

The strength of humoral and cellular immune responses is regulated by the properties of mesoporous silica.
Abstract
Subunit vaccines generally proceed through a 4-step in vivo cascade—the DUMP cascade—to generate potent cell-mediated immune responses: (1) drainage to lymph nodes; (2) uptake by dendritic cells (DCs); (3) maturation of DCs; and (4) Presentation of peptide-MHC I complexes to CD8+ T cells. How the physical properties of vaccine carriers such as mesoporous silica nanoparticles (MSNs) influence this cascade is unclear. We fabricated 80-nm MSNs with different pore sizes (7.8 nm, 10.3 nm, and 12.9 nm) and loaded them with ovalbumin antigen. Results demonstrated these MSNs with different pore sizes were equally effective in the first three steps of the DUMP cascade, but those with larger pores showed higher cross-presentation efficiency (step 4). Consistently, large-pore MSNs loaded with B16F10 tumor antigens yielded the strongest antitumor effects. These results demonstrate the promise of our lymph node-targeting large-pore MSNs as vaccine-delivery vehicles for immune activation and cancer vaccination.
INTRODUCTION
Vaccination, one of the most cost-effective strategies to control infectious diseases, has saved countless lives (1, 2). Vaccines based on protein subunits are safer and easier to produce (3). These exogenous antigens are usually internalized, processed by antigen-presenting cells (APCs), and presented via major histocompatibility complex (MHC) class II molecules to CD4+ T cells. However, elimination of viral infections and tumors requires generation of CD8+ T cell responses. The presentation of exogenous antigens on MHC class I molecules, known as cross-presentation, is essential for the initiation of CD8+ T cell responses (4). In vivo, cross-presentation is mainly carried out by specific dendritic cell (DC) subsets such as CD8+ DCs and CD103+ DCs (5, 6).
Lymph nodes are the main sites for immune activation and surveillance, which contain abundant DCs, including lymph node–resident CD8a+ DCs with cross-presentation ability (4–7). To generate potent cell-mediated immune responses, subunit vaccines need to go through an in vivo cascade of four steps: drainage to lymph nodes, uptake by DCs, maturating DCs, and presenting peptide–MHC I complexes to CD8+ T cells (the DUMP cascade for short). Thus, the overall efficacy to induce potent cellular immune response is a multiplying product of efficiencies of each step (8–11). Consequently, it is critical to maximize the efficiency of each step and to ensure none of the four is close to zero.
Recent studies have shown that nanovesicles with a size between 10 and 100 nm can preferably drain to the lymph nodes through the gap among the lymphatic endothelial cells (12–14). In addition to size, vesicles with negative and hydrophilic surface have been reported to be more efficiently drained to the lymph nodes (15, 16). After the drainage to lymph nodes, antigen-loaded vehicles need to be internalized by DCs; otherwise, vehicles may simply pass around the outside of the lymph node via the subcapsular sinus and leave directly via the efferent lymph vessel. Subsequent activation of these DCs and antigen cross-presentations are also crucial for triggering powerful cellular adaptive immunity.
Mesoporous silica nanoparticles (MSNs) have been extensively studied as vaccine delivery system because of their advantages including ultrahigh specific surface area, easiness for surface modification, adjustable particle size, and excellent biocompatibility (17–22). In addition, MSNs contain negatively charged and hydrophilic silanol groups (Si-OH) on their surface, making MSNs a potential lymph node–targeting carrier. However, the MSNs used in most of previous studies could not proceed through the DUMP cascade smoothly, as they are usually too large (>100 nm) to efficiently migrate to the lymph nodes. As a result, additional adjuvants are generally used to achieve an effective response (23, 24). The other limitation of reported MSNs is that their pore size is relatively small, making it challenging to encapsulate sufficient amount of antigen molecules (25–28). In addition, the antigen cross-presentation ability of MSNs has rarely been investigated.
Here, we specifically designed MSNs that can efficiently go through the DUMP cascade. They have a small size of around 80 nm and a negatively charged surface, which ensure their ability to drain to lymph nodes. They can be efficiently taken up by DCs, mainly through scavenger receptor–mediated endocytosis and subsequently maturate these DCs. We observed that their ability of antigen cross-presentation can be tuned by adjusting the pore size of MSNs. We fabricated MSNs with three different pore sizes ranging from 7.8 nm (small; MSNs-S), 10.3 nm (medium; MSNs-M), to 12.9 nm (large; MSNs-L) (Fig. 1). The obtained MSNs showed the same ability in the first three steps in the DUMP cascade, but different abilities in presenting peptide–MHC I complexes to CD8+ T cells. Consequently, these MSNs with different pore sizes generated different levels of cellular immune responses. To our knowledge, this is the first analysis of the effects of pore size on the ability of lymph node–targeting MSNs to elicit immune responses.
Fig. 1. Schematic illustration of the DUMP cascade of antigen-loaded lymph node–targeting MSNs to induce adaptive immune response.
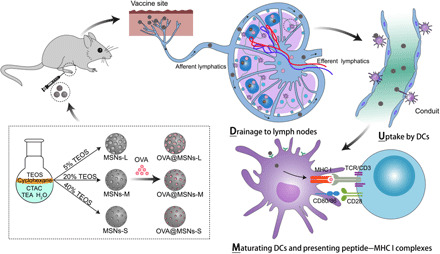
MSNs with different pore sizes were synthesized by adjusting the concentration of tetraethyl orthosilicate (TEOS) in cyclohexane, after which the MSNs were loaded with ovalbumin (OVA) antigen (OVA@MSNs). CTAC, cetyltrimethylammonium chloride; TEA, triethanolamine; TCR, T cell receptor. After subcutaneous injection, OVA@MSNs efficiently accomplished the DUMP cascade: drainage to lymph nodes, uptake by DCs, maturating DCs, and presenting peptide–MHC I complexes to CD8+ T cells.
RESULTS
Fabrication and characterization of MSNs and OVA@MSNs
We prepared MSNs with different pore sizes by adjusting the concentration of tetraethyl orthosilicate (TEOS) in cyclohexane to 5, 20, and 40%, respectively. Dynamic light scattering showed a hydrodynamic diameter of 82.6 ± 0.7 nm for MSNs-L, 84.3 ± 0.4 nm for MSNs-M, and 86.6 ± 2.4 nm for MSNs-S (Fig. 2A). Transmission electron microscopy (Fig. 2B) and scanning electron microscopy (fig. S1A) showed that all three formulations were monodispersed and spherical in shape. Nitrogen absorption and desorption tests (Fig. 2C) and Brunauer-Emmett-Teller analyses showed that the pore sizes of MSNs-L, MSNs-M, and MSNs-S were 12.9, 10.3, and 7.8 nm (Fig. 2D), respectively. Correspondingly, the surface area and pore volume decreased when the pore size decreased, as shown in table S1. The different pore sizes in the three MSN formulations were also confirmed by small-angle x-ray scattering, in which MSNs with larger pores showed diffraction peaks in the lower-angle region, while MSNs with smaller pores showed peaks in the higher-angle region (Fig. 2E) according to the Bragg equation. Together, these results showed that we successfully synthesized three types of small MSNs with a similar particle size but with different mesoporous pore sizes.
Fig. 2. Characterization of MSNs with large (MSNs-L), medium (MSNs-M), and small (MSNs-S) pores.
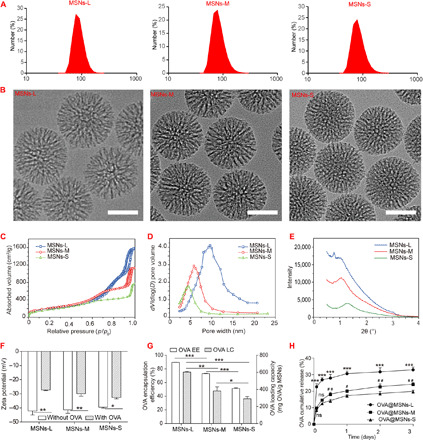
(A) Hydrodynamic diameter distribution. (B) Transmission electron micrographs. Scale bars, 50 nm. (C) Nitrogen absorption-desorption isotherms. (D) Pore size distribution. (E) Small-angle x-ray scattering. (F) Zeta potential of MSNs loaded with or without OVA. (G) Encapsulation efficiency (EE) and loading capacity (LC) of OVA. (H) Release of OVA from OVA@MSNs in PBS. Asterisks indicate P values associated with comparisons between OVA@MSNs-L and OVA@MSNs-M or OVA@MSNs-S; pound signs indicate the same P ranges for comparisons between OVA@MSNs-M with OVA@MSNs-S. The significance of the results was analyzed by using one-way analysis of variance (ANOVA). *P < 0.05, **P < 0.01, and ***P < 0.001; #P < 0.05 and ##P < 0.01; ns, not significant. Data are shown as means ± SD (n = 3). All experiments were repeated two to three times.
The antigen ovalbumin (OVA) was incorporated into MSNs with a probe-sonication method. The antigen-loaded MSNs were referred to as OVA@MSNs hereafter. The particle size and z potential of OVA@MSNs were measured by dynamic light scattering and laser Doppler velocimetry, respectively. Encapsulation of OVA did not significantly alter the size of any of the three MSN formulations (fig. S1B), but the antigen encapsulation tuned the particle surface to less electronegative (Fig. 2F). OVA encapsulation efficiency was 89.6% for OVA@MSNs-L, 73.2% for OVA@MSNs-M, and 51.5% for OVA@MSNs-S when the OVA/MSNs mass ratio was 1:4 (Fig. 2G), suggesting that MSNs with larger pores can load higher amounts of OVA. The maximum OVA loading capacities of the three MSN formulations were found to be 604 mg (MSNs-L), 382 mg (MSNs-M), and 290 mg (MSNs-S) per gram of MSNs. The release rate of OVA from OVA@MSNs in phosphate-buffered saline (PBS) was found to depend on mesoporous pore size: Around 33, 24, and 20% OVA were released from MSNs-L, MSNs-M, and MSNs-S, respectively, on day 3 (Fig. 2H). However, most of the antigen still remained inside of the pores, indicating the strong interaction between MSNs and antigen. The OVA release curves of three MSNs were well correlated with the first-order release kinetic equation (table S2).
Cellular toxicity, uptake, and cross-presentation properties of OVA@MSNs in vitro
After initial antigen loading assays, we investigated the toxicity of MSNs, as well as uptake and cross-presentation properties of OVA@MSNs in vitro. The viability of DC2.4 cells was found to rely on the dose and pore size of MSNs (Fig. 3A). The toxicity of both MSNs-M and MSNs-S significantly increased with higher concentration of MSNs, while the viability of cells treated by MSNs-L remained relatively high (above 70%) even with the highest dose, indicating the better safety of MSNs-L. As cell viabilities exceeded 80% for all MSNs formulations at 20 μg/ml, this dose was used for all subsequent cell experiments.
Fig. 3. Cellular uptake and antigen cross-presentation of OVA@MSNs in vitro.
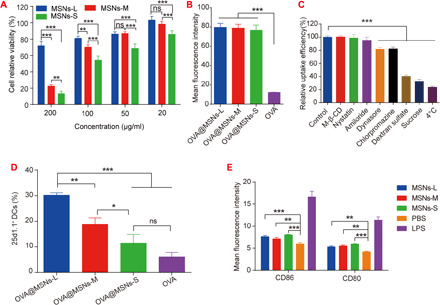
(A) Relative viability of DC2.4 cells exposed to different concentrations of MSNs. (B) Uptake efficiency of OVA@MSNs by DC2.4 cells. (C) Screening of potential mechanisms of OVA@MSNs internalization by DC2.4 cells. M-β-CD, methyl-β-cyclodextrin. (D) Cross-presentation of OVA in DC2.4 cells. (E) Expression of costimulatory molecules CD86 and CD80 on bone marrow–derived DCs (BMDCs) after 18 hours of incubation with MSNs. LPS, lipopolysaccharide. Data are shown as means ± SD (n = 3). Significance of results was analyzed by using one-way ANOVA. *P < 0.05, **P < 0.01, and ***P < 0.001. All experiments were repeated two to three times.
Next, we labeled OVA with Cy5 (cyanine5) dye and assessed the internalization efficiency of OVA@MSNs in DC2.4 cells. The mass ratio of Cy5-OVA to MSNs was adjusted to achieve an antigen loading capacity of 20% in all three MSN formulations. The results showed that the encapsulation of OVA in MSNs significantly increased the internalization of OVA, and the pore size of MSNs did not affect the internalization efficiency (Fig. 3B).
To explore the internalization mechanism of OVA@MSNs, DC2.4 cells were pretreated with different inhibitors to block specific endocytic pathways. The results showed that the uptake efficiency of OVA@MSNs significantly decreased when the cells were incubated at 4°C (reduced by 80%) or pretreated with sucrose (reduced by 65%) as a nonselective endocytic inhibitor (29), suggesting that the internalization of OVA@MSNs was energy dependent (Fig. 3C). Uptake was reduced by about 60% in the presence of dextran sulfate, suggesting that OVA@MSNs were internalized mainly through scavenger receptor–mediated endocytosis. Dynasore and chlorpromazine also inhibited the uptake although to a less extent, suggesting that OVA@MSN internalization also involved dynamin- and clathrin-dependent endocytic pathways. In contrast, methyl-β-cyclodextrin, nystatin, and amiloride did not significantly affect OVA@MSNs uptake, indicating that caveolae/lipid raft–mediated endocytosis or micropinocytosis were not involved (30, 31).
We then investigated the ability of OVA@MSNs to deliver antigen into the cytosol of DC2.4 and induce cross-presentation. OVA@MSNs-treated DC2.4 cells produced more reactive oxygen species (ROS) compared to medium solution (fig. S2A), which is related to endosomal rupture (32). The separation of green fluorescence of OVA and red fluorescence from endosomes at 6 hours after incubation confirmed the escape of OVA@MSNs from endolysosomes (fig. S2B). Next, flow cytometry was used to assess antigen cross-presentation by examining H-2Kb-SIINFEKL complexes on DC surface, which were labeled by monoclonal antibody 25d1.16. Results showed that OVA@MSNs-L, OVA@MSNs-M, and OVA@MSNs-S increased the proportion of 25d1.16-positive cells by 5-, 3-, and 1.8-fold compared to free OVA, respectively (Fig. 3D). These results together suggested that the encapsulation of OVA in MSNs enhanced DC internalization, lysosomal escape, and cross-presentation.
Last, the expression of costimulatory factors CD80 and CD86 on bone marrow–derived DCs (BMDCs) was measured to investigate the ability of MSNs to induce the maturation of BMDCs. All three MSN formulations significantly up-regulated the expression of both factors compared to PBS (Fig. 3E), suggesting their potential adjuvanticity.
Targeting of draining lymph nodes and activation of lymph node–resident DCs by OVA@MSNs
As adaptive immune responses are initiated in the lymph nodes, we assessed the ability of OVA@MSNs to target them. Cy5-OVA@MSNs or free Cy5-OVA was injected subcutaneously into the hind footpad of mice, and the mice were visualized at different time points using an IVIS Spectrum system. Fluorescence signal of free Cy5-OVA was observed initially in popliteal lymph nodes and began to decay immediately after injection. In contrast, fluorescence intensity of Cy5-OVA@MSNs continued to increase and reached a peak at 10 hours after injection (Fig. 4A). Ex vivo images of popliteal and inguinal lymph nodes (Fig. 4B) and semiquantification of the fluorescence intensity in popliteal lymph nodes (Fig. 4C) obtained at 36 hours after injection showed that all three Cy5-OVA@MSNs formulations accumulated to a much greater extent in these lymph nodes than free Cy5-OVA.
Fig. 4. Targeting of draining lymph nodes and activation of lymph node–resident DCs by OVA@MSNs.
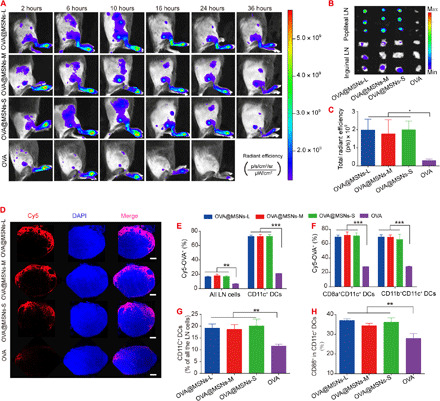
(A) Migration of Cy5-OVA@MSNs from the injection site to draining lymph nodes in vivo, as observed using an IVIS Spectrum system. p, photon. (B) Popliteal and inguinal lymph nodes (LNs) were isolated and visualized at 36 hours after Cy5-OVA@MSNs injection. (C) Total radiant efficiency of popliteal lymph nodes at 36 hours after injection. (D) Popliteal lymph nodes were isolated at 10 hours after OVA@MSNs injection, and frozen sections were prepared and analyzed by confocal fluorescence microscopy. Scale bars, 200 μm. DAPI, 4′,6-diamidino-2-phenylindole. (E and F) Percentages of (E) CD11c+ DCs, (F) CD8a+CD11c+, or CD11b+CD11c+ DCs that were Cy5-OVA positive. (G and H) OVA@MSNs promoted expansion and activation of DCs in lymph nodes. The percentages of CD11c+ DCs in all lymph node cell populations (G) and percentage of CD86+ in CD11c+ DCs (H) at 3 days after immunization. Data are shown as means ± SD (n = 3 to 5). All experiments were repeated two to three times. Photo credit (A): Xiaoyu Hong, Key Laboratory of Drug Targeting and Drug Delivery System of the Education Ministry and Sichuan Province, West China School of Pharmacy, Sichuan University.
To further verify that Cy5-OVA@MSNs migrated to popliteal lymph nodes, these lymph nodes were collected at 10 hours after injection, frozen-sectioned, stained with a nuclear dye (bisbenzimide Hoechst 33342), and examined by confocal fluorescence microscopy. Results showed that Cy5-OVA localized primarily to the superficial cortex of lymph node. The fluorescence of Cy5-OVA was much stronger in lymph nodes from animals injected with Cy5-OVA@MSNs than that of free Cy5-OVA (Fig. 4D). These results confirmed that Cy5-OVA@MSNs reached the popliteal lymph nodes after subcutaneous injection in the footpad. The colocalization of OVA and MSNs in the lymph nodes was also verified. The red fluorescence of Cy5-OVA was almost completely overlapped with the green fluorescence of MSNs-FITC (fluorescein isothiocyanate) (fig. S3), indicating that OVA was not released before reaching the lymph nodes.
To explore the potential of OVA@MSNs to be internalized by lymph node–resident cells, especially DCs, we subcutaneously injected mice with Cy5-OVA@MSNs. At 10 hours after injection, popliteal lymph nodes were removed and analyzed by flow cytometry. All three Cy5-OVA@MSNs formulations significantly promoted Cy5-OVA uptake by lymph node cells compared with free Cy5-OVA (Fig. 4E). Almost 70% of CD11c+ DCs in lymph nodes were positive for Cy5-OVA after treatment with any of the three Cy5-OVA@MSNs formulations, which was significantly higher than that with free Cy5-OVA (Fig. 4E). To assess whether DC subsets differ in their uptake of Cy5-OVA@MSNs, we examined CD8a+CD11c+ population, which can cross-present exogenous antigen to CD8+ T cell, and CD11b+CD11c+ population to prime CD4+ T helper cells (6). Both proportions of CD8a+CD11c+ and CD11b+CD11c+ DCs positive for OVA were similar to the proportion of CD11c+ DCs that were positive for OVA (Fig. 4, E and F). Together, these results showed that MSNs efficiently delivered cargos to lymph node cells such as CD8a+CD11c+ DCs and CD11b+CD11c+ DCs in the popliteal lymph nodes after footpad subcutaneous injection.
Antigen uptake by DCs in the lymph node is just the first step in inducing T cell responses. DCs also need to be activated to express costimulatory molecules during antigen presentation to prime T cells. Therefore, we analyzed the potential of OVA@MSNs to expand and activate DCs in lymph nodes. A greater proportion of DCs in popliteal lymph nodes was detected in animals injected with OVA@MSNs than that injected with free OVA (Fig. 4G). All three OVA@MSNs led to similar higher expression of costimulatory CD86 by DCs in popliteal lymph nodes than free OVA (Fig. 4H).
Ability of OVA@MSNs to activate immune responses
On days 0, 14, and 28, mice were subcutaneously injected with 10 μg of OVA or 40 μg of MSNs loaded with 10 μg of OVA. First, we detected the antibody response on days 21, 28, and 35 to evaluate the elicited humoral immune response. Immunoglobulin G (IgG) and IgG1 antibodies reached a plateau in all groups on day 28, while IgG2a levels kept increasing after each immunization (Fig. 5, A to C). All the three OVA@MSNs induced significantly higher antibody responses than free OVA solution, with one exception that MSNs-S did not increase IgG1 or IgG2a titers compared to free OVA. OVA@MSNs-L elicited the strongest IgG, IgG1, and IgG2a antibody titers, followed by OVA@MSNs-M and OVA@MSNs-S (Fig. 5, A to C). These results suggested that OVA encapsulated in MSNs significantly increased the immunogenicity of OVA, and this enhancement depended on mesoporous pore size.
Fig. 5. Ability of OVA@MSNs to activate immune responses.
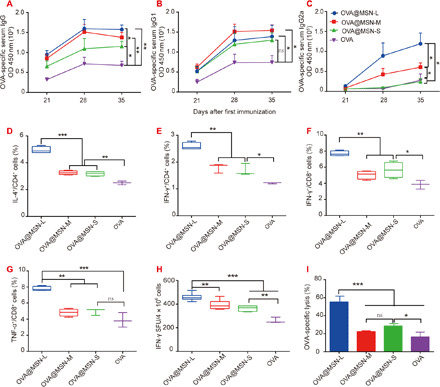
On days 0, 14, and 28, C57BL mice were vaccinated in the footpad with 10 μg of free OVA or 40 μg of MSNs loaded with 10 μg of OVA. Immune responses were measured on days 21, 28, and 35. (A to C) Levels of anti-OVA IgG, IgG1, and IgG2a antibodies in serum were assayed (105 dilution). OD, optical density. (D to G) Splenocytes were restimulated with OVA (100 μg/ml) or SIINFEKL (2 μg/ml) for 6 hours at 37°C; then, flow cytometry was used to determine percentages of OVA-specific (D) IL-4–producing CD4+ T cells, (E) IFN-γ–producing CD4+ T cells, (F) IFN-γ–producing CD8+ cells, and (G) TNF-α–producing CD8+ T cells. (H) Numbers of IFN-γ–secreting CD8+ T cells in the splenocytes were measured using IFN-γ ELISpot assay. SFU, spot-forming unit. (I) Percentage of OVA-specific CD8+ T cell lysis. *P < 0.05, **P < 0.01, and ***P < 0.001. Data are shown as means ± SD (n = 4 to 5).
We next evaluated CD4+ and CD8+ T cell responses induced by OVA@MSNs. Splenocytes of vaccinated mice were harvested on day 35 for ex vivo culture and restimulation. We examined the proportions of CD4+ T cells positive for interferon-γ (IFN-γ) and interleukin-4 (IL-4) and proportions of CD8+ T cells positive for IFN-γ and tumor necrosis factor–α (TNF-α) using flow cytometry. Similar to the enhanced antibody responses, OVA@MSNs significantly increased the excretion of IFN-γ and IL-4 by CD4+ cells and the excretion of IFN-γ and TNF-α by CD8+ cells. Furthermore, OVA@MSNs-L induced the highest proportions of these cells producing the cytokines, followed by OVA@MSNs-M and OVA@MSNs-S (Fig. 5, D to G, and fig. S4). Enzyme-linked immunospot (ELISpot) assay showed a similar trend as the results mentioned above (Fig. 5H and fig. S5).
These experiments revealed that OVA@MSNs can activate an effector phenotype in CD4+ and CD8+ T cells. However, these experiments did not reveal whether the activated immune cells were enough to recognize and kill antigen-bearing target cells. Therefore, we evaluated the cellular immune response by measuring OVA-specific cytotoxic T lymphocyte activity, expressed as the percentage of OVA-specific cytotoxic lysis in vaccinated mice relative to the percentage in untreated mice. OVA@MSNs-L caused a significantly higher percentage of target cell lysis than OVA@MSNs-M and OVA@MSNs-S, while there was no significant difference between OVA@MSNs-M and OVA@MSNs-S (Fig. 5I and fig. S6).
Ability of MSNs to inhibit tumor growth
Our finding that OVA@MSNs induced strong IFN-γ–secreting CD8+ T cells (Fig. 5, F and H), which is critical for tumor inhibition, inspired us to test the potential of OVA@MSNs to function as an antitumor vaccine. C57BL/6 mice were immunized on days 0, 14, and 28 with 10 μg of free OVA or 40 μg of MSNs containing 10 μg of OVA. On day 35, animals were inoculated subcutaneously in the right posterior side with 5 × 105 B16F10-OVA cells (Fig. 6A). All three OVA@MSNs formulations showed significantly stronger tumor growth inhibition potency than free OVA (Fig. 6B). The mice of OVA@MSNs-L and OVA@MSNs-M groups also had significantly longer survival time than that of free OVA (Fig. 6C). The mice of OVA@MSNs-L group also showed significantly longer survival time than that of OVA@MSNs-M and OVA@MSNs-S (Fig. 6C). In summary, the encapsulation of OVA in MSNs significantly enhanced the antitumor immunity of antigen, and mice vaccinated with OVA@MSNs-L were most resistant to tumor challenge.
Fig. 6. Tumor growth inhibition potency of MSN loading antigen and survival curve of mice.
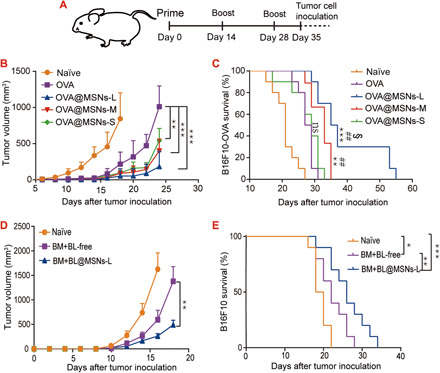
(A) Schematic of the experiment design. (B) Tumor growth curves and (C) Kaplan-Meier survival curves of mice immunized with free OVA and OVA@MSNs. (D) Tumor growth curves and (E) Kaplan-Meier survival curves of mice immunized with free BM+BL and BM+BL@MSNs. Results in (B) and (D) were analyzed using one-way ANOVA. Survival results in (C) and (E) were analyzed using a log-rank test. *P < 0.05, **P < 0.01, and ***P < 0.001 versus free OVA; #P < 0.05, ##P < 0.01, and ###P < 0.001 versus OVA@MSNs-S; §P < 0.05, §§P < 0.01, and §§§P < 0.001 versus OVA@MSNs-M. Data are shown as means ± SD (n = 9 to 10).
We then assessed whether MSNs-L was still effective to deliver B16F10 cell antigen. We extracted cell lysis and cell membrane from in vitro incubated B16F10 cells, which were referred to as BL and BM, respectively. The BL and BM were simultaneously loaded into MSNs-L (BM+BL@MSNs) and then subcutaneously injected on days 0, 14, and 28. On day 35, 1 × 105 B16F10 cells were inoculated subcutaneously. BM+BL@MSNs-L-immunized mice showed significantly slower tumor growth and longer survival time compared with naïve mice or free BM+BL-immunized mice (Fig. 6, D and E). Those results indicated that MSNs-L could be used as a tumor antigen vector, such as tumor cell lysis and tumor cell membrane.
In vivo safety of MSNs
To observe the degradation ability of MSNs in vivo, we isolated the popliteal lymph nodes of mice injected with MSNs. The ultrathin sections of the lymph nodes were made and observed by transmission electron microscopy on days 1, 4, and 7 after injection. Lots of MSNs were trapped in popliteal lymph nodes and showed intact morphology on day 1 after injection (fig. S7A). The particles started to degrade and showed blurred structure on day 4 (fig. S7B). The degradation of all three MSNs was further aggravated on day 7 (fig. S7C). MSNs with larger pore size showed faster degradation rate. The nanoparticle structure of MSNs-L was almost completely lost on day 7, while MSNs-M and MSNs-S still showed a relatively clear nanoparticle structure.
We then assessed the systemic and local toxicity of MSNs. Seven days after the last immunization, we separated the foot, lung, liver, spleen, kidney, and heart of immunized mice for hematoxylin and eosin staining (fig. S8). Compared with the naïve group, the skin and subcutaneous tissues of the OVA-, OVA@MSNs-L–, OVA@MSNs-M–, and OVA@MSNs-S–immunized mice were intact, while small amounts of inflammatory cells infiltrated occasionally, most of which are lymphocytes. The inflammatory cell infiltration may be related to the injection operations, as the same phenomenon occurs in mice injected with OVA. The lung and liver tissues of the immunized mice and the naïve mice were normal. The ratio of spleen red pulp and white pulp was normal, and no abnormal changes were observed in all groups. The number and volume of the kidney glomeruli and renal tubular epithelium were normal in all groups. Myocardium and epicardial abnormalities were not seen in the heart tissue for all groups. These results together indicate that our mode of administration and amount administered will not lead to local or systemic toxicity.
DISCUSSION
Subunit vaccines generally only induce a humoral immune response, but proper delivery vesicles can promote the cellular immune response by successfully navigating the DUMP cascade. Maximizing the efficiency of each step is important for achieving high overall immune responses.
MSNs have long been used as delivery vehicles for small-molecule nucleic acid and protein drugs. In recent years, MSNs also gained attention as vaccine delivery systems. However, MSNs that are currently being investigated have weak lymph node–targeting ability because of their relatively large particle size. In this study, we constructed three types of MSNs, all with sizes of around 80 nm in diameter but with three different pore sizes, and evaluated the ability of each MSN type to traffic model antigen OVA to draining lymph nodes. We found that the synthesized MSNs significantly enhanced each step of the DUMP cascade, resulting in enhanced both antibody and T cellular responses. Furthermore, OVA@MSNs with larger pores induced stronger immune responses and tumor suppression effects than OVA@MSNs with smaller pores. The superior performance of MSNs-L was found to be correlated with their better antigen cross-presentation ability, the last step in the DUMP cascade.
In our study, the internalization of OVA@MSNs was found to be mediated primarily via scavenger receptor (Fig. 3C), which is highly expressed on DCs. Most of the ligands for scavenger receptor are polyanions, and this endocytic pathway was reported to transfer internalized particles to lysosomes for MHC II–restricted presentation (33, 34). We found that our synthesized MSNs helped OVA escape from lysosomal degradation for MHC I–restricted presentation by causing rupture of the endosomal/lysosomal membrane by generating ROS (fig. S2A) (19, 32, 35, 36). The results also showed that the ROS production increased when mesoporous pore size decreased. This is consistent with the results of cell toxicity (Fig. 3A), as ROS is often regarded as a key factor in toxicity (37, 38).
Our synthesized OVA@MSNs elicited both potent humoral and cellular immune responses, in line with their superior ability to target lymph nodes, uptake by lymph node–resident DCs, and maturation of these DCs. DCs are the most powerful and are the only type of APCs that activate naïve T cells and initiate the primary immune responses. CD8a+CD11c+ and CD11b+CD11c+ DCs are the two most important types of DCs in lymph nodes (4, 5, 7, 39). CD8a+CD11c+ DCs were much more efficient in presenting extracellular antigens with MHC I molecules to CD8+ T cells via cross-presentation, while CD11b+CD11c+ DCs mainly present exogenous antigens with MHC II molecules to prime CD4+ T cell (5, 6, 39). In our study, both CD8a+CD11c+ and CD11b+CD11c+ DCs in the lymph nodes could efficiently internalize OVA@MSNs (Fig. 4F). This superior lymph node targeting and internalization by DCs ultimately led to balanced humoral and cellular immune responses and resisted the B16F10-OVA tumor challenge without the need for additional adjuvants. These findings are different from previous reports that MSNs could only induce antigen-specific antibody response. We assume that the underlying reason is that the relatively large particle size of previously reported MSNs were less efficient to migrate to lymph nodes, the first step in the DUMP cascade, resulting in limited levels of the following three steps, including uptake by DCs, maturating DCs, and presenting peptide–MHC I complexes to CD8+ T cells (40, 41).
MSNs with larger pore size showed stronger immune activation and antitumor efficacy than MSNs with smaller pore size (Figs. 5 and 6), although our results showed that pore size did not affect the first three steps in the DUMP cascade, giving the fact that all three kinds of OVA@MSNs showed no difference in lymph node targeting and internalization efficiency of the antigen by lymph node–resident DCs, activation, and proliferation of lymph node–resident DCs (Fig. 4). The stronger antitumor efficacy of MSNs with larger pore size could be caused for two reasons. First, the larger pore size induced stronger cross-presentation of the antigen, the last step in the DUMP cascade, as shown studies of DC2.4 cells in vitro (Fig. 3D). Second, our results showed that MSNs with larger pores degraded faster in the lymph nodes, which may result in faster release of OVA inside the lymph nodes, providing stronger exposure of antigen to DCs (fig. S7).
Our finding that MSNs with larger pore sizes degraded faster is consistent with previous findings from literature. It has been reported that the greater the pore size, the faster the degradation of MSNs and, therefore, the faster the release of the encapsulated drug (42, 43). Rapid degradation of MSNs-L can also help improve their safety in vivo. It has been reported that MSNs administered by intravenous injection could be excreted in their intact form from the liver and kidney, which led to bile secretion disorder of the liver and affected glomerular filtration function of the kidney, respectively (44, 45). However, silicic acid, which is the degradation product of MSNs, is a safe and harmless product that could be absorbed and used by the body. Therefore, the fast degradation rate of MSNs-L could help improve biosafety and biocompatibility.
We improved the OVA-specific immune response by enhancing the DUMP cascade efficiency by controlling the properties of MSNs, including humoral and cellular immune response. There is no doubt that the cellular immune response is the main weapon against tumors; but antibodies, the most important effector molecules for humoral immunity, can medicate tumor lysis by antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity. Consistent with this fact, increasing B cell–derived IgG in serum is associated with a significant tumor regression in murine xenograft models of human lung squamous cell carcinoma (46). In our study, OVA@MSNs-L–vaccinated mice showed significant tumor growth delay compared with OVA@MSNs-M and OVA@MSNs-S, which may be, in part, associated with the increase of OVA-specific antibody. MSNs-L can be used not only as a protein vaccine delivery vehicle but also as a delivery complex antigen such as tumor cell lysate and tumor cell membrane (Fig. 6), indicating the potential clinical value of MSNs-L for cancer vaccination.
In conclusion, we improved the immune response intensity by means of enhancing the DUMP cascade efficiency by controlling the properties of MSNs, with the result that MSNs-L–loaded antigens induce significant antitumor effect without combination with other adjuvants. To our knowledge, no previous reports have shown that mesoporous silica could induce an antitumor effect without additional adjuvants. In future studies, MSNs-L could be tested for use with the many existing tumor-associated antigens and a growing number of tumor neoantigens.
MATERIALS AND METHODS
Materials
TEOS was purchased from Aladdin (Shanghai, China). OVA (albumin from chicken egg white) was purchased from Sigma-Aldrich (MO, USA), and endotoxin in OVA was removed with Pierce High Capacity Endotoxin Removal Spin Columns (Thermo Fisher Scientific, IL, USA). Recombinant mouse granulocyte-macrophage colony-stimulating factor (GM-CSF) was purchased from R&D Systems (Minneapolis, MN, USA). All the other chemicals used are of analytical grade, and Milli-Q water (18 megohm/cm; Millipore Co.) was used for the preparation of all solutions.
Cell
DC2.4 cells (obtained from the Third Military Medical University) and B16F10-OVA cells (provided by the researcher of the Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences) were maintained in RPMI 1640 medium (HyClone; Life Technologies, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA) and 1% penicillin-streptomycin liquid (Solarbio, Beijing, China) at 37°C with 5% CO2 incubator.
Animals
Healthy female C57BL/6 mice (6 to 8 weeks) were purchased from the Experimental Animal Center of Sichuan Provincial People’s Hospital (Chengdu, China) and housed in a specific pathogen–free, light-cycled, and temperature-controlled facility. All animal experiments were performed in accordance to the guidelines approved by the Institutional Animal Care and Use Committees of Sichuan University.
Synthesis and characterization of MSNs with different pore sizes
The MSNs-L, MSNs-M, and MSNs-S were synthesized by a previously reported procedure with modification (43). To prepare MSNs-L, briefly, 6 g of cetyltrimethylammonium chloride (CTAC) and 0.18 g of triethanolamine (TEA) were added to 60 ml of water and stirred gently at 60°C for 1 hour in a 100-ml round-bottom flask. Then, 20 ml of TEOS in cyclohexane (5 v/v %) was carefully added to the water-CTAC-TEA solution and kept at 60°C in an oil magnetics stirrer for 12 hours. The products were collected by centrifugation and washed with ethanol three times. Then, the collected products were extracted with 80 ml of 0.6 weight % ammonium nitrate (NH4NO3) ethanol solution at 60°C for 6 hours thrice to remove the template. The final products were washed with ethanol and water three times and lyophilized for the subsequent experiments. MSNs-M and MSNs-S were synthesized in the same manner as MSNs-L except that the concentration of TEOS in cyclohexane used were 20 and 40%, respectively.
Size and zeta potential were measured on a Malvern ZEN3690 Zetasizer apparatus. Nitrogen adsorption-desorption measurements were conducted at 77 K with a Kubo-X1000 (China). Scanning electron microscope images were taken with an S-4800 (Japan) microscope operating at 15 kV with no metal coating. Transmission electron microscopy images were obtained on a Tecnai G2 F20 S-TWIN (The Netherlands) microscope operating at 200 kV. Small-angle x-ray scattering were conducted on Empyrean (The Netherlands).
Protein loading and release
The MSN aqueous solution (0.8 ml) was dispersed by probe sonication for 5 min (180 W, 5 s on, 5 s off) in an ice bath. Then, 200 μl of OVA aqueous solution was added into the dispersed MSN solution, and the OVA was loaded into the mesoporous MSNs with probe sonication for 5 min (150 W, 5 s on, 5 s off) in an ice bath. The OVA-loaded MSNs were referred to as OVA@MSNs. The excess OVA was removed by ultrafiltration. The filtrate in the outer tube was used to determine encapsulation efficiency of OVA with Micro BCA Protein Assay Kit (Thermo Scientific, USA). The OVA@MSNs in the inner tube were diluted 10 times with PBS (pH 7.4) followed by incubation in a 37°C shaker at 150 rpm to detect the release ability of OVA@MSNs. At specific time points, 0.5 ml of samples of the solution were obtained and centrifuged to collect the supernatant, and the pellet was resuspended in 0.5 ml of PBS and added back into the release system. The protein concentration in the centrifuge supernatant was measured as mentioned above, and the cumulative release percentage of OVA was calculated.
Toxicity of MSNs to DC2.4 cells
DC2.4 cells were seeded at 1 × 104 cells per well in 96-well plates. The followed operation was performed until the cells reached 80% confluence. After washing with PBS three times, the cells were incubated with 100 μl of cell culture medium containing different concentrations of MSNs-L, MSNs-M, or MSNs-S. All MSNs stock solution were prepared in Milli-Q water by sonication and diluted with RPMI 1640 medium. After incubation for 24 hours, 10 μl of Cell Counting Kit-8 (Dojindo, China) were added in each well, followed by additional incubation for 2 hours. The viability of cells was measured by detecting absorbance at 450 nm using a Tecan Spark 10M multipurpose microplate reader (Switzerland).
In vitro cellular uptake and mechanisms studies
To obtain Cy5-labeled OVA, 20 mg of OVA was dissolved in PBS containing 25 mM dithiothreitol (DTT), stirred under nitrogen for 4 hours to break up disulfide bonds. Excess DTT was removed using G50 dextran column. Next, 3.5 mg of sulfo-Cy5 maleimide was added into the purified OVA. The mixture was stirred at 4°C overnight. Last, free sulfo-Cy5 maleimide was removed by dialysis, and the purified Cy5-OVA was stored at 4°C after lyophilization.
Cy5-OVA@MSNs were prepared by sonication. DC2.4 cells were seeded at 1 × 105 cells per well in 12-well plates, and uptake experiments were performed until cells were 80% confluent. Cells were incubated for 1 hour in 1 ml of serum-free RPMI 1640 medium containing 5 μg of Cy5-OVA or 20 μg of MSNs loaded with 5 μg of Cy5-OVA. Then, the cells were collected, centrifuged, and washed three times with PBS, and the uptake of antigen was detected using an FC 500 flow cytometer.
To explore the uptake mechanisms of OVA@MSNs in DC2.4 cells, cultured DC2.4 cells were pretreated with selected inhibitors of different internalization pathway for 1 hour. Then, Cy5–OVA@MSNs-L containing 5 μg of Cy5-OVA was added and incubated for another 1 hour. The following uptake inhibitors were used: methyl-β-cyclodextrin (65 μg ml−1), nystatin (25 μg ml−1), amiloride (26 μg ml−1), dynasore (16 μg ml−1), chlorpromazine (5 μg ml−1), dextran sulfate (100 μg ml−1), and sucrose (154 mg ml−1). After incubation, the cells were collected by centrifugation and washed three times with PBS, and the uptake of OVA was detected using an FC 500 flow cytometer.
Intracellular process of OVA@MSNs
DC2.4 cells were seeded at 1 × 105 cells per well in 12-well plates. When cells reached 60% confluence, they were incubated in 1 ml of complete RPMI 1640 medium containing 5 μg of OVA or 20 μg of MSNs loaded with 5 μg of OVA for 2 hours. The cells were washed, and OVA was removed; the cells were incubated for another 34 hours. Afterward, the cells were collected, washed twice with PBS, stained with phycoerythrin (PE)–conjugated anti-mouse SIINFEKL/H-2Kb antibody (eBioscience, CA, USA), and the cross-presentation was analyzed by flow cytometry.
DC2.4 cells were incubated in 1 ml of serum-free RPMI 1640 medium containing 20 μg of MSNs. After 1 hour of incubation, the MSNs were removed. Cells were washed with PBS and incubated with 1 ml of fresh RPMI 1640 medium at 37°C with 5% CO2 incubator for 6 hours, and cells were incubated with 10 μM 2′,7′-dichlorodihydrofluorescein diacetate. After washing with PBS, cells were detected by flow cytometry.
BMDCs were obtained from mouse bone marrow, as described previously (47). Briefly, bone marrow cells obtained from the femur and tibia of 6-week-old male C57BL/6 mice were resuspended in RPMI 1640 medium (Gibco, USA) supplemented with 10% heat-inactivated FBS (Gibco, USA), 1% penicillin-streptomycin liquid (Solarbio, Beijing, China), and 50 μM 2-mercaptoethanol. Then, bone marrow cells were seeded at 5 × 106 per 100-mm bacteriological petri dishes in 10 ml of R10 containing GM-CSF (20 ng/ml; R&D Systems, USA). At day 3, another 10 ml of R10 containing GM-CSF (20 ng/ml) was added to the plate. At day 6, half of the culture supernatant was replaced with fresh R10 containing GM-CSF (20 ng/ml). At day 8, the cells were collected and incubated with PBS containing 50% FBS at 37°C for 30 min to remove residual granulocytes. The purity of BMDCs was assessed by measuring the percentage of CD11c-positive cells using an FC 500 flow cytometer (Beckman Coulter, USA). The percentage of CD11c-positive cells in BMDC used in all experiments was higher than 85%.
BMDCs were seeded at 2 × 106 cells per well in 12-well plates and incubated for 18 hours with 20 μg of MSNs-L, MSNs-M, or MSNs-S. Cells incubated with PBS and lipopolysaccharide were used as negative and positive control, respectively. Then, cells were collected, washed twice with PBS, and stained using FITC-conjugated anti-mouse CD80 antibody and APC-conjugated anti-mouse CD86 antibody (eBioscience, CA, USA). The expression of costimulatory molecules CD80 and CD86 on the surface of BMDCs was detected using flow cytometry.
DC2.4 cells were seeded at 1 × 105 cells per well in 12-well plates. Then, cells were incubated in 1 ml of serum-free RPMI 1640 medium containing 5 μg of Cy5-OVA or 20 μg of MSNs loaded with 5 μg of Cy5-OVA. After 1 hour of incubation, the Cy5-OVA@MSNs were removed. Cells were washed three times with PBS and incubated in 1 ml of fresh RPMI 1640 medium at 37°C with 5% CO2 incubator. Then, the cells were taken out at specific time points (1, 3, and 6 hours) and incubated with 1 ml of RPMI 1640 medium containing 75 nM LysoTracker Red for 30 min at 37°C, washed with PBS, and then incubated with Bisbenzimide H 33342 (10 μg/ml) (Sigma-Aldrich) in PBS for 10 min. After washing by centrifugation, cells were placed in a confocal dish and observed using confocal laser scanning microscope (ZEISS LSM 800; Carl Zeiss, Germany).
Lymph node distribution of OVA@MSNs and activation of lymph node–resident DCs in vivo
To investigate the targeting ability of OVA@MSNs, 10 μg of Cy5-OVA or 40 μg of MSNs loaded with 10 μg of Cy5-OVA were subcutaneously injected into the hind footpad of mice (n = 3 animals per group). The popliteal lymph nodes were visualized by using an IVIS Spectrum system (Caliper, Hopkinton, MA, USA) at designed time points. After 36 hours, the popliteal and inguinal lymph nodes were isolated for ex vivo imaging (n = 3 animals per group). The fluorescence intensity at the popliteal lymph nodes was semiquantified by using Living Image 4.0 software.
To observe the distribution of OVA@MSNs in lymph nodes, Cy5-OVA@MSNs were subcutaneously injected into the hind footpad of mice (n = 3 animals per group). Mice were sacrificed at 10 hours, and popliteal lymph nodes were collected for frozen section study. The lymph nodes were washed with PBS, embedded with tissue freezing medium (Sakura Tissue-Tek, USA) and sectioned using freezing microtome (Leica CM1950; Germany). They were fixed with 4% paraformaldehyde for 10 min, washed with PBS, and stained with Bisbenzimide H 33342 (5 μg/ml) for 15 min. After mounting with antifluorescence quencher, they were observed using a confocal laser scanning microscope.
To verify that OVA@MSNs were internalized by DC cells resident in lymph nodes, Cy5-OVA@MSNs were subcutaneously injected into the hind footpad of mice (n = 4 animals per group). Mice were sacrificed at 10 hours, and the popliteal lymph nodes were isolated. Lymph node single-cell suspension was obtained by grinding the lymph nodes through a 70-μm cell sieve. Cells were washed with PBS; stained with PE-conjugated anti-mouse CD11c antibody (eBioscience, CA, USA), FITC-conjugated anti-mouse CD8a antibody, or FITC-conjugated anti-mouse CD11b antibody; and analyzed using flow cytometer.
Colocation of OVA and MSNs in popliteal lymph node was as follows: FITC (10 mg) in 5 ml of absolute ethanol was added into 5 ml of absolute ethanol containing 10 μl of 3-aminopropyltriethoxysilane and stirred at 60°C for 2 hours; then, 40 mg of MSNs dispersed with 10 ml of absolute ethanol was added and stirred for another 12 hours. After centrifugation, precipitation was washed two times with ethanol, and MSNs-FITC was freeze-dried. Cy5-OVA@MSNs-FITC containing 10 μg of Cy5-OVA and 40 μg of MSNs were subcutaneously injected into the hind footpad of mouse (n = 3 animals per group). Mice were sacrificed at 10 hours, and popliteal lymph nodes were collected for frozen section study. The lymph nodes were washed with PBS, embedded with tissue freezing medium (Sakura Tissue-Tek, USA), and sectioned afterward using freezing microtome (Leica CM1950, Germany). They were fixed with 4% paraformaldehyde for 10 min, washed with PBS, and stained with Bisbenzimide H 33342 (5 μg/ml) for 15 min. Once mounted with antifluorescence quencher, they were observed using a confocal laser scanning microscope (ZEISS LSM 800; Carl Zeiss, Germany).
To investigate the ability of OVA@MSNs to activate DC cells in vivo, OVA@MSNs were subcutaneously injected into the hind footpad of mouse (n = 4 animals per group). Three days after administration, popliteal lymph nodes were isolated and homogenized into a single-cell suspension. Then, the obtained cells were stained with PE-conjugated anti-mouse CD11c antibody and APC-conjugated anti-mouse CD86 antibody and analyzed using a flow cytometer.
Animal immunization and detection
All immunizations were administered by hind footpad subcutaneous injection using a BD 29-gauge insulin syringe needle. Each formulation contained 10 μg of OVA or 40 μg of MSNs loaded with 10 μg of OVA. After primary immunization on day 0, booster immunizations were given on days 14 and 28. Blood samples (n = 4 to 5 animals per group) were obtained from the angular vein on days 21, 28, and 35, and serum was obtained by centrifugation for detection of antibody. When mice were sacrificed on day 35, spleens were collected for analysis of cellular immune responses.
Transplant plates (96-well) were coated with 100 μl of OVA (10 μg ml−1) in carbonate-bicarbonate buffer (0.1 M, pH 9.6) per well overnight at 4°C. Afterward, the plates were washed three times with phosphate buffer solution containing 0.05% Tween 20 (PBST) and incubated with PBS containing 1% bovine serum albumin at 37°C for 2 hours. The plates were washed with PBST and then incubated with a 10-fold serially diluted serum sample starting from 1:100 dilution in PBST at 37°C for 2 hours. Next, the plates were washed five times with PBST and incubated at 37°C for 1 hour with 100 μl of horseradish peroxidase–conjugated anti-mouse IgG, IgG1, or IgG2a (diluted 1:10,000). After washing, the plates were incubated at room temperature for 30 min with 100 μl of trimethylboron (InnoReagents, China). Last, the reaction was stopped by 50 μl of 1 M H2SO4, and the absorbance at 450 nm was measured using a Tecan Spark 10M multimode microplate reader (Switzerland).
Animals were sacrificed on day 7 after the last immunization (n = 5 animals per group), and spleens were collected and homogenized to obtain a single-cell suspension. Splenocytes were incubated with ACK lysis buffer to remove the red cells and used for the following experiments. For intracellular cytokine staining of CD4+ T cells, splenocytes were incubated at 37°C for 6 hours with OVA (100 μg ml−1) and brefeldin A (5 μg ml−1). Then, cells were stained with FITC-conjugated anti-mouse CD4, APC-conjugated anti-mouse IFN-γ, and PE-conjugated anti-mouse IL-4 and analyzed using a flow cytometer. For intracellular cytokine staining of CD8+ T cells, splenocytes were incubated at 37°C for 6 hours with SIINFEKL (2 μg ml−1) and brefeldin A (5 μg ml−1). Then, cells were stained with FITC-conjugated anti-mouse CD8a, APC-conjugated anti-mouse IFN-γ, and PC7-conjugated anti-mouse TNF-α and analyzed using a flow cytometer.
The spleen lymphocytes of immunized mice (n = 5 animals per group) were isolated on day 7 after the last immunization using a mouse spleen lymphocyte separation solution (Dakewe Biotech Co. Ltd.) according to the manufacturer’s instruction. The obtained lymphocytes were counted using a cell counter (Countstar, ALIT Life Science, China), and the cell concentration was adjusted to 4 × 107 cells/ml. The cells were seeded at a density of 4 × 106 cells per well into mouse IFN-γ precoated 96-well plates (Dakewe, Biotech Co. Ltd.) with SIINFEKL (10 μg ml−1), and the following operations were completed according to the manufacturer’s procedure.
On day 7 after the last immunization, mice (n = 4 to 5 animals per group) were injected with 1 × 107 carboxyfluorescein diacetate succinimidyl ester (CFSE)–labeled splenocytes through the tail vein. Half of the splenocytes were incubated with SIINFEKL (2 μg ml−1) and stained with 4 μM CFSE (denoted as CFSEhigh). The other half cells were incubated without SIINFEKL and stained with 0.4 μM CFSE (denoted as CFSElow). Mice were sacrificed at 20 hours after injection, and splenocytes were collected and analyzed using a flow cytometer. The percentage of specific sacrificing was calculated using the following formula
Tumor challenge
On day 7 after the last immunization, 5 × 105 B16F10-OVA were inoculated subcutaneously in the right posterior side of mice (n = 9 to 10 animals per group). The length and width of the tumor were measured using a vernier caliper every 2 days. Tumor volume was calculated by following equation: V = length × width2/2. Mice were sacrificed when the tumor length exceeded 2 cm or tumor volume exceeded 2000 mm3.
B16F10 cell lysis (BL) and B16F10 cell membrane (BM) were extracted from in vitro incubated B16F10 cells and then loaded into MSNs-L by sonication, which was referred as to BM+BL@MSNs-L. Mice were immunized on days 0, 14, and 28 by hind footpad subcutaneous injection. Each formulation contained 10 μg of BL, 10 μg of BM, and 40 μg of MSNs-L. On day 35, 1 × 105 B16F10 were inoculated subcutaneously in the right posterior side of mice (n = 9 to 10 animals per group). The flowing tumor detection operation refers to B16F10-OVA.
In vivo safety of MSNs
MSNs (40 μg) were subcutaneously injected into the hind footpad of mouse. The popliteal lymph nodes were isolated on days 1, 4, and 7 after MSNs injection and immediately fixed at 4°C in 2.5% glutaraldehyde. Further preparation and slicing of the samples were performed by Servicebio Technology Company (Wuhan, China). After fixing in OsO4, the samples were dehydrated in alcohol and then embedded in resin, and the lymph nodes with thickness of 50 to 80 nm were placed on the copper grids and viewed on a Tecnai G2 F20 S-TWIN microscope.
On day 7 after the last immunization, the mice were sacrificed, and the heart, liver, spleen, lungs, kidneys, and foot (injection side) were separated and immediately placed in 4% paraformaldehyde. The fixed tissues were embedded in paraffin for section and then stained with hematoxylin and eosin. The inflammation and pathological changes were observed by microscopy.
Statistical analysis
All results were expressed as means ± SD. Experiments were assessed for significance using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test, unless noted otherwise. All statistical analyses were performed with GraphPad Prism 6.0 (GraphPad Software). Levels of significant differences were expressed as follows: *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Acknowledgments
We acknowledge the use of the Instrumentation Facility at Sichuan University. Funding: This work was supported by the National Natural Science Foundation of China (no. 81925036 and 81872814), the Science and Technology Major Project of Sichuan Province (2018SZDZX0018), and the Fundamental Research Funds for the Central Universities. Author contributions: X.H., X.S., Z.Z., T.G., and L.Z. designed the project. X.H. drafted the manuscript and performed/analyzed all experiments. Y.H. and Y.Z. performed animal experiments. X.H., X.S., X.Z., L.Z., and G.D. revised the manuscript. Competing interests: X.S., Z.Z., X.H., and T.G. are inventors on a patent application related to this work filed by Sichuan University (filed under number 201910508439.8). The authors declare no other competing interests. Data availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/25/eaaz4462/DC1
REFERENCE AND NOTES
- 1.Clemens J., Holmgren J., Kaufmann S. H. E., Mantovani A., Ten years of the global alliance for vaccines and immunization: Challenges and progress. Nat. Immunol. 11, 1069–1072 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Pulendran B., Ahmed R., Immunological mechanisms of vaccination. Nat. Immunol. 12, 509–517 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bachmann M. F., Jennings G. T., Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 10, 787–796 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Joffre O. P., Segura E., Savina A., Amigorena S., Cross-presentation by dendritic cells. Nat. Rev. Immunol. 12, 557–569 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Shortman K., Heath W. R., The CD8+ dendritic cell subset. Immunol. Rev. 234, 18–31 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Eisenbarth S. C., Dendritic cell subsets in T cell programming: Location dictates function. Nat. Rev. Immunol. 19, 89–103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Segura E., Amigorena S., Cross-presentation in mouse and human dendritic cells. Adv. Immunol. 127, 1–31 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Thomas S. N., Vokali E., Lund A. W., Hubbell J. A., Swartz M. A., Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials 35, 814–824 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Qian Y., Jin H., Qiao S., Dai Y., Huang C., Lu L., Luo Q., Zhang Z., Targeting dendritic cells in lymph node with an antigen peptide-based nanovaccine for cancer immunotherapy. Biomaterials 98, 171–183 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Jiang H., Wang Q., Sun X., Lymph node targeting strategies to improve vaccination efficacy. J. Control. Release 267, 47–56 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Kim S.-Y., Noh Y.-W., Kang T. H., Kim J.-E., Kim S., Um S. H., Oh D.-B., Park Y.-M., Lim Y. T., Synthetic vaccine nanoparticles target to lymph node triggering enhanced innate and adaptive antitumor immunity. Biomaterials 130, 56–66 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Swartz M. A., The physiology of the lymphatic system. Adv. Drug Deliv. Rev. 50, 3–20 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Kang S., Ahn S., Lee J., Kim J. Y., Choi M., Gujrati V., Kim H., Kim J., Shin E.-C., Jon S., Effects of gold nanoparticle-based vaccine size on lymph node delivery and cytotoxic T-lymphocyte responses. J. Control. Release 256, 56–67 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Quach Q. H., Ang S. K., Chu J.-H. J., Kah J. C. Y., Size-dependent neutralizing activity of gold nanoparticle-based subunit vaccine against dengue virus. Acta Biomater. 78, 224–235 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Rao D. A., Forrest M. L., Alani A. W. G., Kwon G. S., Robinson J. R., Biodegradable PLGA based nanoparticles for sustained regional lymphatic drug delivery. J. Pharm. Sci. 99, 2018–2031 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stylianopoulos T., Poh M.-Z., Insin N., Bawendi M. G., Fukumura D., Munn L. L., Jain R. K., Diffusion of particles in the extracellular matrix: The effect of repulsive electrostatic interactions. Biophys. J. 99, 1342–1349 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tasciotti E., Liu X., Bhavane R., Plant K., Leonard A. D., Price B. K., Cheng M. M.-C., Decuzzi P., Tour J. M., Robertson F., Ferrari M., Mesoporous silicon particles as a multistage delivery system for imaging and therapeutic applications. Nat. Nanotechnol. 3, 151–157 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Xuan M., Shao J., Dai L., He Q., Li J., Macrophage cell membrane camouflaged mesoporous silica nanocapsules for in vivo cancer therapy. Adv. Healthc. Mater. 4, 1645–1652 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Durfee P. N., Lin Y.-S., Dunphy D. R., Muñiz A. J., Butler K. S., Humphrey K. R., Lokke A. J., Agola J. O., Chou S. S., Chen I.-M., Wharton W., Townson J. L., Willman C. L., Brinker C. J., Mesoporous silica nanoparticle-supported lipid bilayers (protocells) for active targeting and delivery to individual leukemia cells. ACS Nano 10, 8325–8345 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Meka A. K., Abbaraju P. L., Song H., Xu C., Zhang J., Zhang H., Yu M., Yu C., A vesicle supra-assembly approach to synthesize amine-functionalized hollow dendritic mesoporous silica nanospheres for protein delivery. Small 12, 5169–5177 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Meng H., Wang M., Liu H., Liu X., Situ A., Wu B., Ji Z., Chang C. H., Nel A. E., Correction to use of a lipid-coated mesoporous silica nanoparticle platform for synergistic gemcitabine and paclitaxel delivery to human pancreatic cancer in mice. ACS Nano 10, 6416 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xuan M., Shao J., Zhao J., Li Q., Dai L., Li J., Magnetic mesoporous silica nanoparticles cloaked by red blood cell membranes: Applications in cancer therapy. Angew. Chem. Int. Ed. Engl. 57, 6049–6053 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Cha B. G., Jeong J. H., Kim J., Extra-large pore mesoporous silica nanoparticles enabling co-delivery of high amounts of protein antigen and toll-like receptor 9 agonist for enhanced cancer vaccine efficacy. ACS Cent. Sci. 4, 484–492 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu Y., Yang Y., Gu Z., Zhang J., Song H., Xiang G., Yu C., Glutathione-depletion mesoporous organosilica nanoparticles as a self-adjuvant and co-delivery platform for enhanced cancer immunotherapy. Biomaterials 175, 82–92 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Demento S. L., Cui W., Criscione J. M., Stern E., Tulipan J., Kaech S. M., Fahmy T. M., Role of sustained antigen release from nanoparticle vaccines in shaping the T cell memory phenotype. Biomaterials 33, 4957–4964 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W., Wang L., Liu Y., Chen X., Liu Q., Jia J., Yang T., Qiu S., Ma G., Immune responses to vaccines involving a combined antigen–nanoparticle mixture and nanoparticle-encapsulated antigen formulation. Biomaterials 35, 6086–6097 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Croissant J. G., Fatieiev Y., Khashab N. M., Degradability and clearance of silicon, organosilica, silsesquioxane, silica mixed oxide, and mesoporous silica nanoparticles. Adv. Mater. 29, 1604634 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Yang Y., Jambhrunkar M., Abbaraju P. L., Yu M., Zhang M., Yu C., Understanding the effect of surface chemistry of mesoporous silica nanorods on their vaccine adjuvant potency. Adv. Healthc. Mater. 6, 1700466 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Guo S., Zhang X., Zheng M., Zhang X., Min C., Wang Z., Cheon S. H., Oak M.-H., Nah S.-Y., Kim K.-M., Selectivity of commonly used inhibitors of clathrin-mediated and caveolae-dependent endocytosis of G protein–coupled receptors. Biochim. Biophys. Acta 1848, 2101–2110 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Nabi I. R., Le P. U., Caveolae/raft-dependent endocytosis. J. Cell Biol. 161, 673–677 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pelkmans L., Secrets of caveolae- and lipid raft-mediated endocytosis revealed by mammalian viruses. Biochim. Biophys. Acta 1746, 295–304 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Morishige T., Yoshioka Y., Inakura H., Tanabe A., Yao X., Narimatsu S., Monobe Y., Imazawa T., Tsunoda S., Tsutsumi Y., Mukai Y., Okada N., Nakagawa S., The effect of surface modification of amorphous silica particles on NLRP3 inflammasome mediated IL-1β production, ROS production and endosomal rupture. Biomaterials 31, 6833–6842 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Harshyne L. A., Zimmer M. I., Watkins S. C., Barratt-Boyes S. M., A role for class A scavenger receptor in dendritic cell nibbling from live cells. J. Immunol. 170, 2302–2309 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Canton J., Neculai D., Grinstein S., Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 13, 621–634 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Hirai T., Yoshioka Y., Takahashi H., Ichihashi K., Yoshida T., Tochigi S., Nagano K., Abe Y., Kamada H., Tsunoda S., Nabeshi H., Yoshikawa T., Tsutsumi Y., Amorphous silica nanoparticles enhance cross-presentation in murine dendritic cells. Biochem. Biophys. Res. Commun. 427, 553–556 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Wang X., Li X., Yoshiyuki K., Watanabe Y., Sogo Y., Ohno T., Tsuji N. M., Ito A., Comprehensive mechanism analysis of mesoporous-silica-nanoparticle-induced cancer immunotherapy. Adv. Healthc. Mater. 5, 1169–1176 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Huang Y.-W., Cambre M., Lee H.-J., The toxicity of nanoparticles depends on multiple molecular and physicochemical mechanisms. Int. J. Mol. Sci. 18, 2702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Refsnes M., Skuland T., Lilleaas E., Øvrevik J., Låg M., Concentration-dependent cytokine responses of silica nanoparticles and role of ROS in human lung epithelial cells. Basic Clin. Pharmacol. Toxicol. 125, 304–314 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Macri C., Pang E. S., Patton T., O’Keeffe M., Dendritic cell subsets. Semin. Cell Dev. Biol. 84, 11–21 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Mercuri L. P., Carvalho L. V., Lima F. A., Quayle C., Fantini M. C. A., Tanaka G. S., Cabrera W. H., Furtado M. F. D., Tambourgi D. V., Matos J. R., Jaroniec M., Sant’Anna O. A., Ordered mesoporous silica SBA-15: A new effective adjuvant to induce antibody response. Small 2, 254–256 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Carvalho L. V., Ruiz R. C., Scaramuzzi K., Marengo E. B., Matos J. R., Tambourgi D. V., Fantini M. C. A., Sant’Anna O. A., Immunological parameters related to the adjuvant effect of the ordered mesoporous silica SBA-15. Vaccine 28, 7829–7836 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Godin B., Gu J., Serda R. E., Bhavane R., Tasciotti E., Chiappini C., Liu X., Tanaka T., Decuzzi P., Ferrari M., Tailoring the degradation kinetics of mesoporous silicon structures through PEGylation. J. Biomed. Mater. Res. A 94, 1236–1243 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen D., Yang J., Li X., Zhou L., Zhang R., Li W., Chen L., Wang R., Zhang F., Zhao D., Biphase stratification approach to three-dimensional dendritic biodegradable mesoporous silica nanospheres. Nano Lett. 14, 923–932 (2014). [DOI] [PubMed] [Google Scholar]
- 44.He Q., Zhang Z., Gao F., Li Y., Shi J., In vivo biodistribution and urinary excretion of mesoporous silica nanoparticles: Effects of particle size and PEGylation. Small 7, 271–280 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Huang X., Li L., Liu T., Hao N., Liu H., Chen D., Tang F., The shape effect of mesoporous silica nanoparticles on biodistribution, clearance, and biocompatibility in vivo. ACS Nano 5, 5390–5399 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Mizukami M., Hanagiri T., Shigematsu Y., Baba T., Fukuyama T., Nagata Y., So T., Ichiki Y., Sugaya M., Yasuda M., So T., Takenoyama M., Sugio K., Yasumoto K., Effect of IgG produced by tumor-infiltrating B lymphocytes on lung tumor growth. Anticancer Res. 26, 1827–1831 (2006). [PubMed] [Google Scholar]
- 47.Lutz M. B., Kukutsch N., Ogilvie A. L. J., Rössner S., Koch F., Romani N., Schuler G., An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 223, 77–92 (1999). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/25/eaaz4462/DC1