

Abstract
Ovarian cancer (OvCa) remains the most common cause of death from gynecological malignancies. Genetically engineered mouse models have been used to study initiation, origin, progression, and/or mechanisms of OvCa. Based on the clinical features of OvCa, we examined a quadruple combination of pathway perturbations including PTEN, TRP53, RB1, and/or CDH1. To characterize the cancer-promoting events in the ovarian surface epithelium (OSE), Amhr2cre/+ mice were used to ablate floxed alleles of Pten, Trp53, and Cdh1, which were crossed with TgK19GT121 mice to inactivate RB1 in KRT19-expressing cells. Inactivation of PTEN, TRP53, and RB1 with or without CDH1 led to the development of type I low-grade OvCa with enlarged serous papillary carcinomas and some high-grade serous carcinomas (HGSCs) in older mice. Initiation of epithelial hyperplasia and micropapillary carcinoma started earlier at 1 month in the triple mutations of Trp53, Pten, and Rb1 mice as compared to 2 months in quadruple mutations of Trp53, Pten, Rb1, and Cdh1 mice, whereas both genotypes eventually developed enlarged proliferating tumors that invaded into the ovary at 3–4 months. Mice with triple and quadruple mutations developed HGSC and/or metastatic tumors, which disseminated into the peritoneal cavity at 4–6 months. In summary, inactivation of PTEN, TRP53, and RB1 initiates OvCa from the OSE. Additional ablation of CDH1 further increased persistence of tumor dissemination and ascites fluid accumulation enhancing peritoneal metastasis.
Keywords: PTEN, TRP53, CDH1, RB1 and epithelial ovarian cancer
Inactivation of TRP53, PTEN, and RB1 initiates ovarian cancer from the ovarian surface epithelium. Additional ablation of CDH1 further increased persistence of tumor dissemination.
Introduction
Ovarian cancer (OvCa) remains the most common cause of death from gynecological malignancies and is the fifth leading overall cause of death from cancer in women. In 2019, approximately 22,500 new OvCa cases and 14,000 deaths were estimated from OvCa in the United States [1]. The majority of OvCa (85–90%) is histologically classified as epithelial ovarian cancer (EOC) [2, 3]. They are further divided into type I, low-grade OvCa and type II, high-grade aggressive malignancies based on the clinical behavior and histopathological types of EOC [3, 4]. Most type II malignancies feature high-grade serous carcinomas (HGSCs), which have a high propensity for metastasis, resulting in low overall survival [3]. The cancer genome landscape of HGSC has unequivocally reported that 96% of HGSCs harbor mutations of TP53 [5]. These studies have also highlighted alterations in the RB1 pathway (67%), the phosphoinositide 3-kinase (PI3K) pathway (45%), and Notch signaling (22%) in HGSC [5]. Although OvCa is a complex heterogeneous disease, a clear understanding of molecular pathogenesis in OvCa is necessary for early stage diagnosis as well as rational design of therapies.
Genetically engineered mouse models of OvCa have been developed to understand the specific functions of oncogenes and/or tumor suppressors and the etiology of OvCa formation [3, 6–8]. Via deletion of regulatory floxed genes by injecting adenovirus-Cre to the ovarian bursa and/or crossing with Amhr2cre/+ mice, the ovarian surface epithelium (OSE) can be transformed into OvCa. Specifically, combination of Pten, KrasG12D, and/or Trp53R172H mutation by Amhr2cre/+ mice can induce EOC such as serous papillary adenocarcinomas, mucinous with serous features of carcinomas and/or HGSC [9–11]. On the other hand, recent evidence, from clinical and animal studies of OvCa, shows that HGSCs appear to arise from the fallopian tube epithelium (FTE) [3]. The use of Pax8 or Ovgp1 promoters to express Cre recombinase enables to target/disrupt genes in FTE [12–15] resulting in the induction of HGSC and serous tubal intraepithelial carcinomas mimicking the development of OvCa [12, 14]. While each model will provide some similarities and differences in the clinical features and molecular aspects of OvCa, genetically engineered mice with deletion or inactivation of tumor suppressor genes and/or exogenous expression of oncogenes in a tissue-specific manner provide a better understanding of the molecular mechanisms in the precise origin of cells in OvCa.
In the present study, we have generated a mouse model of OvCa in which Trp53, Pten, and/or RB1, well-known altered signaling pathways in OvCa, were conditionally ablated or inactivated in the OSE using Amhr2cre/+ mice. We have also induced disruption of epithelial cells by deleting Cdh1 with a combination of Trp53, Pten, and/or RB1 in OSE to examine the importance of epithelial structure of OSE. While CDH1 mutation and/or deletion is not common (~5%) in OvCa based on datasets from The Cancer Genome atlas Program (TCGA), physical disruption of epithelial cells in the OSE can be routinely induced by ovulation. Increased ovulations with damaged OSE could serve as the origin of OvCa [3, 16]. In addition, CDH1 ablation is one of the progressive phenomena in cancer metastasis [17–19]. Inactivation of TRP53, PTEN, and RB1 in the OSE was predicted to induce type I low-grade OvCa such as papillary features of adenocarcinomas with some HGSC phenotype and metastasis. The combined inactivation of TRP53, PTEN, and RB1 in the OSE resulted in loss of normal epithelial characteristics. Interestingly, loss of Cdh1 seemed to cause disruption in the epithelial histoarchitecture that delayed the onset of OvCa and elicited poorly differentiated ovarian tumors. However, after OvCa establishment, these mice exhibited poor survivability and all succumbed to the disease by 6.5 months.
Materials and methods
Animals and tissue collection
Mice were maintained in the vivarium at Southern Illinois University according to the institutional guidelines for the care and use of laboratory animals (protocol #16–038). Health of mice was continuously monitored by investigators and vivarium staff according to National Research Council guidelines [20]. Mice with tumor burden precluding normal movement and access to food/water were euthanized and the day of sacrifice recorded. B6.129-Amhr2tm3(cre)Bhr (aka Amhr2cre/+) mice were provided by Dr. Richard R. Behringer (Univ. Texas MD Anderson Cancer Center [21]). B6.129-Cdh1tmKem2/J (aka Cdh1flox, Jax #005319) and B6.129S4-Ptentm1Hwu (aka Ptenflox, Jax #006440) mice were obtained from the Jackson Laboratory. FVB.129-Trp53tm1Brn (aka Trp53flox, #01XC2) mice were obtained from the Mouse Models of Human Cancers Consortium, NIH. B6.D2-Tg(K19GT121)Tvd (aka TgK19GT121) mice were obtained from Drs. Yurong Song and Terry van Dyke [22, 23]. Tissues were collected at 1, 2, 3, 4, and 6 months, and were fixed in fresh 4% paraformaldehyde in PBS at room temperature and embedded in paraffin, or homogenized with Trizol (Thermo Fisher, Waltham, MA) for RNA extraction.
Histology and immunohistochemistry
Tissue sections were stained with haematoxylin and eosin (H&E) and evaluated by Marilène Paquet, a board-certified pathologist according to current ovarian cancer pathogenesis [3, 4, 24]. At least five sections were examined for each genotype or time point. Immunolocalization of Ki67, KRT14, CDH1, PAX8, WT1, SV40 T Antigen (RB1), and PTEN was determined in cross-sections (5 μm) of paraffin-embedded tissue sections using specific primary antibodies and a Vectastain Elite ABC Kit (Vector laboratories, Burlingame, CA, USA). Primary antibodies used in these analyses were anti-Ki67 (1:250 dilution, 550609, BD Biosciences, San Jose, CA); KRT14 (1:200 dilution, PIPA532460, Invitrogen, Waltham, MA); anti-CDH1 (1:250 dilution, 610181, BD Biosciences); PAX8 (1:200 dilution, 10336–1-AP, Proteintech, Rosemont, IL); anti-WT1 (1:200 dilution, ab89901, Abcam, Cambridge, MA); anti-SV40 T Antigen (1:150 dilution, DP02, Millipore Sigma, St. Louis, MO); and PTEN (1:250 dilution, 9552, Cell Signaling Technology, Danvers, MA).
Quantitative real-time PCR analysis (QPCR)
Total RNA was isolated from the tissues, and cDNA was synthesized from total RNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher, USA). Relative gene expression was determined by SYBR green (Bio Rad, USA) incorporation using a Bio-Rad CFX as described previously [25]. Primer sequences were determined using NCBI’s design tools and are provided in Supplementary Table S1.
Statistical analysis
Graphs were constructed and statistics were analyzed with GraphPad Prism 5.0 (GraphPad, San Diego, CA). Overall survival rates were determined by Kaplan–Meier analysis and P-value was determined by log-rank test. Student’s t-test was used to compare the relative Trp53 mRNA expression levels between control and Trp53d/d mice. Tests of significance were performed using the appropriate error terms according to the expectation of the mean squares for error. A P-value of 0.05 or less was considered significant.
Results
Generation of mice with Pten, Trp53, and Cdh1 ablation and RB1 inactivation in the mouse ovary
To characterize the cancer-promoting events in the ovary, Amhr2Cre/+ mice were crossed with Ptenf/f, Trp53f/f, Cdh1f/f, and/or TgK19GT121 mice to provide a tissue-specific ablation/inactivation of PTEN, TRP53, CDH1, and/or RB1 in Amhr2-expressing cells. TgK19GT121 mice can Cre-dependently inactivate RB1 in KRT19 (cytokeratin 19)-expressing cells. T121 is comprised of the first 121 amino acids of the SV40 large T antigen and inactivates RB1 and its functionally redundant proteins RBL1 (p107) and RBL2 (p130) (see details in [22, 23]). Amhr2+/+Ptenf/f Trp53f/f Cdh1f/f TgK19GT121 = Control, Amhr2Cre/+Ptenf/f Trp53+/+ Cdh1+/+ = Ptend/d, Amhr2Cre/+Pten+/+ Trp53f/f Cdh1+/+ = Trp53d/d, Amhr2Cre/+Pten+/+ Trp53+/+ Cdh1f/f = Cdh1d/d, Amhr2Cre/+Pten+/+ Trp53+/+ Cdh1+/+ TgK19GT121 = TgK19GT121, Amhr2Cre/+Ptenf/f Trp53f/f Cdh1+/+ = Ptend/d Trp53d/d, Amhr2Cre/+Ptenf/f Trp53f/f Cdh1f/f = Ptend/d Trp53d/d Cdh1d/d, Amhr2Cre/+Ptenf/f Trp53f/f Cdh1+/+ TgK19GT121 = Ptend/d Trp53d/d TgK19GT121, and Amhr2Cre/+Ptenf/f Trp53f/f Cdh1f/f TgK19GT121 = Ptend/d Trp53d/d Cdh1d/d TgK19GT121. Genotyping by PCR is shown in Figure 1A, and the primers for genotyping are shown in Supplementary Table S1. Cre recombinase in Amhr2Cre/+ mice is active in the OSE and granulosa cells in the ovary [26], and it is known that Amhr2Cre/+ mice are able to induce EOC and/or granulosa tumors depending on the target genes [9, 27, 28]. The ability of Amhr2-Cre recombinase to mediate ablation of Trp53, Cdh1, and Pten in the ovary was confirmed by Trp53 mRNA analysis, as well as PTEN and CDH1 (black arrow) immunoreactivity (Figure 1B–D). A floxed eGFP-stop-T121 cassette is inserted into exon 1 start codon of KRT19, and RB1 inactivation in KRT-expressing cells has been confirmed in the OSE [22]. Our results also showed that SV40 T antigen positive cells were observed in the OSE (black arrows) of TgK19GT121 mice and EOC (black arrows) of Ptend/d Trp53d/d TgK19GT121 and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice (Figure 1E).
Figure 1.
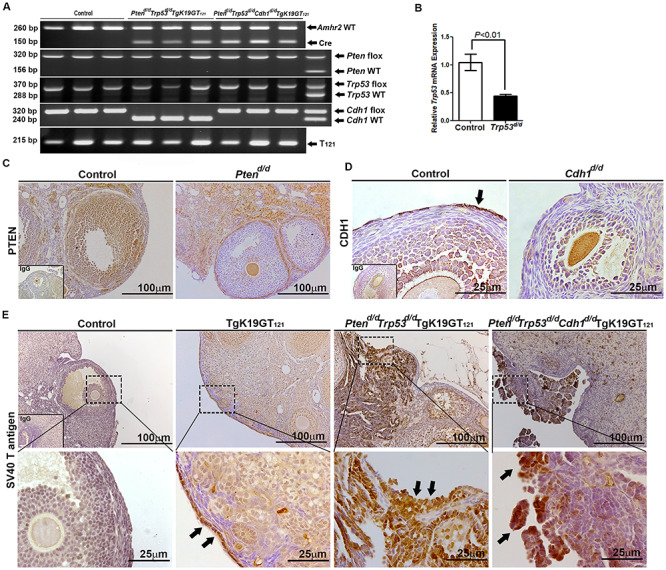
Analysis of conditional ablation/inactivation of Pten, Trp53, Cdh1, and Rb1 induced by Amhr2Cre/+ mice in mouse ovary. (A) Genotyping PCR of tail DNA to detect the Amhr2Cre/+ transgene (three representative mice per each genotype and heterozygous, f/+). (B) QPCR analysis of Trp53 normalized to Rpl19 in the ovary of control and Trp53d/d mice. Immunolocalization of (C) PTEN in the ovary of control and Ptend/d mice; (D) CDH1 in the ovary of control and Cdh1d/d mice; and (E) SV40 T antigen in the ovary of control, TgK19GT121, Ptend/d Trp53d/d TgK19GT121, and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice. Black arrows show positive immunostaining.
Impact of conditional inactivation of TRP53, PTEN, CDH1, and RB1 in the ovary
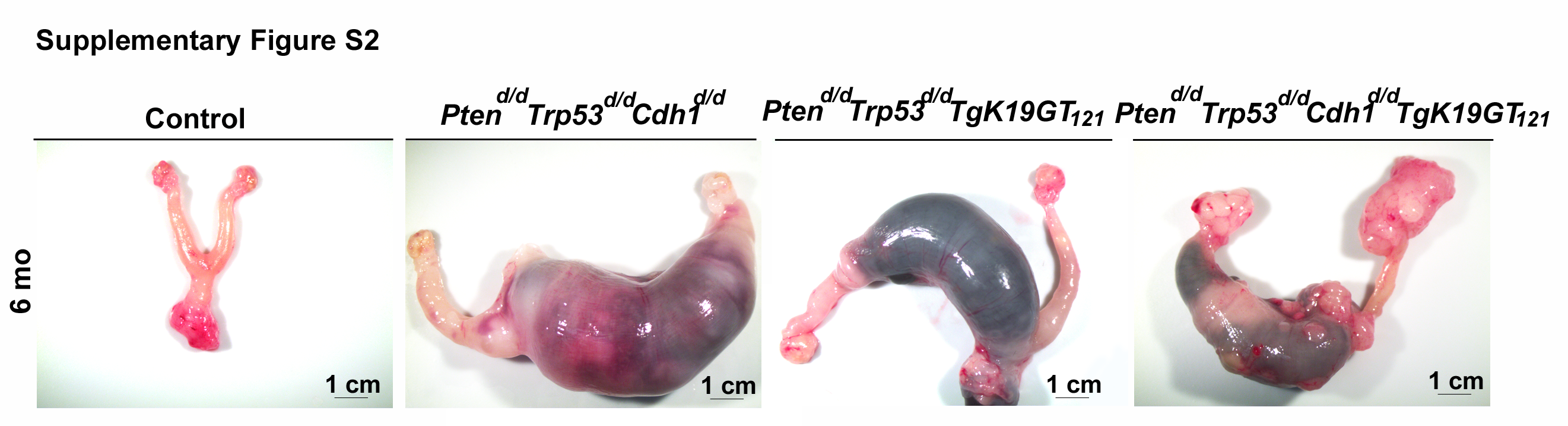
Nine different genotypes of mice (control, Ptend/d, Trp53d/d, Cdh1d/d, TgK19GT121, Ptend/d Trp53d/d, Ptend/d Trp53d/d Cdh1d/d, Ptend/d Trp53d/d TgK19GT121, and Ptend/d Trp53d/d Cdh1d/d TgK19GT121) were generated (n = 4–13 at each time point). Reproductive tract gross morphology was assessed at 6 month, Ptend/d, Trp53d/d, Cdh1d/d, TgK19GT121, Ptend/d Trp53d/d, and Ptend/d Trp53d/d Cdh1d/d mice appeared to have normal ovaries based on the gross morphology of ovaries (Figure 2A and B). Histological analysis indicates that Ptend/d Trp53d/d Cdh1d/d mice did not show abnormal cancerous ovaries (Supplementary Figure S1), but an enlarged uterus (Figure 2B, arrow) was commonly (6 of 8 mice) observed in these mice at 6 months. Cre recombinase in Amhr2Cre/+ mice is active in the uterine stroma and myometrium [26], potentially eliciting this abnormal morphology, but our focus was to characterize the role of these genes in OvCa. Because normal ovarian histology in Ptend/d mice has been reported [26] and mice with a double ablation of Ptend/d Trp53d/d possess ovaries with normal gross morphology (Figure 2A) and histology at 6 months (Supplementary Figure S1), Trp53d/d, Ptend/d, and Ptend/d Trp53d/d mice were removed from further analysis. Ptend/d Trp53d/d TgK19GT121 and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice (n = 4–13, Table 1) exhibited enlarged ovarian tumors starting at 3 months (Figure 2B). Ptend/d Trp53d/d TgK19GT121 mice (2 of 6 mice) and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice (8 of 13 mice) also showed abnormally enlarged uteri at 6 months (Supplementary Figure S2 and Table 1). Those putative uterine tumors were largely necrotic and contained large amount of putrid fluid. Ascites fluid accumulation and extensive peritoneal carcinomatosis with clusters of tumor nodules (black arrows, Figure 3A) and primary ovarian tumors (white arrows, Figure 3A) were observed in one Ptend/d Trp53d/d TgK19GT121 mouse at 4 months (Figure 3 and Table 1). Metastatic tumors were detected in Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice at 4 months (2 of 5 mice) and 6 months (5 of 13 mice) (Figure 3A and Table 1).
Figure 2.
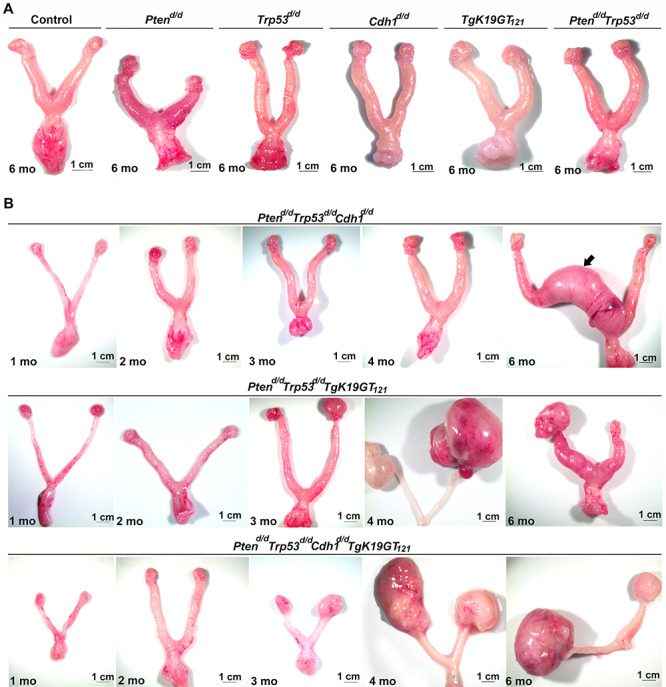
Gross morphology of the female reproductive tract in (A) control, Ptend/d, Trp53d/d, Cdh1d/d, TgK19GT121, and Ptend/d Trp53d/d at 6 months of age; and (B) Ptend/d Trp53d/d Cdh1d/d, Ptend/d Trp53d/d TgK19GT121, and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice at 1–6 months of age. Arrow indicates uterine enlargement observed in 75% of Ptend/d Trp53d/d Cdh1d/d mice at 6 months.
Table 1.
Characteristics of Ptend/d Trp53d/d TgK19GT121 and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice.
| Genotype | Phenotype | Age | ||||
|---|---|---|---|---|---|---|
| 1 month | 2 months | 3 months | 4 months | 6 months | ||
| Ptend/d Trp53d/d TgK19GT121 | OSE hyperplasia | 71.4% (5/7) | 11.1% (1/9) | 0% | 0% | 50.0% (3/6) |
| Serous papillary carcinomas | 57.1% (4/7) | 44.4% (4/9) | 71.4% (5/7) | 100.0% (5/5) | 50.0% (3/6) | |
| HGSC | 0% | 0% | 14.3% (1/7) | 60.0% (3/5) | 16.7% (1/6) | |
| Peritoneal metastasis | 0% | 0% | 0% | 60.0% (3/5) | 0% | |
| Ascites fluid accumulation | 0% | 0% | 0% | 20.0% (1/5) | 0% | |
| Granulosa cell tumors | 0% | 11.1% (1/9) | 0% | 0% | 33.3% (2/6) | |
| Necrotic tumors | 0% | 0% | 14.3% (1/7) | 0% | 0% | |
| Uterine tumors | 0% | 0% | 0% | 0% | 16.7% (2/6) | |
| OSE hyperplasia | 0% (0/8) | 30.0% (3/9) | 25.0% (1/4) | 0% | 0% | |
| Serous papillary carcinomas | 0% | 44.4% (4/9) | 75.0% (3/4) | 80.0% (4/5) | 69.2% (9/13) | |
| Pten d/d Trp53 d/d | HGSC | 0% | 0% | 0% | 20.0% (1/5) | 38.5% (5/13) |
| Cdh1 d/d | Peritoneal metastasis | 0% | 0% | 0% | 40.0% (2/5) | 38.5% (5/13) |
| TgK19GT 121 | Ascites fluid accumulation | 0% | 0% | 0% | 40.0% (2/5) | 38.5% (5/13) |
| Granulosa cell tumors | 0% | 0% | 0% | 0% | 0% | |
| Necrotic tumors | 0% | 0% | 0% | 20.0% (1/5) | 0% | |
| Uterine tumors | 0% | 0% | 0% | 0% | 61.5% (8/13) | |
Figure 3.
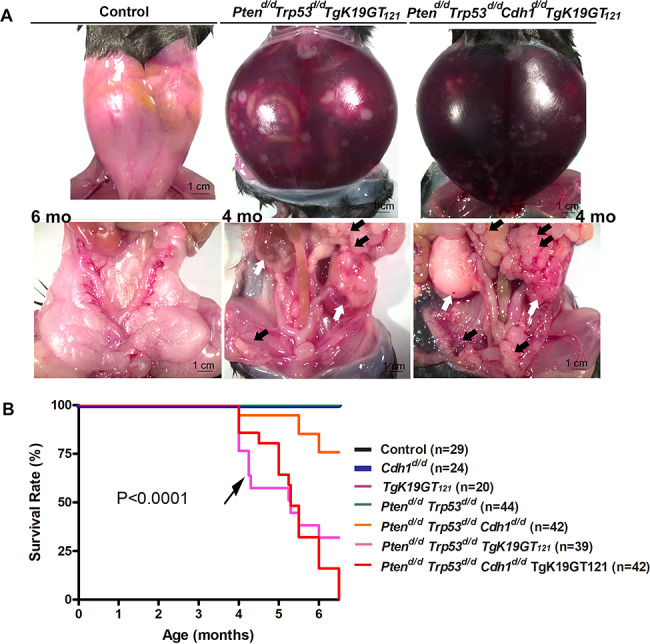
(A) Ascites fluid accumulation and peritoneal carcinomatosis tumors were observed in Ptend/d Trp53d/d TgK19GT121 and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice. White arrows show primary ovarian tumors and black arrows show extensive peritoneal carcinomatosis with clusters of tumor nodules. (B) Overall survival rate for control, Cdh1d/d, TgK19GT121, Ptend/d Trp53d/d, Ptend/d Trp53d/d Cdh1d/d, Ptend/d Trp53d/d TgK19GT121, and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice was determined by Kaplan–Meier analysis (P < 0.0001). All mice were monitored for any sign of sickness or discomfort. When present, those mice were euthanized, and the day of sacrifice was recorded. Black arrow shows that more mice with Ptend/d Trp53d/d TgK19GT121 died by 4.5 months compared to mice with Ptend/d Trp53d/d Cdh1d/d TgK19GT121.
Kaplan–Meier analysis revealed that survival rates of Ptend/d Trp53d/d TgK19GT121 and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice were significantly reduced (P < 0.0001, Figure 3B). While Ptend/d Trp53d/d TgK19GT121 and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice exhibited a median survival of 5.3 months, 71.8% of Ptend/d Trp53d/d TgK19GT121 and 100% of Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice died or reached our tumor burden euthanasia criteria by 6.5 months. We observed that more mice with Ptend/d Trp53d/d TgK19GT121 compared to mice with Ptend/d Trp53d/d Cdh1d/d TgK19GT121 died prior to 4.5 months (black arrow in Figure 3B). Some Ptend/d Trp53d/d TgK19GT121 mice were still alive at 6–6.5 months, whereas no Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice survived to 6.5 months. The uterine tumor burden observed in a few Ptend/d Trp53d/d Cdh1d/d mice led to death/euthanasia between 4 and 6.5 months (Figure 3B), whereas median survival was undefined by Kaplan–Meier analysis. Control, Cdh1d/d, TgK19GT121, and Ptend/d Trp53d/d mice appeared healthy until at least 6.5 months.
Histopathology of OvCa in Ptend/d, Trp53d/d TgK19GT121, and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice
Histopathological examination demonstrated that single ablation of Pten, Trp53, or Cdh1, and inactivation of RB1, double ablation of Pten and Trp53, as well as triple ablation of Pten, Trp53, and Cdh1 in the ovary by 6 months did not show in any abnormalities (Supplementary Figure S1). In contrast, Ptend/d Trp53d/d TgK19GT121 and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice developed similar features of EOC (Figure 4). However, initiation of EOC occurs earlier in Ptend/d Trp53d/d TgK19GT121 mice (Figure 4a and Table 1). Ptend/d Trp53d/d TgK19GT121 mice developed epithelial hyperplasia (5 of 7 mice) and low-grade serous micropapillary carcinomas (4 of 7 mice) that originated from the OSE at 1 months (Figure 4a and Table 1). The first signs of epithelial hyperplasia (3 of 9 mice) and micropapillary features (4 of 9 mice) in the OSE of Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice were observed at 2 months (Figure 4f and Table 1). No Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice (0 of 8 mice) showed abnormal OSE at 1 months (Figure 4e and Table 1). However, around 3–4 months, both Ptend/d Trp53d/d TgK19GT121 and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice exhibited invasion of serous carcinomas into the ovary with papillary and some HGSC features (Figure 4c and g and Table 1). When some of these mice started dying at ~ 4 months in both genotypes, we observed that EOC had already invaded into the entire ovary and the peritoneal cavity. The features of HGSC were first observed at 3 months in Ptend/d Trp53d/d TgK19GT121 mice and at 4 months in Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice (Table 1). At 6 months (Figure 4d and h and Table 1), Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice showed increased features of serous papillary carcinoma (9 of 13 mice) and HGSC (5 of 13 mice), as well as ascites fluid accumulation (5 of 13 mice), and peritoneal carcinomatosis (5 of 13 mice). In contrast, Ptend/d Trp53d/d TgK19GT121 mice exhibited less HGSC (1 of 6 mice), but showed some granulosa cell tumors (2 of 6 mice).
Figure 4.
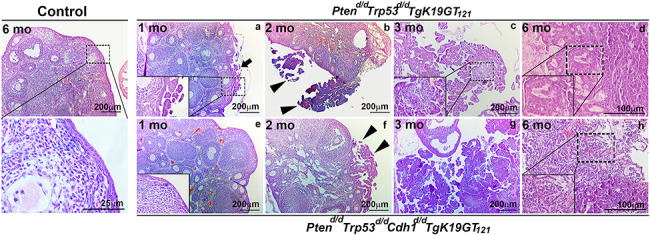
Ovarian histopathology of control, Ptend/d Trp53d/d TgK19GT121, and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice at 1–6 months of age (a–h). Tissues were stained using H&E. Black arrow indicates ovarian surface epithelial hyperplasia and arrowheads show serous micropapillary carcinomas.
As shown in Table 1, both genotypes of mice developed heterogeneous OvCa with papillary, HGSC, granulosa cell tumors, and/or necrotic tumors, whereas other types of EOC such as endometrioid, clear cell, and mucinous carcinomas were not observed. In contrast, serous papillary carcinomas were enlarged during the progression of OvCa in both genotypes. While there was some variability within animals of each group, several histopathological OvCa features were commonly identified in both genotypes including EOC hyperplasia (Figure 5a, arrow), progression of micropapillary and papillary carcinomas originating from the OSE (Figure 5b and c), papillary structure with slit-like spaces (Figure 5d), and papillary architecture with cytosolic atypia (Figure 5e). In addition, HGSC was also observed (Figure 5f), especially in the later stages of OvCa progression, characterized by nuclear atypia (Figure 5g, arrows), apoptotic cells (Figure 5h, arrow), abnormal mitosis (Figure 5i, arrows), and cell clumps (Figure 5j, arrow). We also observed cribriform architecture (Figure 5k), granulosa cell tumors (Figure 5l), papillary architecture with necrosis (Figure 5m), necrotic tumors (Figure 5n), papillary carcinoma with cystic features (Figure 5o), and dissemination of tumors into the peritoneal cavity (Figure 5p). However, oviducts of mice from both genotypes showed normal histology (Figure 5q).
Figure 5.
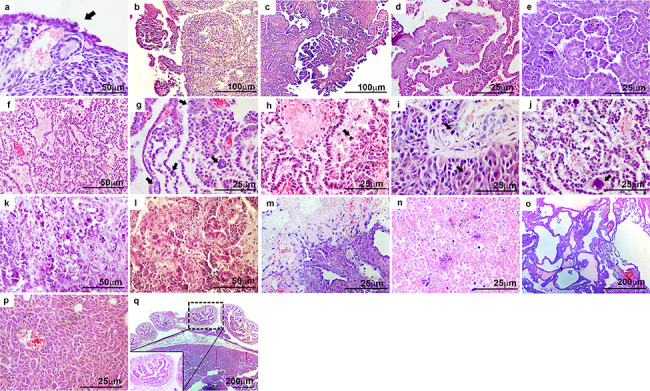
Ptend/d Trp53d/d TgK19GT121 and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice possess many histopathological features of OvCa in common. Representative images are presented for: (a) epithelial hyperplasia (black arrow), (b, c) progression of micropapillary and papillary carcinomas, (d) papillary structure with slit-like spaces, (e) papillary architecture with cytosolic atypia, (f) high-grade serous carcinoma (HGSC), (g–j) representative features of HGSC with (g) nuclear atypia (black arrows), (h) apoptotic cells (black arrow), (i) abnormal mitosis (black arrows), (j) cell clumps (black arrow), (k) cribriform architecture, (l) granulosa cell tumor, (m) papillary architecture with necrosis, (n) necrotic tumor, (o) papillary with cystic features, (p) peritoneal metastatic tumor, and (q) normal oviduct.
Strong staining of Ki67 positive cells, as a marker of cell proliferation, was observed within hyperplastic OSE (black arrows) at 2 months and OvCa tumors at 2–3 months in Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice, suggesting abnormal proliferation was present (Figure 6A). In contrast, control OSE lacked Ki67 staining, normal granulosa cells within growing follicles serve as a positive control for staining in healthy follicles. Those hyperplastic OSE (black arrows) at 2 months and OvCa at 2–3 months showed strong expression of KRT14, a marker of epithelial cells, and WT1, a marker of cancer cells in EOC (Figure 6A). Note: WT1 is known to be detectable in normal granulosa cells [29, 30]. PAX8, a marker of oviductal epithelial cells, was not detected within OvCa tumors, but was not altered in oviductal cells of ovarian sections of mice with EOC (Figure 6B). Similar staining was observed in Ptend/d Trp53d/d TgK19GT121 (data not shown). CDH1 was positive in OSE in control mice (white arrow) and EOC in Ptend/d Trp53d/d TgK19GT121 mice, but not present in Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice (Figure 6C). Note: CDH1 is positive in oocytes, and granulosa cells show some background fluorescence (see IgG control). Disseminated tumors retrieved from the peritoneal cavity displayed strong Ki67, KRT14, and WT1 expression throughout the tissue section (Figure 6D).
Figure 6.

Immunohistochemical analysis of (A) Ki67 as a marker of cell proliferation, KRT14 as a marker of epithelial cells, and WT1 as a marker of cancer cells (black arrows show hyperplastic OSE at 2 months), (B) PAX8 as a marker of oviductal epithelial cells in Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice. Similar staining was observed in Ptend/d Trp53d/d TgK19GT121 (data not shown). (C) CDH1 (white arrows show CDH1 positive cells) in control, Ptend/d Trp53d/d TgK19GT121, and Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice. (D) Immunohistochemical analysis of Ki67, KRT14, and WT1 in metastatic tumors.
Discussion
The Cancer Genome Atlas (TCGA) dataset indicates that HGSC harbor TP53 mutations in at least 96% of tumors [5]. Deregulation of the RB1 and PI3K pathways has been reported in 67 and 45% of HGSC, respectively [5]. While homozygous PTEN mutations are found in 7% of HGSC [5], total or partial loss of PTEN protein was reported in 15 or 50% of HGSC, respectively [31], implying that loss of PTEN function could be an induction of aberrant AKT activation in HGSC. Loss of CDH1 function has been implicated in the progression and metastasis of numerous malignancies [17–19]. Disruption of epithelial cells routinely occurs in the OSE during ovulation. Damaged OSE can result in the formation of a cyst with an epithelial lining inside and can be the origin of EOC [3]. In fact, the increased number of ovulations is considered as one of the risk factors of EOC [16]. Owing to those clinical features of OvCa, we have generated and characterized a mouse model in which TRP53, PTEN, RB1, and/or CDH1 are conditionally inactivated using Amhr2cre/+. Based on the specificity of RB1 inactivation in KRT19-expressing cells [22] and the results of initial epithelial hyperplasia in the OSE, the present study indicates that EOC originates from the OSE. Although histopathological analysis revealed that most tumors developed type I low-grade OvCa with the features of serous papillary carcinomas, some of the tumors became HGSCs exhibiting nuclear atypia and peritoneal metastasis. It has been reported that the OSE can develop into serous papillary adenocarcinomas, when Amhr2cre/+ mice are used to mutate Pten and express KrasG12D [9]. With an addition of Trp53R172H mutant expression to disruption of Pten gene and KrasG12D, OSE forms mucinous with serous features of carcinomas [11]. Pten deletion and Trp53R172H mutation are also able to develop HGSC [10]. Adenovirus-Cre-mediated ablation of Trp53 and Rb1 [32] in the OSE induces poorly differentiated carcinomas. Inactivation of TRP53, RB1, and BRCA1/2 in the OSE leads to the development of ovarian tumors with HGSC phenotype [23]. These results suggest that altered combinations of TRP53, RB1, and PTEN functions in the OSE are most likely critical to induce low- and high-grade features of EOC.
In our model, inactivation of TRP53, PTEN, and RB1 with or without CDH1 ablation showed similar pathogenesis including enlarged focal areas of papillary differentiation as shown in Figure 4. On the other hand, Ptend/d Trp53d/d TgK19GT121 mice initiated OSE hyperplasia and micropapillary differentiation at the age of 1 month (Figure 4a). Interestingly, the initiation of EOC in Ptend/d Trp53d/d TgK19GT121 mice started earlier than that in Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice (Figure 4a and e). Although we do not have a direct answer to address this observation, disruption of epithelial cells by loss of CDH1 in the OSE might lead to insufficient epithelial ablation/inactivation of TRP53, PTEN, and RB1, slowing down the progression of carcinogenesis. One possibility is that the loss of CDH1 leads to sloughing and death of abnormal epithelial cells that might otherwise lead to cancer. In fact, survival rate of Ptend/d Trp53d/d TgK19GT121 mice dropped 50% around 4 months, whereas ~ 75% of Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice survived until 5 months. However, a few more Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice developed HGSC, serous papillary carcinomas, and/or metastatic tumors at 6 months of age, and all mice died by 6.5 months. We observed that some of Ptend/d Trp53d/d TgK19GT121 mice survived past the time-point of 4 months, and those mice showed less invasive and aggressive features. Indeed, we did not find any signs of metastasis in Ptend/d Trp53d/d TgK19GT121 mice that survived at 6 months. Thus, TRP53, PTEN, and RB1 inactivation is enough to initiate and progress early tumor development, whereas loss of CDH1 eventually accelerates invasive and metastatic progression of tumorigenesis in OvCa. On the other hand, HGSC was observed in older mice at 3–6 months for Ptend/d Trp53d/d TgK19GT121 mice, or 4–6 months for Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice, whereas low-grade serous papillary carcinomas have been observed from the early EOC development. These results let us speculate that HGSC could be differentiated from serous papillary carcinomas. However, the initiation and development of HGSC might be separately considered differentiation from other types of EOC, because HGSC has been detected in early stages of OvCa in clinical studies [3].
We have occasionally observed granulosa cell tumors in Ptend/d Trp53d/d TgK19GT121 mice. This observation is surprising, because RB1 inactivation via Cre-dependent induction of the Krt19 promoter in Ptend/d Trp53d/d TgK19GT121 should be limited only to OSE. Thus, these animals would possess granulosa-specific deletion for only TRP53 and PTEN. However, Ptend/d Trp53d/d mice did not show any abnormal histopathology in the ovary, indicating that inactivation of RB1 in the OSE and loss of Trp53 and Pten either in OSE or granulosa cells somehow could associate to induce granulosa cell tumors.
Based on the results of PAX8 expression, our model did not induce any tumors derived from the stroma of the oviduct where Amhr2-Cre recombinase is active. Since inactivation of RB1 and CDH1 is limited to epithelial cells, this result indicates that Amhr2-Cre-driven ablation of TRP53 and PTEN in the stroma of the oviduct is not able to induce tumorigenesis. The use of Pax8 or Ovgp1 promoters to express Cre recombinase is able to target in oviductal epithelial cells of oviduct [12–15]. Targeting Brca1/2, Trp53R172H, and Pten genes using Pax8cre induces HGSC originated from the oviduct [12]. Brca1, Trp53, Rb1, Nf1, and/or Pten defects using Ovgp1-iCre-ERT2 result in serous tubal intraepithelial carcinomas and HGSC [14]. These results suggest that inactivation of genes in the epithelial cells of the oviduct is crucial to develop OvCa. On the other hand, we observed uterine tumor burden in some Ptend/d Trp53d/d Cdh1d/d, Ptend/d Trp53d/d TgK19GT121, or Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice at 6 months of age. Within the uterus, AMHR2 expression is limited only to the stroma and myometrium [26]. Thus, inactivation of TRP53 and PTEN is only likely to be induced in the uterus. However, Ptend/d Trp53d/d mice did not show any abnormal uterine morphology. It has been reported that loss of PTEN by Amhr2cre/+ mice fails to induce endometrial cancer, but generates abnormal myometrial adipogenesis [26]. Although the reasons for the development of necrotic uterine tumor burden observed in Ptend/d Trp53d/d Cdh1d/d, Ptend/d Trp53d/d TgK19GT121, or Ptend/d Trp53d/d Cdh1d/d TgK19GT121 mice are still unclear, those tumors might be endometrial adenosarcoma, which might warrant further investigation in the future to determine the suitability of this model for uterine cancer studies.
Collectively, the results of the present study suggest that the impact of inactivation of TRP53, PTEN, and RB1 in the OSE causes development of EOC with low-grade serous papillary carcinomas and HGSC. Ablation of CDH1 further accelerates the development of aggressive metastatic tumors at later stages. In contrast, EOC from the OSE did not initiate HGSC at early stage of OvCa development.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. Richard R. Behringer at The University of Texas, MD Anderson Cancer Center for providing us Amhr2cre/+ mice.
Conflict of Interest
The authors have nothing to disclose.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019; 69:7–34. [DOI] [PubMed] [Google Scholar]
- 2. Cho KR, Shih Ie M. Ovarian cancer. Annu Rev Pathol 2009; 4:287–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim J, Park EY, Kim O, Schilder JM, Coffey DM, Cho CH, Bast RC Jr. Cell origins of high-grade serous ovarian cancer. Cancers (Basel) 2018; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kurman RJ, Shih Ie M. Pathogenesis of ovarian cancer: lessons from morphology and molecular biology and their clinical implications. Int J Gynecol Pathol 2008; 27:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cancer Genome Atlas Research Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bobbs AS, Cole JM, Cowden Dahl KD. Emerging and evolving ovarian cancer animal models. Cancer Growth Metastasis 2015; 8:29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lengyel E, Burdette JE, Kenny HA, Matei D, Pilrose J, Haluska P, Nephew KP, Hales DB, Stack MS. Epithelial ovarian cancer experimental models. Oncogene 2014; 33:3619–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morin PJ, Weeraratna AT. Genetically-defined ovarian cancer mouse models. J Pathol 2016; 238:180–184. [DOI] [PubMed] [Google Scholar]
- 9. Fan HY, Liu Z, Paquet M, Wang J, Lydon JP, DeMayo FJ, Richards JS. Cell type-specific targeted mutations of Kras and Pten document proliferation arrest in granulosa cells versus oncogenic insult to ovarian surface epithelial cells. Cancer Res 2009; 69:6463–6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim J, Coffey DM, Ma L, Matzuk MM. The ovary is an alternative site of origin for high-grade serous ovarian cancer in mice. Endocrinology 2015; 156:1975–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ren YA, Mullany LK, Liu Z, Herron AJ, Wong KK, Richards JS. Mutant p53 promotes epithelial ovarian cancer by regulating tumor differentiation, metastasis, and responsiveness to steroid hormones. Cancer Res 2016; 76:2206–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perets R, Wyant GA, Muto KW, Bijron JG, Poole BB, Chin KT, Chen JY, Ohman AW, Stepule CD, Kwak S, Karst AM, Hirsch MS et al. . Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 2013; 24:751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Russo A, Czarnecki AA, Dean M, Modi DA, Lantvit DD, Hardy L, Baligod S, Davis DA, Wei JJ, Burdette JE. PTEN loss in the fallopian tube induces hyperplasia and ovarian tumor formation. Oncogene 2018; 37:1976–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhai Y, Wu R, Kuick R, Sessine MS, Schulman S, Green M, Fearon ER, Cho KR. High-grade serous carcinomas arise in the mouse oviduct via defects linked to the human disease. J Pathol 2017; 243:16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu R, Zhai Y, Kuick R, Karnezis AN, Garcia P, Naseem A, Hu TC, Fearon ER, Cho KR. Impact of oviductal versus ovarian epithelial cell of origin on ovarian endometrioid carcinoma phenotype in the mouse. J Pathol 2016; 240:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Purdie DM, Bain CJ, Siskind V, Webb PM, Green AC. Ovulation and risk of epithelial ovarian cancer. Int J Cancer 2003; 104:228–232. [DOI] [PubMed] [Google Scholar]
- 17. Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, Warda A, Lochner D, Birchmeier W. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol 1991; 113:173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998; 392:190–193. [DOI] [PubMed] [Google Scholar]
- 19. Vleminckx K, Vakaet L Jr, Mareel M, Fiers W, van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 1991; 66:107–119. [DOI] [PubMed] [Google Scholar]
- 20. Burkholder T, Foltz C, Karlsson E, Linton CG, Smith JM. Health evaluation of experimental laboratory mice. Curr Protoc Mouse Biol 2012; 2:145–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jamin SP, Arango NA, Mishina Y, Hanks MC, Behringer RR. Requirement of Bmpr1a for Mullerian duct regression during male sexual development. Nat Genet 2002; 32:408–410. [DOI] [PubMed] [Google Scholar]
- 22. Song Y, Gilbert D, O'Sullivan TN, Yang C, Pan W, Fathalizadeh A, Lu L, Haines DC, Martin PL, Van Dyke T. Carcinoma initiation via RB tumor suppressor inactivation: a versatile approach to epithelial subtype-dependent cancer initiation in diverse tissues. PLoS One 2013; 8:e80459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Szabova L, Yin C, Bupp S, Guerin TM, Schlomer JJ, Householder DB, Baran ML, Yi M, Song Y, Sun W, McDunn JE, Martin PL et al. . Perturbation of Rb, p53, and Brca1 or Brca2 cooperate in inducing metastatic serous epithelial ovarian cancer. Cancer Res 2012; 72:4141–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cho KR. Ovarian cancer update: Lessons from morphology, molecules, and mice. Arch Pathol Lab Med 2009; 133:1775–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hayashi K, Erikson DW, Tilford SA, Bany BM, Maclean JA 2nd, Rucker EB 3rd, Johnson GA, Spencer TE. Wnt genes in the mouse uterus: Potential regulation of implantation. Biol Reprod 2009; 80:989–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Daikoku T, Jackson L, Besnard V, Whitsett J, Ellenson LH, Dey SK. Cell-specific conditional deletion of Pten in the uterus results in differential phenotypes. Gynecol Oncol 2011; 122:424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mullany LK, Fan HY, Liu Z, White LD, Marshall A, Gunaratne P, Anderson ML, Creighton CJ, Xin L, Deavers M, Wong KK, Richards JS. Molecular and functional characteristics of ovarian surface epithelial cells transformed by KrasG12D and loss of Pten in a mouse model in vivo. Oncogene 2011; 30:3522–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Richards JS, Fan HY, Liu Z, Tsoi M, Lague MN, Boyer A, Boerboom D. Either Kras activation or Pten loss similarly enhance the dominant-stable CTNNB1-induced genetic program to promote granulosa cell tumor development in the ovary and testis. Oncogene 2012; 31:1504–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen M, Zhang L, Cui X, Lin X, Li Y, Wang Y, Wang Y, Qin Y, Chen D, Han C, Zhou B, Huff V et al. . Wt1 directs the lineage specification of sertoli and granulosa cells by repressing Sf1 expression. Development 2017; 144:44–53. [DOI] [PubMed] [Google Scholar]
- 30. Gao F, Zhang J, Wang X, Yang J, Chen D, Huff V, Liu YX. Wt1 functions in ovarian follicle development by regulating granulosa cell differentiation. Hum Mol Genet 2014; 23:333–341. [DOI] [PubMed] [Google Scholar]
- 31. Hanrahan AJ, Schultz N, Westfal ML, Sakr RA, Giri DD, Scarperi S, Janakiraman M, Olvera N, Stevens EV, She QB, Aghajanian C, King TA et al. . Genomic complexity and AKT dependence in serous ovarian cancer. Cancer Discov 2012; 2:56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Flesken-Nikitin A, Choi KC, Eng JP, Shmidt EN, Nikitin AY. Induction of carcinogenesis by concurrent inactivation of p53 and Rb1 in the mouse ovarian surface epithelium. Cancer Res 2003; 63:3459–3463. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.