered radial distribution functions are shown in Figure 8 together with the experimental data.91 The positions of the classical peaks are not altered significantly by the filtration, but they become broadened and get closer to the experimental curves. Note that the 1PT and 2PT methods do not correct the structural properties.
Figure 8.
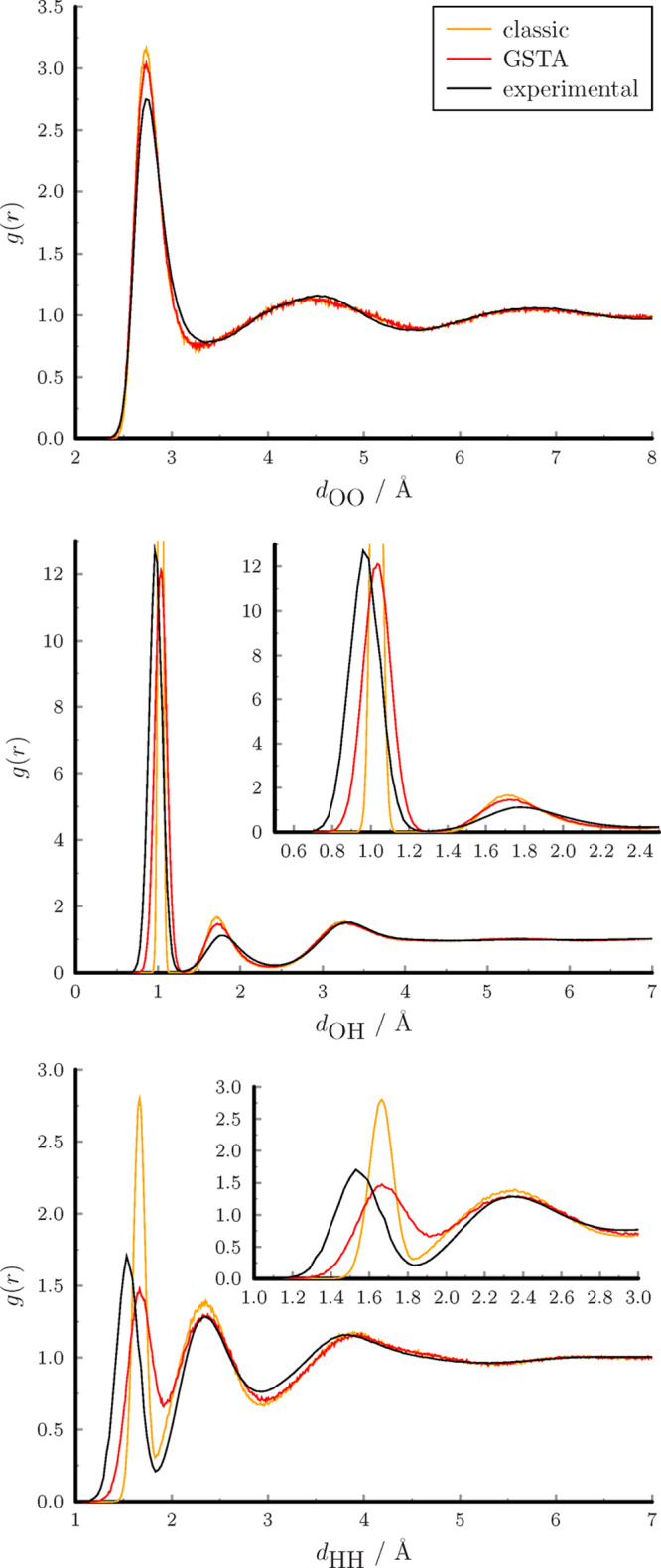
Radial distribution functions of atom pairs in liquid water.
Static Dielectric Constant
The static dielectric constant is another equilibrium property which exhibits nuclear quantum effects. Although, for liquid water the effect of deuterium substitution is small, the experimental dielectric constants are 78.39 vs 78.06 for H2O and D2O;92 in ice, the isotope effect is the opposite and more enhanced. The static dielectric constants of ordinary and heavy ice are 110 and 124 parallel to the c axis of the crystal at −25 °C, and the difference is even larger at lower temperature.
We have calculated the dielectric constant for both the classical and filtered structures at 298.15 K using the following relationship:
| 73 |
where ϵ0 is the vacuum permittivity, V is the volume of the simulation box, and M is the total dipole moment of the system. We computed the dielectric constant for SPC/Fw and AMOEBA14 water models. In Table 3, we collected our GSTA results along with previous classic and PIMD values from the literature. Our classical values are in good agreement with the literature values. GSTA slightly increases the dielectric constant with about 0.5, which is smaller than its uncertainty. The differences between the classical and filtered trajectories can be calculated pairwise, resulting in a more accurate estimation 0.47 ± 0.03 and 0.6 ± 0.3 for AMOEABA14 and SPC/Fw, respectively. The good agreement with the experimental value is due to the fact that the parameters of the SPC/Fw and AMOEBA14 water models were optimized to reproduce the dielectric constant. The filtered dielectric constant is only slightly larger than the classical value, which is in line with the small difference between the dielectric constants of H2O and D2O (78.39 vs 78.06, respectively).92
Table 3. Static Dielectric Constant of Liquid Water.
| model | classic | GSTA | PIMD |
|---|---|---|---|
| SPC/Fw | 80 ± 2a; 79.1 ± 6 | 79.7 ± 6 | 64 ± 4b |
| AMOEBA14 | 79.4 ± 1.6c; 83.6 ± 7 | 84.1 ± 7 | |
| SPC/F | 80 ± 8d | 67 ± 9d | |
| SPC/F2 | 94 ± 10d | 74 ± 11d | |
| TTM2.1-F | 67 ± 2e | 74 ± 2e | |
| TTM3-F | 94 ± 1f | 81 ± 2g | |
| q-SPC/Fw | 86 ± 4b; 90 ± 3h | ||
| q-TIP4P/F | 58 ± 1g | 60 ± 3h | |
| fm-TIP4P/F-TPSS-D3 | 48.38i | 45.69i | |
| experimentj | D2O: 78.06; H2O: 78.39 | ||
For the SPC/Fw water model, PIMD gives a smaller dielectric constant by 16 than the classical value. Thus, GSTA predicts a significantly lower quantum effect for the dielectric constant than PIMD (+0.6 vs −16), so even the effect is the opposite. The quantum effect determined by PIMD strongly depends on the water model. In most cases, the dielectric constant decreases drastically, but in some cases, it increases. This implies that the overall effect comes from competing quantum effects, as Habershon et al. proposed for the self-diffusion coefficient of water.24 They also mentioned that this may affect equilibrium properties such as the melting point of ice.
In order to investigate the possible source of these competing effects, we calculated the molecular dipole moment μ and the Kirkwood g-factor GK for the SPC/Fw water model. (For the AMOEBA14 model, it is not so straightforward to calculate the molecular dipole moment because it is a polarizable water model.) The molecular dipole moment is calculated as a root-mean-square of the individual dipole vectors:
| 74 |
where Nmol is the number of the molecules. The Kirkwood g-factor is defined as
| 75 |
The dielectric constant can be expressed from the molecular dipole moment and the GK factor.
| 76 |
The molecular dipole moments and the Kirkwood g-factor are collected in Table 4 as well as the static dielectric constants.
Table 4. Components of Static Dielectric Constant of Water.
| model | classic | GSTA | difference | |
|---|---|---|---|---|
| ϵs | AMOEBA14 | 83.6 ± 7 | 84.1 ± 7 | 0.47 ± 0.03 |
| ϵs | SPC/Fw | 79.1 ± 6 | 79.7 ± 6 | 0.58 ± 0.33 |
| μ/D | SPC/Fw | 2.3727 ± 0.0003 | 2.3993 ± 0.0007 | 0.0266 ± 0.0005 |
| GK | SPC/Fw | 4.06 ± 0.31 | 4.00 ± 0.31 | –0.059 ± 0.017 |
The overall effect is a product of both the molecular dipole moment and the Kirkwood g-factor. The sign of these effects can be positive or negative depending on the water model, but in experiments, the overall effect is small. Most of the PIMD simulations indicate large quantum effects on the static dielectric constant which implies a large effect on the molecular dipole moment and/or on the Kirkwood g-factor as well. This does not mean that PIMD would be inaccurate, just indicates that these force fields were not designed to reproduce the experimental NQE on dielectric constant with PIMD simulations. GSTA predicts small effects on these properties, which is in line with the experiments. The q-TIP4P/F force field predicts the smallest NQE in PIMD simulations, and the difference between the dielectric constants is smaller than the uncertainty (Table 3).
4.3.2. Dynamical Properties
The investigation of NQEs on dynamical properties is less straightforward than that on static properties. Even classical simulations show drastic differences for H2O and D2O simply because the deuterium moves slower than protium due to the mass difference. Standard PIMD calculations cannot model time-dependent processes, but there are several approximate PIMD-based methods such as RPMD, CMD, or LSC-IVR simulations which can imitate quantum dynamics.13,18,97 Hence, it is challenging to separate quantum and classical effects in isotope substitutions, and this makes it difficult to validate new methods like GSTA on dynamical NQEs.
Self-Diffusion Coefficient
The self-diffusion coefficient Ds can be determined from the zero frequency value of VDOS:
| 77 |
Since the weight function wU(ν) is always 1 at zero frequency, the self-diffusion coefficient does not change with the application of the GSTA method (see eqs 8 and 77). The self-diffusion coefficient can be calculated from the mean square displacement of the atoms using the Einstein equation:
| 78 |
From this equation it is also evident that one obtains the same self-diffusion coefficient after filtering the classical trajectory, since GSTA perturbs the classical coordinates with a few tenths of Å which becomes negligible compared to the movement of the atoms at a sufficiently long time. Thus, previous classical self-diffusion coefficients are collected in Table 5 which are identical with the GSTA values as well. PIMD-based approximate quantum dynamics methods like CMD, RPMD, or LSC-IVR change the classical value of self-diffusion coefficient. The quantum effect (relative change to the classical value) varies between 180% and −30% depending on the water model and quantum dynamics method.
Table 5. Calculated Self-Diffusion Coefficients of Liquid Water in Å2/ps.
| classic/GSTA |
quantum |
||||||
|---|---|---|---|---|---|---|---|
| model | H2O | D2O | method | H2O | D2O | quantum effect | isotope effect |
| SPC/Fw | 0.232a | CMD | 0.32b | 38% | |||
| q-SPC/Fw | 0.18b | CMD | 0.24b | 39% | |||
| q-SPC/Fw | 0.18b | LSC-IVR | 0.50c | 178% | |||
| SPC/F | 0.30d | CMD | 0.42d | 40% | |||
| SPC/F2 | 0.22d | CMD | 0.38d | 73% | |||
| q-TIP4P/F | 0.192e | RPMD | 0.221e | 0.172e | 15% | –29% | |
| MB-pol | 0.23f | CMD | 0.22g | –4% | |||
| revPBE-D3 | 0.222h | TRPMD | 0.16h | –28% | |||
| revPBE0-D3 | 0.267h | TRPMD | 0.229h | –14% | |||
| fm-TIP4P/F-TPSS-D3 | 0.3i | RPMD | 0.288i | –4% | |||
| TTM2.1-F | 0.150j | 0.140j | CMD | 0.225j | 0.185j | 50%; 32% | –7%; −18% |
| TTM3F | 0.27k | CMD | 0.30k | 11% | |||
| vdW-cx | 0.23k | CMD | 0.34k | 48% | |||
| MCY | 0.25l,m | 0.198m | –21% | ||||
| SPC rigid | 0.44n | 0.33n | –25% | ||||
| SPC flexible | 0.61n | 0.59n | –3% | ||||
| SPC/E | 0.224o | 0.201o | –10% | ||||
| SPC-MPG | 0.275p | 0.244p | –11% | ||||
| TIP4P/2005F | 0.22p | 0.182p | –17% | ||||
| experiment | 0.230q | 0.177r | –23% | ||||
Although GSTA does not show any nuclear quantum effect on the self-diffusion coefficient, classical simulations can also reproduce the experimental isotope effect. The experimental self-diffusion of heavy water is 23% slower than that of the light water, and in classical simulations, this varies between 3% and 25%. We have found only two quantum simulations on heavy water with q-TIP4P/F and TTM2.1-F water models, and the calculated isotope effects (−29% and −18%) are in good agreement with the experiment as well as the absolute values.
Infrared Absorption Spectrum
In the classical simulation, the infrared spectrum of the SPC/Fw water model failed to reproduce the experimental spectrum qualitatively because of the harmonic model and the fixed point charges.28,106 This is why we calculated the infrared absorption spectrum of the AMOEBA14 water model which is polarizable, and the OH bonds are anharmonic. We determined the infrared absorption spectra both from the classical and filtered trajectory at different levels of theory (see details in Appendix B).
For classical simulation, the absorption cross section is
| 79 |
Using the harmonic quantum correction factor on the classical line shape function we obtain107,108
| 80 |
The absorption cross section is calculated from the filtered trajectories by the expression
| 81 |
Interestingly, almost identical spectra were obtained from the filtered trajectories and from the classical simulation applying the harmonic quantum correction factor (see orange and blue lines in Figure 9). This implies that the dipole moment changes linearly with the coordinates during the filtration. The absorption spectrum of the classical trajectory without quantum correction shows much lower intensities (green line). The relative intensities are also different, but the positions of the peaks are the same as in the quantum corrected spectra. Comparison with the experimental spectrum (black line) shows that the quantum corrected peaks deviate with ±100 cm–1, and the relative intensities are reproduced qualitatively.109 The peaks of the symmetric and antisymmetric OH stretches are partially overlapped due to the anharmonicity of the AMOEBA14 water model. The calculated spectrum also reproduces the smaller peaks at around 200 and 2200 cm–1 which are completely missing from the spectrum of nonpolarizable water models.
Figure 9.

Infrared absorption spectrum of AMOEBA14 water model at 298.15 K. Calculated intensities are scaled by a factor of 0.417.
In previous dynamic simulations, the quantum spectra deviated significantly from the ones computed from classical simulations. The classical peaks are always blue-shifted compared to the peaks of quantum simulations, the largest difference is in the OH stretching being shifted by ∼100 cm–1.9,24,28,64,97,110−114 Assuming that GSTA does not change the position of the peaks, the spectra of the filtered trajectories are always blue-shifted relative to the spectra of quantum simulations independently from the water model. The justification is that in classic simulation the atoms vibrate at the bottom of the potential well and experience less anharmonicity than in real quantum dynamics where the atoms reach higher potential energies because of the zero-point energy. As mentioned in the Self-Diffusion Coefficient section, filtration does not change the frequencies of the vibrations but their intensities. Since GSTA is based on a harmonic approximation, it cannot reproduce the exact position of the vibrational frequencies as Marx already showed for quantum harmonic correction.107
In previous simulations, imaginary frequencies were also identified in the vibrational spectrum of water.112,115 GSTA cannot reproduce these imaginary frequencies, because in classical simulations, the VACF functions are even. Therefore, only real frequencies appear in VDOS after Fourier transform.
Real time autocorrelation functions can also be determined by the maximum entropy analytic continuation (MEAC) method for the calculation of quantum properties.116−119 In MEAC, the imaginary time correlation function is converted into a real time correlation function, and the former is generated from PIMD simulations. MEAC needs an approximate real time function so-called prior which can be a general function (i.e., flat prior) or a more specific one which comes from a CMD, RPMD, or LSC-IVR calculation. The input of GSTA is only a classical trajectory in the traditional sense that it propagates in real time as a single bead. MEAC is useful well below room temperature; therefore, it is not used for liquid water. Typically, it is applied for liquid para-hydrogen.117
4.4. Computational Cost and Accuracy
According to the convolution theorem, the same results can be obtained for the calculation of thermodynamic properties both in the time and frequency domains. The Fourier transform should not affect the results. This is true for continuous periodic signals, but a finite number of data are represented in discrete points in MD simulations. This is why the calculated quantities carry numerical errors, and different estimators of the same property can give different values. Here, we illustrate that the same correction of the VDOS and VACF can give significantly different results.
The VDOS function is generally determined via the Fourier transformation of the VACF function. The Fourier transform can be carried out with different numerical integrators. The most common technique corresponds to the left Riemann integral, which is probably the default algorithm in most of the simulation programs:
| 82 |
Using the trapezoidal rule for discrete Fourier transform we get
| 83 |
The
calculated 1PT heat capacities are collected in Table 6. In the generally applied method,
the VDOS correction using the left Riemann sum has a large error.
It overestimates the classical 1PT heat capacity 9R = 74.826 J/mol/K by 15%. The corrected 1PT heat capacity is overestimated
by 4%, but if it is normalized with the classical value (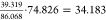 ), then the underestimation
is 9%. Using
the trapezoidal rule for integration, all methods give the same results
within 0.01%.
), then the underestimation
is 9%. Using
the trapezoidal rule for integration, all methods give the same results
within 0.01%.
Table 6. 1PT Heat Capacities of Liquid Water in J/mol/K.
| VDOS correction |
VACF correction |
||||
|---|---|---|---|---|---|
| left Riemann sum | trapezoidal rule | left Riemann sum | trapezoidal rule | trajectory smoothing | |
| classical | 86.068 | 74.821 | 74.826 | 74.826 | 74.826 |
| quantum | 39.319 | 37.759 | 39.290 | 37.760 | 37.757 |
| normalized quantum | 34.183 | 37.762 | 39.290 | 37.760 | 37.757 |
In order to sample the low frequency motions for the calculation of the VDOS, a long simulation time is necessary, and as a rule of thumb, the length of the VACF should be at least as long as the time period of the lowest frequency motion. As was already shown in the Theory2.1 section, the calculation and the correction of the VDOS are performed with a double integral (eqs 2 and 16), which is more CPU-demanding than the correction of the VACF which requires only a single integral (eq 23). One can expect that the calculations will be more accurate if all data are used; i.e., the length of the VACF equals the simulation length. The γcV weight function and the gcV kernel function converges exponentially to zero; therefore, the contribution of the long times are negligible to the thermodynamic properties. The question is this: what is the maximum time separation which has a significant contribution to the particular property?
We can give an upper limit for the maximum time separation (tmax) with the calculation of the heat capacity of ideal monatomic gases. The velocities can be considered as constant between two collisions, and its autocorrelation function is 1. In this case, there is no quantum effect, and the exact heat capacity is 3/2kB. The heat capacity with the filtration of the velocity considering the integration up to tmax is
| 84 |
where tanh denotes the hyperbolic tangent function. For the VACF correction
| 85 |
Both estimators lead to the exact classical result with a long tmax. The convergence of the functions are shown in Figure 10 at T = 298.15 K. The exact result is reached within 50 fs with an error smaller than 0.01%. This means that a 50 fs long VACF is enough for the quantum correction, and it is not necessary to calculate the VACF for several ps long. Calculations of the VACF consume considerable amounts of memory which correlate with tmax, and the number of the mathematical operations correlate with the square of tmax.
Figure 10.
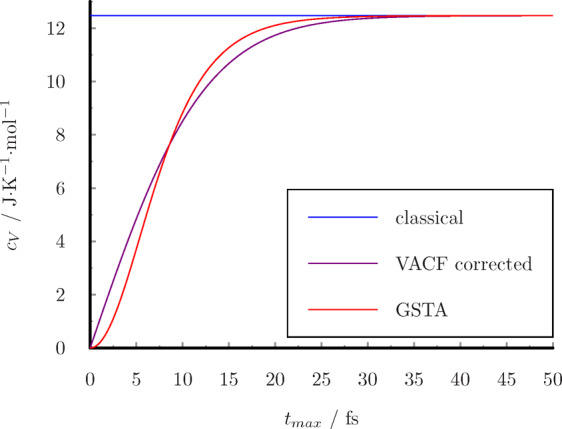
Convergence of heat capacity of ideal monatomic gas with maximum time separation.
5. Conclusions
In summary, we have proposed a new method, GSTA for quantum correction of classical and BOMD simulations. Our qualitative findings are summarized in Table 7 regarding the capability and accuracy of the GSTA method compared to other methods. A clear advance of GSTA compared to 1PT or 2PT methods is that the effect of anharmonicity can be determined rigorously using the work of smoothing defined by eq 53. Another novelty is that structural NQEs can be investigated with the filtration of the coordinates. In more advanced methods, where anharmonicity can be described, the classical dynamics is modified to incorporate NQEs (e.g., ZPEs are added to the different normal modes of vibrations). The good agreement with the experiments indicates the plausibility of our smoothing technique. Zero-point vibrations are automatically taken into account by the proper enhancement of high frequency motions from the classical trajectories. The necessary simulations are orders of magnitude faster than with the golden standard technique, PIMD.
Table 7. Reliability of Different Methods for Estimation of Nuclear Quantum Effectsa.
| static properties |
dynamical properties |
||||
|---|---|---|---|---|---|
| method | heat capacity | structure | dielectric constant | diffusion | absorption spectrum |
| classical/BOMD | – | – | – | – | – |
| 1PT | + | – | – | – | – |
| 2PT | + | – | – | – | – |
| 1PT+AC | ++ | – | – | – | – |
| GSTA | ++ | ++ | + | – | + |
| PIMD | +++ | +++ | +++ | – | – |
| CMD, RPMD, LSC-IVR | +++ | +++ | +++ | ++ | ++ |
–, no effect; +, rough estimation; ++, good approximation; +++, exact for a particular potential.
GSTA reproduced large NQEs for heat capacity and structural properties. In contrast to PIMD computations, GSTA does not change the IR absorption spectrum or the dielectric constant significantly, and the self-diffusion coefficient remains exactly the same as the classical value. The main reason is that the input of GSTA is a classical trajectory. The quantum fluctuations are added after the simulation, so it does not change what the initial and final states were and how long it took to get there. In contrast, in PIMD calculations, these properties deviate notably from the classical values. While PIMD requires reparametrized or ab initio models to avoid double counting, GSTA can be applied on empirically derived force fields or model potentials.
Our method offers an alternative way to estimate NQEs routinely in theoretical investigations. In addition, force fields and water models can be improved using GSTA. The proposed method can be easily combined with molecular modeling programs to perform simulations and analyze the trajectories for which our source code is available at GitHub.120
Here, we have determined the heat capacity and some structural and dynamical properties for various systems as a proof of concept, but in subsequent studies, we show that other thermodynamic quantities, such as entropy and free energy, which are strongly related to the heat capacity, can be estimated by GSTA. We also intend to test the applicability and the limitation of our method on other NQEs, like tunneling, and further spectroscopic and dynamic properties.
Acknowledgments
This work was supported by the National Research, Development and Innovation Office of Hungary (NKFI, Grants No. K-124885 and No. PD-121070). A.M. acknowledges the János Bolyai Scholarship from the Hungarian Academy of Sciences. The authors are grateful to Dr. Tibor Nagy and Dr. András Stirling for insightful discussions. D.B. thanks Fahim Faizi and Adam Kells for their comments.
Appendix A
Change of Potential and Kinetic Energy upon Smoothing
A trajectory can be described as a Fourier series with the coefficients an:
| 86 |
| 87 |
| 88 |
Without loss of generality, we assume that the x(t) function is an even function, the periodicity of the trajectory is 2π, and a0 = 0. The an coefficients:
| 89 |
Then, the filtered trajectories can be written with the w weight functions:
| 90 |
| 91 |
| 92 |
Here,  is the actual frequency
as the first argument
of the w weight function for the Fourier series.
is the actual frequency
as the first argument
of the w weight function for the Fourier series.
The smoothed kinetic energy can be written as
| 93 |
The classic kinetic energy:
| 94 |
The work of smoothing at a specific time:
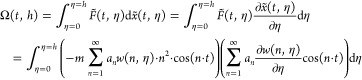 |
95 |
The average work of smoothing:
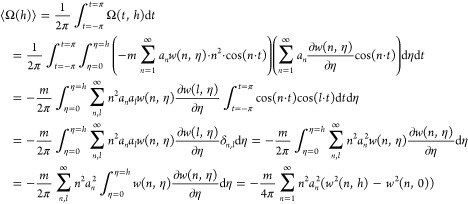 |
96 |
After some trivial transformations and using eqs 93 and 60 we get
| 97 |
Appendix B
Calculation of Infrared Spectrum
The classical line shape function is the Fourier transform of the autocorrelation of the classical total dipole moment:
| 98 |
The classical infrared spectrum can be obtained as the classical absorption cross section:
| 99 |
This classical absorption spectrum can be seen in Figure 9.
The simplest incorporation of quantum effect is the application of the harmonic quantum correction factor:107,108
| 100 |
The quantum corrected absorption cross section is the product of eqs 99 and 100:
| 101 |
After simplification we get
| 102 |
One can estimate the real part of the line shape function as
| 103 |
The complete line shape function is in the following relationship with its real part:107
| 104 |
The absorption cross section can be expressed as
 |
105 |
The penultimate term in the equation above is the weight function in eq 8, so the final expression for the absorption cross section:
| 106 |
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jctc.9b00703.
Author Present Address
§ D.F.: Institute of Chemistry, ELTE, Eötvös Loránd University, Pázmány Péter Sétány 1/A, H-1117 Budapest, Hungary.
The authors declare no competing financial interest.
Supplementary Material
References
- Cheng G.-J.; Zhang X.; Chung L. W.; Xu L.; Wu Y.-D. Computational Organic Chemistry: Bridging Theory and Experiment in Establishing the Mechanisms of Chemical Reactions. J. Am. Chem. Soc. 2015, 137, 1706–1725. 10.1021/ja5112749. [DOI] [PubMed] [Google Scholar]
- Bao J. L.; Xing L.; Truhlar D. G. Dual-level method for estimating multi-structural partition functions with torsional anharmonicity. J. Chem. Theory Comput. 2017, 13, 2511–2522. 10.1021/acs.jctc.7b00232. [DOI] [PubMed] [Google Scholar]
- Skouteris D.; Calderini D.; Barone V. Methods for Calculating Partition Functions of Molecules Involving Large Amplitude and/or Anharmonic Motions. J. Chem. Theory Comput. 2016, 12, 1011–1018. 10.1021/acs.jctc.5b01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprowl L. H.; Campbell C. T.; Árnadóttir L. Hindered Translator and Hindered Rotor Models for Adsorbates: Partition Functions and Entropies. J. Phys. Chem. C 2016, 120, 9719–9731. 10.1021/acs.jpcc.5b11616. [DOI] [Google Scholar]
- Duan L.; Liu X.; Zhang J. Z. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein–Ligand Binding Free Energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. 10.1021/jacs.6b02682. [DOI] [PubMed] [Google Scholar]
- Chong S.-H.; Ham S. New Computational Approach for External Entropy in Protein–Protein Binding. J. Chem. Theory Comput. 2016, 12, 2509–2516. 10.1021/acs.jctc.6b00174. [DOI] [PubMed] [Google Scholar]
- Li Y.-P.; Bell A. T.; Head-Gordon M. Thermodynamics of Anharmonic Systems: Uncoupled Mode Approximations for Molecules. J. Chem. Theory Comput. 2016, 12, 2861–2870. 10.1021/acs.jctc.5b01177. [DOI] [PubMed] [Google Scholar]
- Aieta C.; Gabas F.; Ceotto M. An Efficient Computational Approach for the Calculation of the Vibrational Density of States. J. Phys. Chem. A 2016, 120, 4853–4862. 10.1021/acs.jpca.5b12364. [DOI] [PubMed] [Google Scholar]
- Marsalek O.; Markland T. E. Quantum Dynamics and Spectroscopy of Ab Initio Liquid Water: The Interplay of Nuclear and Electronic Quantum Effects. J. Phys. Chem. Lett. 2017, 8, 1545–1551. 10.1021/acs.jpclett.7b00391. [DOI] [PubMed] [Google Scholar]
- Moberg D. R.; Straight S. C.; Knight C.; Paesani F. Molecular Origin of the Vibrational Structure of Ice Ih. J. Phys. Chem. Lett. 2017, 8, 2579–2583. 10.1021/acs.jpclett.7b01106. [DOI] [PubMed] [Google Scholar]
- Tuckerman M. E. On the Quantum Nature of the Shared Proton in Hydrogen Bonds. Science 1997, 275, 817–820. 10.1126/science.275.5301.817. [DOI] [PubMed] [Google Scholar]
- Guo J.; Lu J.-T.; Feng Y.; Chen J.; Peng J.; Lin Z.; Meng X.; Wang Z.; Li X.-Z.; Wang E.-G.; Jiang Y. Nuclear quantum effects of hydrogen bonds probed by tip-enhanced inelastic electron tunneling. Science 2016, 352, 321–325. 10.1126/science.aaf2042. [DOI] [PubMed] [Google Scholar]
- Ceriotti M.; Fang W.; Kusalik P. G.; McKenzie R. H.; Michaelides A.; Morales M. A.; Markland T. E. Nuclear Quantum Effects in Water and Aqueous Systems: Experiment, Theory, and Current Challenges. Chem. Rev. 2016, 116, 7529–7550. 10.1021/acs.chemrev.5b00674. [DOI] [PubMed] [Google Scholar]
- Feynman R. P.; Hibbs A. R.; Styer D. F.. Quantum Mechanics and Path Integrals; Amended ed.; Dover Books on Physics; Dover Publications, 2010. [Google Scholar]
- Berne B. J.; Thirumalai D. On the Simulation of Quantum Systems: Path Integral Methods. Annu. Rev. Phys. Chem. 1986, 37, 401–424. 10.1146/annurev.pc.37.100186.002153. [DOI] [Google Scholar]
- Liu J.; Li D.; Liu X. A simple and accurate algorithm for path integral molecular dynamics with the Langevin thermostat. J. Chem. Phys. 2016, 145, 024103 10.1063/1.4954990. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Liu X.; Chen Z.; Zheng H.; Yan K.; Liu J. A unified thermostat scheme for efficient configurational sampling for classical/quantum canonical ensembles via molecular dynamics. J. Chem. Phys. 2017, 147, 034109 10.1063/1.4991621. [DOI] [PubMed] [Google Scholar]
- Markland T. E.; Ceriotti M. Nuclear quantum effects enter the mainstream. Nature Rev. Chem. 2018, 2, 0109 10.1038/s41570-017-0109. [DOI] [Google Scholar]
- Ceriotti M.; Bussi G.; Parrinello M. Langevin Equation with Colored Noise for Constant-Temperature Molecular Dynamics Simulations. Phys. Rev. Lett. 2009, 102, 020601 10.1103/PhysRevLett.102.020601. [DOI] [PubMed] [Google Scholar]
- Ceriotti M.; Bussi G.; Parrinello M. Nuclear Quantum Effects in Solids Using a Colored-Noise Thermostat. Phys. Rev. Lett. 2009, 103, 030603 10.1103/PhysRevLett.103.030603. [DOI] [PubMed] [Google Scholar]
- Dammak H.; Chalopin Y.; Laroche M.; Hayoun M.; Greffet J.-J. Quantum Thermal Bath for Molecular Dynamics Simulation. Phys. Rev. Lett. 2009, 103, 190601. 10.1103/PhysRevLett.103.190601. [DOI] [PubMed] [Google Scholar]
- Ceriotti M.; Bussi G.; Parrinello M. Colored-Noise Thermostats à la Carte. J. Chem. Theory Comput. 2010, 6, 1170–1180. 10.1021/ct900563s. [DOI] [Google Scholar]
- Brieuc F.; Bronstein Y.; Dammak H.; Depondt P.; Finocchi F.; Hayoun M. Zero-Point Energy Leakage in Quantum Thermal Bath Molecular Dynamics Simulations. J. Chem. Theory Comput. 2016, 12, 5688–5697. 10.1021/acs.jctc.6b00684. [DOI] [PubMed] [Google Scholar]
- Habershon S.; Markland T. E.; Manolopoulos D. E. Competing quantum effects in the dynamics of a flexible water model. J. Chem. Phys. 2009, 131, 024501 10.1063/1.3167790. [DOI] [PubMed] [Google Scholar]
- Spura T.; John C.; Habershon S.; Kühne T. D. Nuclear quantum effects in liquid water from path-integral simulations using an ab initio force-matching approach. Mol. Phys. 2015, 113, 808–822. 10.1080/00268976.2014.981231. [DOI] [Google Scholar]
- Ceriotti M.; Manolopoulos D. E.; Parrinello M. Accelerating the convergence of path integral dynamics with a generalized Langevin equation. J. Chem. Phys. 2011, 134, 084104 10.1063/1.3556661. [DOI] [PubMed] [Google Scholar]
- Elton D. C.; Fritz M.; Fernández-Serra M. Using a monomer potential energy surface to perform approximate path integral molecular dynamics simulation of ab initio water at near-zero added cost. Phys. Chem. Chem. Phys. 2019, 21, 409–417. 10.1039/C8CP06077K. [DOI] [PubMed] [Google Scholar]
- Paesani F.; Zhang W.; Case D. A.; Cheatham T. E.; Voth G. A. An accurate and simple quantum model for liquid water. J. Chem. Phys. 2006, 125, 184507. 10.1063/1.2386157. [DOI] [PubMed] [Google Scholar]
- McBride C.; Vega C.; Noya E. G.; Ramírez R.; Sesé L. M. Quantum contributions in the ice phases: The path to a new empirical model for water—TIP4PQ/2005. J. Chem. Phys. 2009, 131, 024506 10.1063/1.3175694. [DOI] [PubMed] [Google Scholar]
- Medders G. R.; Babin V.; Paesani F. Development of a “First-Principles” Water Potential with Flexible Monomers. III. Liquid Phase Properties. J. Chem. Theory Comput. 2014, 10, 2906–2910. 10.1021/ct5004115. [DOI] [PubMed] [Google Scholar]
- Habershon S.; Manolopoulos D. E. Free energy calculations for a flexible water model. Phys. Chem. Chem. Phys. 2011, 13, 19714–19727. 10.1039/c1cp21520e. [DOI] [PubMed] [Google Scholar]
- McBride C.; Aragones J. L.; Noya E. G.; Vega C. A study of the influence of isotopic substitution on the melting point and temperature of maximum density of water by means of path integral simulations of rigid models. Phys. Chem. Chem. Phys. 2012, 14, 15199–15205. 10.1039/c2cp42393f. [DOI] [PubMed] [Google Scholar]
- Pamuk B.; Soler J. M.; Ramírez R.; Herrero C. P.; Stephens P. W.; Allen P. B.; Fernández-Serra M.-V. Anomalous Nuclear Quantum Effects in Ice. Phys. Rev. Lett. 2012, 108, 193003. 10.1103/PhysRevLett.108.193003. [DOI] [PubMed] [Google Scholar]
- Geerke D. P.; Luber S.; Marti K. H.; Van Gunsteren W. F. On the direct calculation of the free energy of quantization for molecular systems in the condensed phase. J. Comput. Chem. 2009, 30, 514–523. 10.1002/jcc.21070. [DOI] [PubMed] [Google Scholar]
- Cheng B.; Engel E. A.; Behler J.; Dellago C.; Ceriotti M. Ab initio thermodynamics of liquid and solid water. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 1110–1115. 10.1073/pnas.1815117116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuarrie D.; Simon J.. Molecular Thermodynamics; University Science Books, 1999. [Google Scholar]
- Ribeiro R. F.; Marenich A. V.; Cramer C. J.; Truhlar D. G. Use of Solution-Phase Vibrational Frequencies in Continuum Models for the Free Energy of Solvation. J. Phys. Chem. B 2011, 115, 14556–14562. 10.1021/jp205508z. [DOI] [PubMed] [Google Scholar]
- Grimme S. Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory. Chem. - Eur. J. 2012, 18, 9955–9964. 10.1002/chem.201200497. [DOI] [PubMed] [Google Scholar]
- Berens P. H.; Mackay D. H. J.; White G. M.; Wilson K. R. Thermodynamics and quantum corrections from molecular dynamics for liquid water. J. Chem. Phys. 1983, 79, 2375–2389. 10.1063/1.446044. [DOI] [Google Scholar]
- Lin S.-T.; Blanco M.; Goddard W. A. The two-phase model for calculating thermodynamic properties of liquids from molecular dynamics: Validation for the phase diagram of Lennard-Jones fluids. J. Chem. Phys. 2003, 119, 11792–11805. 10.1063/1.1624057. [DOI] [Google Scholar]
- Lin S.-T.; Maiti P. K.; Goddard W. A. Two-Phase Thermodynamic Model for Efficient and Accurate Absolute Entropy of Water from Molecular Dynamics Simulations. J. Phys. Chem. B 2010, 114, 8191–8198. 10.1021/jp103120q. [DOI] [PubMed] [Google Scholar]
- Laury M. L.; Wang L.-P.; Pande V. S.; Head-Gordon T.; Ponder J. W. Revised Parameters for the AMOEBA Polarizable Atomic Multipole Water Model. J. Phys. Chem. B 2015, 119, 9423–9437. 10.1021/jp510896n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt M.; Hirshberg M.; Sharon R.; Laidig K. E.; Daggett V. Calibration and Testing of a Water Model for Simulation of the Molecular Dynamics of Proteins and Nucleic Acids in Solution. J. Phys. Chem. B 1997, 101, 5051–5061. 10.1021/jp964020s. [DOI] [Google Scholar]
- Waheed Q.; Edholm O. Quantum Corrections to Classical Molecular Dynamics Simulations of Water and Ice. J. Chem. Theory Comput. 2011, 7, 2903–2909. 10.1021/ct2003034. [DOI] [PubMed] [Google Scholar]
- Yeh K.-Y.; Huang S.-N.; Chen L.-J.; Lin S.-T. Diffusive and quantum effects of water properties in different states of matter. J. Chem. Phys. 2014, 141, 044502 10.1063/1.4890572. [DOI] [PubMed] [Google Scholar]
- Pascal T. A.; Schärf D.; Jung Y.; Kühne T. D. On the absolute thermodynamics of water from computer simulations: A comparison of first-principles molecular dynamics, reactive and empirical force fields. J. Chem. Phys. 2012, 137, 244507. 10.1063/1.4771974. [DOI] [PubMed] [Google Scholar]
- Naserifar S.; Goddard W. A. The quantum mechanics-based polarizable force field for water simulations. J. Chem. Phys. 2018, 149, 174502. 10.1063/1.5042658. [DOI] [PubMed] [Google Scholar]
- Pascal T. A.; Lin S.-T.; Goddard W. A. III Thermodynamics of liquids: standard molar entropies and heat capacities of common solvents from 2PT molecular dynamics. Phys. Chem. Chem. Phys. 2011, 13, 169–181. 10.1039/C0CP01549K. [DOI] [PubMed] [Google Scholar]
- Caleman C.; van Maaren P. J.; Hong M.; Hub J. S.; Costa L. T.; van der Spoel D. Force Field Benchmark of Organic Liquids: Density, Enthalpy of Vaporization, Heat Capacities, Surface Tension, Isothermal Compressibility, Volumetric Expansion Coefficient, and Dielectric Constant. J. Chem. Theory Comput. 2012, 8, 61–74. 10.1021/ct200731v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.-N.; Pascal T. A.; Goddard W. A.; Maiti P. K.; Lin S.-T. Absolute Entropy and Energy of Carbon Dioxide Using the Two-Phase Thermodynamic Model. J. Chem. Theory Comput. 2011, 7, 1893–1901. 10.1021/ct200211b. [DOI] [PubMed] [Google Scholar]
- Chen P.; Nishiyama Y.; Wohlert J.; Lu A.; Mazeau K.; Ismail A. E. Translational Entropy and Dispersion Energy Jointly Drive the Adsorption of Urea to Cellulose. J. Phys. Chem. B 2017, 121, 2244–2251. 10.1021/acs.jpcb.6b11914. [DOI] [PubMed] [Google Scholar]
- Chen M.; Pendrill R.; Widmalm G.; Brady J. W.; Wohlert J. Molecular Dynamics Simulations of the Ionic Liquid 1-n-Butyl-3-Methylimidazolium Chloride and Its Binary Mixtures with Ethanol. J. Chem. Theory Comput. 2014, 10, 4465–4479. 10.1021/ct500271z. [DOI] [PubMed] [Google Scholar]
- Doherty B.; Zhong X.; Gathiaka S.; Li B.; Acevedo O. Revisiting OPLS Force Field Parameters for Ionic Liquid Simulations. J. Chem. Theory Comput. 2017, 13, 6131–6145. 10.1021/acs.jctc.7b00520. [DOI] [PubMed] [Google Scholar]
- Wohlert J. Vapor Pressures and Heats of Sublimation of Crystalline β-Cellobiose from Classical Molecular Dynamics Simulations with Quantum Mechanical Corrections. J. Phys. Chem. B 2014, 118, 5365–5373. 10.1021/jp501839k. [DOI] [PubMed] [Google Scholar]
- Lai P.-K.; Hsieh C.-M.; Lin S.-T. Rapid determination of entropy and free energy of mixtures from molecular dynamics simulations with the two-phase thermodynamic model. Phys. Chem. Chem. Phys. 2012, 14, 15206–15213. 10.1039/c2cp42011b. [DOI] [PubMed] [Google Scholar]
- Pascal T. A.; Goddard W. A. Interfacial Thermodynamics of Water and Six Other Liquid Solvents. J. Phys. Chem. B 2014, 118, 5943–5956. 10.1021/jp410861h. [DOI] [PubMed] [Google Scholar]
- Desjarlais M. P. First-principles calculation of entropy for liquid metals. Phys. Rev. E 2013, 88, 062145 10.1103/PhysRevE.88.062145. [DOI] [PubMed] [Google Scholar]
- Sun T.; Xian J.; Zhang H.; Zhang Z.; Zhang Y. Two-phase thermodynamic model for computing entropies of liquids reanalyzed. J. Chem. Phys. 2017, 147, 194505. 10.1063/1.5001798. [DOI] [PubMed] [Google Scholar]
- Persson R. A. X.; Pattni V.; Singh A.; Kast S. M.; Heyden M. Signatures of Solvation Thermodynamics in Spectra of Intermolecular Vibrations. J. Chem. Theory Comput. 2017, 13, 4467–4481. 10.1021/acs.jctc.7b00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannir Sivajothi S. S.; Lin S.-T.; Maiti P. K. Efficient Computation of Entropy and Other Thermodynamic Properties for Two-Dimensional Systems Using Two-Phase Thermodynamic Model. J. Phys. Chem. B 2019, 123, 180–193. 10.1021/acs.jpcb.8b07147. [DOI] [PubMed] [Google Scholar]
- Dietschreit J. C. B.; Peters L. D. M.; Kussmann J.; Ochsenfeld C. Identifying Free Energy Hot-Spots in Molecular Transformations. J. Phys. Chem. A 2019, 123, 2163–2170. 10.1021/acs.jpca.8b12309. [DOI] [PubMed] [Google Scholar]
- Tiwari A.; Honingh C.; Ensing B. Accurate calculation of zero point energy from molecular dynamics simulations of liquids and their mixtures. J. Chem. Phys. 2019, 151, 244124. 10.1063/1.5131145. [DOI] [PubMed] [Google Scholar]
- Bernhardt M. P.; Dallavalle M.; Van der Vegt N. F. A. Application of the 2PT model to understanding entropy change in molecular coarse-graining. Soft Mater. 2020, 0, 1–16. 10.1080/1539445X.2020.1737118. [DOI] [Google Scholar]
- Cisneros G. A.; Wikfeldt K. T.; Ojamäe L.; Lu J.; Xu Y.; Torabifard H.; Bartók A. P.; Csányi G.; Molinero V.; Paesani F. Modeling Molecular Interactions in Water: From Pairwise to Many-Body Potential Energy Functions. Chem. Rev. 2016, 116, 7501–7528. 10.1021/acs.chemrev.5b00644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. W.The Scientist & Engineer’s Guide to Digital Signal Processing; California Technical Publishing, 1997. [Google Scholar]
- Hase W. L.; Duchovic R. J.; Hu X.; Komornicki A.; Lim K. F.; Lu D.-h.; Peslherbe G. H.; Swamy K. N.; Van de Linde S. R.; Varandas A. J. C.; Wang H.; Wolf R. J. VENUS96: A General Chemical Dynamics Computer Program. QCPE Bull. 1996, 16, 43. [Google Scholar]
- Perdew J. P.; Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B: Condens. Matter Mater. Phys. 1992, 45, 13244–13249. 10.1103/PhysRevB.45.13244. [DOI] [PubMed] [Google Scholar]
- Krishnan R.; Binkley J.; Seeger R.; Pople J. Self-consistent molecular-orbital methods. Basis set for correlated wave-functions. J. Chem. Phys. 1980, 72, 650–654. 10.1063/1.438955. [DOI] [Google Scholar]
- Xu X.; Goddard W. A. Bonding Properties of the Water Dimer: A Comparative Study of Density Functional Theories. J. Phys. Chem. A 2004, 108, 2305–2313. 10.1021/jp035869t. [DOI] [Google Scholar]
- Swope W. C.; Andersen H. C.; Berens P. H.; Wilson K. R. A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: Application to small water clusters. J. Chem. Phys. 1982, 76, 637–649. 10.1063/1.442716. [DOI] [Google Scholar]
- Ponder J. W.TINKER: Software Tools for Molecular Design. https://dasher.wustl.edu/tinker (accessed April 2020).
- Bernal J. D.; Fowler R. H. A Theory of Water and Ionic Solution, with Particular Reference to Hydrogen and Hydroxyl Ions. J. Chem. Phys. 1933, 1, 515–548. 10.1063/1.1749327. [DOI] [Google Scholar]
- Bussi G.; Zykova-Timan T.; Parrinello M. Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J. Chem. Phys. 2009, 130, 074101 10.1063/1.3073889. [DOI] [PubMed] [Google Scholar]
- Beeman D. Some multistep methods for use in molecular dynamics calculations. J. Comput. Phys. 1976, 20, 130–139. 10.1016/0021-9991(76)90059-0. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Berens P. H.; Wilson K. R. Molecular dynamics and spectra. I. Diatomic rotation and vibration. J. Chem. Phys. 1981, 74, 4872–4882. 10.1063/1.441739. [DOI] [Google Scholar]
- Case D. A.; Darden T. A.; Iii T. E. C.; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Walker R. C.; Zhang W.; Merz K. M.; Roberts B.; Hayik S.; Roitberg A.; Seabra G.; Swails J.; Götz A. W.; Kolossváry I. W.; Wong K. F.; Paesani F.; Vanicek J.; Wolf R. M.; Liu J.; Wu X.; Brozell S. R.; Steinbrecher T.; Gohlke H.; Cai Q.; Ye X.; Hsieh M.-J.; Cui G.; Roe D. R.; Mathews D. H.; Seetin M. G.; Salomon-Ferrer R.; Sagui C. A.; Babin V.; Luchko T.; Gusarov S.; Kovalenko A.; Kollman P. A.; Cheatham T. E.; Goetz A. W.; Kolossvai I.. AMBER12; University of California: San Francisco, 2012.
- Furtenbacher T.; Szidarovszky T.; Hrubý J.; Kyuberis A. A.; Zobov N. F.; Polyansky O. L.; Tennyson J.; Császár A. G. Definitive Ideal-Gas Thermochemical Functions of the H216O Molecule. J. Phys. Chem. Ref. Data 2016, 45, 043104 10.1063/1.4967723. [DOI] [Google Scholar]
- Feistel R.; Wagner W. A New Equation of State for H2O Ice Ih. J. Phys. Chem. Ref. Data 2006, 35, 1021–1047. 10.1063/1.2183324. [DOI] [Google Scholar]
- Chase M. Jr... NIST-JANAF Themochemical Tables, Fourth ed.; Journal of Physical and Chemical Reference Data Monographs; NIST, 1998; p 1325. [Google Scholar]
- Shvab I.; Sadus R. J. Atomistic water models: Aqueous thermodynamic properties from ambient to supercritical conditions. Fluid Phase Equilib. 2016, 407, 7–30. 10.1016/j.fluid.2015.07.040. [DOI] [Google Scholar]
- Wu Y.; Tepper H. L.; Voth G. A. Flexible simple point-charge water model with improved liquid-state properties. J. Chem. Phys. 2006, 124, 024503 10.1063/1.2136877. [DOI] [PubMed] [Google Scholar]
- Vega C.; Sanz E.; Abascal J. L. F. The melting temperature of the most common models of water. J. Chem. Phys. 2005, 122, 114507. 10.1063/1.1862245. [DOI] [PubMed] [Google Scholar]
- Shinoda W.; Shiga M. Quantum simulation of the heat capacity of water. Phys. Rev. E 2005, 71, 041204 10.1103/PhysRevE.71.041204. [DOI] [PubMed] [Google Scholar]
- Shiga M.; Shinoda W. Calculation of heat capacities of light and heavy water by path-integral molecular dynamics. J. Chem. Phys. 2005, 123, 134502. 10.1063/1.2035078. [DOI] [PubMed] [Google Scholar]
- Vega C.; Conde M. M.; McBride C.; Abascal J. L. F.; Noya E. G.; Ramirez R.; Sesé L. M. Heat capacity of water: A signature of nuclear quantum effects. J. Chem. Phys. 2010, 132, 046101 10.1063/1.3298879. [DOI] [PubMed] [Google Scholar]
- Dang L. X.; Pettitt B. M. Simple intramolecular model potentials for water. J. Phys. Chem. 1987, 91, 3349–3354. 10.1021/j100296a048. [DOI] [Google Scholar]
- González M. A.; Abascal J. L. F. A flexible model for water based on TIP4P/2005. J. Chem. Phys. 2011, 135, 224516. 10.1063/1.3663219. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Abascal J. L. F.; Vega C. A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 2005, 123, 234505. 10.1063/1.2121687. [DOI] [PubMed] [Google Scholar]
- Soper A. The radial distribution functions of water and ice from 220 to 673 K and at pressures up to 400 MPa. Chem. Phys. 2000, 258, 121–137. 10.1016/S0301-0104(00)00179-8. [DOI] [Google Scholar]
- Vidulich G. A.; Evans D. F.; Kay R. L. The dielectric constant of water and heavy water between 0 and 40.degree. J. Phys. Chem. 1967, 71, 656–662. 10.1021/j100862a028. [DOI] [Google Scholar]
- Lobaugh J.; Voth G. A. A quantum model for water: Equilibrium and dynamical properties. J. Chem. Phys. 1997, 106, 2400–2410. 10.1063/1.473151. [DOI] [Google Scholar]
- Paesani F.; Iuchi S.; Voth G. A. Quantum effects in liquid water from an ab initio-based polarizable force field. J. Chem. Phys. 2007, 127, 074506 10.1063/1.2759484. [DOI] [PubMed] [Google Scholar]
- Elton D. C.; Fernández-Serra M.-V. Polar nanoregions in water: A study of the dielectric properties of TIP4P/2005, TIP4P/2005f and TTM3F. J. Chem. Phys. 2014, 140, 124504. 10.1063/1.4869110. [DOI] [PubMed] [Google Scholar]
- Elton D. C.Understanding the Dielectric Properties of Water. Ph.D. Thesis, Stony Brook University, 2016. [Google Scholar]
- Liu J.; Miller W. H.; Paesani F.; Zhang W.; Case D. A. Quantum dynamical effects in liquid water: A semiclassical study on the diffusion and the infrared absorption spectrum. J. Chem. Phys. 2009, 131, 164509. 10.1063/1.3254372. [DOI] [PubMed] [Google Scholar]
- Reddy S. K.; Straight S. C.; Bajaj P.; Huy Pham C.; Riera M.; Moberg D. R.; Morales M. A.; Knight C.; Götz A. W.; Paesani F. On the accuracy of the MB-pol many-body potential for water: Interaction energies, vibrational frequencies, and classical thermodynamic and dynamical properties from clusters to liquid water and ice. J. Chem. Phys. 2016, 145, 194504. 10.1063/1.4967719. [DOI] [PubMed] [Google Scholar]
- Rapaport D.; Scheraga H. Structure and dynamics of the “configuration interaction” model of liquid water. Chem. Phys. Lett. 1981, 78, 491–494. 10.1016/0009-2614(81)85244-X. [DOI] [Google Scholar]
- Impey R.; Madden P.; McDonald I. Spectroscopic and transport properties of water. Mol. Phys. 1982, 46, 513–539. 10.1080/00268978200101361. [DOI] [Google Scholar]
- Teleman O.; Jönsson B.; Engström S. A molecular dynamics simulation of a water model with intramolecular degrees of freedom. Mol. Phys. 1987, 60, 193–203. 10.1080/00268978700100141. [DOI] [Google Scholar]
- Svishchev I. M.; Kusalik P. G. Dynamics in liquid water, water-d2, and water-t2: a comparative simulation study. J. Phys. Chem. 1994, 98, 728–733. 10.1021/j100054a002. [DOI] [Google Scholar]
- Damian J. I. M.; Malaspina D. C.; Granada J. R. Vibrational spectra of light and heavy water with application to neutron cross section calculations. J. Chem. Phys. 2013, 139, 024504 10.1063/1.4812828. [DOI] [PubMed] [Google Scholar]
- Price W. S.; Ide H.; Arata Y. Self-Diffusion of Supercooled Water to 238 K Using PGSE NMR Diffusion Measurements. J. Phys. Chem. A 1999, 103, 448–450. 10.1021/jp9839044. [DOI] [Google Scholar]
- Price W. S.; Ide H.; Arata Y.; Söderman O. Temperature Dependence of the Self-Diffusion of Supercooled Heavy Water to 244 K. J. Phys. Chem. B 2000, 104, 5874–5876. 10.1021/jp0015372. [DOI] [Google Scholar]
- Wang L.-P.; Head-Gordon T.; Ponder J. W.; Ren P.; Chodera J. D.; Eastman P. K.; Martinez T. J.; Pande V. S. Systematic Improvement of a Classical Molecular Model of Water. J. Phys. Chem. B 2013, 117, 9956–9972. 10.1021/jp403802c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez R.; López-Ciudad T.; Kumar P. P.; Marx D. Quantum corrections to classical time-correlation functions: Hydrogen bonding and anharmonic floppy modes. J. Chem. Phys. 2004, 121, 3973–3983. 10.1063/1.1774986. [DOI] [PubMed] [Google Scholar]
- Lawrence C. P.; Skinner J. L. Quantum corrections in vibrational and electronic condensed phase spectroscopy: Line shapes and echoes. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 6720–6725. 10.1073/pnas.0408813102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertie J. E.; Lan Z. Infrared Intensities of Liquids XX: The Intensity of the OH Stretching Band of Liquid Water Revisited, and the Best Current Values of the Optical Constants of H2O(l) at 25°C between 15,000 and 1 cm–1. Appl. Spectrosc. 1996, 50, 1047–1057. 10.1366/0003702963905385. [DOI] [Google Scholar]
- Habershon S.; Fanourgakis G. S.; Manolopoulos D. E. Comparison of path integral molecular dynamics methods for the infrared absorption spectrum of liquid water. J. Chem. Phys. 2008, 129, 074501 10.1063/1.2968555. [DOI] [PubMed] [Google Scholar]
- Paesani F.; Xantheas S. S.; Voth G. A. Infrared Spectroscopy and Hydrogen-Bond Dynamics of Liquid Water from Centroid Molecular Dynamics with an Ab Initio-Based Force Field. J. Phys. Chem. B 2009, 113, 13118–13130. 10.1021/jp907648y. [DOI] [PubMed] [Google Scholar]
- Liu J.; Miller W. H.; Fanourgakis G. S.; Xantheas S. S.; Imoto S.; Saito S. Insights in quantum dynamical effects in the infrared spectroscopy of liquid water from a semiclassical study with an ab initio-based flexible and polarizable force field. J. Chem. Phys. 2011, 135, 244503. 10.1063/1.3670960. [DOI] [PubMed] [Google Scholar]
- Rossi M.; Liu H.; Paesani F.; Bowman J.; Ceriotti M. Communication: On the consistency of approximate quantum dynamics simulation methods for vibrational spectra in the condensed phase. J. Chem. Phys. 2014, 141, 181101. 10.1063/1.4901214. [DOI] [PubMed] [Google Scholar]
- Ruiz Pestana L.; Marsalek O.; Markland T. E.; Head-Gordon T. The Quest for Accurate Liquid Water Properties from First Principles. J. Phys. Chem. Lett. 2018, 9, 5009–5016. 10.1021/acs.jpclett.8b02400. [DOI] [PubMed] [Google Scholar]
- Liu X.; Liu J. Critical role of quantum dynamical effects in the Raman spectroscopy of liquid water. Mol. Phys. 2018, 116, 755–779. 10.1080/00268976.2018.1434907. [DOI] [Google Scholar]
- Paesani F.; Voth G. A. Nonlinear quantum time correlation functions from centroid molecular dynamics and the maximum entropy method. J. Chem. Phys. 2008, 129, 194113. 10.1063/1.3013365. [DOI] [PubMed] [Google Scholar]
- Liu J.; Miller W. H. Linearized semiclassical initial value time correlation functions with maximum entropy analytic continuation. J. Chem. Phys. 2008, 129, 124111. 10.1063/1.2981065. [DOI] [PubMed] [Google Scholar]
- Habershon S.; Braams B. J.; Manolopoulos D. E. Quantum mechanical correlation functions, maximum entropy analytic continuation, and ring polymer molecular dynamics. J. Chem. Phys. 2007, 127, 174108. 10.1063/1.2786451. [DOI] [PubMed] [Google Scholar]
- Rabani E.; Reichman D. R.; Krilov G.; Berne B. J. The calculation of transport properties in quantum liquids using the maximum entropy numerical analytic continuation method: Application to liquid para-hydrogen. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 1129–1133. 10.1073/pnas.261540698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berta D.; Ferenc D.; Madarasz A.. GSTA-heatcap. https://github.com/bertadenes/GSTA-heatcap (accessed April 2020).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.