

Abstract
Competence in muscle biopsy evaluation is a core component of neuropathology practice. The practicing neuropathologist should be able to prepare frozen sections of muscle biopsies with minimal artifacts and identify key histopathologic features of neuromuscular disease in hematoxylin and eosin-stained sections as well as implement and interpret a basic panel of additional histochemical, enzyme histochemical, and immunohistochemical stains. Important to everyday practice is a working knowledge of normal muscle histology at different ages, muscle motor units, pitfalls of myotendinous junctions, nonpathologic variations encountered at traditional and nontraditional muscle sites, the pathophysiology of myonecrosis and regeneration, and approaches to distinguish muscular dystrophies from inflammatory myopathies and other necrotizing myopathies. Here, we provide a brief overview of what every neuropathologist needs to know concerning the muscle biopsy.
Keywords: Inflammatory myopathy, Muscle biopsy, Muscular dystrophy, Myopathy, Neurogenic atrophy
INTRODUCTION
Basic skills and knowledge to process and evaluate muscle biopsies are essential to the practicing neuropathologist. The current article outlines aspects of the muscle biopsy that every neuropathologist needs to know. These include procedures aimed at optimizing the muscle biopsy quality. The classic histopathological findings of select muscle diseases are illustrated along with a brief discussion of motor units, common pitfalls, myonecrosis and regeneration, muscular dystrophies, and inflammatory myopathies. Finally, common myths surrounding the muscle biopsy are addressed. This article does not aim to be all-inclusive in its breadth of scope but rather to stand as a practical overview of the muscle biopsy.
Optimizing the Muscle Biopsy
The 2 most common methods of procuring muscle biopsies are the needle biopsy and the open biopsy. The open biopsy requires a larger incision than the Bergström needle or alternative types of needle-core biopsies but offers the advantage of obtaining a larger sample (1, 2). In 35 years of experience at the University of Iowa, open biopsies with a muscle clamp have been the preferred method due to the technical ease of obtaining an adequate sample. The following will briefly outline the steps involved in processing a muscle biopsy (Fig. 1), though different techniques and protocols may be similarly successful (3).
FIGURE 1.

Open muscle biopsy freezing methodology. Isopentane is prepared by filling a small metal cup about 3-quarters full (A) and suspending it in liquid N2 within a Dewar (B). A muscle biopsy is received in a clamp (C). Tissue is cut from the clamp and mounted on a preassembled plastic coverslip/cork leaving a 0.2-cm gap between the upper surface of the cork and lower end of the biopsy (D). Muscle fibers are oriented at 90 degrees to the cork surface and parallel to the plane of the coverslip. The gap between cork and biopsy is filled with optimal cutting temperature (OCT) (E); a dissecting needle inserted into the cork behind the coverslip serves as a handle for manipulating the mounted biopsy. When the isopentane is nearly −160°C (stir the isopentane to assure a uniform temperature), the biopsy is submersed for snap freezing (F). After placing the frozen muscle inside a cryostat, the coverslip and cork are removed (G); the buffer of OCT below the biopsy is evident. This OCT buffer prevents thawing and refreezing of the biopsy when it is mounted to a chuck for sectioning (H). Before storage in a −80°C freezer, the biopsy is wrapped completely in precooled foil (I) and placed in a polycon (J), that when closed is airtight. The foil and polycon prevent desiccation of the biopsy.
The first decision in the processing and evaluation of muscle is how to distribute the sample. Frozen tissue is the best preparation for diagnostic purposes as it allows for routine histology and enzyme histochemistry, immunostains, and biochemistry, including Western blots and molecular studies of DNA or mRNA. As such, freezing tissue should be prioritized over additional tissue allocations. In cases with adequate tissue, a portion of tissue may be fixed in formalin for paraffin embedment and an additional portion fixed in glutaraldehyde for embedment in epoxy resin (e.g. epon). Formalin-fixed tissue allows for routine histological evaluation, special stains such as Congo red, and immunohistochemistry. Glutaraldehyde-fixed tissue is used for plastic section light microscopy (“thicks”) and electron microscopy (“thins”).
Once the specimen arrives in the laboratory, the sample is cut from the muscle clamp. Assuming an adequate sample, a 0.1- to 0.2-cm diameter strip of muscle along the full length of the biopsy is trimmed for glutaraldehyde fixation. The remainder of the specimen is mounted on a combination plastic coverslip and cork, this apparatus being easily constructed using a prep blade to cut a slit in the cork for the coverslip. The advantage of this method is that it uses common, replaceable items, is cost effective, and maintains the orientation of the sample throughout the freezing process. A dissecting needle is placed into the cork behind the coverslip and functions as a handle for the cork. Next, the gap between the top surface of the cork and the bottom of the muscle sample is filled with optimal cutting temperature compound (OCT). Of note, it is not advisable to cover the whole muscle with OCT but only to apply enough OCT to allow adequate adherence to a cryostat chuck when it comes time to cut cryosections.
An important step in specimen processing is the use of isopentane (2-methyl butane) for freezing the biopsy. First, obtain a metal cup that will fit into a Dewar of liquid nitrogen. A flexible wire may be used as a handle for the cup. Next, pour isopentane into the metal cup and lower the metal cup into the liquid nitrogen filled Dewar. Stir the isopentane continuously before introduction of the specimen, keeping in mind that the isopentane will unavoidably freeze at the edges of the metal cup. Once the isopentane reaches −155°C to −160°C, submerge the muscle sample in the isopentane. Remove the muscle from the isopentane after freezing, detach the plastic coverslip, and cut away the cork. Mount the biopsy in the cryostat and cut frozen sections. For storage of the muscle biopsy, wrap the sample in precooled foil before placing it in a prelabeled and precooled polycon (airtight) container for freezer storage. At −80°C, muscle biopsies prepared in this way can be stored 30 years and longer without noticeable degradation.
Despite care and experience in the processing and freezing of muscle specimens, there will inevitably be occasions where evaluation of the frozen specimen is compromised by ice crystal artifact, sometimes severe, or lack of proper orientation. In cases with extensive ice crystals, it may be advisable to thaw and refreeze the specimen. First, cut and store unstained sections of the suboptimal specimen in case the tissue is subsequently damaged or destroyed. Next, thaw the tissue, remove excess OCT (or other mounting medium), and process it as if it were a fresh sample received from the operating room as outlined above. Such reprocessing may create rounded fibers in the sample; however, proper orientation and the elimination of ice crystals are far preferable to attempting to identify histopathology in tissue that contains severe freezing artifacts (Fig. 2).
FIGURE 2.

Normal muscle in cryosections and salvaging biopsies from extreme ice crystal artifacts. (A) This hematoxylin and eosin photomicrograph shows the morphology of normal muscle in a biopsy that was prepared, sectioned, and stained optimally. (B, C) A muscle biopsy without diagnostic abnormality arrived with poor orientation and severe ice crystal artifacts. Thawing, reorienting, and refreezing restored the morphology, including the polygonal shapes of individual muscle fibers. (D, E) This biopsy is from a pediatric patient with autophagic vacuolar myopathy due to a mutation in VMA21 (X-linked myopathy with excess autophagy). After thawing and refreezing, the muscle fibers are rounded, but they have almost no ice crystals and small autophagic vacuoles are evident in some muscle fibers (arrows). Size scale bars: A–D = 100 µm; E = 50 µm.
After frozen sections are obtained, the next steps in the processing and evaluation of the muscle biopsy depend on the diagnostic needs of the case, pathologist preference and experience, and strengths/skills of the processing laboratory. At the University of Iowa, the following standard protocol is performed. Muscle biopsy cryosections are processed for hematoxylin and eosin (H&E) staining, fiber typing using slow myosin and fast myosin immunohistochemistry, enzyme histochemistry for nicotinamide adenine dinucleotide (NADH) tetrazolium reductase, succinate dehydrogenase (SDH), and cytochrome C oxidase/succinate dehydrogenase (COX/SDH) combination, and histochemistry for Gomori trichrome. Additional studies are tailored to best fit the individual biopsy and relevant clinical history. Many other laboratories prefer a much broader initial approach to every biopsy by including additional histochemical, enzyme histochemical, and/or immunohistochemical stains in the initial workup of the specimen.
A final note on the processing of muscle biopsies is the importance of H&E balance. Too much eosin or too little hematoxylin compromises the ability to evaluate finer histological details. An optimal H&E stain facilitates reliable histology for critical decisions in the subsequent evaluation of the muscle biopsy.
Evaluating H&E-Stained Frozen or Paraffin Sections
Evaluating the muscle biopsy begins with clinical history. For example, simply knowing the age and physical conditioning of the patient is important to the expected muscle fiber diameter. Fiber diameters range from approximately 10 µm in a neonate to 40–60 µm in a normal teen or adult to 80–100 µm in well-trained athletes.
A well-balanced H&E section is the essential starting point for muscle biopsy histopathologic evaluation, though with the limitation of not being able to distinguish fiber types or visualize nemaline rods. The following selected cases exemplify the utility of an initial H&E evaluation for diagnosis.
Neurogenic Disease
Angulated, atrophic fibers in groups represent the denervation of scattered motor units and define neurogenic atrophy (Fig. 3A); these groups are smaller early in the progression of disease and larger later when reinnervated motor units are larger. In acute denervation, target fibers are often evident (Fig. 3B).
FIGURE 3.

A good quality, cryosection hematoxylin and eosin (H&E) slide is your “best friend.” When muscle biopsies are processed in an optimal fashion, a wide range of neuromuscular diseases and systemic disorders are readily recognized in H&E-stained sections. (A) Neurogenic atrophy. (B) Target fibers in acute denervation. (C) Spinal muscular atrophy (SMA), in this patient with a non-5q form of SMA. (D) Perifascicular pattern pathology common to dermatomyositis-spectrum disorders and antisynthetase spectrum disorders. (E) Inclusion body myositis. (F) Sarcoid myopathy. (G) Centronuclear myopathy, in this patient due to a dominant mutation in DNM2. (H) Central core disease (myopathy), in this patient due to recessive RYR1 mutations. (I) Danon disease, an autophagic vacuolar myopathy due to an X-linked mutation in LAMP2. (J) Pompe disease, an autophagic vacuolar myopathy due to recessive mutations in GAA. (K) Ragged-red fibers in mitochondrial myopathy. (L) Lipid storage myopathy. The scale bar in panel L is 50 µm for panels G–J and L; 100 µm for panels A–, E, F, and K; 200 µm for panel D.
The alternating pattern of grouped large (hypertrophic) fibers and grouped small (hypotrophic) fibers (Fig. 3C) is classic for spinal muscular atrophy (SMA), which includes a number of genetic disorders affecting the spinal motor neurons (4). Molecular testing in hypotonic, or “floppy,” infants has largely replaced muscle biopsies for the diagnosis of SMA (5), with the most common cause of SMA being mutations of the survival of motor neuron 1 (SMN1) gene located at 5q13.2 (6). However, muscle biopsies continue to be useful in cases of non-5q SMA. In the featured example (Fig. 3C), the patient was found after the muscle biopsy evaluation to have a form of non-5q SMA due to mutations in EXOSC9 (7).
Inflammatory Myopathies
Dermatomyositis is an idiopathic inflammatory myopathy that affects both adult and pediatric populations and often features skin manifestations, such as papules (Gottron papules) or erythema over the dorsal digits, heliotrope erythema surrounding the orbit, and erythematous macules, among other dermatological manifestations. Muscle weakness, often proximal in distribution, and myalgia are also present (8). On H&E evaluation, an inflammatory infiltrate may be found predominantly in a perimysial, perivascular distribution (9). Perifascicular atrophy with or without myonecrosis and regeneration is the signature histopathologic change readily evident in H&E-stained sections (Fig. 3D). Additional autoimmune disorders share the perifascicular and perimysial pattern of pathology and fall into the categories of dermatomyositis spectrum disorders or the antisynthetase myositis spectrum (10).
Inclusion body myositis (IBM) is an idiopathic inflammatory myopathy that typically affects middle-aged to older individuals. Clinical signs include difficulty with ambulation and standing, handgrip, and swallowing (11). Myofibers with rimmed vacuoles, eosinophilic cytoplasmic inclusions, endomysial inflammation, and atrophic fibers (sometimes mimicking neurogenic atrophy) are seen on H&E-stained sections in IBM (Fig. 3E) (12).
The finding of granulomas on H&E sections of a muscle biopsy (Fig. 3F) should raise the possibility of myopathic involvement by sarcoidosis, a multisystem disorder of unknown etiology and diagnosis of exclusion (13, 14).
Congenital Myopathies
Centronuclear myopathies are a group of genetic diseases classified under the larger category of congenital myopathies and include X-linked recessive, autosomal dominant, and autosomal recessive patterns of inheritance. Symptoms, severity of disease, etiology, and age of onset vary, but central nuclei are seen on H&E sections (Fig. 3G) (15).
Central core disease is a congenital myopathy typically affecting infants and children, often manifesting with hypotonia and abnormalities in motor development, and caused by mutations in the skeletal muscle ryanodine receptor (RYR1) gene at chromosome 19q13.1 (16). Central cores are seen on H&E sections (Fig. 3H) (17).
Autophagic Vacuolar Myopathies
Danon disease is caused by a mutation in the X-linked lysosome-associated membrane protein 2 (LAMP2) gene and can present in infants, children, or adults (18, 19). Manifestations are much more severe in males and include cardiomyopathy along with autophagic vacuolar myopathy. Skeletal muscle biopsy reveals basophilic autophagic vacuoles, numerous internalized nuclei, and complex muscle fiber splitting on H&E sections (Fig. 3I). Another X-linked autophagic vacuolar myopathy is illustrated in Figure 2E.
Pompe disease, often clinically diagnosed in infants, is an autosomal recessive metabolic disease caused by deficiency in acid α-glucosidase (20). As acid α-glucosidase is necessary for lysosomal metabolism of glycogen, lysosomes accumulate glycogen and are often seen as large, irregular vacuoles in muscle fibers on H&E sections (Fig. 3J) (21).
Metabolic Myopathies
Ragged-red fibers are commonly associated with primary mitochondrial disorders (mitochondrial myopathies), though small numbers are often seen in older, healthy individuals (22). Ragged-red fibers are well-visualized with Gomori trichrome and SDH stains but are also readily seen on H&E sections (Fig. 3K).
Lipid storage myopathies are caused by abnormalities in fatty acid metabolism. Presentation and pathological manifestations vary based on the particular disease and gene mutations. Frequently, the accumulation of lipid droplets within muscle fibers can be visualized on H&E-stained sections (Fig. 3L) (23, 24).
Vasculitis
The muscle biopsy is a common diagnostic tissue for many forms of vasculitis. Shown here is eosinophilic granulomatosis with polyangiitis (formerly Churg-Strauss syndrome), an antineutrophil cytoplasmic antibody-associated vasculitis that typically features asthma, peripheral eosinophilia, and symptoms of small-vessel vasculitis (25). H&E sections of muscle biopsies in eosinophilic granulomatosis with polyangiitis may reveal necrotizing vasculitis, granulomas, and infiltration by eosinophils (Fig. 4A) (26).
FIGURE 4.
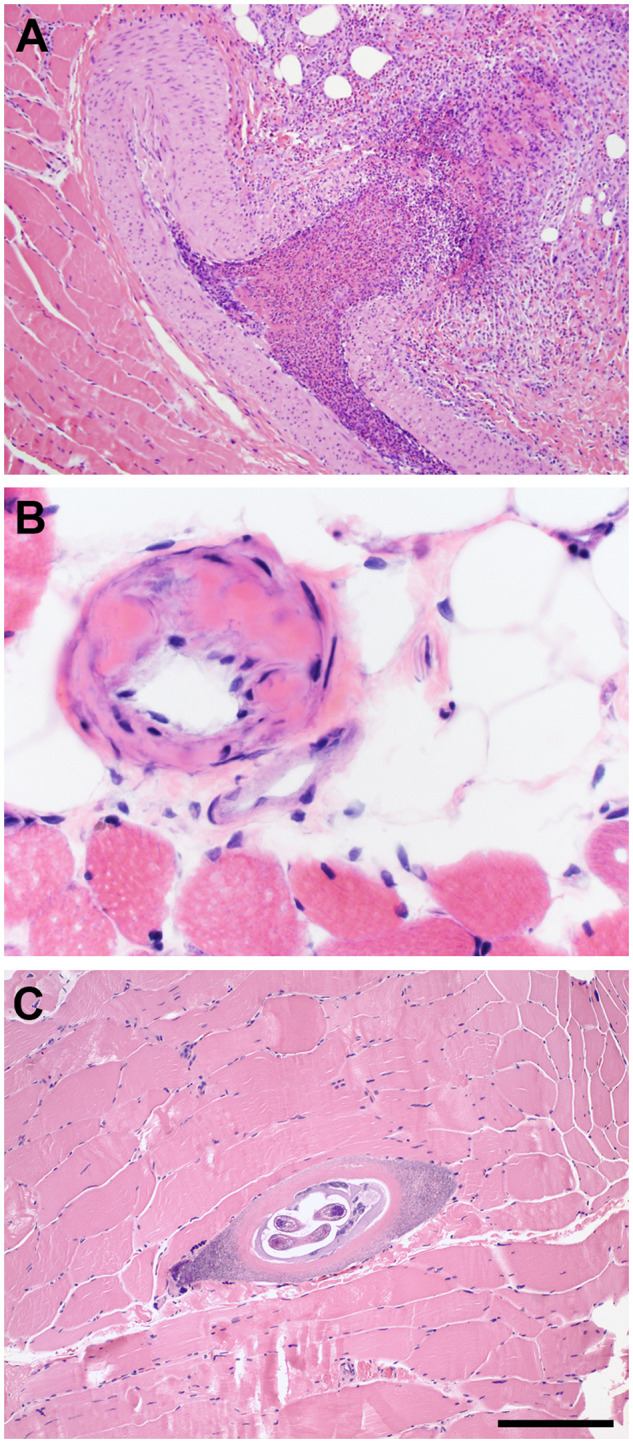
Paraffin section hematoxylin and eosin (H&E) slides. While recognizable in cryosections, a variety of systemic and infectious diseases may be more readily detected in H&E-stained paraffin sections of muscle biopsies. (A) Vasculitis, in this case eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome). (B) Amyloidosis in a patient with monoclonal gammopathy. (C) Trichinella myopathy (Trichinellosis). The scale bar in panel C is 200 µm for panels A and C; 50 µm for panel B.
Amyloidosis (Amyloid Myopathy)
Amyloid-light chain amyloidosis is a systemic disease resulting from extracellular deposits of immunoglobulin light chains (27). Clinical amyloid myopathy is typified by muscle weakness and may feature pseudohypertrophy of skeletal muscles. Muscle biopsies in such cases may reveal amyloid accumulations on H&E sections (Fig. 4B) (28).
Trichinellosis
Trichinellosis is caused by the roundworm parasite Trichinella spiralis. Trichinella newborn larvae penetrate into the vasculature of the gut wall, circulate widely, and enter individual muscle fibers, converting them to nurse cells where the larvae further develop and lie dormant within human skeletal muscle (29). They are readily evident in standard H&E sections, particularly in formalin-fixed, paraffin-embedded tissue (Fig. 4C).
Motor Units, Fiber Typing, and Other Light Microscopic Characteristics
After the initial assessment of the sample on H&E sections, the next step is the evaluation of motor units by fiber typing, requiring enzyme histochemistry, and/or immunohistochemistry (Table). Muscle fibers are arranged in motor units with characteristics of either type I (slow-twitch) or type II (fast-twitch) fibers. A useful mnemonic (as taught to the senior author by Robert “Bob” Schelper) to recall features of type I fibers is: “One mighty slow fat red ox.” That is, type “one” fibers are “mighty” (contain more mitochondria), “slow” twitch, “fat” (contain more neutral lipid), appear “red” (as in dark meat, especially limb muscles of domestic fowl), and rely primarily on “ox”-idative metabolism. Type I fibers are darker than type II fibers on NADH, SDH, COX, and ATPase pH 4.2 staining and contain slow myosin heavy chain (MHC). In contrast, type II fibers are fast-twitch, appear white, and contain less neutral lipid and more abundant glycogen. Type II fibers are darker on ATPase pH 9.4 staining and contain fast MHC.
TABLE.
Distinguishing Type I From Type II Muscle Fibers
| Muscle Fiber Type | Type I | Type II |
|---|---|---|
| Predominant actions |
|
|
| Physiology | Slow twitch | Fast twitch |
| Predominant myosin (gene) |
|
|
| Typically stain darker |
|
ATPase pH 9.4 |
| Glycogen | Less | More (glycolytic metabolism) |
| Mitochondria | More numerous | Less numerous |
| Neutral lipid | More (oxidative metabolism) | Less |
| Meat color (e.g. domestic fowl) | Red (dark) | White |
“One mighty slow fat red ox.” Mnemonic used to recall features of type I muscle fibers first learned from mentor Robert Schelper, circa 1984.
Differentiation of type I and type II fibers based on glycogen and neutral lipid content is less reliable. Glycogen is leached during tissue processing or the staining of sections. Oil-Red O can be performed to evaluate lipid in cryosections of muscle, but staining quality is typically inconsistent and often contains significant artifacts leading to inaccurate interpretation. A more reliable approach to the evaluation of lipid and glycogen content is to utilize glutaraldehyde-fixed tissue embedded in epoxy resin (e.g. epon). While light microscopy of “thick sections” may be sufficient in many instances, electron microscopy is sometimes necessary.
Myotendinous Junctions and Other Confounding Factors in Muscle Biopsy Evaluation
It is important to be aware of confounding factors in muscle biopsy interpretation, such as myotendinous junctions and other normal structures. The myotendinous junction is located where the muscle fibers interface (interdigitate) with tendon or fascia (Fig. 5A–D). In this location one normally encounters complex splitting of muscle fibers, numerous internal nuclei, and occasional cytoplasmic inclusion bodies or nemaline rods. Other normal structures that may cause confusion in the evaluation of the muscle biopsy are terminal branches of intramuscular nerve twigs, neuromuscular junctions, and muscle spindles (Fig. 5F, G). Not recognizing normal variations in these structures may lead to misdiagnosis.
FIGURE 5.

Normal structures may cause confusion. (A–D) Complex muscle fiber splitting and increased internally placed nuclei are normal at myotendinous junctions and interfaces of muscle with fascia. Dystrophin immunofluorescence (C) and electron microscopy (D) best illustrate the extreme feathering of muscle fibers where they interface with tendon or fascia. (E, F) Muscle spindles are encapsulated structures with myelinated axons and intrafusal muscle fibers in a wide range of sizes depending on the level at which the spindle is sectioned. The number and orientation of internal nuclei also vary widely. The scale bar in panel C is 100 µm for panels A and C; 50 µm for panels B, E, and F. The scale bar in panel D is 1.5 µm.
In addition, it is important for the neuropathologist to be aware of the site of the muscle biopsy. Nontraditional biopsy sites, such as the cricopharyngeus muscle or extraocular muscles, may have misleading site-specific features that are normal for that particular muscle. The fiber type distribution, mitochondrial content (including the presence of ragged-red fibers), and endomysial fibrous tissue vary among different muscles. As such, it is important to be cognizant of site-specific features to avoid misdiagnosis.
A Brief Word on Histopathologic Biomarkers of Disease
An in-depth discussion of classic biomarkers of disease is outside the scope of this article; nonetheless, it is important to roughly divide muscle diseases into neurogenic versus myopathic disorders. Various histopathologic biomarkers of disease tend to segregate with either neurogenic or myopathic disease. First, one must consider atrophy and hypertrophy of muscle fibers. A scattered distribution of atrophic and hypertrophic fibers suggests myopathic disease. Angulated, atrophic fibers in groups, pyknotic nuclear clusters, and an SMA-like appearance favor neurogenic disease as do the presence of target fibers. Fiber type grouping is an abnormality of fiber type distribution that typically occurs in neurogenic disease as a consequence of reinnervation. However, a fiber type predominance, cores, rods, central nuclei, internal nuclei with splits, internalized capillaries, ragged red fibers, COX-negative fibers, myonecrosis, regeneration, autophagic vacuoles, inflammatory cell infiltrates, MHC class I expression, and complement C5b-9 deposition, among other histopathologic features, favor a myopathic disease process. It is important to interpret findings in the context of the whole biopsy. Attributing too much importance to a single pathologic feature may lead to an incorrect diagnosis. For example, targetoid fibers may look very similar to cores; identifying angulated, atrophic fibers in small groups within the same biopsy provides for greater accuracy in interpreting the true nature of the targetoid fibers.
Myonecrosis and Regeneration
It is important to understand the underlying pathophysiology of myonecrosis and regeneration as such changes are seen in many muscle diseases, including muscular dystrophies, inflammatory myopathies, toxic and metabolic myopathies, and genetic, nondystrophic myopathies. In normal muscle, there are satellite cells that go largely unnoticed until there is an inciting pathology. Regardless of the underlying etiology, myonecrosis tends to occur segmentally in individual or clusters of muscle fibers. Early in this process, the histopathology is simply coagulation necrosis (Fig. 6A, fibers labeled #1). Macrophages then traverse the muscle fiber basement membrane to phagocytize the dead sarcoplasm, eventually filling the entire space formerly occupied by sarcomeres and other sarcoplasmic elements (Fig. 6A, fibers labeled #2 and #3). The macrophage infiltration is an injury repair process rather than myositis. In response to the injury, viable satellite cells proliferate to generate new myoblasts which eventually bridge the gap created by the myonecrosis (30). The processes of macrophage phagocytosis, satellite cell proliferation, and myoblast regeneration can occur simultaneously and within close proximity (Fig. 6A, fibers labeled #4); the histopathology may be confusing if not recognized as a repair process. During the process of myonecrosis and regeneration, the original muscle fiber basement membrane remains largely intact (Fig. 6B) to support satellite cell proliferation, myoblast migration, and myoblast fusion.
FIGURE 6.
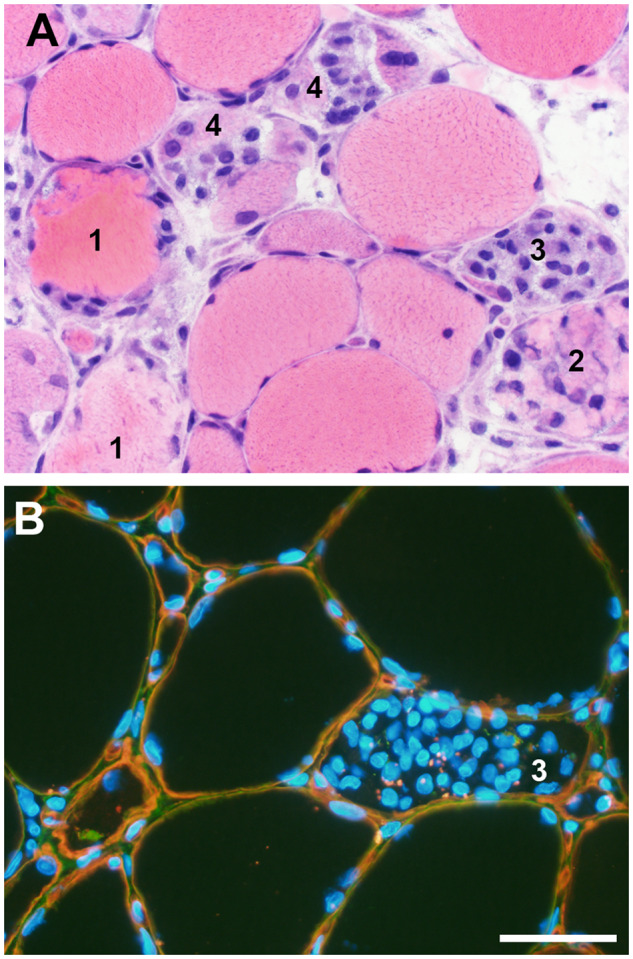
Myonecrosis and regeneration. A wide range of histopathology representing the entire spectrum of acute muscle fiber necrosis through regeneration may be seen in any necrotizing myopathy regardless of the etiology. (A) In a single high-power field, this muscle photomicrograph illustrates acute necrosis (#1), early invasion of a necrotic fiber by phagocytic macrophages (#2), numerous macrophages nearly filling the space originally occupied by sarcoplasm (#3), and early regeneration at the periphery of muscle fibers still containing macrophages (#4). (B) Dual label immunofluorescence for collagen VI (green) and perlecan (red) illustrates that the muscle fiber basal lamina (yellow) is largely intact during myonecrosis/regeneration in this dystrophic muscle biopsy. Macrophages clustered inside a necrotic muscle fiber (#3) are very similar to those illustrated in the hematoxylin and eosin-stained cryosection in panel A, #3; some DAPI-positive nuclei may be myoblasts as seen in panel A, fibers labeled #4. Nuclei are blue (DAPI). The scale bar is 50 µm for both A and B.
Dystrophinopathy—The Prototypical Muscular Dystrophy
Muscular dystrophies are caused by genetic deficiencies of proteins necessary for normal muscle function. One such protein, dystrophin, is located at the sarcolemma of striated muscle fibers where it stabilizes the surface membrane by anchoring the dystrophin-glycoprotein complex that bridges the cytoskeleton to the extracellular matrix (31). The dystrophin gene (DMD), a 2.6 million base pair gene with 79 coding exons, encodes for dystrophin. Abnormal dystrophin expression results in the dystrophinopathies, X-linked recessive muscular dystrophies that include Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), a milder allelic variant of DMD, and female carriers, both clinically manifesting and nonmanifesting (32).
Nearly all dystrophinopathy patients (approximately 95%) can be diagnosed by readily available molecular genetic testing (33), and a muscle biopsy is not needed. However, there continue to be a variety of situations where a muscle biopsy is obtained from dystrophinopathy patients (34). The scenarios include a lack of clinical suspicion, a failure of DMD deletion or duplication testing and complete sequencing to identify a pathogenic DMD variant, identifying DMD variants of unknown significance (see section on VUS, below), and dystrophinopathy patients whose clinical courses are discordant with the DMD variant identified by molecular testing. Among the approximately 6600 muscle biopsies evaluated at the University of Iowa between January 1998 and December 2019, 278 (4.2%) were dystrophinopathies.
The classic histopathologic findings in dystrophinopathies (Figs. 7A–C and 9B, E) include atrophy and hypertrophy, myonecrosis and regeneration, endomysial fibrosis, and fatty replacement (35). To recognize that a dystrophic muscle biopsy is a dystrophinopathy requires immunostaining for dystrophin. Dystrophin Western blots may be helpful in select cases, but they are largely no longer relevant for diagnostic testing.
FIGURE 7.

Duchenne muscular dystrophy (DMD). The severity of dystrophic pathology may vary extensively depending on the DMD mutation, the age of the patient at the time of biopsy, and the muscle biopsied. (A) Early, mildly dystrophic pathology with active myonecrosis and regeneration is illustrated in panel. (B, C) Later (chronic), more severe dystrophic pathology is shown. Immunofluorescence evaluation of DMD muscle shows a loss of dystrophin (cf. panels D and G) and upregulation of utrophin (cf. panels E and H). Utrophin positivity in the upper right corner (E) is a nerve twig and in the upper left corner (H) is a small artery. Endomysial capillary endothelia are normally positive as seen in both E and H. Spectrin immunostaining assures that the DMD muscle fibers are non-necrotic and free of nonspecific proteolytic degradation (F, I). Dystrophin-positive revertant fibers and Schwann cells are commonly encountered in muscle biopsies from DMD patients (K). The spectrin immunofluorescence of the same cryosection (staining performed by dual label methodology) is shown in panel J; muscle fibers “a,” “b,” and “c” are the same in each panel (J, K). The scale bar in panel K is 50 µm for panels D–H and I; 100 µm for panels A–C, J, and K.
In the diagnostic approach to dystrophinopathies, it is important to use multiple antibodies. Antidystrophin antibodies should interrogate the carboxy terminus, rod domain, and amino terminus regions of full-length dystrophin. Antibodies that evaluate the expression of utrophin and nNOS provide useful ancillary information. The classic abnormal pattern of immunostaining in DMD is the loss of dystrophin and gain (upregulation) of utrophin (Fig. 7D–I). The typical DMD muscle biopsy is also negative for nNOS (not shown). Relatively frequently, one may encounter dystrophin-positive revertant fibers in DMD muscle biopsies (Fig. 7J, K). If peripheral nerve twigs are included in a DMD muscle biopsy, Schwann cells may be dystrophin-positive (Fig. 7K). This is because the start site for Schwann cell dystrophin (Dp116) is located within intron 55 of the DMD gene (32). Whenever the pathogenic DMD variant is proximal to intron 55, Schwann cells are able to produce Dp116.
The abnormal patterns of immunostaining in BMD include an overall reduced expression of dystrophin, relative reduction in the intensity of staining for some dystrophin epitopes, and complete loss of some dystrophin epitopes (Figs. 8B, C and 9C, D, F, G). Hot spot in frame deletions in DMD occur in many patients with BMD. To illustrate this point, data from France indicate that the most common hot spot in frame deletions involve various combinations of exons from 3 to 7 or combinations of exons from 45 to 51 ([36] and online database: http://umd.be/DMD/W_DMD/index.html). Approximately 90% of the 650 patients in the French data with these hot spot in frame deletions have deletions in the exon 45–51 region of the DMD gene. Importantly, the most commonly used antidystrophin antibodies (DYS1, DYS2, and DYS3) do not interrogate the exons of either hot spot region. If the overall expression of truncated (mutant) dystrophin approximates normal, these in frame deletion cases will be missed by immunostaining with DYS1, DYS2, and DYS3. To mitigate this failure to recognize in frame deletion BMD cases, the panel of anti-dystrophin antibodies should include 1 or more antibodies that detect hot spot epitopes. For example, antibodies directed at epitopes in exons 46 and 50 will detect >80% of hot spot deletions by 1 or both being negative (Fig. 8A–D). Because the nNOS localization site in dystrophin is in the same exon 45–51 hot spot region, many BMD muscle biopsies will fail to stain for nNOS (Fig. 8E). As seen in DMD cases, utrophin is often expressed at the sarcolemma in BMD cases (Fig. 8F).
FIGURE 8.

Becker muscular dystrophy and female dystrophinopathy carriers. In frame deletions in the hot spot region of the DMD gene may show normal immunofluorescence intensity for dystrophin outside of the deleted exons. In this patient with an exons 45–51 in frame DMD deletion (A–F), the carboxy terminus antibody and exon 43 antibody appear normal, while antibodies directed at exons 46 and 50 are negative. This deletion includes the binding site for nNOS, and nNOS fails to localize at the sarcolemma (E). Utrophin is upregulated at the sarcolemma of many muscle fibers (F). Female carriers are recognized in muscle biopsies by the loss of dystrophin immunostaining from non-necrotic muscle fibers (G–K). Serial sections stained for dystrophin, hematoxylin and eosin, and spectrin are shown here. The artery and muscle fibers #1–4 are the same in each panel. The scale bar in panel I is 50 µm for panels I–K; 100 µm for panels A–F.
An additional consideration for the dystrophinopathies is the evaluation of muscle biopsies to identify female carriers. In the 21-year experience at the University of Iowa aforementioned, approximately 9% of dystrophinopathy cases were female carriers. In such cases, the specific diagnostic finding is non-necrotic, dystrophin-negative muscle fibers. These are most readily recognized by comparing serial sections stained with H&E and immunostained for dystrophin and spectrin (Fig. 8G–I). Clinical history may provide important clues that raise suspicion for dystrophinopathy, such as a Gowers sign, elevated creatine kinase, or male relatives with signs of dystrophinopathy. The clinical severity of carriers identified by muscle biopsy evaluation at the University of Iowa ranged from DMD-like muscular dystrophy to persistent hyperCKemia without weakness (unpublished data).
In summary, approximately 95% of dystrophinopathy patients can be diagnosed by readily available molecular testing. However, dystrophinopathy patients still undergo muscle biopsies for a variety of reasons. The practicing neuropathologist should be aware of variations in dystrophinopathy presentation, limitations of various dystrophin antibodies, and the importance of being mindful of hot spot in frame deletions in cases of BMD.
Variants of Unknown Significance
Increasingly, muscle biopsies are performed to determine whether VUS are truly pathogenic. In such cases, it is important to keep in mind classic biomarkers of disease. Some genetic neuromuscular diseases have highly specific histopathologic features that are evident in H&E-stained cryosections. In these instances, VUS resolution may be as simple as evaluation of the H&E slide. Figure 9A illustrates an example of a severely hypotonic male infant with a VUS identified in MTM1 and a family history of an older brother who died in utero. The muscle biopsy showed classic H&E histopathology for myotubular myopathy, including central nuclei, central localization of organelles, such as mitochondria, and glycogen aggregates that manifest as vacuoles in paraffin or frozen sections (37).
FIGURE 9.

Resolving variants of unknown significance (VUS). (A) The histopathologic phenotype of X-linked myotubular myopathy is sufficiently specific that a VUS in MTM1 may be resolved by simply evaluating a hematoxylin and eosin-stained cryosection. In the patient illustrated here, a small in-frame deletion in the MTM1 gene was classified a VUS and the muscle biopsy revealed central nuclei, central vacuoles, central consolidation of sarcoplasmic organelles, and numerous, abnormally small muscle fibers. Scale bar: 50 µm. (B–D) After identifying a VUS in DMD (c.79G>C, p. A27P), this 2-year-old boy underwent a muscle biopsy to clarify the pathogenicity of the variant. The histopathology is dystrophic (B). Dystrophin immunofluorescence intensity is mildly to moderately reduced using antibodies directed at the carboxy terminus and rod domains; the C-terminus is shown (C). No staining was observed with an actin binding domain antibody directed at an epitope in exons 7/8 near the amino terminus (D). (E–G) After identifying a VUS in DMD (c.76A>C, p. N26H), this 11-year-old boy underwent a muscle biopsy to clarify the pathogenicity of the variant. The histopathology is dystrophic (E). Dystrophin immunofluorescence intensity is mildly to moderately reduced using antibodies directed at the carboxy terminus and rod domains; exon 46 is shown here (F). No staining was observed with an actin binding domain antibody directed at an epitope in exons 7/8 near the amino terminus (G). The scale bar in panel D is 100 µm for panels B–D. The scale bar in panel G is 100 µm for panels E–G.
More commonly, the resolution of a VUS may require the integration of histopathology, immunostaining panels, and/or Western blotting. For example, a 2-year-old male patient and an 11-year-old male patient were found to have missense variants in DMD, c.79G>C, p. A27P and c.76A>C, p. N26H, respectively. While considered VUS in the molecular genetic testing reports, these variants are predicted to possibly alter biophysical properties of the actin binding domain near the amino terminus of dystrophin (38). Though full-length dystrophin is produced and dystrophin immunostaining is positive in their muscle biopsies using carboxy terminus and rod domain antibodies, immunostaining with an antibody directed at the exons 7/8 of the actin binding domain is negative. The binding of some antibodies in this region of dystrophin appears to be dependent on tertiary protein structure. Thus, the dystrophic histopathology and abnormal immunohistochemical pattern strongly suggest that the p. A27P (Fig. 9B–D) and p. N26H (Fig. 9E–G) variants are both pathogenic. Similar to the in frame deletion BMD cases discussed above, these missense variant examples illustrate the importance of understanding the binding locations and properties of antibodies, particularly those directed at dystrophin.
Inflammatory Myopathies and Immune-Mediated Necrotizing Myopathy
The topic of inflammatory myopathies is an evolving one with different perspectives on the diseases and their classifications, see for example Pestronk (39), Allenbach et al (10), and Tanboon and Nashino (40). Older terms, such as dermatomyositis and polymyositis, are now largely replaced by terms like immune myopathy with perimysial pathology, dermatomyositis spectrum disorders, antisynthetase syndrome myositis, sporadic inclusion body myositis (sIBM), and immune-mediated necrotizing myopathy (IMNM).
sIBM is the most common inflammatory myopathy and tends to have a strong clinical-pathological correlation. At the time of muscle biopsy (Fig. 10A–C), sIBM often shows evidence of chronic necrotizing disease with variable degrees of inflammatory cell infiltrates, rimmed vacuoles, and COX-negative fibers (not shown). Diffuse MHC class I expression is common. Cytoplasmic protein aggregates, such as TDP-43, may also present and can be demonstrated with immunohistochemistry.
FIGURE 10.

Myositis and immune-mediated necrotizing myopathy (IMNM). The more common forms of inflammatory myopathy are readily distinguished by hematoxylin and eosin staining and immunofluorescence. (A–C) In this patient with inclusion body myositis, the biopsy manifests lymphocytic infiltrates, endomysial fibrosis, fatty replacement, rimmed-vacuoles, and diffuse expression of myosin heavy chain (MHC) class I. (D–F) A perifascicular pattern of myonecrosis, regeneration, and MHC class I expression define this example of dermatomyositis spectrum disorder. Embryonic myosin heavy chain is a marker of regeneration (E). IMNM is not easily distinguishable from other types of necrotizing myopathy (G–I). The patient illustrated in panels G and I became symptomatic while taking a statin. The patient shown in panel H is statin-naïve and was followed for several years with a clinical diagnosis of limb-girdle muscular dystrophy. Both patients have highly elevated anti-HMGCR antibodies. The pattern of MHC class I expression (I) closely follows the pattern of myonecrosis. The scale bar in panel I is 50 µm for panels B and H; 100 µm for panels A, C, G, and I; 200 µm for panels D–F.
There are a variety of autoantibodies, clinical phenotypes, and histological findings within the inflammatory myopathies, and a particularly useful paper in summarizing these findings was published by Allenbach et al (10). Keeping this in mind, it is recommended that one approach inflammatory myopathy muscle biopsies by first dividing cases into big categories based on clinical and histological findings, such as cases associated with dermatomyositis and cases associated with antisynthetase syndrome. Both will typically have a perifascicular pattern of histopathology as illustrated in Figure 10D–F. In brief, dermatomyositis spectrum disorders have autoantibodies to Mi-2, melanoma differentiation-associated gene 5, and transcriptional intermediary factor-1 gamma, among others (10). Antisynthetase syndrome myositis features autoantibodies to Jo-1, antithreonyl-tRNA synthetase (PL-7), and others (10).
In contrast, IMNM features clinical and pathological overlap with limb girdle muscular dystrophy, histologically demonstrates myonecrosis, and regeneration with minimal lymphocyte infiltrates, and most often manifests a patchy MHC class I upregulation that typically follows the distribution of necrotic or regenerating muscle fibers (Fig. 10G–I). Patients with IMNM often have anti-signal recognition particle (SRP) or antihydroxy-3-methylglutaryl-CoA reductase antibodies (41).
There is certain degree overlap among the inflammatory myopathies. An illustrative case example is that of an elite, teenage athlete who became bedbound and was unable to move her limbs against gravity. The biopsy histologically strongly suggested a dermatomyositis spectrum disorder (Fig. 10D–F), but the patient was found to have autoantibodies to SRP, which most often manifests as IMNM.
Muscle Biopsy Myths
There are many myths surrounding muscle biopsies that are worth dispelling. A common myth is that numerous ring fibers and numerous internally placed nuclei strongly favor the diagnosis of myotonic dystrophy. In reality, both abnormalities are entirely nonspecific. Another common myth is that biopsies must be frozen within 30 minutes of sample acquisition, and a related myth is that autopsy muscle tissue is unfit for evaluation. Neither of these myths is universally true. While certain enzyme histochemical or immunohistochemical stains may be suboptimal (e.g. cytochrome C oxidase) and certain ultrastructural evaluations may be compromised (e.g. mitochondrial cristae), the majority of histopathologic abnormalities used to diagnose neuromuscular diseases are adequately preserved when biopsy freezing is delayed or when muscle is obtained at autopsy. Of course, shorter postmortem intervals are preferable to longer postmortem intervals.
Regarding the evaluation of neuromuscular disease at autopsy, muscles such as the diaphragm, those from the abdominal and pelvic walls, and even the deltoid muscle may be sampled under a standard complete autopsy permission. Specific permissions allow the evaluation of skeletal muscles of the limbs, including standard sites like the quadriceps, deltoid, biceps, and gastrocnemius muscles. The same methodologies normally used for muscle biopsies may be successfully employed with autopsy material.
In conclusion, this article briefly touches on what every neuropathologist needs to know regarding the muscle biopsy. Neuropathologists should know how to prepare pristine, frozen muscle sections, be familiar with and comfortable evaluating a core panel of histological stains and immunostains, recognize and interpret major biomarkers of disease, understand the immunohistochemical tools for diagnosing dystrophinopathies, recognize and interpret patterns of inflammatory and other immune-mediated myopathies, and understand the importance of working with the clinical care team for clinical-pathological correlation. These skills and knowledge will facilitate high quality, everyday neuropathological practice and will help guide additional avenues of investigation that may be required to reach definitive diagnoses.
ACKNOWLEDGMENTS
Thank you to Joel Carl for his expertise in assembling and formatting the figures for publication.
This work was supported by NIH (NINDS) (U54, NS053672 to S.A.M.) that funds the Iowa Wellstone Muscular Dystrophy Cooperative Research Center.
The authors have no duality or conflicts of interest to declare.
REFERENCES
- 1. Meola G, Bugiardini E, Cardani R.. Muscle biopsy. J Neurol 2012;259:601–10 [DOI] [PubMed] [Google Scholar]
- 2. Joyce NC, Oskarsson B, Jin L-W.. Muscle biopsy evaluation in neuromuscular disorders. Phys Med Rehabil Clin N Am 2012;23:609–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meng H, Janssen PML, Grange RW, et al. Tissue triage and freezing for models of skeletal muscle disease. J Vis Exp 2014;e51586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zalneraitis EL, Halperin JJ, Grunnet ML, et al. Muscle biopsy and the clinical course of infantile spinal muscular atrophy. J Child Neurol 1991;6:324–8 [DOI] [PubMed] [Google Scholar]
- 5. Arnold WD, Kassar D, Kissel JT.. Spinal muscular atrophy: Diagnosis and management in a new therapeutic era. Muscle Nerve 2015;51:157–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kolb SJ, Kissel JT.. Spinal muscular atrophy: A timely review. Arch Neurol 2011;68:979–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burns DT, Donkervoort S, Müller JS, et al. Variants in EXOSC9 disrupt the RNA exosome and result in cerebellar atrophy with spinal motor neuronopathy. Am J Hum Genet 2018;102:858–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iaccarino L, Ghirardello A, Bettio S, et al. The clinical features, diagnosis and classification of dermatomyositis. J Autoimmun 2014;48–49:122–7 [DOI] [PubMed] [Google Scholar]
- 9. Dalakas MC, Hohlfeld R.. Polymyositis and dermatomyositis. Lancet 2003;362:971–82 [DOI] [PubMed] [Google Scholar]
- 10. Allenbach Y, Benveniste O, Goebel HH, et al. Integrated classification of inflammatory myopathies. Neuropathol Appl Neurobiol 2017;43:62–81 [DOI] [PubMed] [Google Scholar]
- 11. Greenberg SA. Inclusion body myositis: Clinical features and pathogenesis. Nat Rev Rheumatol 2019;15:257–72 [DOI] [PubMed] [Google Scholar]
- 12. Dimachkie MM, Barohn RJ.. Inclusion body myositis. Semin Neurol 2012;32:237–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wolfe SM, Pinals RS, Aelion JA, et al. Myopathy in sarcoidosis: Clinical and pathologic study of four cases and review of the literature. Semin Arthritis Rheum 1987;16:300–6 [DOI] [PubMed] [Google Scholar]
- 14. Silverstein A, Siltzbach LE.. Muscle involvement in sarcoidosis: Asymptomatic, myositis, and myopathy. Arch Neurol 1969;21:235–41 [DOI] [PubMed] [Google Scholar]
- 15. Romero NB, Bitoun M.. Centronuclear myopathies. Semin Pediatr Neurol 2011;18:250–6 [DOI] [PubMed] [Google Scholar]
- 16. Jungbluth H. Central core disease. Orphanet J Rare Dis 2007;2:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sewry CA, Müller C, Davis M, et al. The spectrum of pathology in central core disease. Neuromuscul Disord 2002;12:930–8 [DOI] [PubMed] [Google Scholar]
- 18. Cenacchi G, Papa V, Pegoraro V, et al. Review: Danon disease: Review of natural history and recent advances. Neuropathol Appl Neurobiol 2020;46:303–22. [DOI] [PubMed] [Google Scholar]
- 19. Sugie K, Yamamoto A, Murayama K, et al. Clinicopathological features of genetically confirmed Danon disease. Neurology 2002;58:1773–8 [DOI] [PubMed] [Google Scholar]
- 20. Dasouki M, Jawdat O, Almadhoun O, et al. Pompe disease: Literature review and case series. Neurol Clin 2014;32:751–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feeney EJ, Austin S, Chien Y-H, et al. The value of muscle biopsies in Pompe disease: Identifying lipofuscin inclusions in juvenile- and adult-onset patients. Acta Neuropathol Commun 2014;2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pfeffer G, Chinnery PF.. Diagnosis and treatment of mitochondrial myopathies. Ann Med 2013;45:4–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liang W-C, Nishino I.. Lipid storage myopathy. Curr Neurol Neurosci Rep 2011;11:97–103 [DOI] [PubMed] [Google Scholar]
- 24. Bruno C, DiMauro S.. Lipid storage myopathies. Curr Opin Neurol 2008;21:601–6 [DOI] [PubMed] [Google Scholar]
- 25. Greco A, Rizzo MI, De Virgilio A, et al. Churg-Strauss syndrome. Autoimmun Rev 2015;14:341–8 [DOI] [PubMed] [Google Scholar]
- 26. Vital A, Vital C, Viallard J-F, et al. Neuro-muscular biopsy in Churg-Strauss syndrome: 24 cases. J Neuropathol Exp Neurol 2006;65:187–92 [DOI] [PubMed] [Google Scholar]
- 27. Desport E, Bridoux F, Sirac C, et al. AL amyloidosis. Orphanet J Rare Dis 2012;7:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gertz MA, Kyle RA.. Myopathy in primary systemic amyloidosis. J Neurol Neurosurg Psychiatry 1996;60:655–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gottstein B, Pozio E, Nöckler K.. Epidemiology, diagnosis, treatment, and control of trichinellosis. CMR 2009;22:127–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dumont NA, Bentzinger CF, Sincennes M-C, et al. Satellite cells and skeletal muscle regeneration. Compr Physiol 2015;5:1027–59 [DOI] [PubMed] [Google Scholar]
- 31. Gao QQ, McNally EM.. The dystrophin complex: Structure, function, and implications for therapy. Compr Physiol 2015;5:1223–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Muntoni F, Torelli S, Ferlini A.. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol 2003;2:731–40 [DOI] [PubMed] [Google Scholar]
- 33. Aartsma-Rus A, Ginjaar IB, Bushby K.. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet 2016;53:145–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carlson CR, Moore SA, Mathews KD.. Dystrophinopathy muscle biopsies in the genetic testing ERA: One center’s data. Muscle Nerve 2018;58:149–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Flanigan KM. Duchenne and Becker muscular dystrophies. Neurol Clin 2014;32:671–88 [DOI] [PubMed] [Google Scholar]
- 36. Tuffery-Giraud S, Béroud C, Leturcq F, et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: A model of nationwide knowledgebase. Hum Mutat 2009;30:934–45 [DOI] [PubMed] [Google Scholar]
- 37. Lawlor MW, Beggs AH, Buj-Bello A, et al. Skeletal muscle pathology in X-linked myotubular myopathy: Review with cross-species comparisons. J Neuropathol Exp Neurol 2016;75:102–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Henderson DM, Lee A, Ervasti JM.. Disease-causing missense mutations in actin binding domain 1 of dystrophin induce thermodynamic instability and protein aggregation. Proc Natl Acad Sci 2010;107:9632–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pestronk A. Acquired immune and inflammatory myopathies: Pathologic classification. Curr Opin Rheumatol 2011;23:595–604 [DOI] [PubMed] [Google Scholar]
- 40. Tanboon J, Nishino I.. Classification of idiopathic inflammatory myopathies: Pathology perspectives. Curr Opin Rheumatol 2019;32:704–14 [DOI] [PubMed] [Google Scholar]
- 41. Pinal-Fernandez I, Casal-Dominguez M, Mammen AL.. Immune-mediated necrotizing myopathy. Curr Rheumatol Rep 2018;20:21. [DOI] [PMC free article] [PubMed] [Google Scholar]