

Significance
Acute myeloid leukemia is a blood cancer that has poor outcomes with conventional treatment. The clinical behavior of the disease is driven in large part by the gene mutations that are responsible for disease development. The combination of mutations in the genes CEBPA and CSF3R is associated with an increased risk of recurrent disease after standard therapy. We have identified a drug combination that blocks signaling through the JAK/STAT pathway and inhibits the epigenetic regulator LSD1. This drug combination leads to maturation of immature leukemia cells and improves survival in mouse models in a synergistic manner. This therapeutic approach is a promising strategy for the treatment of AML patients with mutations in CEBPA and CSF3R.
Keywords: acute myeloid leukemia, LSD1, CSF3R, epigenetics, targeted
Abstract
Acute myeloid leukemia (AML) is a deadly hematologic malignancy with poor prognosis, particularly in the elderly. Even among individuals with favorable-risk disease, approximately half will relapse with conventional therapy. In this clinical circumstance, the determinants of relapse are unclear, and there are no therapeutic interventions that can prevent recurrent disease. Mutations in the transcription factor CEBPA are associated with favorable risk in AML. However, mutations in the growth factor receptor CSF3R are commonly co-occurrent in CEBPA mutant AML and are associated with an increased risk of relapse. To develop therapeutic strategies for this disease subset, we performed medium-throughput drug screening on CEBPA/CSF3R mutant leukemia cells and identified sensitivity to inhibitors of lysine-specific demethylase 1 (LSD1). Treatment of CSF3R/CEBPA mutant leukemia cells with LSD1 inhibitors reactivates differentiation-associated enhancers driving immunophenotypic and morphologic differentiation. LSD1 inhibition is ineffective as monotherapy but demonstrates synergy with inhibitors of JAK/STAT signaling, doubling median survival in vivo. These results demonstrate that combined inhibition of JAK/STAT signaling and LSD1 is a promising therapeutic strategy for CEBPA/CSF3R mutant AML.
Acute myeloid leukemia (AML) is a hematologic malignancy driven by the combination of two or more oncogenic mutations. For the majority of patients with AML, the standard of care of chemotherapy with cytarabine and anthracycline has remained unchanged for the last 40 y (1). The response to this therapy varies based on genomic subtype. Favorable-risk disease can often be cured with conventional chemotherapy alone. However, even in this subgroup ∼50% of patients relapse and require bone marrow transplantation (2). Unfortunately, the mechanistic underpinnings of relapse in favorable-risk disease are unknown, and specific therapeutic strategies to prevent relapse are lacking.
The transcription factor CCAAT enhancer binding protein alpha (CEBPA) is recurrently mutated in ∼10% of AML (3). During normal myelopoiesis, CEBPA is required for the production of mature myeloid cells, and C-terminal mutations disrupt this process, blocking myeloid differentiation (4). CEBPA is a relatively unusual gene in that it has only a single exon but has two translational start sites. N-terminal mutations typically produce a frameshift and premature stop codon, disrupting production of the long (p42) isoform of the protein with sustained production of the short (p30) isoform (5). The p30 isoform lacks a transactivation domain responsible for interaction with E2F and cell cycle regulation (6). Thus, alterations in the p42/p30 isoform ratio result in cell cycle dysregulation and uncontrolled growth (7). In contrast, C-terminal mutations occur in the basic leucine zipper domain and block interaction with DNA, disrupting myeloid maturation (8). When an N-terminal CEBPA mutation is present on one allele and a C-terminal mutation is present on the other (biallelic CEBPA mutation), the resultant AML is associated with favorable prognosis (9). Interestingly, decreased CEBPA expression also drives a similar gene expression profile, arguing that decreased CEBPA activity is a critical feature in the oncogenic activity of mutant CEBPA (10). Like other categories of favorable-risk disease, many patients can be cured with conventional therapy. However, approximately one-half of patients will ultimately relapse, and the determinates of this recurrent disease are unknown.
Recently, mutations in the granulocyte colony stimulating factor receptor (CSF3R) have been identified as one of the most commonly co-occurring mutations in CEBPA mutant AML (11 –13). The presence of CSF3R mutations in CEBPA mutant AML is associated with an increased risk of relapse and unfavorable prognosis (14). Membrane proximal CSF3R mutations disrupt an O-linked glycosylation site, leading to ligand-independent receptor dimerization and activation of downstream JAK/STAT signaling (15). When present in isolation, these mutations are highly associated with chronic neutrophilic leukemia. Mutant CSF3R activates both proliferative and differentiative transcriptional programs driving the proliferation of neutrophil precursors and the overproduction of mature neutrophils that are the hallmark of this disease (15, 16). However, when a C-terminal CEBPA mutation is present, differentiation downstream of CSF3R is blocked, but proliferation is unabated (17). In mouse models, only the combination of mutations leads to a completely penetrant, rapidly lethal AML, while single mutations produce long latency disease in only a fraction of animals (17). Mutant CEBPA blocks the activation of a key subset of enhancers that are crucial for myeloid differentiation, driving AML initiation. Based on this disease mechanism, we propose that therapeutic strategies targeting the leukemic epigenome will be particularly effective in this disease subtype.
Herein, we describe a medium-throughput drug screen that identifies inhibitors of lysine-specific demethylase 1 as highly active in CEBPA/CSF3R mutant AML. Inhibition of LSD1 reactivates myeloid lineage enhancers, restores expression of differentiation-associated genes, and drives morphologic neutrophil lineage differentiation in AML cells. Although inhibition of LSD1 alone fails to improve survival in mice harboring CEBPA/CSF3R mutant AML, combined inhibition of the JAK/STAT pathway and LSD1 produces complete hematologic disease control and doubles median survival. These data identify combined JAK/STAT and LSD1 blockade as an exciting therapeutic approach for patients with CEBPA/CSF3R mutant AML.
Results
We previously reported that retroviral expression of mutant CSF3R (CSF3RT618I) and C-terminal mutant CEBPA (CEBPAV314VW) in mouse bone marrow is sufficient to permit indefinite culture in cytokine-free media (17). To identify therapies active against this subtype of AML, we performed a functional screen utilizing a library of compounds for which we have accumulated sensitivity data on a large number of primary patient samples (Fig. 1A ) (15, 18). This allows us to plot the inhibitory concentration 50 (IC50) as a percentage of the median IC50 of previously screened samples, with those inhibitors with the lowest percentage having the highest relative efficacy. As expected, numerous inhibitors of the JAK/STAT pathway were identified. Interestingly, the CSF3RT618I/CEBPAV314VW cells were also highly sensitive to GSK2879552, an inhibitor of the histone demethylase LSD1. During normal blood development, LSD1 is present in numerous chromatin-remodeling complexes that decommission enhancers, leading to gene silencing (19). As mutant CEBPA produces a unique epigenetic landscape in AML and enhancer activation is crucial to the oncogenic activity of CEBPA mutant AML (17, 20), we investigated the efficacy of multiple targeted epigenetic inhibitors against our CEBPA/CSF3R mutant cell line. We assessed the in vitro cytotoxicity of inhibitors of LSD1 (GSK2979552 and GSK-LSD1), Jumonji histone demethylase (KDM4C, JHDM inhibitor VIII), disruptor of telomeric silencing 1-like (DOT1L, EPZ004777), enhancer of zeste 2 (EZH2, UNC1999), histone deacetylases (HDAC, panobinostat), and DNA methyl transferases (DNMT, azacitidine) (Fig. 1B ). GSK-LSD1 was the most potently cytotoxic of all of the epigenetic agents evaluated, suggesting that LSD1 inhibitors might be particularly effective against CSF3R-CEBPA mutant AML.
Fig. 1.

CEBPA/CSF3R mutant AML demonstrates sensitivity to JAK/STAT and LSD1 inhibition. (A) Mouse bone marrow cells immortalized through retroviral transduction with CSF3RT618I and CEBPAV314VW were screened against a panel of 175 inhibitors with established sensitivities in primary patient samples. Drugs with an IC50 < 20% median IC50 of all prior samples are shown. (B) CEBPA/CSF3R mutant AML cells were screened against a panel of epigenetic inhibitors with cytotoxicity assessed after 72 h of culture.
LSD1 inhibitors are known to reactivate differentiation-associated enhancers in MLL-rearranged AML (21 –24). To assess whether a similar mechanism occurs in CSF3R/CEBPA mutant AML, we performed RNA-sequencing (RNA-seq) on CEBPA/CSF3R mutant AML cells treated with GSK-LSD1. This revealed the up-regulation of numerous genes associated with maturing myeloid cells, including Clu and members of the Mmp and S100 families (Fig. 2A and SI Appendix, Table S1) (25). LSD1 is known to interact with growth factor independent 1 (GFI1) family members, and LSD1 inhibition displaces GFI1 from chromatin (24). Indeed, we found that Gfi1 and Gfi1b were among the most highly up-regulated genes identified with LSD1 inhibition, consistent with an autofeedback loop. To confirm that LSD1 inhibition was not altering the expression of either transduced oncogene, we assessed human CEBPA and CSF3R expression in response LSD1 inhibitor treatment and found no significant changes ( SI Appendix, Fig. S1A ). To validate the observed expression changes, we confirmed the differential expression of key differentiation-associated genes by real-time PCR ( SI Appendix, Fig. S1B ). Gene ontology analysis revealed that genes up-regulated in response to LSD1 inhibition were associated with multiple neutrophil processes, including activation, neutrophil-mediated immune responses, and degranulation (Fig. 2B and SI Appendix, Table S2). In addition, we observed an enrichment of genes coding for neutrophil granule proteins. We next compared genes that were differentially expressed in response to LSD1 inhibition with RNA-seq on mouse bone marrow expressing mutant CSF3R alone or mutant CSF3R and mutant CEBPA (Fig. 2C ) (17). LSD1 inhibition up-regulated numerous genes that were expressed in the setting of CSF3RT618I alone but suppressed when CEBPAV314VW was coexpressed. Thus, treatment of CEBPA/CSF3R mutant AML cells with GSK-LSD1 leads to reactivation of differentiation-associated genes suppressed by mutant CEBPA.
Fig. 2.
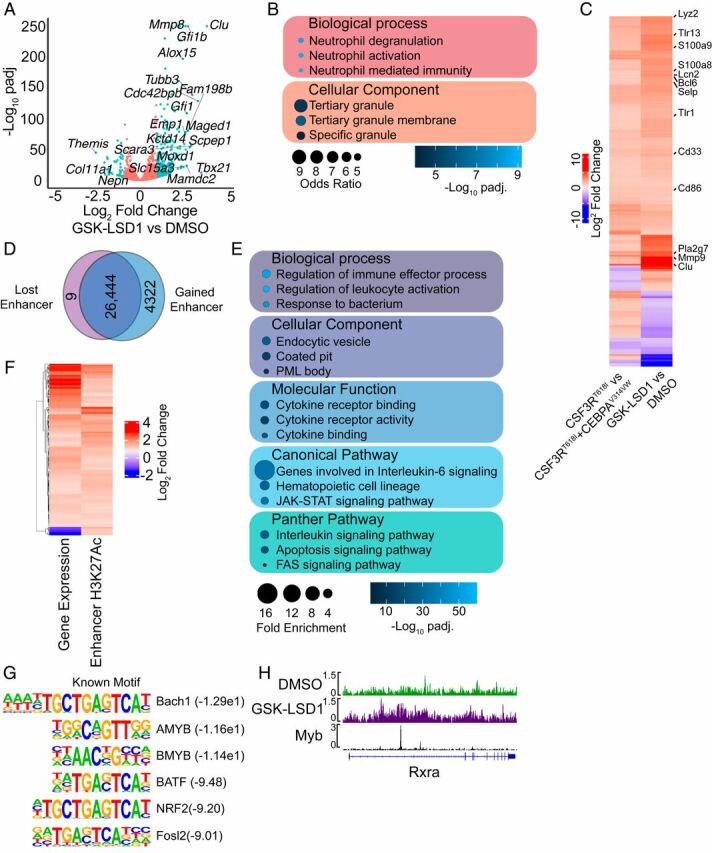
LSD1 inhibition reactivates differentiation-associated enhancers. (A) Differential gene expression assessed by RNA-seq in CEBPA/CSF3R mutant AML cells treated with GSK-LSD1 (4 nM) or DMSO for 48 h (n = 3/group). (B) Gene ontology analysis performed on genes with increased expression after LSD1 inhibitor treatment. (C) Correlation of genes differentially expressed in mouse bone marrow cells expressing CSF3RT618I or CSF3RT618I/CEBPAV314VW or upon LSD1 inhibitor treatment. (D) CEBPA/CSF3R AML cells were treated with GSK-LSD1 (4 nM) or DMSO for 48 h and subjected to histone mark ChIP-seq (n = 2/group). Enhancers were defined as the presence of H3K4me1 and H3K27Ac along with the absence of H3K4me3. Differential H3K27Ac signal at enhancers in response to GSK-LSD1 treatment. (E) Gene ontology analysis on enhancers with increased H3K27Ac in response to GSK-LSD1 treatment. (F) Correlation between differential gene expression and differential enhancer H3K27Ac in response to GSK-LSD1 treatment. (G) Motif enrichment analysis at enhancers with gained H3K27Ac in response to GSK-LSD1 treatment. (H) Example of gained enhancer at the Rxra locus with an overlapping Myb peak.
To assess whether LSD1 inhibition produces reactivation of differentiation-associated enhancers, we performed chromatin immunoprecipitation sequencing (ChIP-seq) for multiple covalent histone modifications, enabling the identification of active enhancers (presence of H3K4me1 and H3K27Ac, absence of H3K4me3). We identified 30,775 active enhancers across both experimental conditions. Assessment of differential H3K27Ac revealed 4,322 enhancers with increased H3K27Ac in response to GSK-LSD1 treatment and 9 with a loss of H3K27Ac (Fig. 2D and SI Appendix, Table S3). Gene ontogeny analysis of the enhancers gained upon LSD1 inhibitor treatment revealed the activation of pathways associated with immune activation, endocytosis, cytokine signaling, and activation of the JAK/STAT pathway (Fig. 2E and SI Appendix, Table S4). These regions of gained H3K27Ac in response to LSD1 inhibition were associated with increased expression of the nearest gene (R 2 = 0.19, P = 0.0027, Fig. 2F ). Motif enrichment analysis revealed an enrichment of motifs for Myb family members, which are known to directly interact with LSD1 and prevent differentiation (Fig. 2G ) (26). To confirm the association of Myb binding with regions of gained H3K27Ac, we analyzed the overlap of Myb ChIP-seq peaks with these differential active enhancers. We found a strong statistically significant enrichment of overlap of Myb peaks with active enhancers with gained H3K27Ac upon LSD1 inhibitor treatment and a depletion of overlap in enhancers without change in H3K27Ac (Fig. 2H and SI Appendix, Fig. S1C ) (27). Collectively, these data demonstrate that LSD1 inhibition activates Myb bound enhancers with up-regulation of genes associated with myeloid differentiation.
As LSD1 inhibition activates a transcriptional and epigenetic program consistent with myeloid differentiation, we assessed whether epigenetic modulation produced immunophenotypic and morphologic differentiation in cells expressing mutant CSF3R and CEBPA. We assessed the ability of select drugs identified on our initial screen to overcome the differentiation blockade observed in HoxB8-ER cells expressing mutant CSF3R and CEBPA (17, 28). Inhibition of LSD1 by GSK2879552 produced a dose-dependent increase in expression of CD11b and GR1 (markers of myeloid maturation) (Fig. 3A ). In contrast, neither JAK/STAT (ruxolitinib) nor BET inhibition (JQ1, OTX-015) promoted differentiation. We then assessed the ability of several LSD1 inhibitors to produce immunophenotypic differentiation in CEBPA/CSF3R AML cells. Irreversible (GSK-LSD1 and GSK2879552) but not reversible (OG-L002 and SP2509) LSD1 inhibitors produced dose-dependent increases in GR1 and CD11b expression and morphologic evidence of neutrophil maturation (Fig. 3 B and C ). In contrast select other epigenetic agents only produced significant immunophenotypic differentiation at markedly higher doses ( SI Appendix, Fig. S2A ).
Fig. 3.

JAK/STAT and LSD1 inhibition fails to improve survival in vivo. (A) GR-1 and CD11b expression as measured by flow cytometry in HoxB8 ER cells expressing CSF3RT618I and CEBPAV314VW after 48 h of estrogen withdrawal and treatment with ruxolitinib, CPI-0610, OTX-015, JQ1, or GSK2879552 (n = 3/dose). (B) GR-1 and CD11b expression by flow cytometry in murine bone marrow cells immortalized through expression of CEBPAV314VW + CSF3RT618I and treated for 72 h with GSK2879552, GSK-LSD1, OG-L002, or SP2509 (n = 3/dose). (C) Representative images from CEBPA/CSF3R AML cells treated with 5 nM GSK-LSD1 for 72 h. (D) Survival posttreatment initiation for mice transplanted with 10,000 cells expressing CSF3RT618I and CEBPAV314VW and treated with GSK2879552 at 1.5 mg/kg/day or ruxolitinib at 90 mg/kg/day by twice daily oral gavage starting at week 2. (E) Weekly WBC count posttreatment initiation. (F) Spleen weight at time of survival endpoint (n = 4 to 5/group). In all cases, values are represented as mean ± SEM. Survival was assessed by the log-rank test. Significance of other comparisons was assessed by Student’s t test for two group comparisons or ANOVA with Sidak’s posttest, as appropriate.
Early data demonstrate that inhibition of JAK/STAT signaling with ruxolitinib is effective in patients with CSF3R-mutant chronic neutrophilic leukemia (15). JAK/STAT inhibition has also been suggested as a therapeutic strategy for CEBPA mutant AML irrespective of CSF3R mutational status (12). Our data above suggest that LSD1 inhibition might also be an effective therapy in this disease subtype. We therefore assessed disease response to GSK289552 and ruxolitinib in mice harboring CEBPA/CSF3R mutant AML. In mice transplanted with CSF3RT618I alone, marked disease control is achieved with ruxolitinib monotherapy (16). We treated mice harboring CSF3RT618I and CEBPAV314VW with ruxolitinib alone or GSK289552 alone. However, despite in vitro efficacy, neither single agent improved survival, controlled white blood cell (WBC) count, or significantly reduced spleen size (Fig. 3 D–F ). To confirm that GSK2879552 was having the expected effect in vivo, we assessed the expression of genes known to be regulated by LSD1 after 24 h of GSK2879552 treatment (0.75 mg/kg every 12 h). Consistent with our in vitro findings, GSK2879552 up-regulated both BCL6 and Pla2g7 in the bone marrow of CSF3R/CEBPA leukemic mice ( SI Appendix, Fig. S2B ).
LSD1 inhibitors demonstrate increased efficacy in combination with other agents (29). As our epigenetic analysis revealed that LSD1 inhibition activates enhancers associated with JAK/STAT signaling, we assessed synergy between inhibitors of these two pathways. Cells treated with the combination of ruxolitinib and GSK-LSD1 showed enhanced differentiation at higher doses compared with cells treated with LSD1 inhibitor alone (Fig. 4A ). Furthermore, Bliss additivity and synergy analysis demonstrated enhanced efficacy of the drug combination compared with single agents (Fig. 4 B and C ). Consistent with the observed cytotoxicity, ruxolitinib and combined ruxolitinib/GSK2879552 eliminated the serial replating of CSF3R/CEBPA AML cells in colony assay ( SI Appendix, Fig. S3E ). GSK2879552 treatment decreased colony number in subsequent replatings and led to exhaustion of replating capacity.
Fig. 4.
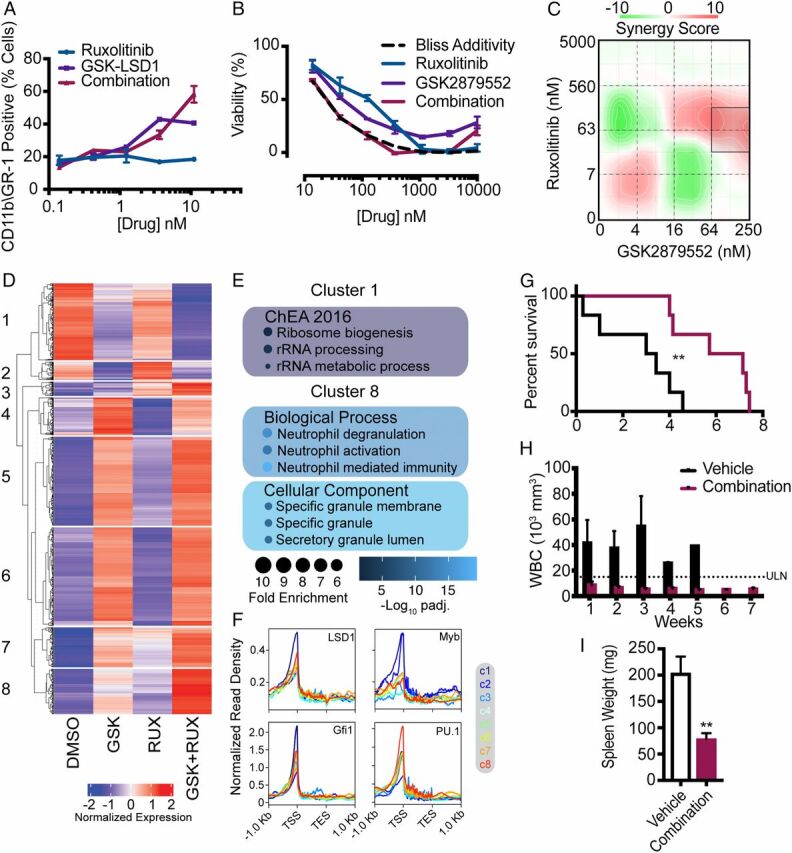
Combined inhibition of JAK/STAT signaling and LSD1 demonstrates synergy in vitro and improves survival in vivo. (A) GR-1 and CD11b expression measured by flow cytometry in murine bone marrow cells immortalized through expression of CEBPAV314VW + CSF3RT618I and treated for 72 h with ruxolitinib, GSK-LSD1, or the combination (n = 3/group). (B) Viability of murine bone marrow cells immortalized through expression of CEBPAV314VW + CSF3RT618I and treated for 72 h with ruxolitinib, GSK-2879552, or the combination. (C) Drug matrix of murine bone marrow cells immortalized through expression of CEBPAV314VW + CSF3RT618I and treated for 72 h with ruxolitinib and GSK-2879552 with synergy assessed by zero interaction potency score. (D) RNA-seq performed on CEBPA/CSF3R mutant AML cells treated with ruxolitinib (50 nM), GSK-LSD1 (2 nM), both drugs in combination, or DMSO (n = 3/group). Gene expression displayed by K-means clustering. (E) Gene ontology analysis of select clusters from D. (F) Overlap of ChIP-seq peaks for LSD1, Myb, Gfi1, and PU.1 with the promoters of differentially expressed genes separated by cluster. (G) Survival posttreatment initiation for mice transplanted with 10,000 cells expressing both oncogenes and treated with GSK2879552 at 1.5 mg/kg/day and ruxolitinib at 180 mg/kg/day or vehicle by oral gavage as a twice daily divided dose starting at week 3. (H) Weekly WBC count post treatment initiation. (I) Spleen weight at time of survival endpoint (n = 5 to 6/group). In all cases, values are represented as mean ± SEM. **P < 0.01. Survival was assessed by log-rank test. Significance of other comparisons was assessed by Student’s t test for two group comparisons or ANOVA with Sidak’s posttest, as appropriate.
To further characterize the mechanism of synergy, we performed RNA-seq on CEBPA/CSF3R mutant AML cells treated with dimethyl sulfoxide (DMSO), ruxolitinib, GSK-LSD1, or the combination (Fig. 4D and SI Appendix, Table S5). To assess patterns of expression across all treatment conditions, we performed unsupervised clustering of all differentially expressed genes. This analysis revealed multiple patterns of differential expression, with GSK-LSD1 being the major determinant of differential expression in most clusters. However, we identified two clusters where GSK-LSD1 and ruxolitinib exerted additive effects. We observed synergistic repression of cluster 1 genes by GSK-LSD1 and ruxolitinib and synergistic induction of cluster 8 genes by the drug combination. Pathway analysis of cluster 1 revealed synergistic repression of genes associated with cellular proliferation (i.e., ribosomal biogenesis) (Fig. 4E and SI Appendix, Table S6). In contrast, cluster 8 contained numerous gene signatures associated with neutrophil maturation and granule proteins.
Collectively, these data demonstrate that GSK-LSD1 and ruxolitinib exhibit synergistic cytotoxicity via down-regulation of proliferative programs and promotion of myeloid differentiation. To correlate these effects with known transcriptional effectors of LSD1, we assessed the overlap of ChIP-seq peaks for LSD1, GFI1, Myb, and PU.1 with each cluster of genes (19, 27, 30, 31). While all transcription factors demonstrated a significant enrichment of overlap with the promoters of all clusters of differentially expressed genes, differential patterns were observed (Fig. 4F and SI Appendix, Table S7). Consistent with the known interplay between Myb and self-renewal programs, we observed the strongest overlap between cluster 1 genes and Myb peaks (32). In contrast, consistent with its known role in myeloid differentiation and previously implicated role in the response to LSD1 inhibitors, we observed the strongest overlap of PU.1 peaks with the cluster 8 genes (neutrophil differentiation). By comparison, LSD1 and GFI1 demonstrated relatively uniform enrichment of overlap with all clusters. This implies that distinct transcription factors control differing aspects of the response to LSD1 inhibition.
To assess toxicity and tolerability of the drug combination in vivo, we first treated healthy mice with the combination of GSK2879552 and ruxolitinib for 2 wk. The drug combination led to a small but significant weight loss over the 2 wk period but did not produce any hematologic abnormalities or overt toxicity in these healthy mice ( SI Appendix, Fig. S3 A–D ). We next investigated the efficacy of the combination of ruxolitinib and GSK2879552 in mice transplanted with CSF3RT618ICEBPAV314VW AML (Fig. 4G ). The combination of GSK2879552 and ruxolitinib normalized peripheral blood WBC counts starting at week 1 and doubled median survival to 6.4 wk up from 3.2 wk in vehicle-treated mice (Fig. 4 G and H ). Spleen weight was substantially reduced by treatment with the combination of ruxolitinib and GSK2879552 (Fig. 4I and SI Appendix, Fig. S3F ). Furthermore, mice treated with the combination of ruxolitinib and GSK2879552 did not experience significant drops in other cell indices, arguing that the therapeutic combination did not produce substantial hematologic toxicity ( SI Appendix, Fig. S3 G and H ). These data demonstrate that combined inhibition of LSD1 and the JAK/STAT pathway is a promising therapeutic strategy in CEBPA mutant AML.
Discussion
In patients with biallelic CEBPA mutant AML, the standard of care treatment involves induction and consolidation with cytarabine-based chemotherapy, followed by close surveillance for disease relapse. Approximately 50% of such patients will relapse with this approach, yet the only intervention that reduces this risk is bone marrow transplantation (33). Given the morbidity associated with bone marrow transplant, coupled with the fact that a second remission is usually achievable in relapsed favorable-risk AML, this approach is typically employed only at the time of relapse (34). Although cure can ultimately be achieved for many patients with favorable-risk disease using bone marrow transplantation, there is still substantial mortality associated with this approach. This is particularly true in older individuals with favorable-risk disease, in whom long-term survival remains less than 20% (35). Therefore, novel therapeutic strategies driven by an understanding of the molecular determinants of relapse would be of high clinical value. Here, we report a combination therapeutic strategy targeting JAK/STAT signaling and LSD1 that demonstrates marked activity in vivo against CEBPA/CSF3R mutant AML, a disease subtype with an increased risk of relapse (14). This approach could be deployed as a component of upfront therapy or utilized in the setting of relapsed disease, potentially offering clinical benefit with reduced toxicity.
Mutations in AML classically occur in two distinct functional categories (36). Class I mutations lead to the activation of signaling pathways, driving proliferation and producing a myeloproliferative disease in isolation. In contrast, class II mutations occur in lineage-determining transcription factors, blocking differentiation and producing myelodysplasia in isolation. When present in combination, the proliferation of immature white blood cells results in AML. Deeper investigation of AML genomics has demonstrated that this class I/class II paradigm is not always present. Indeed, it appears that AML often results from the co-occurrence of multiple class II mutations (18, 37, 38).
Interestingly, favorable-risk disease is typically defined by the presence of specific class II mutations such as biallelic CEBPA mutations, translocations involving core binding factor, and the presence of mutations in nucleophosmin (NPM1). However, it is increasingly evident that the co-occurrence of class I mutations reverses the favorable prognosis associated with these class II mutations. For example, mutations in FMS-like tyrosine kinase 3 (FLT3) frequently co-occur with NPM1 mutations and drive worsened prognosis (2). Class I mutations in KIT frequently co-occur in core binding factor mutant AML and similarly produce adverse outcomes (39). The recently discovered high rate of occurrence of CSF3R mutations in CEBPA mutant AML appear consistent with this paradigm (11 –13, 40). Individuals with biallelic CEBPA mutant AML and mutant CSF3R have inferior outcomes compared to those without mutant CSF3R. Given the similarities between these three disease subtypes, it is possible that an analogous strategy of combination therapy with agents targeting class I and class II mutations will have broad applicability.
LSD1 is a member of multiple chromatin complexes associated with both gene silencing and gene activation. As a histone demethylase, it removes activating H3K4me marks, decommissioning enhancers, and also removes repressive H3K9 marks (41). Recent work, however, suggests that LSD1 blocks myeloid differentiation in MLL-rearranged AML through alternate mechanisms (24). LSD1 is present in multiple repressive complexes that contain growth factor independent 1 (GFI1) (24). GFI1 is responsible for repressing genes associated with alternate cell fates (i.e., monocytes) in developing neutrophils (42). Consistent with this, Gfi1 knockout mice have neutropenia and accumulation of abnormal cells with an intermediate phenotype between monocytes and neutrophils (42). In addition, point mutations in GFI1 in humans are associated with congenital neutropenia (43). Genetic or pharmacologic inhibition of LSD1 disrupts these repressive complexes, displacing GFI1 from chromatin, leading to enhancer activation (24). Consistent with this, we observed marked up-regulation of Gfi1 gene expression in response to LSD1 inhibitor treatment, consistent with displacement of GFI1 from its own promoter and loss of autoregulatory inhibition. Prior work has demonstrated that LSD1 inhibition reactivates genes with enhancers and promoters occupied by known differentiation-promoting transcription factors such as PU.1 and CEBPA (23). Consistent with this, our data for CSF3R/CEBPA mutant AML demonstrated marked reactivation of enhancers in response to LSD1. Additionally, we noted a cluster of genes that show the greatest increase in expression upon combination drug treatment. These genes were associated with a myeloid differentiation signature and showed the strongest enrichment of overlap with PU.1 peaks. Whether LSD1 or JAK/STAT inhibition changes the localization of PU.1 binding is an interesting area for further investigation.
Our data also suggest that LSD1 plays an important role in gene activation. We identified a cluster of genes that demonstrated strong down-regulation in response to combined drug treatment. These genes showed enrichment for proliferative regulators and demonstrated the strongest overlap with Myb peaks. Myb is known to interact with LSD1 in other contexts, and Myb knockdown in MLL-rearranged AML cells produces a gene expression profile similar to LSD1 inhibition (24). In contrast to GFI1, which exerts repressive activity in complexes with LSD1, these proliferative Myb target genes demonstrate decreased expression when treated with the drug combination, suggesting that, in certain contexts, Myb–LSD1 complexes exert transcriptional activating activity, potentially via the modification of repressive H3K9 methylation status.
A major issue noted in early clinical trials of LSD1 inhibitors is development of dose-dependent thrombocytopenia (44). Although we did not observe hematologic toxicity in healthy mice treated with GSK2879552 either alone or in combination with ruxolitinib and the combination was also well tolerated in leukemic mice, this remains a serious concern. Interestingly, we also observed up-regulation of Gfi1b, a transcription factor closely related to GFI1, which is important for gene repression in erythroid and megakaryocytic lineages (45). Therefore, it is possible that the thrombocytopenia observed with LSD1 inhibition may be related to inhibition of Gfi1b, suggesting that the development of strategies that spare this factor may avoid much of the observed hematologic toxicity.
It has been proposed that in AML, epigenetic drugs will likely be most effective in the setting of combination therapy (46). With nearly limitless combinations, preclinical prioritization based on mutational profile and a detailed mechanistic understanding of single agents will be necessary. One possible approach is to combine multiple epigenetic agents with the notion that they will target complementary pathways involved in differentiation arrest. Indeed, a recent study demonstrated synergistic differentiation and cytotoxicity with the combination of LSD1 inhibitors and azacytidine (29). An alternate approach is to target vulnerabilities exposed by one drug with a second agent. Our analysis of LSD1 inhibitor activated enhancers revealed activation of pathways associated with cytokine receptor and JAK/STAT signaling. Thus, LSD1 inhibition may render cells more dependent on JAK/STAT signaling, increasing sensitivity to targeted inhibition of this pathway. One interesting finding in our study was that ruxolitinib augmented the differentiation observed with GSK-LSD1. Given that differentiation downstream of mutant CSF3R appears to be dependent on JAK/STAT signaling, this finding appears to be paradoxical. Prior work suggests that the majority of the prodifferentiative signaling downstream of CSF3R is dependent on Stat3 signaling, while Stat5 prevents differentiation (47). Thus, differential modulation of Stat3 and Stat5 activity could be responsible for the observed enhancement LSD1 inhibitor mediated differentiation produced by ruxolitinib. In future studies, genome-wide assessment of Stat3 and Stat5 binding in response to ruxolitinib and LSD1 inhibition will provide insight into this potential mechanism.
Collectively, our results demonstrate that combination therapy with LSD1 and JAK/STAT inhibitors is highly active in CEBPA/CSF3R mutant AML. This result suggests that the addition of ruxolitinib and GSK2879552 to postremission therapy may reduce the risk of relapse in this unfavorable disease subtype. For patients with relapsed disease, this combination may represent an alternative reinduction strategy for elderly patients in whom reduced toxicity would be of high value.
Materials and Methods
Mice.
Wild-type Balb/cJ mice (JAX# 000651), were obtained from the Jackson Laboratories. All animals were maintained on a normal 12:12 h light:dark cycle and provided ad libitum access to water and food (Purina rodent diet 5001; Purina Mills). Female mice were used for experimentation between 6 and 20 wk of age and were age and weight matched in all experiments. All experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals (48) and approved by the Institutional Animal Care and Use Committee of Oregon Health & Science University.
Cell Lines.
The 293T17 cells were obtained from ATCC and were cultured in Rosewell Park Memorial Institute media (RPMI) (Gibco) supplemented with 20% fetal calf serum (FCS, HyClone). Murine HoxB8-ER cells were a generous gift from David Sykes (Massachusetts General Hospital, Boston, MA) and were cultured in RPMI (Gibco) with 10% FCS (HyClone) and Chinese Hamster Ovary-Stem Cell Factor cell conditioned media (final concentration of ∼100 ng/mL) (28). Mouse bone marrow cell lines were immortalized through retroviral transduction with CSF3RT618I and CEBPAV314VW and were cultured in Iscove Modified Dulbecco Media (Gibco) supplemented with 20% FCS (HyClone). All cells were cultured at 37 °C and 5% CO2. Wild-type HoxB8-ER cells were cultured and differentiated as previously described (28). Cell lines were tested monthly for mycoplasma contamination.
Cloning and Retrovirus Production.
The following retroviral plasmids were utilized: pMSCV-IRES GFP (15), pMSCV-IRES-mCherry FP (a gift from Dario Vignali, (Pittsburgh, PA), Addgene plasmid #52114). The CSF3RT618I mutation was generated as previously described (15). Full-length CEBPA cDNA was obtained from GeneCopoeia. CEBPAV314VW was generated using the Quikchange Site Directed Mutagenesis Kit (Agilent). To produce retrovirus, 293T17 cells were transfected with EcoPac helper plasmid (a gift from Rick Van Etten, Irvine, CA) and the appropriate transfer plasmid. Conditioned media was harvested 48 to 72 h after transduction.
Retroviral Transduction for Bone Marrow Transplant.
Donor mice were injected with 5-FU (100 mg/kg) 5 d prior to sacrifice. Bone marrow was harvested and subjected to ammonium chloride red blood cell lysis (Gibco). Cells were cultured overnight in IL-3 (14 ng/mL), IL-6 (24 ng/μL), and SCF (112 ng/μL) and Wehi conditioned media at 3 × 106/mL, then spinoculated at 0.25E6/mL with 2 mL retroviral conditioned media in a six-well plate. Spinoculation was repeated again on the following day, and transduced cells were isolated using a FACSAria III (BD Biosciences). Recipient mice were lethally irradiated (450 Gy × 2 at least 3 h apart) using an Xstrahl X-ray irradiator (Radsource). Cells were administered via retro-orbital injection under isoflurane anesthesia. Each recipient was administered 10,000 CSF3R/CEBPA positive cells (GFP/RFP positive cells) along with 190,000 fresh whole bone marrow cells (support cells). Mice were maintained on water containing 1.1 mg/mL neomycin trisulfate and 167 μg/mL polymyxin B sulfate for 3 wk posttransplant. Complete blood counts were obtained weekly starting at day 10 using an automated counter (Scil Vet abc). Mice were monitored for leukemia development, and drug treatment was initiated when the majority of transplanted mice had developed leukocytosis. Ruxolitinib (Selleckchem) and GSK2879552 (Selleckchem) were suspended in 5% dimethylacetamide, 0.5% methylcellulose, and 2.5% DMSO. Mice were treated with GSK2879552 at 1.5 mg/kg/day, ruxolitinib at 180 mg/kg/day, both drugs in combination, or vehicle by oral gavage. Survival endpoints included WBC > 100 and moribund appearance as mice were frequently moribund without substantial weight loss. At time of sacrifice, smears were prepared of blood and bone marrow. Spleen weight was recorded, and spleens were fixed for 24 h in 10% zinc formalin (Fisher Scientific), then transferred to 70% ethanol and stored at 4 °C. One tibia was also fixed and stored in a similar fashion.
Medium-Throughput Drug Screening.
IC50 values were compared to all prior samples (AML, CML, CLL) treated (18). Sensitivity was defined as an IC50 below 20% of the median IC50. Drugs that were subsequently found to have more than a 10-fold change in IC50 in confirmatory studies were removed from the final screen results. Compounds that displayed a U-shaped drug response curve were eliminated as follows: Any compound that reached an IC50 but subsequently had two or more points >75% viability at higher drug concentrations was excluded from further analysis. Compounds that failed to drop viability below 25% were excluded. Synergy analysis was performed on an 8 × 8 matrix of drug concentrations. Synergy analysis was performed using SynergyFinder (49).
Real-Time PCR.
Depending on the amount of starting material, RNA was extracted using an RNeasy Micro Kit (Qiagen). Subsequently, cDNA was synthesized using a High Capacity cDNA Synthesis Kit (Thermo Fisher). Real-time PCR was performed using the QuantStudio7 Real Time PCR System (Thermo Fisher) and Taqman primer probes (Thermo Fisher).
Flow Cytometry.
For assessment of myeloid differentiation, phycoerythrin rat anti-mouse GR1 (BD) and PE-Cy7 rat anti-mouse CD11b (BD) were used according to the manufacturer’s protocol. Stained cells were analyzed on a FACSAria III flow cytometer (BD). For differentiation studies, drugs were added using a Digital Dispenser drug printer (HP). All drugs were dissolved in DMSO with a final DMSO concentration of <1%. Cells were incubated for 48 to 72 h in drug and stained with antibodies to GR-1 and CD11b. Stained cells were run on a CytoFLEX flow cytometer (Beckman Coulter) with a 96-well plate loader housed in the Oregon Health & Science University Flow Cytometry Shared Resource.
RNA Sequencing.
CEBPA/CSF3R mutant murine AML cells were treated with 4 nM GSK-LSD1, 2 nM GSK-LSD1, 50 nM ruxolitinib, 2 nM GSK-LSD1/50 nM ruxolitinib, or DMSO for 48 h. Total RNA was extracted using a RNA micro Kit (Qiagen). Library preparation and sequencing was performed by BGI at 50 base pair (BP) paired end for the GSK-LSD1 monotherapy study and 50 BP single end for the combination therapy study.
Chromatin Immunoprecipitation Sequencing.
CEBPA/CSF3R mutant AML cells were cultured for 48 h in 4 nM GSK-LSD1. Chromatin preparation and immunoprecipitation were performed using a SimpleChIP Enzymatic Kit (Cell Signaling Technologies) according to the manufacturer’s instructions. Approximately 5 μg of chromatin from 4 × 106 cells was utilized per histone mark (n = 2 immunoprecipitations/condition). The following antibodies were utilized: rabbit anti-H3K27Ac (Abcam, ab4729), rabbit anti-H3K4me1 (Cell Signaling, D1A9), and rabbit anti-H3K4me3 (Cell Signaling, C32D8). Sequencing libraries for each ChIP and input were generated using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs) according to the manufacturer's instructions. All ChIP-seq libraries were sequenced using PE 150 bp on an Illumina HiSeq4000.
Bioinformatic Analysis.
RNA-seq analysis: Murine samples.
Raw reads were trimmed with BBDuk and aligned with STAR (50). Differential expression analysis was performed using DESeq2 (51). Raw P values were adjusted for multiple comparisons using the Benjamini–Hochberg method. Gene ontology and pathway analysis was performed using Enrichr (52). Genes that were differentially expressed upon treatment with GSK-LSD1 were compared with genes differentially expressed between mouse bone marrow expressing CSF3RT618I alone and CSF3RT618I/CEBPAV314VW (GSE122166). Expression profiles of genes differentially expressed in both datasets are displayed side by side.
ChIP-seq analysis.
Reads were aligned to the mouse reference genome (mm10) using Bowtie with default settings (53). We used the ChromHMM software (54) to characterize and annotate the genomes of each treatment group according to six chromatin states, based on different combinations of H3K4me1, H3K4me3, and H3K27ac marks. Active enhancers were identified through the presence of H3K4me1 and H3K27Ac and the absence of H3K4me3. A catalog of all identified active enhancers was assembled across both treatment conditions. The differential H3K27Ac signal at these enhancers was determined using the getDifferentialPeaksReplicates.pl command in HOMER (55).
Renanalysis of published transcription factor ChIP seq data.
Processed bed files containing called peaks and bigwig files were downloaded from the Cistrome Data browser (56): LSD1(35751), GFI1 (39926), Myb (46473), and PU.1 (90318) (19, 27, 30, 31). Gene coordinates were obtained using the University of California Santa Cruz table browser to download the Refseq known gene catalog. Heat maps were generated for each cluster of differentially expressed genes using deepTools (57). Overlap between either differential enhancers and Myb peaks was performed using bedtools fisher (58). Overlap between the promoters of differentially expressed genes and candidate transcription factors was performed using bedtools fisher and a catalog of regions ±1 kilobase surrounding the transcriptional start site.
Gene ontology analysis for ChIP-seq.
Gene ontology analysis for histone mark ChIP-seq was performed by the Genomic Regions Enrichment of Annotations Tool (59) using the basal plus extension model to annotate enhancer coordinates to nearby genes.
Quantification and Statistical Analysis.
Data are expressed as mean ± SEM. Statistical analysis was performed using Prism software (version 7.0; Prism Software Corp.) or RStudio. Statistical analyses are described in figure legends. All data were analyzed with either an unpaired Student’s t test or ANOVA followed by post hoc analysis using Sidak’s corrected t test. For RNA-seq data, P values were adjusted for repeated testing using a false discovery rate by the method of Benjamini–Hochberg (60). Survival analysis was conducted using the method of Kaplan–Meier, and statistical significance was assessed using a log-rank test.
Data Availability.
The Gene Expression Omnibus (GEO) accession number for all genomic data for RNA-seq and ChIP-seq reported in this paper is GSE138388.
Supplementary Material
Acknowledgments
We thank the following core facilities for their assistance: Histopathology Shared Resource, Flow Cytometry Shared Resource, ExaCloud Cluster Computational Resource, and the Advanced Computing Center. Funding was provided by an American Society of Hematology Research Training Award for Fellows, a Medical Research Foundation Early Clinical Investigator Award, NIH KL2 Grant 5 KL2 TR 2370-3, and Collins Medical Trust Award to T.P.B.; the OHSU School of Medicine Faculty Innovation Fund to B.J.D. and J.E.M.; the Howard Hughes Medical Institute to B.J.D.; and National Cancer Institute Grant #R00-CA190605, an American Society of Hematology Scholar Award, American Cancer Society RSG-19-184-01-LIB, and an MRF New Investigator Grant to J.E.M.
Footnotes
Competing interest statement: B.J.D. declares the following potential competing interests: Monojul, Patient True Talk; SAB: Aileron Therapeutics, ALLCRON, Cepheid, Gilead Sciences, Vivid Biosciences, Celgene & Baxalta (inactive); SAB and stock: Aptose Biosciences, Blueprint Medicines, Beta Cat, GRAIL, Third Coast Therapeutics, CTI BioPharma (inactive); scientific founder and stock: MolecularMD; board of directors and stock: Amgen; board of directors: Burroughs Wellcome Fund, CureOne; joint steering committee: Beat AML LLS; clinical trial funding: Novartis, Bristol-Myers Squibb, Pfizer; royalties: OHSU #606-Novartis exclusive license, OHSU #2573; Dana-Farber Cancer Institute #2063 - Merck exclusive license. J.E.M. receives grant funding from the Gilead Research Scholars Program. The remaining authors declare no competing interests.
Data deposition: The genomic data for RNA-seq and ChIP-seq reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, https://www.ncbi.nlm.nih.gov/geo (accession no. GSE138388).
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1918307117/-/DCSupplemental.
References
- 1. Yates J. W., Wallace H. J. Jr., Cytosine arabinoside (NSC-63878) and daunorubicin (NSC-83142) therapy in acute nonlymphocytic leukemia. Cancer Chemother. Rep. 57, 485–488 (1973). [PubMed] [Google Scholar]
- 2. Döhner H.et al., Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129, 424–447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Avellino R., Delwel R., Expression and regulation of C/EBPα in normal myelopoiesis and in malignant transformation. Blood 129, 2083–2091 (2017). [DOI] [PubMed] [Google Scholar]
- 4. Zhang P.et al., Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity 21, 853–863 (2004). [DOI] [PubMed] [Google Scholar]
- 5. Pabst T.et al., Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat. Genet. 27, 263–270 (2001). [DOI] [PubMed] [Google Scholar]
- 6. Porse B. T.et al., E2F repression by C/EBPalpha is required for adipogenesis and granulopoiesis in vivo. Cell 107, 247–258 (2001). [DOI] [PubMed] [Google Scholar]
- 7. Kirstetter P.et al., Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell 13, 299–310 (2008). [DOI] [PubMed] [Google Scholar]
- 8. Bereshchenko O.et al., Hematopoietic stem cell expansion precedes the generation of committed myeloid leukemia-initiating cells in C/EBPalpha mutant AML. Cancer Cell 16, 390–400 (2009). [DOI] [PubMed] [Google Scholar]
- 9. Dufour A.et al., Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J. Clin. Oncol. 28, 570–577 (2010). [DOI] [PubMed] [Google Scholar]
- 10. Figueroa M. E.et al., Genome-wide epigenetic analysis delineates a biologically distinct immature acute leukemia with myeloid/T-lymphoid features. Blood 113, 2795–2804 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maxson J. E.et al., CSF3R mutations have a high degree of overlap with CEBPA mutations in pediatric AML. Blood 127, 3094–3098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lavallée V.-P.et al., Chemo-genomic interrogation of CEBPA mutated AML reveals recurrent CSF3R mutations and subgroup sensitivity to JAK inhibitors. Blood 127, 3054–3061 (2016). [DOI] [PubMed] [Google Scholar]
- 13. Zhang Y.et al., CSF3R mutations are frequently associated with abnormalities of RUNX1, CBFB, CEBPA, and NPM1 genes in acute myeloid leukemia. Cancer 124, 3329–3338 (2018). [DOI] [PubMed] [Google Scholar]
- 14. Su L.et al., CSF3R mutations were associated with an unfavorable prognosis in patients with acute myeloid leukemia with CEBPA double mutations. Ann. Hematol. 98, 1641–1646 (2019). [DOI] [PubMed] [Google Scholar]
- 15. Maxson J. E.et al., Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N. Engl. J. Med. 368, 1781–1790 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fleischman A. G.et al., The CSF3R T618I mutation causes a lethal neutrophilic neoplasia in mice that is responsive to therapeutic JAK inhibition. Blood 122, 3628–3631 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Braun T. P.et al., Myeloid lineage enhancers drive oncogene synergy in CEBPA/CSF3R mutant acute myeloid leukemia. Nat. Commun. 10, 5455 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tyner J. W.et al., Functional genomic landscape of acute myeloid leukaemia. Nature 562, 526–531 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kerenyi M. A.et al., Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. eLife 2, e00633 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Assi S. A.et al., Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nat. Genet. 51, 151–162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McGrath J. P.et al., Pharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypes. Cancer Res. 76, 1975–1988 (2016). [DOI] [PubMed] [Google Scholar]
- 22. Harris W. J.et al., The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 21, 473–487 (2012). [DOI] [PubMed] [Google Scholar]
- 23. Cusan M.et al., LSD1 inhibition exerts its antileukemic effect by recommissioning PU.1- and C/EBPα-dependent enhancers in AML. Blood 131, 1730–1742 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maiques-Diaz A.et al., Enhancer activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep. 22, 3641–3659 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Graaf C. A.et al., Haemopedia: An expression atlas of murine hematopoietic cells. Stem Cell Reports 7, 571–582 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ahmed M., Streit A., Lsd1 interacts with cMyb to demethylate repressive histone marks and maintain inner ear progenitor identity. Development 145, dev160325 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yue F.et al.; Mouse ENCODE Consortium , A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang G. G.et al., Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat. Methods 3, 287–293 (2006). [DOI] [PubMed] [Google Scholar]
- 29. Duy C.et al., Rational targeting of cooperating layers of the epigenome yields enhanced therapeutic efficacy against AML. Cancer Discov. 9, 872–889 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Khandanpour C.et al., Growth factor independence 1 antagonizes a p53-induced DNA damage response pathway in lymphoblastic leukemia. Cancer Cell 23, 200–214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Link V. M.et al., Analysis of genetically diverse macrophages reveals local and domain-wide mechanisms that control transcription factor binding and function. Cell 173, 1796–1809.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramsay R. G., Gonda T. J., MYB function in normal and cancer cells. Nat. Rev. Cancer 8, 523–534 (2008). [DOI] [PubMed] [Google Scholar]
- 33. Pastore F.et al., Long-term follow-up of cytogenetically normal CEBPA-mutated AML. J. Hematol. Oncol. 7, 55 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burnett A. K.et al., Curability of patients with acute myeloid leukemia who did not undergo transplantation in first remission. J. Clin. Oncol. 31, 1293–1301 (2013). [DOI] [PubMed] [Google Scholar]
- 35. Döhner H.et al.; European LeukemiaNet , Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 115, 453–474 (2010). [DOI] [PubMed] [Google Scholar]
- 36. Kelly L. M., Gilliland D. G., Genetics of myeloid leukemias. Annu. Rev. Genomics Hum. Genet. 3, 179–198 (2002). [DOI] [PubMed] [Google Scholar]
- 37. Ley T. J.et al.; Cancer Genome Atlas Research Network , Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Papaemmanuil E.et al., Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 374, 2209–2221 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cairoli R.et al., Prognostic impact of c-KIT mutations in core binding factor leukemias: An Italian retrospective study. Blood 107, 3463–3468 (2006). [DOI] [PubMed] [Google Scholar]
- 40. Su L.et al., CSF3R mutations were associated with an unfavorable prognosis in patients with acute myeloid leukemia with CEBPA double mutations. Ann. Hematol. 98, 1641–1646 (2019). [DOI] [PubMed] [Google Scholar]
- 41. Kozub M. M., Carr R. M., Lomberk G. L., Fernandez-Zapico M. E., LSD1, a double-edged sword, confers dynamic chromatin regulation but commonly promotes aberrant cell growth. F1000 Res. 6, 2016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Karsunky H.et al., Inflammatory reactions and severe neutropenia in mice lacking the transcriptional repressor Gfi1. Nat. Genet. 30, 295–300 (2002). [DOI] [PubMed] [Google Scholar]
- 43. Person R. E.et al., Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat. Genet. 34, 308–312 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bauer T. M.et al., Phase I, open-label, dose-escalation study of the safety, pharmacokinetics, pharmacodynamics, and efficacy of GSK2879552 in relapsed/refractory SCLC. J. Thorac. Oncol. 14, 1828–1838 (2019). [DOI] [PubMed] [Google Scholar]
- 45. Möröy T., Vassen L., Wilkes B., Khandanpour C., From cytopenia to leukemia: The role of Gfi1 and Gfi1b in blood formation. Blood 126, 2561–2569 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wouters B. J., Delwel R., Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 127, 42–52 (2016). [DOI] [PubMed] [Google Scholar]
- 47. Dwivedi P., Greis K. D., Granulocyte colony-stimulating factor receptor signaling in severe congenital neutropenia, chronic neutrophilic leukemia, and related malignancies. Exp. Hematol. 46, 9–20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. National Research Council , Guide for the Care and Use of Laboratory Animals, (National Academies Press, Washington, DC, ed. 8, 2011). [Google Scholar]
- 49. Ianevski A., He L., Aittokallio T., Tang J., SynergyFinder: A web application for analyzing drug combination dose-response matrix data. Bioinformatics 33, 2413–2415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dobin A.et al., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kuleshov M. V.et al., Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ernst J., Kellis M., Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 12, 2478–2492 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Heinz S.et al., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mei S.et al., Cistrome Data Browser: A data portal for ChIP-seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 45, D658–D662 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ramírez F.et al., deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McLean C. Y.et al., GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Gene Expression Omnibus (GEO) accession number for all genomic data for RNA-seq and ChIP-seq reported in this paper is GSE138388.