

Abstract
A novel coronavirus associated with acute respiratory disease (named SARS‐CoV‐2) is recently identified in Wuhan city, China, spread rapidly worldwide. Early identification of this novel coronavirus by molecular tools is critical for surveillance and control of the epidemic outbreak. We aimed to establish a simple method for the detection of SARS‐CoV‐2 in differentiating with SARS‐CoV. Primers of our in‐house reverse transcription polymerase chain reaction (RT‐PCR) assays were designed to target conserved regions of the RdRP gene and E gene, selected restriction enzymes EcoRI, Tsp45I, and AluI to distinguish between SARS‐CoV‐2 and SARS‐CoV. In this report, a 396‐bp fragment of the RdRp gene and 345‐bp fragment of the E gene were amplified by one‐step RT‐PCR. Enzyme Tsp45I cuts the RdRP‐amplified product of SARS‐CoV‐2 generating three fragments of 45, 154, and 197 bp, but it did not cut the amplicon of SARS‐CoV. In contrast, the amplified product of SARS‐CoV was digested with EcoRI producing two fragments of 76 and 320 bp, whereas the amplicon of SARS‐CoV‐2 was undigested by Tsp45I help to distinguish clearly SARS‐CoV‐2 from SARS‐CoV on gel electrophoresis. In addition, AluI cut the amplicon of the E gene of SARS‐CoV‐2 generating two fragments of 248 and 97 bp without cutting to SARS‐CoV. The accuracy of the assay was confirmed by sequencing and phylogenetic analysis. When evaluated on clinical samples showed a high sensitivity of 95%, specificity of our assay was 100% and clinical performance for detection of SARS‐CoV‐2 in comparison with other reference assays. In conclusion, in the present study, we successfully developed a simple method for molecular detection of SARS‐CoV‐2 in differentiating with SARS‐CoV.
Keywords: coronavirus disease: SARS‐CoV‐2, RT‐PCR‐RFLP, SARS‐CoV
Highlights
We developed a simple method for molecular detection of SARS‐CoV‐2 in differentiating with SARS‐CoV
The assay showed comparable analytical, clinical performance and overall agreement to other reference assays on clinical samples
1. INTRODUCTION
An outbreak of an acute respiratory disease caused by a 2019 novel coronavirus (2019‐nCoV) in Wuhan, a central city of Hubei, China, has spread rapidly worldwide and posed a global challenge for public health. 1 So far, there were more than 78 000 people infected with 2019‐nCoV confirmed and 2445 death have been reported in 28 countries and territory regions. 2 The transmission from human to human through close contact confirmed among infected cases, but secondary spread has been limited in returning travelers from epidemic region. 3 , 4 Based on complete genome sequence and phylogenetic analysis indicated that this nCoV is associated with human severe acute respiratory syndrome coronavirus (SARS‐CoV) and Middle East respiratory syndrome coronavirus (MERS‐CoV), 5 and recently, it was designated as SARS‐CoV‐2. 6 It shares 79% identity with SARS‐CoV and might also use angiotensin‐converting enzyme 2 as a cell receptor. 7 Similar to SARS‐CoV and MERS‐CoV, SARS‐CoV‐2 is an enveloped RNA coronavirus belonging to betacoronavirus subfamily, sarbecovirus subgenus of Coronaviridae family, but it appears the less pathogenic and more contagious and the mortality rate of SARS‐CoV‐2 is lower than that of MERS‐CoV (∼35%) and SARS‐CoV (∼10%). 8 To respond at the potential risk of pandemic outbreak spread worldwide, the World Health Organization (WHO) declared the outbreak of SARS‐CoV‐2 in China as “Public health emergency of international concern” and called substantial global response for the control of this epidemic. There has been no available vaccine or specific antiviral drugs for prevention and treatment of COVID‐19. 5 An important point of SARS‐CoV‐2 infection has a long period of incubation up to 14 days, longer than SARS‐CoV that may lead to a high rate of asymptomatic cases and it may also facilitate the rapid spread of SARS‐CoV‐2 around the world. 9 Therefore, early detection of suspected cases plays a critical role for management, surveillance, and control of this deadly epidemic. WHO and Centers for Disease Control and Prevention recommended the use of real‐time reverse transcription polymerase chain reaction (RT‐PCR) assay for confirmatory detection of SARS‐CoV‐2 in clinical virology laboratory. 10 , 11 However, commercial real‐time RT‐PCR kits approved by the FDA are not available, whereas isolation and viral culture method must be performed under Biosafety level 3 (BSL‐3) facility and RNA‐based metagenomic next‐generation sequencing analysis require modern equipment and technical expertise. 12 , 13 , 14 Therefore, a simple, rapid assay with affordable cost in resource‐limiting countries is very essential for the early detection of suspected cases on inactivated clinical specimens. This study aims to establish a simple method for detection of SARS‐CoV‐2 using one‐step RT‐PCR followed by restriction fragment length polymorphism for detection of SARS‐CoV‐2 in Vietnam.
2. MATERIALS AND METHODS
For assay design, sequences of SARS‐CoV (AY508724.1), bat‐SARS‐like coronavirus strains (MG772933.1 and MG772934.1) retried from GenBank, SARS‐CoV‐2 sequences released on GISAID (https://www.gisaid.org/EPI_ISL_402119; 402120; 402125; 402128; 402132; 403932; 403962; 404228; 406844; 406596; 406597) and SARS‐CoV‐2 sequences from Genbank (MN908947.3, MN985325.1; MN988713.1; LC522975.1) for alignment using BioEdit 7.0 and Clustal W to select highly conserved regions for design primer to amplify a part of the RdRP gene and the E gene of SARS‐CoV‐2 and SARS‐CoV using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/). Primer sequences were: Forward Pr‐GGCCTCACTTGTTCTTGCTC; Reverse Pr‐CACACAACAGCATCGTCAGA for the RdRP gene; Forward primer‐E: CACACAATCGACGGTTCATC; Reverse primer‐E: CGTTTAGACCAGAAGATCAGGAA for the E gene. Primers were purchased from IDT (Coralville, IA).
The viral RNA was extracted from 140 μL of cell culture supernatant of infected cells inactivated with AVL buffer and Ethanol (each 560 µL) using QIAamp Viral RNA Mini Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's instruction. The RNA was finally eluted in a final volume of 60 μL of AE buffer and was stored at −80°C until use. These materials including SARS‐CoV and SARS‐CoV‐2 RNA samples were provided as kind gifts by Prof Dang Duc Anh, National Institute of Hygiene and Epidemiology, Hanoi, Vietnam, and coronavirus RNA specificity panel obtained by the European virus archive global (EVAg), https://www.european-virus-archive.com. In addition, SARS‐CoV‐2 RNA in vitro transcripts of the RdRP gene and the E gene were synthesized from plasmid containing a part of the E and the RdRp gene using Transcript Aid T7 High Yield Transcription Kit (Thermo Fisher Scientific, Waltham, MA). The synthetic viral RNA transcripts were purified using the GeneJET RNA Purification Kit (Thermo Fisher Scientific) according to the manufacturer's instructions. The RNA transcript concentrations were measured by a Nanodrop ND1000 spectrophotometer (Thermo Fisher Scientific) then converted to the number of copies per μL according to the concentrations and sizes of each single‐stranded RNA fragment. Transcribed RNAs were estimated as 4.23 × 1012 copies/μL for the E gene and 2.7 × 1012 copies/μL for the RdRp gene. Then, a serial dilution from 100 to 106 copies/μL of purified viral RNA transcripts were used as positive controls for development and validation of our molecular assays to identify SARS‐CoV‐2.
One‐step RT‐PCR reaction was optimized in a total volume of 30 μL using One‐Step RT‐PCR Kit (QIAGEN, Germany) containing 6 μL of RNA template, 3 μL of each primer, 12.6 μL of molecular grade water, 1.2 μL RT enzyme, 1.2 μL dNTPs, and 6 μL of ×5 Buffer. Cycling conditions were: 50°C for 30 minutes, 95°C for 15 minutes, followed by 35 cycles of 94°C for 30 seconds, 50°C for 30 seconds, and 72°C for 20 seconds) and a final extension at 72°C for 10 minutes. All reactions were run on the Eppendorf Mastercycler pro PCR System (Eppendorf, Hamburg, Germany). Amplified products were separated by electrophoresis on 1.2% agarose gel and visualized under UV light after staining with ethidium bromide for expected sizes of 396 and 344 bp for the fragments of the RdRp gene and the E gene, respectively.
Restriction fragment length polymorphism (RFLP) analysis: the digestions were performed separately in a total volume of 10 µL containing 5 µL of the purified amplification products were added to 3.5 µL water, 1 µL Buffer (×10), and 0.5 µL of each enzyme EcoRI and Tsp45I for digestion of RdRP gene fragment; besides, the amplified fragment of E gene was digested with AluI (New England BioLabs, Hitchin, UK). The reactions were incubated at 37°C for 2 hours, after which digested products were run on 3% agarose gel electrophoresis.
Real‐time RT‐PCR: WHO RT‐PCR targeting E gene was performed as the protocol published by Corman et al. 15 Realstar SARS‐CoV‐2 RT‐PCR 1.0 (RUO) was provided by Altona Diagnostics (Hamburg, Germany). 16 Briefly, the RT‐PCR containing 5 µL of Master A, 15 µL of Master B, 1 µL of internal control, and 10 µL of RNA template for a final volume of 30 µL. Cycling conditions were according to the manufacturer's instructions. All PCRs were run on a LightCycler 480 II (LC480II; Roche).
Statistical analyses were done with SPSS 20.0 (IBM, Armonk, NY). The diagnostic accuracy was analyzed to estimate confidence intervals (95% confidence interval) for sensitivity, specificity. Cohen's κ values were calculated for evaluating overall agreement and comparing assays. P values of <.05 were considered statistically significant.
3. RESULTS
In our method, we designed primer sets to amplify the RdRp gene fragment of 396 bp of both SARS‐CoV‐2 and SARS‐CoV. Similarly, a 345‐bp fragment of the E gene was amplified simultaneously for both SARS‐CoV and SARS‐CoV‐2. Results are as shown in Figure 4 and there was no nonspecific amplification detected with primer sets used in this study.
Figure 4.
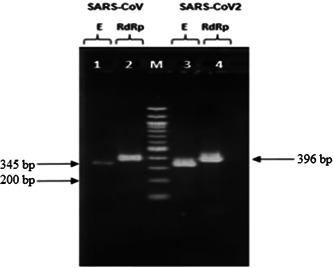
Agarose gel electrophoresis of one‐step RT‐PCR on in vitro transcribed RNAs. Lanes 1 and 2: amplicons of E and RdRp gene of SARS‐CoV; lane M: DNA ladder 100 bp; lanes 3 and 4: amplicons of E and RdRp gene of SARS‐CoV‐2. RT‐PCR, reverse transcription polymerase chain reaction; SARS‐CoV, severe acute respiratory syndrome coronavirus
The restriction enzymes are selected with the aid of NEBcutter V2.0 (www.tools.neb.com/NEBcutter2), which are able to distinguish between SARS‐CoV‐2 and SARS‐CoV by restriction fragments. RFLP strategy for RdRP product is illustrated in Figure 3.
Figure 3.
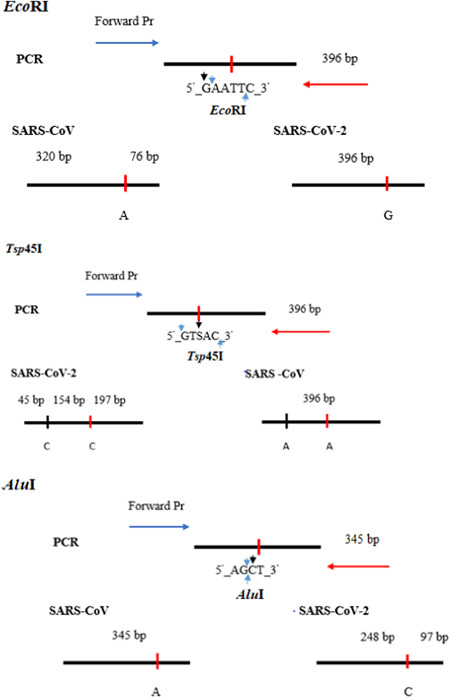
RFLP patterns for differentiating SARS‐CoV‐2 with SARS‐CoV. RFLP, restriction fragment length polymorphism; SARS‐CoV, severe acute respiratory syndrome coronavirus
As described above, for the RdRP gene, enzyme Tsp45I cut PCR amplification of SARS‐CoV‐2 generating three fragments of 45, 154, and 197 bp, and it did not cut RT‐PCR product of SARS‐CoV. In contrast, amplified product of SARS‐CoV was digested with EcoRI producing two fragments of 76 and 320 bp, whereas amplified product of SARS‐CoV‐2 was undigested by EcoRI. Analysis results are illustrated in Figures 1‐3 and 7.
Figure 1.
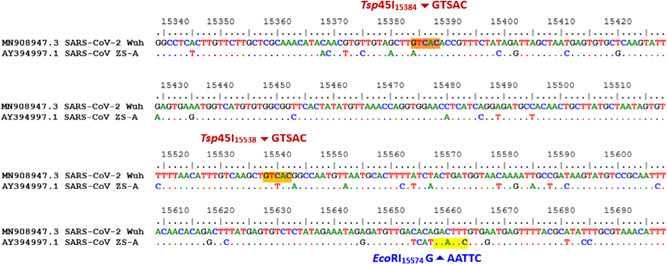
The location cut by restriction enzymes on the RdRp gene, including Tsp45I and EcoRI
Figure 7.
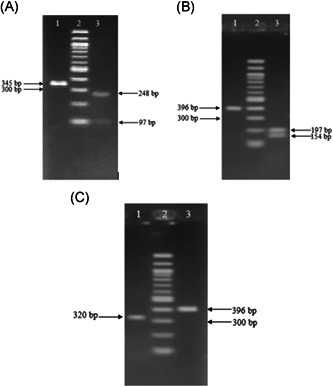
Restriction fragments for differentiating SARS‐CoV‐2 with SARS‐CoV on agarose gel electrophoresis. (A) Lanes 1 and 3: amplicons of E gene of SARS‐CoV and SARS‐CoV‐2 were digested with AluI; lane 2: DNA ladder 100 bp. (B) Lanes 1 and 3: amplicons of RdRp gene of SARS‐CoV and SARS‐CoV‐2 were digested with Tsp45I; lane 2: DNA ladder 100 bp. (C) Lanes 1 and 3: amplicons of RdRp gene of SARS‐CoV and SARS‐CoV‐2 were digested with EcoRI; lane 2: DNA ladder 100 bp. SARS‐CoV, severe acute respiratory syndrome coronavirus
Figure 2.
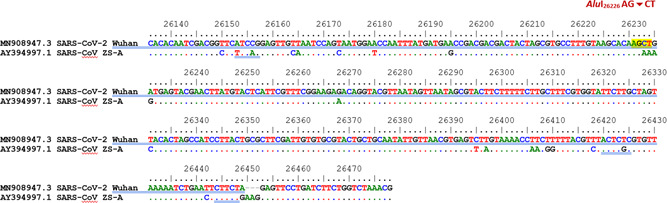
The location cut by restriction enzymes AluI on the E gene
Furthermore, for more reliable identification of SARS‐CoV‐2, the amplified product of the E gene for coronavirus was cut by AluI. Theoretically, there was only a restriction site on the E gene of SARS‐CoV‐2 recognized by AluI producing two fragments of 248 and 97 bp. The E gene amplicons of SARS‐CoV, on the contrary, was not cut by AluI (Figures 3 and 7).
To confirm the method proposed, PCR products were purified and cloned in pGEM‐T Easy vector (Promega, Madison, WI) and then sequenced using a 3130xl sequencer. Sequences obtained were aligned with reference sequences as presented in Table 1 using Bioedit 7.0 and MEGA 7.0 to construct a phylogenetic tree and identified by the BLASTn tool with default parameters at NCBI. The study results showed PCR‐RFLP was concordant with PCR‐sequencing phylogenetic analysis.
Table 1.
Reference sequences used in this study
| Access. No | Species | Reference source |
|---|---|---|
| AY508724.1 | SARS‐CoV | Genbank |
| AY394997.1 | SARS‐CoV | Genbank |
| MG772933.1 | bat‐SARS like coronavirus | Genbank |
| MG772934.1 | bat‐SARS like coronavirus | Genbank |
| MN908947.3 | SARS‐CoV‐2 | Genbank |
| MN985325.1 | SARS‐CoV‐2 | Genbank |
| MN988713.1 | SARS‐CoV‐2 | Genbank |
| LC522975.1 | SARS‐CoV‐2 | Genbank |
| NC019843.3 | Middle East respiratory syndrome coronavirus | Genbank |
| NC005831.2 | Human Coronavirus NL63 | Genbank |
| NC002645.1 | Human Coronavirus 229E | Genbank |
| AY391777.1 | Human Coronavirus OC43 | Genbank |
| NC006577.2 | Human Coronavirus HKU1 | Genbank |
| EPI_ISL_402119 | SARS‐CoV‐2 | GISAID |
| EPI_ISL_402120 | SARS‐CoV‐2 | GISAID |
| EPI_ISL_402125 | SARS‐CoV‐2 | GISAID |
| EPI_ISL_402128 | SARS‐CoV‐2 | GISAID |
| EPI_ISL_402132 | SARS‐CoV‐2 | GISAID |
| EPI_ISL_403932 | SARS‐CoV‐2 | GISAID |
| EPI_ISL_403962 | SARS‐CoV‐2 | GISAID |
| EPI_ISL_404228 | SARS‐CoV‐2 | GISAID |
| EPI_ISL_406844 | SARS‐CoV‐2 | GISAID |
| EPI_ISL_406596 | SARS‐CoV‐2 | GISAID |
| EPI_ISL_406597 | SARS‐CoV‐2 | GISAID |
Next, we evaluated the analytical sensitivity of our method and optimized WHO RT‐PCR were run simultaneously using serial 10‐fold dilutions of in vitro transcribed RNA ranging from 100 to 106 copies/µL. The limit of detection (LOD) was defined as the lowest RNA concentration detected at which 95% of all runs of the triplicates for RT‐PCR and eight replicates for WHO RT‐PCR by probit analysis. The result calculated the LOD of RT‐PCR for the E and the RdRp genes were at 204 copies/reaction and 70 copies/reaction, respectively, whereas LOD of WHO RT‐PCR estimated at 10.15 copies/reaction for the RdRp gene and 9.2 copies/reaction for the E gene in our conditions. We also evaluated the specificity of the assay on the coronavirus RNA specificity panel of four human coronaviruses (MERS‐CoV, human coronavirus [HCOV]‐OC43, HCOV‐229E, and HCOV‐NL63) obtained by the EVAg (https://www.european-virus-archive.com). 17 None of these coronaviruses were detected by our method, found to be 100% specific (Figure 5).
Figure 5.
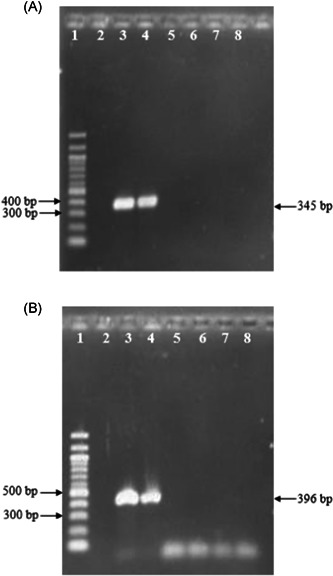
Specificity evaluation of one‐step RT‐PCR using coronavirus specificity panel. (A) Amplicons of E gene; lane 1: DNA ladder 100 bp; lane 2: negative control; lane 3: SARS‐CoV‐2; lane 4: SARS‐CoV; lane 5: MERS‐CoV; lane 6: HCOV‐OC43; lane 7: HCOV‐229E; lane 8: HCOV‐NL63. (B) Amplicons of RdRp gene; lane 1: DNA ladder 100 bp; lane 2: negative control; lane 3: SARS‐CoV‐2; lane 4: SARS‐CoV; lane 5: MERS‐CoV; lane 6: HCOV‐OC43; lane 7: HCOV‐229E; lane 8: HCOV‐NL63. HCOV, human coronavirus; MERS, Middle East respiratory syndrome; RT‐PCR, reverse transcription polymerase chain reaction; SARS‐CoV, severe acute respiratory syndrome coronavirus
To assess the diagnostic sensitivity and clinical performance of our assay, we tested on 20 specimens of patients with COVID‐19 and 50 specimens of suspected cases infected with SARS‐CoV‐2. All specimens were run simultaneously with both three methods including WHO RT‐PCR, Realstar SARS‐CoV‐2, and our assay. The results showed the diagnostic sensitivity of 95% and positive percent agreement of 95.8% when tested on clinical samples. Results are presented in Table 2. There was a sample negative with our method, but it was positive with both WHO RT‐PCR and Realstar SARS‐CoV‐2 assay at the Ct values of 38.56 and 31.76, respectively (Figures 6, 7, 8).
Table 2.
Comparative performance of molecular assays on clinical samples
| Molecular assays | WHO RT‐PCR | κ (SE) | PPA (%) | NPA (%) | ||
|---|---|---|---|---|---|---|
| Positive | Negative | |||||
| Realstar SARS‐CoV‐2 | Positive | 20 | 0 | 1 (<0.001) | 100 | 100 |
| Negative | 0 | 30 | ||||
| RT‐PCR | Positive | 19 | 0 | .958 (0.042) | 95 | 96.77 |
| Negative | 1 | 30 | ||||
Abbreviations: NPA, negative percent agreement; PPA, positive percent agreement; RT‐PCR, reverse transcription polymerase chain reaction; SARS‐CoV, severe acute respiratory syndrome coronavirus; SE, standard error; WHO, World Health Organization.
Figure 6.
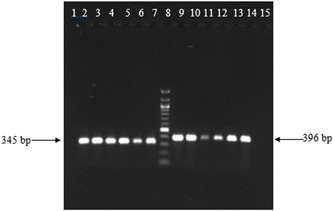
Agarose gel electrophoresis of one‐step RT‐PCR on clinical samples. Lanes 1 and 15: negative control; lanes 2 to 7: amplicons of E gene of SARS‐CoV‐2; lane 8: DNA ladder 100 bp; lanes 9 to 14: amplicons of RdRp gene of SARS‐CoV‐2. RT‐PCR, reverse transcription polymerase chain reaction; SARS‐CoV, severe acute respiratory syndrome coronavirus
Figure 8.
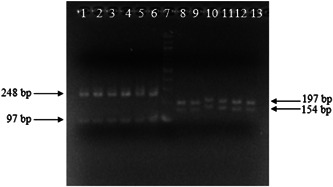
Agarose gel electrophoresis. lanes 1 to 6: amplicons of E gene of SARS‐CoV‐2 were digested with AluI; lanes 8 to 13: amplicons of RdRp gene of SARS‐CoV‐2 were digested with Tsp45I; lane 7: DNA ladder 100 bp. SARS‐CoV, severe acute respiratory syndrome coronavirus
4. DISCUSSION
Acute respiratory disease caused by a nCoV (SARS‐CoV‐2) has been recently discovered in China (December 2019). It posed a public health emergency, affecting both socioeconomic issues and global security. Until now, there is no vaccine and specific antivirals for the treatment of SARS‐CoV‐2. Therefore, it is crucial to establish a diagnostic method for suspected cases identification and surveillance of SARS‐CoV‐2. Several studies have shown that SARS‐CoV‐2 shares 79% to 82% identical to SARS‐CoV genome sequence. 7 , 18 This is a challenging problem for the selection of highly conserved regions to design primers and probes of molecular diagnostic assays of SARS‐CoV‐2. Recently, molecular assays based on real‐time RT‐PCR were developed by authors, but they were optimized on synthetic transcribed RNAs of SARS‐CoV‐2. 15 , 19 Therefore, it needs to be validated on sample panels of RNA extracted from the inactivated cell culture supernatant of SARS‐CoV‐2 or clinical specimens collected of confirmed patients before launching for clinical virology laboratories.
In this study, based on sequences analysis retrieved from GenBank, GISAID (Table 1) using bioinformatics tools to design primers and analysis restriction enzymes, we established a simple method for molecular detection of SARS‐CoV‐2 in differentiating with SARS‐CoV using RNA extracted from inactivated cell culture supernatant of SARS‐CoV‐2 and SARS‐CoV. This technique is rapid, effective‐cost, and does not require gene sequencing or real‐time RT‐PCR. In addition, it is a specific and definitive discriminatory test for SARS‐CoV‐2 and SARS‐CoV, providing a useful molecular tool for screening of suspected cases.
When assessed on clinical samples in comparison with other reference assays demonstrated a high sensitivity of 95% and clinical diagnostic performance for detection of SARS‐CoV‐2 with high κ values observed, confirming a high concordance with reference assays (Table 2). Interestingly, the specificity of the assay was 100% and no false‐positive results observed with other coronaviruses were tested in this study (Figure 5). To the best of our knowledge, this is the first report presented an in‐house RT‐PCR allowed for differential detection of SARS‐CoV‐2 with SARS‐CoV in Vietnam at the LOD of 70 copies/reaction for E gene and 204 copies/reaction for the RdRp gene, but it had higher LOD values as compared with the WHO RT‐PCR assay were 10.15 copies/reaction for the RdRp gene and 9.2 copies/reaction for the E gene. However, when evaluated on clinical specimens, there was only one discordant result observed, but this sample had the highest Ct values of 38.56 and 31.76 for WHO RT‐PCR and Realstar SARS‐CoV‐2 assays, respectively. This could be explained that this specimen had a low concentration of SARS‐CoV‐2 and it could be below the LOD of our assay. Recently, there was a publication that introduced an RT‐PCR assay for the detection of coronaviruses from four genera with the LOD ranged from 4 to 4 × 102 copies/reaction, depending on the CoV species tested. This assay detected all previously known CoVs, including SARS‐CoV‐2, but it could not distinguish SARS‐CoV with SARS‐CoV‐2. 20
In conclusion, in the present study, we successfully developed a simple and effective‐cost method for molecular detection of SARS‐CoV‐2 in differentiating with SARS‐CoV. This helps us to effectively control of COVID‐19 in resource‐challenged settings.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
HAS, DTTH, NDT, and HXS conceived, designed, and supervised the study and drafted and critically reviewed the manuscript. LTBQ, LTHT, and VTN performed the experiments, contributed to the acquisition of data and analysis and interpretation of data. NTS, LVN, and TVT contributed to material support, acquisition of data, and commented on the manuscript. LBQ, TTH, NTL, NVB, HVL, and DQ critically reviewed the manuscript.
ACKNOWLEDGMENTS
The authors would like to thank Dr Van Dinh Trang of the National Hospital of Tropical Diseases for excellent laboratory assistance with this study. The authors would also like to thank Dr Mark Bertram from Altona Diagnostics for providing the reagents used in this study. This study was supported by the Ministry of Science and Technology of Vietnam (Gran: 29/20‐ĐTĐL.CN‐CNN). The study was also supported by the European Virus Archive goes Global (EVAg) project that is supported by the European Union's Horizon 2020 research and the innovation program under Grant Agreement No. 653316.
Son HA, Hang DTT, Thuan ND, et al. A simple method for detection of a novel coronavirus (SARS‐CoV‐2) using one‐step RT‐PCR followed by restriction fragment length polymorphism. J Med Virol. 2020;92:2839–2846. 10.1002/jmv.26171
Ho Anh Son, Dinh Thi Thu Hang, and Nghiem Duc Thuan contributed equally to this study and are co‐first authors.
REFERENCES
- 1. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727‐733. 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. 2020.https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200223-sitrep-34-covid-19.pdf?sfvrsn=44ff8fd3_2. Accessed February 24, 2020.
- 3. Phan LT, Nguyen TV, Luong QC, et al. Importation and human‐to‐human transmission of a novel coronavirus in Vietnam. N Engl J Med. 2020;382:872‐874. 10.1056/NEJMc2001272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chan JF‐W, Yuan S, Kok K‐H, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person‐to‐person transmission: a study of a family cluster. The Lancet. 2020;395:514‐523. 10.1016/S0140-6736(20)30154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eurosurveillance Editorial Team . Note from the editors: World Health Organization declares novel coronavirus (2019‐nCoV) sixth public health emergency of international concern. Euro Surveill. 2020;25(5):200131e. 10.2807/1560-7917.ES.2020.25.5.200131e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang S, Shi Z, Shu Y, et al. A distinct name is needed for the new coronavirus. The Lancet. 2020;395:P949. 10.1016/S0140-6736(20)30419-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu S, Li A, Liu Y, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet Lond Engl. 2020;216:267‐274. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020;92:418‐423. 10.1002/jmv.25681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang X, Rayner S, Luo M‐H. Does SARS‐CoV‐2 has a longer incubation period than SARS and MERS? J Med Virol. 2020;92:476‐478. 10.1002/jmv.25708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Laboratory testing for 2019 novel coronavirus (2019‐nCoV) in suspected human cases. 2020. https://www.who.int/publications-detail/laboratory-testing-for-2019-novel-coronavirus-in-suspected-human-cases-20200117. Accessed February 3, 2020.
- 11. National Health Commission of the People's Republic of China . Technical guidance for laboratory testing of 2019‐nCoV infection (third edition). Biosaf Health. 2020;2:3‐5. 10.1016/j.bsheal.2020.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clinical Specimens: Novel Coronavirus 2019 (2019‐nCoV) | CDC. 2020. https://www.cdc.gov/coronavirus/2019-ncov/lab/guidelines-clinical-specimens.html. Accessed February 3, 2020.
- 13. Department of Health Science, Technology and Education, National Health Commission of People's Republic of China . Laboratory biosafety guide for 2019‐nCoV (Second Edition). Biosafety and Health. 2020;2(1):1‐2. 10.1016/j.bsheal.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen L, Liu W, Zhang Q, et al. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak. Emerg Microbes Infect. 2020;9(1):313‐319. 10.1080/22221751.2020.1725399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Corman VM, Landt O, Kaiser M, et al. Detection of 2019 novel coronavirus (2019‐nCoV) by real‐time RT‐PCR. Euro Surveill. 2020;25(3):2000045. 10.2807/1560-7917.ES.2020.25.3.2000045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. 2020.https://altona-diagnostics.com/files/public/Content%20Homepage/-%2002%20RealStar/INS%20-%20RUO%20-%20EN/RealStar%20SARS-CoV-2%20RT-PCR%20Kit%201.0_WEB_RUO_EN-S02.pdf. Accessed May 15, 2020.
- 17. Romette JL, Prat CM, Gould EA, et al. The European Virus Archive goes global: a growing resource for research. Antiviral Res. 2018;158:127‐134. 10.1016/j.antiviral.2018.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gralinski LE, Menachery VD. Return of the coronavirus: 2019‐nCoV. Viruses. 2020;12(2):135. 10.3390/v12020135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chu DKW, Pan Y, Cheng SMS, et al. Molecular diagnosis of a novel coronavirus (2019‐nCoV) causing an outbreak of pneumonia. Clin Chem. 2020;66:549‐555. 10.1093/clinchem/hvaa029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xiu L, Binder RA, Alarja NA, et al. A RT‐PCR assay for the detection of coronaviruses from four genera. J Clin Virol. 2020;128:104391. 10.1016/j.jcv.2020.104391 [DOI] [PMC free article] [PubMed] [Google Scholar]