

Abstract
Cells confront DNA damage in every cell cycle. Among the most deleterious types of DNA damage are DNA double-strand breaks, which can cause cell lethality if unrepaired or cancers if improperly repaired. In response to DNA double-strand breaks, cells activate a complex DNA damage checkpoint (DDC) response that arrests the cell cycle, reprograms gene expression and mobilizes DNA repair factors to prevent the inheritance of unrepaired and broken chromosomes. Here we examine the DDC, induced by DNA double-strand breaks, in the budding yeast model system and in mammals.
Keywords: Checkpoint, DNA Double-Strand Break, DNA Repair, Kinases, Cell Cycle
1. INTRODUCTION
DNA damage is a ubiquitous fact of life. Human cells encounter up to 100,000 instances of DNA damage per day (1), caused by a range of endogenous and exogenous insults. Even unperturbed DNA replication is intrinsically genotoxic and a major source of mutations and chromosome breakages (2). Failure to respond to DNA damage has dire consequences, and can result in mutations, gross chromosomal rearrangements and/or aneuploidy, which lead to disease, loss of fitness and death. In eukaryotes, cellular responses to most types of DNA damage involve a signaling transduction pathway, referred as the DNA damage checkpoint (DDC), which is responsible for sensing DNA damage and coordinating DNA repair transactions with the cell cycle and other key cellular processes.
1.1. Types of DNA Damage
Chromosomes are subjected to different forms of DNA damage, and the type of damage incurred dictates the subsequent repair mechanism. When damage is limited to one strand, the lesion is often repaired by excision of the damaged base followed by DNA synthesis using the opposite strand as a template. These single-stranded forms of DNA damage – chemical base modifications or misincorporation of ribonucleotides – are frequent and are efficiently corrected by the cell using either base excision repair or nucleotide excision repair mechanisms (reviewed in (3, 4)). However, photodimers, intra- and inter-strand crosslinks, among other obstacles, can block replication and may result in double strand breaks (DSBs) (5) (Figure 1). Moreover, single-strand nicks can be converted into one-ended DSBs upon replication fork passage. DSBs can arise directly from many additional sources, including ionizing radiation, the excision of transposable elements, failures of type II topoisomerases, and the action of site-specific endonucleases. Vertebrate cells suffer a dozen or more chromatid breaks every replication cycle. This is evident from the high frequency of chromatid breaks when the key Rad51 repair protein is depleted from chicken DT40 cells (6) and from the frequency of sister-chromatid exchanges in humans when the BLM helicase is absent (7). Unlike single stranded DNA damage, a DSB cannot be repaired by using the complementary strand as a template and instead, one of two repair mechanisms are employed; nonhomologous end joining (NHEJ) or homologous recombination (HR) (8). Site-specific endonucleases, which make DSBs at specific locations within the genome, have been the workhorses used to detailed mechanisms behind HR and NHEJ.
Figure 1: Sources of DSBs and the responses mediated by DDC kinases.
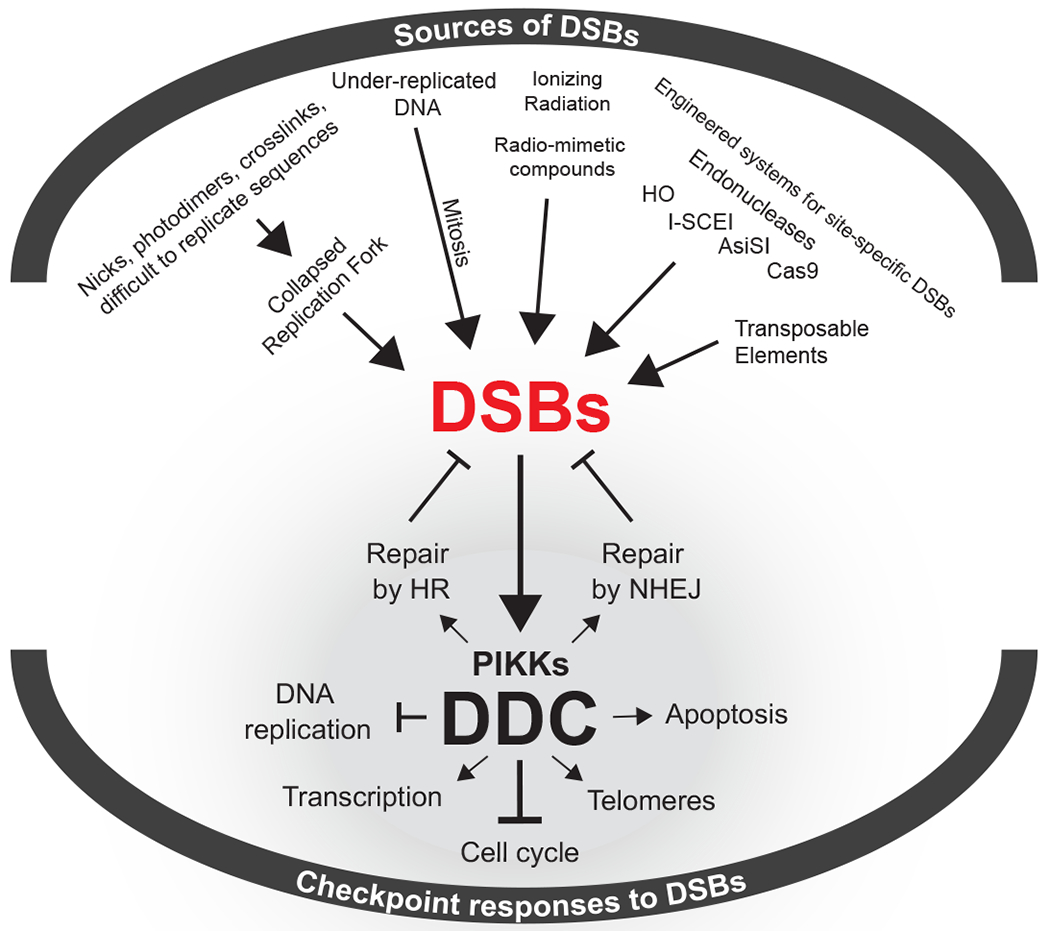
DSBs originating from endogenous or exogenous sources trigger the activation of DDC kinases that coordinate an intricate cellular response that includes cell cycle arrest and the mobilization of DSB-repair pathways.
1.1.1. Site-specific endonucleases as tools to study responses to DSBs.
Our understanding of the DNA damage response has also greatly been aided by site-specific endonucleases. The budding yeast Saccharomyces cerevisiae possess two endogenous endonucleases, HO and I-SceI, that have been the mainstay of experiments creating site-specific DSBs. I-SceI recognizes an 18 bp sequence and triggers a gene-drive event in mitochondria to spread an intron encoding the endonuclease (9). The creation of a synthetic I-SceI gene compatible with nuclear gene expression and mRNA translation has led to its use in many organisms, including mammals. The nuclear-encoded HO gene controls mating type switching in which a DSB in the MAT locus is repaired by homologous recombination using a donor sequence that introduces sequences of the opposite mating-type (reviewed in (10)). HO requires a 24-bp sequence that can be moved to other locations to study various types of DSB repair, including nonhomologous end-joining (NHEJ), microhomology-mediated end-joining (MMEJ), and various forms of homologous recombination (HR), including single-strand annealing (SSA), gene conversions with and without an accompanying crossover and break-induced replication (reviewed in (11, 12)). Creation of an inducible HO gene fused to a galactose-inducible promoter makes it possible to induce synchronous cleavage of one or several chromosomal sites in less than 60 min. A special advantage of HO is that it is rapidly degraded, allowing the damage to be inflicted transiently (13). Induction of I-SceI is less efficient and the protein is not rapidly degraded. Recently, CRISPR/Cas9-mediated DSBs have also been employed in yeast (14).
In mammals, site-specific endonucleases including I-SceI have also been used to study both repair and DNA damage responses. AsiSI, which cleaves about 100 sites in the mammalian genome has proven to be particularly useful in distinguishing roles of different DNA damage associated kinases, ATM and DNA-PKcs (15). The attachment of an auxin-induced degron to AsiSI has also allowed the rapid depletion of the nuclease after cleavage (16). The advent of Cas9-directed cleavage has stimulated an explosion of papers concerned with gene editing both by NHEJ and HR mechanisms (reviewed by (17)). Cas9 has also been used to demonstrate that even a small number of such DSBs are sufficient to retard cell cycle progression, by activation of DNA damage signaling (18).
1.2. Repair of Double-Strand Breaks
The reader is referred to several recent reviews that explore DSB repair in detail (8, 11, 12, 19, 20). Here we will confine our introduction to identifying key processes that are implicated in the generation and regulation of the DDC. .
The core machinery both in NHEJ and HR are evolutionarily conserved, although mammalian repair processes are overlaid with a number of proteins absent in budding yeast (e.g. DNA-PKcs in NHEJ and BRCA1 and BRCA2 (among others) in HR). In G1 cells, DSBs are predominantly repaired by NHEJ, because the 5’ to 3’ resection of DSB ends that is required for HR and for MMEJ is inhibited. DSB ends are recognized both by the MRE11-RAD50-NBS1 complex (Mre11-Rad50-Xrs2 in budding yeast), which recruits the ATM (Tel1) kinase, and by KU70-KU80 (hereafter Ku) proteins. Ku binding recruits DNA ligase 4 and its associated XRCC4 (yeast Lif1) and XLF (yeast Nej1) proteins to the break to effect NHEJ. NHEJ continues to function throughout the cell cycle, but once 5’ to 3’ resection is initiated in cells that have entered S phase, HR is enabled, principally to repair replication-induced breaks by recombination with a sister chromatid, but also between homologous chromosomes or with a homologous sequence located at an ectopic site. 5’ to 3’ resection leads to 3’-ended single-strand DNA (ssDNA) that is initially coated by the 3-member ssDNA binding protein complex, RPA. The activation of ATR (Mec1) depends on the binding of its obligate heterodimer partner, ATRIP (Ddc2) to RPA. Rad51 displaces RPA on ssDNA then initiates strand exchange between the broken end(s) and a homologous donor sequence. In budding yeast, Rad51 is loaded on to the DNA by the mediator Rad52 and a set of Rad51 paralogues. In mammals the principal loader of RAD51 is BRCA2, along with a set of RAD51 paralogs.
2. THE DNA DAMAGE CHECKPOINT
The first mutants found to be defective in mitotic delay following UV damage were uncovered in the fission yeast Schizosaccharomyces pombe by Hannan, Miller and Nasim (21). Painter and Young (22) later reported that cell lines derived from ataxia-telangiectasia patients underwent DNA synthesis after x-radiation while healthy cells did not. Weinert and Hartwell began dissecting the DDC in S. cerevisiae using a UV-sensitive mutation of RAD9 that failed to arrest in the G2/M phase of the cell cycle before completing DNA repair (23). Subsequent work by many investigators over the following three decades has revealed the general mechanism of checkpoint activation, maintenance, and deactivation. In this review we focus on studies in budding yeast and in mammals. Important contributions have also been made using fission yeast, especially in the study of checkpoint responses to DNA replication stress (24), which is beyond the scope of this review.
2.1. The Cell Cycle and the DNA Damage Checkpoint
In budding yeast, the DDC can be activated in three phases of the cell cycle. DNA damage incurred in G1 activates a transient DNA damage response that temporarily delays S-phase onset, providing extra time for DNA repair before replication (25, 26). Damage that arises during S-phase slows replication and triggers a coordinated effort between replication fork and DNA repair machinery to resolve the damage (27). DNA lesions still present after completion of DNA replication activate the G2/M checkpoint, which stalls cell division until the damage is repaired (23). If repair is successful, the DDC is extinguished, and cells proceed through mitosis in a process known as recovery (28). However, sustained DNA damage does not prevent cell division indefinitely, as both budding yeast and metazoans will deactivate the checkpoint and proceed through cell division without repairing DNA, a process called adaptation (29, 30).
DDC in yeast is quite sensitive, with a single DSB being capable of activating the DDC and evoking a robust G2/M arrest (31). In mammalian cells, a few DSBs (1-4 breaks) can mildly activate the DDC and result in only minor effects in cell cycle progression (18). It has been documented that mammalian cells can often carry DNA lesions induced by replication stress to the following G1 cycle (32, 33), likely reflecting the higher DNA damage thresholds required for DDC activation and imposition of cell cycle arrest in mammals. As a consequence, vertebrates are likely more prone to encounter unrepaired DNA in the following G1 phase, and could be more reliant on G1 checkpoints for inducing cell cycle arrest upon low levels of DNA damage (34).
2.2. Primary Control of the DNA Damage Checkpoint: PIKK kinases
In mammals, DNA damage signaling is initiated by two members of the phosphoinositide 3-kinase-related kinase (PIKK) family of proteins, ATM (ataxia telangiectasia mutated) and ATR (ATM and rad3-related). A third PIKK, DNA-PKcs, is also involved (35), although its role in the control of DDC signaling is less well understood. Both ATM and ATR phosphorylate SQ/TQ residues in effector proteins to launch a cascade of signals that establishes a transcriptional program and prevents cell division (Figure 2) (36, 37). Budding yeast homologs of ATM (Tel1) and ATR (Mec1) carry out similar roles, although, as discussed below, they have exchanged some roles compared to their mammalian homologues. Budding yeast lack DNA-PKcs. Key proteins that control the DNA damage response are listed in Table 1.
Figure 2: The DDC signaling network in (A) budding yeast and (B) mammals.
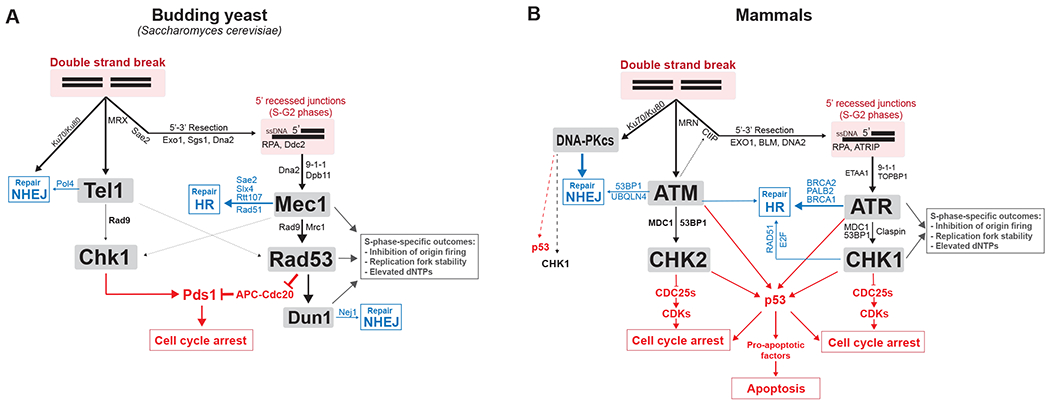
Simplified network view of the role of DDC kinases with their main co-factors, regulators, adaptors and substrates. Blue indicates key effectors through which DDC kinases regulate DNA repair of DSBs. Red indicates key effectors through which DDC kinases mediate cell cycle arrest and apoptosis. More information about substrates involved in DNA repair can be found here (197).
Table 1:
Selection of DNA damage checkpoint and repair proteins.
| Category | S. cerevisiae | Humans | Note |
|---|---|---|---|
| PI3K-like kinases | Mec1 | ATR | PIKK-like kinase; initiates checkpoint signaling |
| Tel1 | ATM | PIKK-like kinase; initiates checkpoint signaling | |
| Clamp loader | Rad24 | Rad17 | 9-1-1 clamp loader in complex with Rfc2-5 |
| 9-1-1 clamp | Ddc1 | Rad9 | Clamps ds/ssDNA junctions |
| Mec3 | Hus1 | ||
| Rad17 | Rad1 | ||
| Adaptor proteins | Rad9 | 53BP1, MDC1 | Facilitates activation of downstream checkpoint kinases |
| Dbp11 | TOPBP1 | Activator of Mec1/ATR. Also functions as adaptor to couple 9-1-1 to checkpoint adaptors and DNA repair proteins | |
| Sensor | Ddc2 | ATRIP | Obligate binding partner of Mec1; recruits Mec1 to RPA coated ssDNA |
| MRX complex | Mre11 | Mre11 | Nuclease |
| Rad50 | Rad50 | ||
| Xrs2 | NBS1 | ||
| Downstream checkpoint kinases | Rad53 | CHK2 | Primary kinase responsible for checkpoint signal propagation |
| Chk1 | CHK1 | Stabilizes Pds1 by phosphorylation | |
| Securin | Pds1 | Securin | Inhibitor of Esp1seperase; degraded by APC-Cdc20 |
| Separase | Esp1 | Separase | Cleaves cohesin rings for anaphase onset |
| Spindle assembly checkpoint | Cdc20 | p55 | Specificity factor for APC E3 ubiquitin ligase |
| Mad2 | MAD2L1 | Mitotic spindle assembly checkpoint protein | |
| Repair Proteins | Yku70/Yku80 | Ku70/Ku80 | DSB sensor; required for NHEJ |
| Rad51 | Rad51 | Recombinase; binds RPA coated ssDNA; performs homology search during HR | |
| Rad52 | BRCA2 | Deposits Rad51; stimulates strand exchange | |
| Resection | Dna2 | DNA2 | 5’ - 3’ endonuclease (budding yeast Dna2 is also an activator of Mec1) |
| Exo1 | EXO1 | 5’ – 3’ endonuclease; cooperates with Sgs1 helicase | |
| Sae2 | CtIP | Endonuclease; cooperates with MRX to initiate end processing | |
| Phosphatases involved in DDC deactivation | Ptc2, Ptc3 | Type 2C phosphatase (PP2C) | |
| Pph3 | PP4 | Pph3 is the catalytic subunit of budding yeast type 4 phosphatase (PP4) | |
| PP1, PP2A | Type 1 and 2A phosphatase complexes | ||
| Adaptation kinase | Cdc5 | PLK1 | Polo-like kinase required for adaptation to DNA damage |
2.3. ATM and ATR structures
ATM and ATR are part of the phosphatidylinositol 3-kinase related kinases, all of which are very large proteins whose structures have only recently been solved at atomic or near-atomic resolution. All of these proteins have a similar amino acid sequence architecture, with a large series of HEAT repeats at the N-terminus followed by a FAT domain, the kinase domain and a FAT-associated C-terminal FATC domain (Figure 3A and 3B). The HEAT repeats consist of units containing two alpha helices joined by a short linker. ATR has 45 such repeats and ATM even more (38). These repeats can form a highly ordered structure that facilitates monomer-monomer interactions and likely the binding of other proteins; but in the case of ATR, a single amino acid change in HEAT repeat 27 results in hyperactivation of the kinase (39). The FAT domain is a conserved ~500 amino acid domain shared by several kinase families (DNA-PKcs, ATM, ATR and TRRAP) (40); its function is poorly defined. In ATM, a conserved serine 1961 is phosphorylated after ATM activation; but how this modification leads to dissociation of the dimer is not known (41). The actual kinase domain of these PIKK kinases constitutes only a small fraction of the total protein; single amino acid mutations replacing a conserved aspartate are kinase-inactive. Within the kinase domain is a PIKK regulatory domain (PRD) that is required for activity and for interaction with ATR’s co-activator, TOPBP1 (42).
Figure 3: ATM and ATR structures.
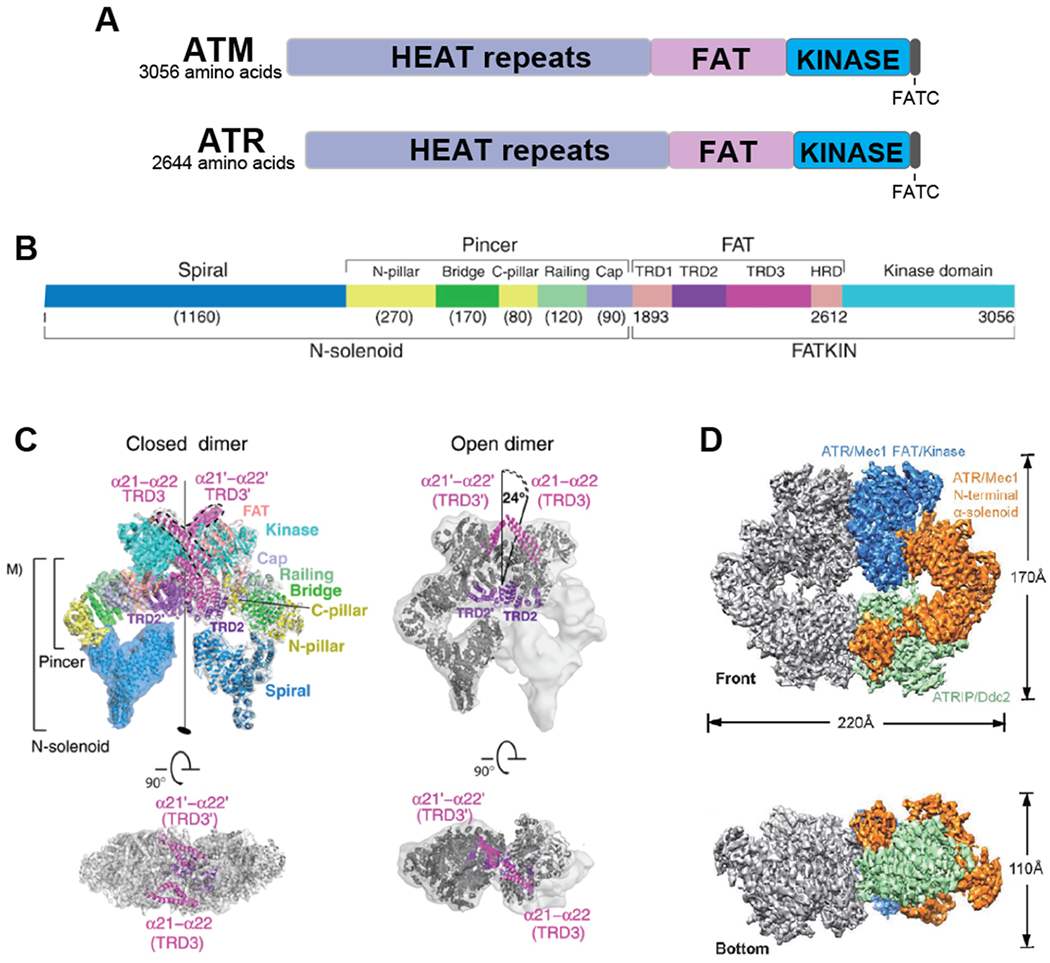
A. Overall domain structure of ATM and ATR. B. A more detailed structure of ATM including structural features identified by cryo-electron microscopy (45). C. Cryo-electron microscope-derived open (presumably active) and closed structures of ATM, as determined by Baretic et al. (45). The FAT and kinase region (FATKIN) has been solved at a higher resolution than the N-terminal solenoid domain. In the closed form, the conformation of the active site is maintained by interaction with a long helical hairpin in the TRD3 (tetratricopeptide repeats domain 3) domain. D. Electron microscope-derived structure of yeast Mec1-Ddc2 (ATR-ATRIP), determined by Wang et al. (47).
The C-terminal of these proteins includes a FATC domain for which several functions have been noted. FATC domains of DNA-PKcs, ATM and TRRAP all interact with the chromatin remodeler, histone acetyltransferase Tip60 (43). In addition, the C-terminus of budding yeast’s ATM homolog, Tel1, appears to be required for the protein’s association with broken chromosome ends and for full kinase activity (44).
Both mTOR and DNA-PKcs have been solved by X-ray crystallography and those data, combined with recent advances in electron microscopy, have yielded our first images of ATM (45, 46) and ATR (47) structures. ATM proves to be a front-to-front homodimer, as illustrated in Figure 3C. The arrangement of the two monomers does not reveal how the kinase domain would contact serine 1961, whose autophosphorylation has been correlated with kinase activation. ATR is found as a dimer of heterodimers, with the ATR-associated ATRIP protein that is required for ATR to bind to broken DNA ends. The most detailed structural information has come from studying the budding yeast proteins, Mec1/ATR and Ddc2/ATRIP (47). This 3.9Å structure reveals the amino acids needed for both ATRIP-ATR association and for dimer stability (Figure 3D). The structure of the active site suggests that several HEAT repeats come into contact (including the repeat analogous to the human repeat 27 whose S1333A mutation was implicated in hyperactivating the kinase).
2.4. Activation of the DDC: Detection of DSBs
Detection of broken DNA ends is the initiating event both for DSB repair and for checkpoint activation (Figure 4). Experimentally, DSBs are created by ionizing radiation, by clastogens such as bleomycin, and by site-specific endonuclease cleavages (Figure 1). Breaks created by ionizing radiation may be more complex, terminating in glycosidic fragments of the deoxyribose-phosphate backbone, while chemically and enzymatically-created ends have 5’ phosphates and 3’ hydroxyls at the sites of cleavage. Hence the recognition of the broken ends may differ between “clean” and “dirty” DSBs (48). In yeast, chromosome breaks created by rupture of a dicentric chromosome proved to have the same pattern of repair as those induced by HO endonuclease (49).
Figure 4: Detection of DSBs by PIKKs and downstream DDC signaling in budding yeast.
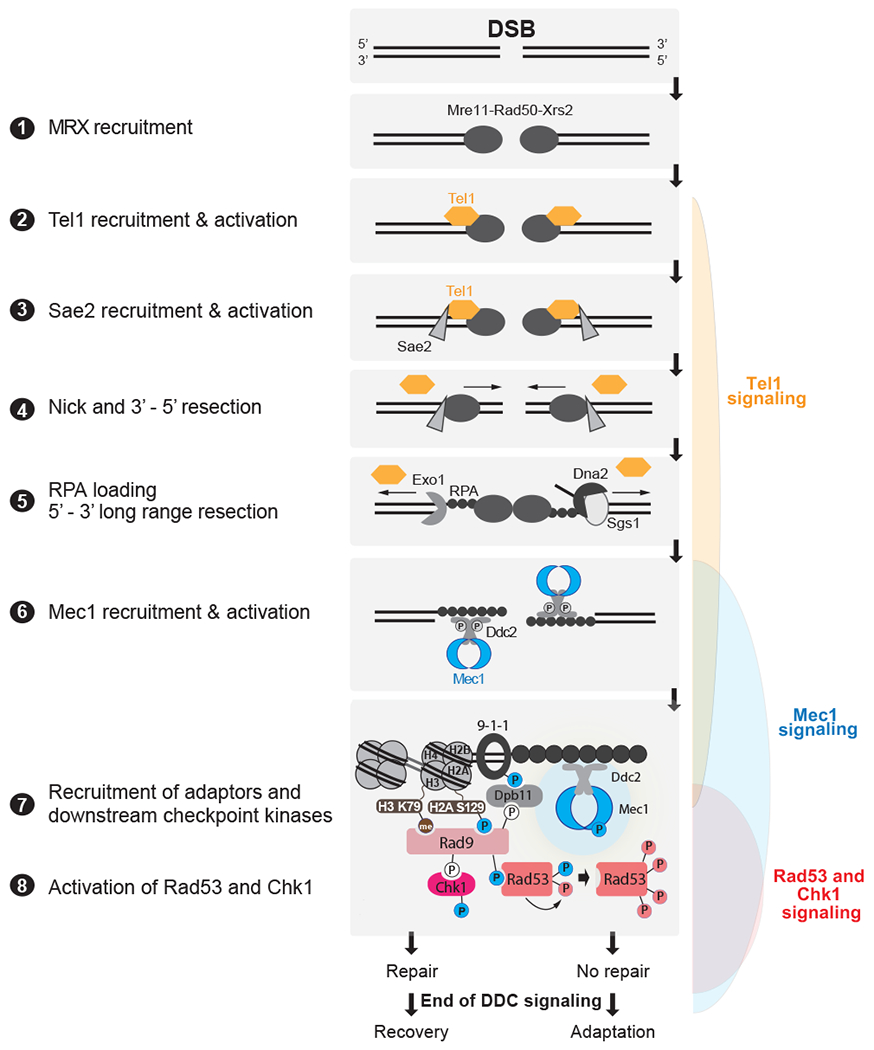
DSBs are first recognized by the MRX complex, which binds to the broken ends. MRX recruits the PIK kinase Tel1 and Sae2 to begin DSB end processing by Exo1 and Dna2. RPA coats ssDNA, leading to the recruitment of Mec1-Ddc2 dimers. The 9-1-1 clamp loader assembles the 9-1-1 clamp at the 5’ recessed end of the dsDNA/ssDNA junction. Dpb11 is recruited to 9-1-1 via a Mec1-dependent phosphorylation in the Ddc1 subunit. Mec1 and Tel1 phosphorylate histone H2A on S129 (Y-H2AX). Rad9 bound to Chkl is recruited to Y-H2AX through Rad9’s BRCT domain and histone H3K79me through Rad9’s TUDOR domain. Rad9 is shown here as monomeric for simplicity. Mec1 then phosphorylates and activates Chk1 and phosphorylates Rad9, priming Rad9 for Rad53 recruitment. Rad53 binds phosphorylated Rad9 allowing for Mec1-dependent phosphorylation and activation. Activated Rad53 phosphorylates and activates other Rad53 molecules leading to full checkpoint activation. Tel1 in yeast contributes a very modest role to activating Rad53 and Chk1. Bottom most panel shows key interactions and phosphorylation events involved in activation of the DDC. Color of phosphorylation sites (circles with “P”) refer to the kinase responsible: blue=Mec1, red=Rad53, white=CDK, gray=unknown. See text for additional details.
A key modulator of repair and damage signaling is the MRN (MRX in budding yeast) complex, which plays multiple roles in DNA end-recognition and end-processing. Dirty ends and ends that are blocked by protein adducts can be “cleaned up” by the action of the Mre11-Rad50 nuclease complex (50). Mre11-Rad50, along with an adaptor/chaperone protein Nbs1/Xrs2 associate with an enhancer of the nuclease activity of the complex, CtIP/Sae2 (51).
The MR complexes also can bridge DSB ends and play an important but not exclusive role in tethering the two ends of a DSB together (52). Consequently, when a DSB in budding yeast is flanked with LacO arrays to which LacI-YFP can bind (53) or when Rad51-GFP binds to resected DSB ends (54), one sees only a single fluorescent focus, indicating that the ends are tethered. Disruption of the MRX complex results in a fraction of the ends forming two distinct foci (55), but there are other, undefined mechanisms that hold broken ends together as well.
MRX also plays a role in displacing tightly bound proteins from DSB ends by nicking the DNA and then resecting in a 3’ to 5’ direction, thus removing a short (20-50 nt) region of one strand and creating a 3’ overhang on the adjacent strand (56). The Ku70/Ku80 heterodimer is displaced in this way, allowing for access to the DSB end by the Exo1 5’ to 3’ exonuclease (57, 58). Both Ku and MRX are rapidly recruited to DSB ends (59). A similar role for Mre11-Rad50-Nbs1 in displacing Ku (and MRN itself) has been shown in S. pombe (60).
2.4.1. Resection of the broken ends.
Activation of PIKKs at DSBs is profoundly impacted by resection of DSB ends. Loss of CtIP in vertebrate cells strongly prevents resection and the formation of RPA foci (61); but in budding yeast, loss of MRX/Sae2 only retards resection near the DSB by a factor of two and doesn’t impair long-range resection (62–64). In G2/M-arrested budding yeast cells, loss of the MRX complex blocks resection profoundly (65), suggesting that there is an MRX-independent resection process in S phase. Long range 5’ – 3’ resection in both budding yeast and mammals is carried out by two partially redundant exonucleases, Exo1 and a complex containing the endonuclease Dna2 and the Sgs1/BLM helicase, which is associated with Top3 and Rmi1 (56, 64, 66). In budding yeast, these two activities act independently to resect DNA at the slow rate of about 1 nt/sec (~4 kb/h) (67). In mammals, it has been difficult to measure a resection rate; one estimate by measuring RPA binding to ssDNA suggests a rate of only 0.2 kb/h (68). However, as in yeast, resection in mammalian cells can continue for tens of kb, as measured by single-strand annealing assays (69). Exo1 removes single nucleotides from the DSB end, whereas the Sgs1/BLM and Dna2-dependent process involves a helicase/endonuclease cleavage that liberates short oligonucleotides (70–72). In budding yeast, deleting either EXO1 or DNA2 results in a slight defect in resection but inactivation of both severely cripples resection, preventing DDC activation (73). In mammalian cells, there is a different relationship of the BLM helicase to resection: BLM promotes both Exo1 and Dna2’s cleavage of DNA (74). RPA stimulates resection activity initially, but later checkpoint kinase-dependent phosphorylation of RPA appears to retard subsequent resection (75).
Resection is also tightly controlled by the cell cycle. Both Sae2 and Dna2 are targets of Cdc28 (budding yeast cyclin-dependent kinase, CDK1) for activation by phosphorylation (66, 76). Alanine substitution of CDK phosphorylation sites within Sae2 markedly reduces resection (66), while similar substitutions of 3 CDK phospho-sites in Dna2 has a mild effect, as Exo1 is still active (76). The G1 block to resection in budding yeast can be overcome by inactivating Ku proteins, enabling Exo1 to access the DSB end and thereby allowing resection even though CDK remains inactive (77).
In mammals, resection is under similar cell cycle regulation, with CDK also being crucial to promote resection by phosphorylating CtIP, the functional ortholog of Sae2 (61). Overall, in both yeast and mammals, cell cycle-dependent control of resection becomes a major determinant of how, and which, PIKKs are activated and the type of DDC response is mounted.
2.4.2. Activation of Tel1/ATM and Mec1/ATR.
MRX/MRN play a key role in the recruitment and activation of Tel1/ATM to a blunt, unresected, DSB end. In both budding yeast and mammals the C-terminus of Xrs2/Nbs1 appears to harbor a Tel1/ATM-interacting domain (78), but recent work suggests that in mammals, ATM activation is a property of MRE11- RAD50 themselves (79). In contrast, Mec1/ATR is not recruited to blunt or nearly blunt ends. Instead, Mec1/ATR recruitment requires that the DSB ends be resected (Figure 4); consequently, in G1-arrested cells and in other conditions where resection of DSB ends is blocked, Mec1/ATR is not activated (80, 81). Mec1 can be activated in the absence of a DSB, in G1-arrested cells, by treating cells with the UV-mimetic drug, 4NQO, when there are multiple RPA-bound single-stranded regions resulting from nucleotide excision repair that could attract Ddc2-Mec1 to these regions and activate the checkpoint (81). In fact, Mec1 can be activated even in the absence of DNA damage by tethering multiple copies of LacI-Ddc2 and LacI-Ddc1 to an array of LacO sequences (82).This activation can occur in G2/M-arrested cells and requires the scaffold protein Rad9 (discussed in more detail below) to mediate phosphorylation of Rad53. Rad9’s recruitment presumably requires that Mec1 phosphorylate nearby histone H2A (γ-H2AX), because the level of phosphorylation of Rad53 was markedly less when cells carry a non-phosphorylatable histone H2A-S129 mutation.
Once resection begins, the checkpoint signaling is switched from Tel1/ATM to Mec1/ATR -dependence (Figure 4) (83). Mec1/ATR recruitment to the break is mediated by its obligate binding partner, Ddc2/ATRIP (also known as Lcd1), which binds to RPA bound to ssDNA (84). Following recruitment, at least two pathways exist for Mec1/ATR activation (reviewed in more detail by (85, 86)). First, Mec1/ATR can be activated by the Dpb11/TOPBP1 scaffold, which is recruited to DNA lesions at ss/dsDNA junctions created by end resection, mediated by the 9-1-1 checkpoint clamp complex (Ddc1-Rad17-Mec3/RAD9-RAD1-HUS1) (87, 88). In budding yeast, both Dpb11 and the 9-1-1 component Ddc1 have Mec1-activating domains, whereas in mammals an ATR-activating domain is present in TOPBP1, but absent in the 9-1-1 complex. A second mode of Mec1/ATR activation has been recently uncovered involving the replication/DNA damage-associated protein Dna2 (yeast) and ETAA1 (mammals) (89–92). Interestingly, differently from TOPBP1, which depends on a ss/dsDNA junction for recruitment (via 9-1-1 loading), ETAA1, which interacts with RPA, was proposed to mediate ATR activation at long stretches of RPA-coated ssDNA, therefore providing a system capable of sensing ssDNA-containing structures that may not efficiently activate ATR through TOPBP1.
2.5. A Cascade of Signaling
The activation of Tel1/ATM and Mec1/ATR launches a broad cellular response, resulting from the phosphorylation of a number of substrates, many of which are themselves kinases with different specificities. In mammals, the principal kinase activated by ATM is CHK2, while ATR activates CHK1 (Figure 2B) and both kinases are important in the damage response. In budding yeast, the division of labor is different; Mec1 (ATR) activates Rad53 (CHK2) and contributes to Chk1 activation while Tel1 (ATM) normally contributes a very modest role to activating the downstream DDC kinases (Figure 2A). In mec1Δ mutants, however, deletion of Sae2 reveals an Mre11/Tel1-dependent checkpoint response (called the TM pathway) that leads to robust Rad53 phosphorylation; this Mec1 bypass has been little studied (93).
2.5.1. Phosphorylation of histone H2A.
In mammals, one very rapidly appearing phosphorylation is that of the C-terminal SQ site of the histone variant H2AX, referred to as γ-H2AX. γ-H2AX appears within minutes of exposure to ionizing radiation and can spread over 1 Mb around the break site (94). All three human checkpoint PIKKs have been shown to carry out this modification. The magnitude of the γ-H2AX modification by ATM is facilitated by the γ-H2AX-binding protein MDC1, but it does not affect the extent of such spreading (95). In mammals, propagation of γ-H2AX appears to transiently inhibit transcription (96); but there are also new transcripts that appear associated with the DSB ends (97). There is no such transient inhibition of transcription in budding yeast, where gene expression around the DSB site continues until 5’ to 3’ resection renders genes single-stranded (98, 99).
In budding yeast, there is no distinct H2AX isoform but core histone H2A is phosphorylated at the equivalent C-terminal SQ site. Spreading of γ-H2AX around a site-specific DSB in budding yeast occurs soon after DSB formation and, in about 30 min, reaches ~50 kb on either side of the DSB (80, 100). Spreading is not uniform: transcriptionally active areas adjacent the DSB are refractory to γ-H2AX modification, but when transcription is turned off, γ-H2AX is quickly established within the locus, specifically by Mec1 (101). How Mec1 and Tel1 coordinate their activity to rapidly spread γ-H2AX is unknown but Mec1 is able to spread γ-H2AX to an adjacent unbroken chromosome in trans (101, 102). A detailed kinetic analysis of γ-H2AX suggests that Tel1 primarily moves down the chromatin (1-dimensional diffusion) whereas Mec1 either diffuses from its initial binding site or comes into contact by looping from the end (K. Li, G. Bronk, J. Kondev and J.E. Haber, unpublished) (101).
2.5.2. DNA damage checkpoint adaptors.
Transduction of signaling from PIKKs to downstream checkpoint kinases Rad53/CHK2 and Chk1/CHK1 requires adaptor proteins that recruit these kinases within close proximity of PIKKs engaged at the DSB ends or nearby ssDNA. The molecular events and determinants of such transduction process are best understood in budding yeast. While attempts have been made to expand the findings from yeast to mammals, many gaps in our understanding of the mammalian system remain, likely due to the increased complexity and redundancy of the proteins and regulatory mechanisms involved.
2.6.2.1. Budding Yeast Rad9: a checkpoint adaptor paradigm.
Rad9 is a large (150 kDa) scaffold protein required for robust G2/M checkpoint activation (Figure 4). Cells lacking RAD9 exhibit a very brief checkpoint arrest after a single DSB, whereas mec1Δ sml1Δ cells have no arrest at all (103, 104).
Rad9 contains two chromatin interacting domains; a TUDOR domain that binds histone H3 trimethylated on lysine 79 (H3K79me3) and tandem BRCT domains that recognize γ-H2AX (105, 106). H3K79me3 is found constitutively throughout the genome, established by the histone methyltransferase Dot1 (107, 108). Full DDC activation requires both Rad9 domains and therefore both histone modifications H3K79me3 and γ-H2AX. It is unclear whether Rad9 binds two histones within the same nucleosome, or adjacent nucleosomes.
Recruitment of Rad9 to DNA lesions is also dependent on the scaffold Dpb11 and the 9-1-1 clamp member Ddc1 (109, 110) (Figure 4). Dpb11 contains a pair of tandem BRCT domains that bind phosphoproteins – one binds Rad9 and other binds Ddc1 (111, 112). Dpb11’s interaction with Rad9 is also regulated by phosphorylation of two S/TP residues in Rad9, S462 and T474, both of which are CDK consensus sites (110). Originally, it was proposed that CDK phosphorylation of these sites restricts Rad9-Dpb11 binding to G2, when CDK activity is high. However, a recent report found that these residues are phosphorylated in Rad9 during G1 even upon direct CDK inhibition (113), suggesting additional kinases are involved and implying more complex cell cycle regulation.
Upon recruitment to a DSB, Rad9 is phosphorylated by Mec1 and Tel1 on multiple SQ/TQ sites. These phosphorylations serve two purposes: to promote Rad9 multimerization mediated through its BRCT domains and to prime docking sites on Rad9 to which the Rad53 kinase binds (114, 115). Mutations that impair Rad9 oligomerization do not prevent Rad53 activation, but checkpoint maintenance is lost, indicating that oligomerization is need to sustain checkpoint signaling (115).
In mammals, the identity and precise roles of DDC adaptors are not as well defined as in yeast and remain somewhat controversial. 53BP1 and MDC1 were shown to participate in signal transduction from ATM and ATR to CHK2 and CHK1 (116–121). Both 53BP1 and MDC1 are large BRCT-domain containing scaffolds that share functional similarities with Rad9, including the ability to directly bind to γ-H2AX (122–124) and to the Dpb11/TOPBP1 scaffold (125–127). 53BP1 is the mammalian ortholog of budding yeast Rad9, and both proteins also share key roles in limiting resection (128–130). A key difference between yeast Rad9 and 53BP1 is that the latter directly binds to N-terminally ubiquitylated H2A – a modification lacking in budding yeast - and uses this chromatin modification as a major recruitment mechanism (131). In both yeast and vertebrates, the Mrc1/Claspin adaptor has a well-established role in transducing Mec1/ATR signaling to Rad53/CHK1 at stalled replication forks (132, 133), and could in principle participate in the DDC response to a DSB occurring at the replication fork.
2.5.3. Yeast Rad53 and Chk1 checkpoint kinases.
Rad53 serves as the primary DDC signal transducer in budding yeast. Like Mec1, Rad53 is an essential gene, whose deletion can be propagated by raising the level of dNTPs (134), either by overexpressing ribonucleotide reductase (RNR) (135), by deleting the RNR inhibitor gene SML1 (136) or by deleting the RNR transcriptional repressor gene CRT1 (137). Upon activation, Rad53 governs a widespread transcriptional response (138), largely through the activation of MBF-dependent transcription (analogous to human E2F) (139, 140) and through activation of the downstream Dun1 kinase, which controls transcription factors such as Crt1 and Ndd1 (137, 141). Rad53 also triggers a Dun1-independent phosphorylation of the repressor, Rph1, allowing upregulation of the Phr1 photolyase as well as several key genes in the autophagy pathway (142, 143). Rad53 helps to restrain mitosis (see section 2.7. below) and rad53Δ mutants display a much-shortened length of checkpoint arrest following a DSB (104). Cells lacking Rad53 signaling are sensitive to numerous DNA damaging agents (144, 145).
After Rad53 is recruited to SQ/TQ phosphorylation sites on Rad9 (114, 146, 147), its proximity to Mec1 allows for initial Mec1-dependent phosphorylation on Rad53’s N-terminal SQ/TQ cluster domain (SCD) (148). Once activated, Rad53 hyperphosphorylates other nearby Rad53 molecules in trans resulting in a population of fully activated Rad53, which then dissociates from Rad9 (149). While Rad9 promotes Rad53 activation, Rad53 also limits Rad9 oligomerization by phosphorylating and disrupting Rad9’s trans BRCT domain interactions thereby limiting its own activation through a negative feedback loop (115). In vitro, Rad53 autophosphorylation and activation can be achieved simply by self-oligomerization, even in the absence of Rad9 and Mec1, suggesting that the role of Rad9 and Mec1 in Rad53 activation is to promote localized accumulation of Rad53 and not necessarily direct activation (150).
Chromatin remodelers, which remove and restructure nucleosomes, have been implicated in regulating Rad53 activation (151, 152). The yeast INO80 chromatin remodeling complex interacts with Rad53 in vitro and in vivo following MMS treatment, dependent on an SQ phosphorylation site within the INO80 subunit Ies4 (153), indicating that INO80 activity is regulated by PIKK-dependent phosphorylation. ies4Δ cells and Ies4 mutants lacking SQ phosphorylation sites display reduced Rad53 phosphorylation after MMS treatment. This defect is further exacerbated in ies4Δ rad9Δ double mutants, suggesting that INO80-dependent Rad53 activation functions in parallel to Rad9’s activation (153).
Budding yeast Chk1 plays a relatively minor role in DNA damage signaling transduction. chk1Δ mutants display only a slight reduction in cell cycle delay after a single DSB and chk1Δ rad53Δ double mutants exhibit similar short G2/M arrest as rad53Δ alone (103, 104). Chk1 activation requires Rad9 but not Rad9’s SQ/TQ cluster, which is required for Rad53 activation (154). Instead, Chk1 activation requires Rad9’s N-terminal Chk1 activation domain (CAD) (154, 155) (156), which is phosphorylated independently of DNA damage during the S, G2, and M phases of the cell.
Some of the initial studies on the activation and subsequent adaptation of the DNA damage checkpoint were performed by using a temperature-sensitive mutation of Cdc13, a component of the telomere end-protection complex in budding yeast (157). Cdc13 inactivation leads to the deprotection of telomeres, resulting in their 5’ to 3’ resection and the activation of the Mec1-dependent DDC. By and large the behavior of cells triggered by unprotecting chromosome ends is similar to that achieved by creating a single site-specific DSB with HO endonuclease; for example, adaptation-defective mutations in casein kinase II (ckb1Δ) or a point mutation in the Cdc5 Polo-like kinase (cdc5-ad) that were identified in the cdc13 system are also adaptation-defective in the HO system. However, there are some differences worth noting. First, whereas Chk1 plays only a minor role in maintaining G2/M arrest in the HO system, it is much more important upon cdc13 inactivation. Second, whereas 2 HO-induced DSBs are sufficient to block adaptation, the de-protection of 32 telomeres in cdc13-1 at its restrictive temperature does not elicit such a strong response; cells still adapt. A likely explanation is that telomeres are constantly being restored by telomerase, and that ends are not nearly as extensively degraded as are HO-induced DSBs (158). Indeed, when resection is impaired by together deleting Sgs1, Exo1 and Rad9, cdc13-1 cells will continue to grow at their restrictive temperature, whereas under these conditions a single HO-induced DSB remains lethal.
2.5.4. Mammalian CHK1 and CHK2 checkpoint kinases.
ATM and ATR activate the downstream checkpoint kinases CHK2 and CHK1, which together mediate key checkpoint outcomes including cell cycle arrest and apoptosis (Figure 2) (159). The early checkpoint signaling response to DSBs is carried out predominantly through the ATM-CHK2 signaling axis. Once ends are resected, ATR and ATRIP are recruited to ssDNA via RPA, leading to activation of CHK1 (160, 161). In the canonical mode of activation, recruitment of CHK2 and CHK1 to DNA lesions via checkpoint adaptors allow their direct phosphorylation by ATM and ATR, causing the relief of the inhibitory domains of CHK2 and CHK1 and their activation (reviewed in (162)). Similar to Rad53, overexpression of CHK2 in bacteria results in its trans-phosphorylation and auto-activation, arguing for oligomerization and a concentration-dependent effect in promoting its activation (163, 164). Whereas insolubility issues have prevented expression of CHK1 in bacteria, overexpression of CHK1 in human cells resulted in only minor activation, much lower compared to activation of CHK2 in a similar system, suggesting differences in the regulatory mechanism of activation (165). DNA damage-induced phosphorylation sites in CHK1 and CHK2 are commonly used as readouts of DDC activation in mammalian cells. Key markers of DDC activation include phosphorylation of CHK2 at threonine 68 (an ATM site) (164, 166) and CHK1 phosphorylation at serine 317 and serine 345 (ATR sites) (167, 168).
Intriguingly, while DNA-PKcs has little role in CHK2 activation, it was recently reported to promote CHK1 activation in cells lacking ATR signaling (169). Activation of DNA-PKcs in the absence of ATR signaling is dependent on ssDNA, structure-specific nucleases and KU70, suggesting that DSBs formed through the processing of stalled replication forks activate DNA-PKcs. This finding reveals unexpected levels of complexity in the crosstalk between PIKKs and downstream checkpoint kinases in mammals. Since inhibitors of DDC kinases are currently in clinical trials, understanding non-canonical mechanisms of signaling crosstalk is relevant for therapeutic purposes. For example, the finding that DNA-PKcs can activate CHK1 helps explain why CHK1 inhibitors are more toxic than ATR inhibitors, and provides rationale for the combined use of inhibitors (169).
2.6. Enforcing Cell Cycle Arrest and Apoptosis
In higher eukaryotes, cells suffering DNA damage are principally under the control of the transcription factor p53, which orchestrates the decision for cells to arrest the cell cycle, enter apoptosis or senesce (reviewed in (170, 171)). p53 is phosphorylated and activated by all DDC kinases, highlighting the centrality of p53 as a target through which the DDC can enforce an arrest in the cell cycle or, upon multiple and/or persistent unrepaired DSBs, apoptosis (Figure 2). DDC kinases CHK2 and CHK1 also promote cell cycle arrest through multiple p53-independent mechanisms, such as inhibition of the CDC25 phosphatase, which drives CDK inhibition (Figure 2). Details of cell cycle regulation by DDC kinases in mammals are the focus of numerous reviews (172–175).
Budding yeast lack a p53-like homologue. Instead, cell cycle arrest triggered by the DDC is achieved largely through regulation of the molecular chaperone protein Pds1 (securin in mammals) and its binding partner, the cohesin protease Esp1 (separase) (176, 177). In undamaged cells, Pds1 binds Esp1, inhibiting its protease activity (178). During the metaphase to anaphase transition, Pds1 is ubiquitinated by the E3 ubiquitin ligase APC-Cdc20 and subsequently degraded by the proteasome (179). Degradation of Pds1 frees Esp1 to cleave cohesin rings, allowing sister chromatids to segregate. Upon DNA damage, Pds1 is phosphorylated by Chk1, blocking its interaction with APC-Cdc20 (176). Cdc20 is also phosphorylated, but instead by Rad53, to prevent APC-Cdc20 from ubiquitinating Pds1 (180). Together, both Pds1 and Cdc20 phosphorylation inhibit mitosis progression by keeping Esp1 inactive. Deletion of PDS1 reduces the duration of G2/M arrest by 60% while arrest is completely abrogated in pds1Δ rad53Δ (104, 181). In addition, DNA damage results in the sequestration of a fraction of Pds1 and Esp1 to the vacuole (182) adding an additional layer of control over cell cycle progression.
2.7. Regulation of DNA Repair via DDC Signaling
Apical checkpoint PIKKs directly phosphorylate and regulate key proteins involved in the repair of DSBs (Figure 2). Such control of DSB repair largely occurs independently of downstream kinases, consistent with the view that Rad53/CHK2 and Chk1/CHK1 are mobile kinases that tend to coordinate global or “distant” responses (such as cell cycle arrest) (183), and are therefore not best suited to control localized DNA repair transactions. Although unbiased proteomic analysis revealed local and distant roles for all DDC kinases (184), primary targets of these PIKKs in budding yeast and mammals are indeed highly enriched in proteins that localize at or close to DNA lesions (184). Among these local targets are numerous proteins involved in DNA repair.
DNA-PKcs and ATM play central roles in NHEJ-mediated repair in mammals, with DNA-PKcs signaling playing the principal role (185–187). Although several NHEJ factors are known to be phosphorylated by DNA-PKcs, the crucial substrates and phosphorylation events through which DNA-PKcs mediates NHEJ remain unknown. The only exception is DNA-PKcs itself, whose autophosphorylation is important to release DNA-PKcs from DNA ends to allow for ligation (188). ATM limits resection to favor NHEJ by two recently discovered mechanisms. First, ATM phosphorylates 53BP1 to assemble the 53BP1-RIF1-REV7-SHIELDIN anti-resection complex (189). Second, ATM promotes the degradation of MRE11, by phosphorylating the proteasome-associated ubiquitin receptor UBQLN4 (190). Paradoxically, ATM also plays a pro-resection function through the phosphorylation of CtIP (191). Nonetheless, inhibition or loss of ATM only mildly impair HR-mediated repair (192, 193), indicating that ATM is not crucial for promoting CtIP function, resection or other key steps in HR.
ATR plays important roles in HR-mediated repair. This was recently demonstrated in human osteosarcomas cells in which Cas9 was used to induce HR-mediated DSB repair (192, 194). Phosphorylation of PALB2 by ATR was recently reported to promote recruitment of the PALB2-BRCA2 complex to damaged sites via BRCA1, to promote RAD51 loading (195). However, the scenario is likely more complex, with ATR controlling multiple steps in HR. For example, chronic inhibition of ATR-CHK1 signaling strongly impairs HR efficiency by inhibiting E2F transcription, which depletes the abundance of key HR factors (196). A more detailed review on the role and mechanisms by which DDC kinases regulate DNA repair machineries in yeast and in mammals is provided by reference (197).
2.8. Checkpoint maintenance
It would appear that all of the sensory apparatus to turn on the DDC has been identified; but it is less certain, once the checkpoint has been activated, how it is “maintained”. Given that Rad53/CHK2 is heavily autophosphorylated, one might imagine that some of the proteins involved in initially detecting the damage would cease to be required. By using conditionally inactive proteins, such as those fused to an auxin-inducible or temperature-sensitive degrons, it is possible to establish the DDC and then inactivate specific elements. For example, degradation of Ddc2 (198) or heat inactivation of Mec1 (199) rapidly extinguishes the checkpoint suggesting that at least some of the proteins needed to establish the signal are needed to also maintain it. The situation is less clear in human cells as the effect of ATM and ATR chemical inhibitors after establishment of a DDC response results in variable results depending on the cell line used, possibly due to the variable levels of expression of phosphatases involved in DDC downregulation in different cell lines (D. Dibitetto, C. Ascençao, J. Badar, M. Smolka; unpublished).
Unexpectedly, the maintenance of the DNA damage signal in budding yeast appears to require another checkpoint: the spindle-assembly checkpoint (SAC) (104, 200). Deletion of MAD2 or other SAC genes markedly shortens the arrest in response to a single DSB and suppresses the permanent arrest induced a variety of adaptation-defective conditions (see below) (104). The SAC appears to be activated by monitoring some change in the centromere region associated with the spread of γ-H2AX slowly down the chromosome until it reaches the kinetochore. Deletion of the SAC can be mimicked by deleting the centromere on the damaged chromosome but not when the damage is on a different chromosome. SAC is not strongly activated when the DSB is located several hundred kilobase pairs from its centromere.
2.9. Checkpoint Deactivation
With or without successful repair, the DDC is eventually terminated by dedicated deactivation mechanisms (Figure 5). Checkpoint deactivation is crucial to allow cell cycle progression and cell proliferation, and mutants that fail to properly deactivate the DDC are sensitive to DNA damage (201–203). Deactivation of the DDC after successful repair is associated with the process of recovery, whereas DDC deactivation upon persistent damage is referred as adaptation (see sections below). At the level of kinases and kinase targets, deactivation is achieved primarily by the action of phosphatases, including, but not limited to, PP2C and PP4 phosphatases in budding yeast (Figure 5) and PP2A phosphatases in mammals (204). These phosphatases remove activating phosphorylations from key checkpoint components, such as downstream checkpoint kinases and γ-H2AX. Since cell cycle arrest is mostly established via the downstream checkpoint kinases Chk1/CHK1 and Rad53/CHK2, deactivation of these kinases allows rapid termination of the pro-arrest signaling. The checkpoint adaptors, such as budding yeast Rad9, offer another key point of regulation, as their degradation or disengagement from DNA lesions prevents new checkpoint kinases from becoming active to maintain the cell cycle arrest. For example, in budding yeast the Slx4-Rtt107 repair factors compete with Rad9 at sites of DNA lesions to disengage Rad9 and dampen the checkpoint (203, 205, 206) (Figure 5). A similar competition-based mechanism of DDC downregulation has yet to be demonstrated in mammals.
Figure 5. Mechanisms of DDC deactivation in budding yeast.
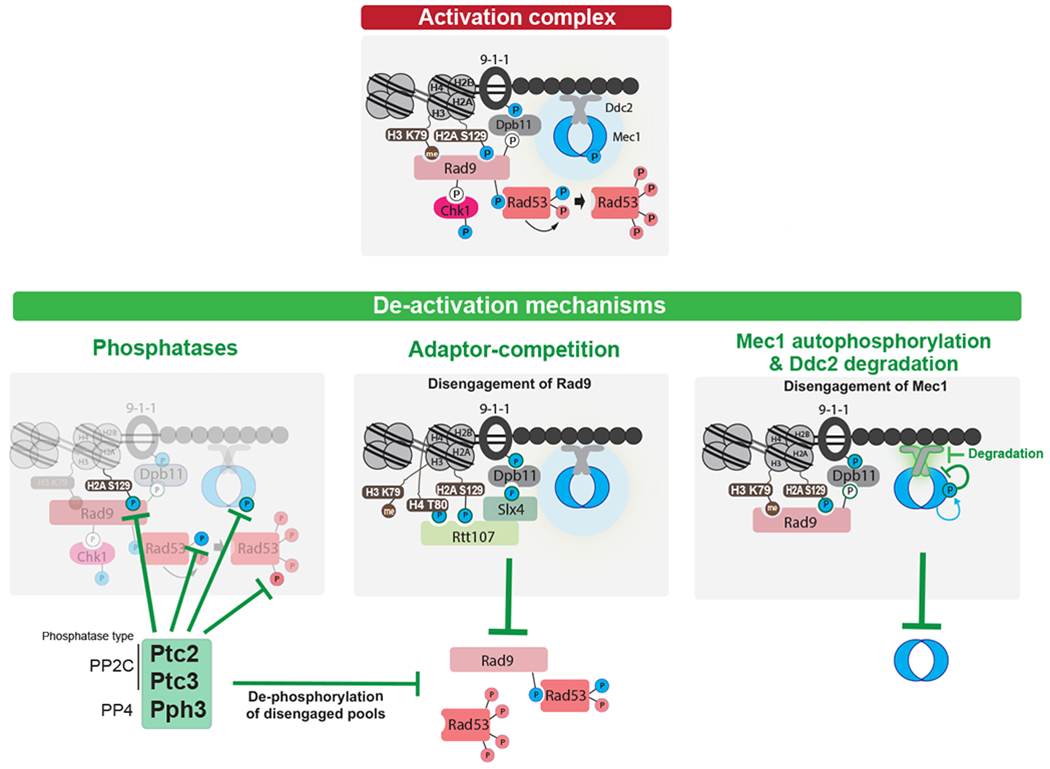
Top: Key interactions and phosphorylation events involved in activation of the DDC. Color of phosphorylation sites (circles with “P”) refer to the kinase responsible: blue=Mec1, red=Rad53, white=CDK. Bottom: three modes of down-regulating the DDC. Note that current evidence supports a model in which the PP4 phosphatase Pph3 removes Mec1 phosphorylation sites involved in Mec1 activation (237), while a later Mec1 autophosphorylation site triggers further Mec1 deactivation (210). For adaptor-competition, recent evidences suggest that Sae2 may also compete with Rad9 (238, 239), although the mechanism is not understood. See text for additional details.
2.9.1. Adaptation to DNA Damage
In budding yeast, a single unrepaired DSB elicits robust DDC activation that persists for at least 9 h, after which, and still without repair, cells adapt to the damage by switching off the DDC and proceed through several rounds of mitosis (31, 157, 207). A key feature of adaptation is that the cell becomes “deaf” to the presence of continued DNA damage; consequently, there is no arrest when cells traverse the next G2/M boundary. While adaptation has traditionally been studied using site-specific endonucleases such as HO, adaptation has also been observed in aneuploid yeast cells, which frequently display Rad52-GFP foci in S-phase, an indicator of DNA damage (208). Moreover, adaptation appears to be a conserved phenomenon. Xenopus egg extracts treated with the replication inhibitor aphidicolin eventually switched off checkpoint signaling and human osteosarcoma cells were seen to enter mitosis despite the presence of γ–H2AX foci following gamma irritation (209).
How the checkpoint signal is extinguished during adaptation is slowly coming into focus. Recently we found that Mec1 autophosphorylation at serine 1964 is required to turn off the signal (Figure 5); mec1-S1964A fails to adapt and Rad53 remains hyperphosphorylated (210). This modification only appears several hours after Mec1 has phosphorylated Ddc2, Rad9 and histone H2A; how S1964 is initially prevented from being phosphorylated remains unknown. Moreover, two serines in Ddc2 that are not SQ/TQ sites are required to adapt; these serines appear to be involved in regulating the stability of Ddc2. Ddc2 is normally a stable protein, but is degraded at the time adaptation occurs (210).
Most adaptation-defective mutants exhibit hyperphosphorylated Rad53 that persists for >24 h after DNA damage, while adaptation results in the loss of Rad53 hyperphosphorylation about 9-12 h after creating the DSB. These mutants include, but are not limited to deletion of the PP2C phosphatases, Ptc2 and Ptc3, which prevents dephosphorylation of Rad53 and presumably other Mec1 kinase targets (211, 212) and abrogation of casein kinase II, which is required to promote the association of Ptc2 with Rad53 via a phosphorylation of a threonine in Ptc2 that is recognized by Rad53’s FHA domain (211). However, the way in which mutations implicated in DSB repair (e.g. rad51Δ or rdh54Δ, but not rad52Δ or rad54Δ) (199, 213) prevent adaptation is not yet understood. Adaptation is also prevented by a mutation in the Cdc5 (Plk1) kinase that has altered activity; but again, how this occurs is not understood (157, 214).
The relationship between the extent of 5’ to 3’ resection of DSB ends and adaptation is complex. The adaptation defect in yku70Δ or yku80Δ cells appeared to be related to the fact that resection is twice as fast as wild type and that the adaptation defect of Ku mutants is suppressible by deleting MRE11, which slows resection (207). These observations led to the idea that the cell might monitor the rate of resection, for example by detecting short deoxyoligonucleotides that would be liberated by Mre11-Rad50 or by the helicase-endonucleases involved in long-range resection; indeed in Xenopus, such short oligonucleotides stimulate the activation of ATM (215). But in yeast, elimination of the long-range helicase-endonuclease complex (e.g., deleting SGS1) or slowing down resection by eliminating the Fun30/SMARCAD1 chromatin remodeler did not shorten arrest, but unexpectedly made cells adaptation-defective (205, 216, 217). Moreover, H2BK123R, which abolishes histone H2BK123 ubiquitination, triggers early adaptation despite an elevated resection rate (D.P. Waterman and J. E. Haber, unpublished). Therefore, resection cannot be the sole signal responsible for adaptation.
Noncanonical adaptation-defective mutants, which fail to resume cell cycle progression despite dephosphorylation of Rad53, have been found in genes involved in processes outside the nucleus. Deletion of components of the Golgi associated retrograde particle (GARP), such as VPS51 or YPT6 (182), display other disruptions of normal adaptation suggestive of a complex crosstalk between nuclear and cytoplasmic processes required for cell cycle progression.
2.9.2. Recovery from DNA Damage
After successfully repairing their DNA, cells rapidly switch off the DDC reenter the cell cycle through a process known as recovery (28). Recovery has been best-studied when there is a long delay between the induction of a DSB and the completion of repair, such as when a deletion between repeated sequences flanking a DSB are separated by 25 kb , such that resection- moving 4 kb/h – will only promote repair by single-strand annealing after 6 h (211). Only a fraction of adaptation defective mutants are also recovery-defective; these include vps51Δ (and other members of GARP), sae2Δ and ptc2Δ ptc3Δ (29, 182), suggesting that cell cycle progression and completion of repair are two separate events.
2.10. DDC and Cancer
The DDC is often activated in pre-neoplastic cells in response to oncogene activation, establishing a barrier cells must overcome to enter a malignancy. Hyperactivation of the DDC, typically triggered by oncogene-induced replication stress (reviewed in (218)) often leads to apoptosis or senescence, but cells may escape this terminal fate by gaining mutations or copy number variations in genes that either abolish checkpoint activation or cause its premature deactivation. Mutations in DDC genes such as ATM and TP53 are a central factor in contributing to genomic instability seen in tumors because they allow for the accumulation of more mutations over many cell generations (219, 220). Heterozygous mutations in in ATM occur in ~1% of the population and like inherited TP53 mutations, predispose individuals to cancer (221, 222). Somatic TP53 mutations are by far the most frequently occurring across all tumor samples, present in nearly 40% (223), and ATM mutations are frequent in several cancers (221).
The role of other components of checkpoint signaling in cancer development is more complex. ATR is an essential kinase and its activity is important for the growth of many cancers (224). CHK1 exhibits both tumor-promoting and -inhibiting behaviors. In agreement with its role as a tumor suppressor, heterozygous deletion of CHK1 in mice together with heterozygous loss of Trp53 (mouse p53) potentiates mammary tumor formation (225). However, the same study found that homozygous loss of CHK1, expressed by a tissue specific driver, inhibited mammary tumor development (225). Moreover, skin-specific homozygous deletions of CHK1 prevented tumor formation in mouse skin cells exposed to chemical carcinogens (226). Therefore, it appears that while loss of some checkpoint factors increases the likelihood of cancer, amplification or gain-of-function mutations in other may help cells handle burdens imposed by oncogene activation and therefore promote tumorigenesis.
2.11. Manipulation of checkpoint responses to DSBs: Applications and Perspectives
The last 15 years have witnessed a revolution in the development of specific inhibitors of DDC kinases. A potent and highly selective inhibitor for ATM (KU-55933) was developed in 2004 (227), ending the dark years of reliance on unspecific inhibitors such as caffeine and Wortmannin. The first potent and selective inhibitor for ATR, VE-821, with minimal cross-reactivity against ATM and DNA-PKcs, was developed in 2011 (228). Since then, inhibitors with better bioavailability, potency and selectivity have been developed for nearly all DDC kinases (229). These agents have allowed more careful studies on the architecture of the DSB-induced signaling network and the mechanisms by which DDC kinases control key functional outputs. Importantly, these inhibitors exhibitstrong synergism with radiation therapy and DSB-inducing drugs, including topoisomerase-inhibitors and PARP-inhibitors, in sensitizing cancers, which led to numerous clinical trials currently underway (230). In addition to cancer therapy, the ability to manipulate the DDC may have a range of other applications, including improved strategies for precision genome editing. The efficiency of CRISPR-Cas9 mediated genome engineering was demonstrated to be compromised by DSB-induction of a p53 response leading to cell cycle arrest or apoptosis, especially in pluripotent stem cells and non-cancerous cells (231, 232) as well as in pigs (233); however, the effect is not seen in other cell lines (234). This suggests that by manipulating the DDC response to Cas9-induced DSB it should be possible to bypass the deleterious p53-dependent outcomes and improve genome editing efficiency. In fact, a transient inhibition of p53 was recently shown to reverse the constrained proliferation of edited pluripotent stem cells (235).
3. SUMMARY AND PERSPECTIVES
Since the first observations of cell cycle delays in response to DNA damage, our understanding of the DDC has increased tremendously, from its activation, maintenance and deactivation to its interplay with other cellular pathways. Broken DNA ends are substrates for recruitment and activation of the apical kinase Tel1/ATM. Once cells have committed to HR, ssDNA generated by 5’ – 3’ resection allows for robust checkpoint activation via Mec1/ATR-mediated signaling. Activation of apical kinases results in the phosphorylation of dozens of substrates within the vicinity of the DSB to create a region of chromatin “inflammation”, orchestrate DNA repair and trigger downstream signaling events. DDC kinases Chk1/CHK1 and Rad53/CHK2 amplify DDC signaling to mediate a response that stalls cell cycle progression, rewires transcriptional programs and impacts several other cellular processes. Upon successful DNA repair, the DDC is inactivated through the concerted action of phosphatases and the disengagement of kinases and DDC adaptors from the site lesion, allowing cells to progress through the cell cycle. But if repair fails, cells adapt to the damage by switching off checkpoint signaling via and resume cell cycle progression despite the continual presence of DNA damage.
Despite many advances in characterizing the DDC in the last four decades, several outstanding questions need to be addressed to complete a detailed picture. In particular, it remains largely unclear how the apical kinases coordinate DNA repair transactions necessary for suppressing genomic instabilities upon DSBs. For example, how Mec1/ATR coordinates homology-directed DNA repair remains a fundamental knowledge gap in the field and an important barrier to better understand how DNA damage-induced signaling prevents chromosomal rearrangements. Multiple targets of Tel1/ATM and Mec1/ATR have been identified in the HR machinery, but we still don’t have a comprehensive and mechanistic understanding of how, and which, defined set of signaling events determine repair pathway commitment and coordinate steps in HR. Solving this question requires defining spatio-temporal signaling dynamics and dissecting the functionality of an extremely complex repertoire of phosphorylation events.
Another question that remains unanswered concerns what fraction of ATM and ATR are activated by one or a few DSBs. In yeast, downstream targets such as Rad53 have well-documented mechanisms of recruitment to the Rad9 scaffold and extensive autophosphorylation that can account for how most of the Rad53 in the cell is hyperphosphorylated when there is only a single DSB, assuming that there is robust turnover. Recent estimates of protein abundance in yeast (236) indicate that there are approximately 300 molecules of Mec1, 1000 Rad9 and 2100 Rad53; yet the great majority of both Rad9 and Rad53 migrate on western blots as hyperphosphorylated forms. Bakkenist and Kastan (41) suggested that mammalian ATM propagated its autoactivation by exchanging monomers (one activated monomer trans-phosphorylating a partner that would then diffuse away and allow another monomer to become associated with a presumably tethered active monomer). Yeast Mec1 has been shown to phosphorylate at least one site (not essential for its activity) in trans (210) and this may be a property of all such apical kinases.
Finally, a major outstanding question about DDC termination concerns exactly how checkpoint signaling is deactivated and phosphatase activity is regulated. In addition to addressing whether phosphatase activity is inducible or constitutive, it will be important to thoroughly explore if phosphatase action is out-competed by checkpoint activation/maintenance. It will also be crucial to explore whether mechanisms of DDC termination found in yeast (Figure 5) are also present in mammals.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health to J.E.H. (R01GM61766 and R35GM127029) and M.B.S. (R01GM097272 and R01HD095296). D.P.W. was a Trainee of NIH Genetics Training Grant TM32 GM007122.
REFERENCES
- 1.Hoeijmakers JH. 2009. N Engl J Med 361: 1475–85 [DOI] [PubMed] [Google Scholar]
- 2.Branzei D, Foiani M. 2005. Curr Opin Cell Biol 17: 568–75 [DOI] [PubMed] [Google Scholar]
- 3.Scharer OD. 2013. Cold Spring Harb Perspect Biol 5: a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krokan HE, Bjoras M. 2013. Cold Spring Harb Perspect Biol 5: a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexander JL, Orr-Weaver TL. 2016. Genes Dev 30: 2241–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, et al. 1998. EMBO J 17: 598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vilenchik MM, Knudson AG. 2003. Proc Natl Acad Sci U S A 100: 12871–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haber JE. 2014. Genome stability : DNA repair and recombination. New York: GS/Garland Science, Taylor & Francis Group; xvi, 399 pages pp. [Google Scholar]
- 9.Jacquier A, Dujon B. 1985. Cell 41: 383–94 [DOI] [PubMed] [Google Scholar]
- 10.Lee CS, Haber JE. 2015. Microbiol Spectr 3: MDNA3-0013-2014 [DOI] [PubMed] [Google Scholar]
- 11.Haber JE. 2016. Annu Rev Genet 50: 1–28 [DOI] [PubMed] [Google Scholar]
- 12.Jasin M, Rothstein R. 2013. Cold Spring Harb Perspect Biol 5: a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaplun L, Ivantsiv Y, Kornitzer D, Raveh D. 2000. Proc Natl Acad Sci U S A 97: 10077–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM. 2013. Nucleic Acids Res 41: 4336–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caron P, Choudjaye J, Clouaire T, Bugler B, Daburon V, et al. 2015. Cell Rep 13: 1598–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, et al. 2014. Nat Struct Mol Biol 21: 366–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jasin M, Haber JE. 2016. DNA Repair (Amst) 44: 6–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van den Berg J,AGM, Kielbassa K, Feringa FM, Freire R, Medema RH. 2018. Nucleic Acids Res 46: 10132–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ceccaldi R, Rondinelli B, D’Andrea AD. 2016. Trends Cell Biol 26: 52–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kakarougkas A, Jeggo PA. 2014. Br J Radiol 87: 20130685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hannan MA, Miller DR, Nasim A. 1976. Radiat Res 68: 469–79 [PubMed] [Google Scholar]
- 22.Painter RB, Young BR. 1980. Proc Natl Acad Sci U S A 77: 7315–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinert TA, Hartwell LH. 1988. Science 241: 317–22 [DOI] [PubMed] [Google Scholar]
- 24.Murray JM, Carr AM. 2018. Curr Opin Cell Biol 52: 120–5 [DOI] [PubMed] [Google Scholar]
- 25.Siede W, Friedberg AS, Dianova I, Friedberg EC. 1994. Genetics 138: 271–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerald JN, Benjamin JM, Kron SJ. 2002. J Cell Sci 115: 1749–57 [DOI] [PubMed] [Google Scholar]
- 27.Paulovich AG, Hartwell LH. 1995. Cell 82: 841–7 [DOI] [PubMed] [Google Scholar]
- 28.Vaze MB, Pellicioli A, Lee SE, Ira G, Liberi G, et al. 2002. Mol Cell 10: 373–85 [DOI] [PubMed] [Google Scholar]
- 29.Harrison JC, Haber JE. 2006. Annu Rev Genet 40: 209–35 [DOI] [PubMed] [Google Scholar]
- 30.Shaltiel IA, Krenning L, Bruinsma W, Medema RH. 2015. J Cell Sci 128: 607–20 [DOI] [PubMed] [Google Scholar]
- 31.Sandell LL, Zakian VA. 1993. Cell 75: 729–39 [DOI] [PubMed] [Google Scholar]
- 32.Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, et al. 2011. J Cell Biol 193: 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, et al. 2011. Nat Cell Biol 13: 243–53 [DOI] [PubMed] [Google Scholar]
- 34.Feng W, Jasin M. 2017. Nat Commun 8: 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blackford AN, Jackson SP. 2017. Mol Cell 66: 801–17 [DOI] [PubMed] [Google Scholar]
- 36.Kim ST, Lim DS, Canman CE, Kastan MB. 1999. J Biol Chem 274: 37538–43 [DOI] [PubMed] [Google Scholar]
- 37.O’Neill T, Dwyer AJ, Ziv Y, Chan DW, Lees-Miller SP, et al. 2000. J Biol Chem 275: 22719–27 [DOI] [PubMed] [Google Scholar]
- 38.Perry J, Kleckner N. 2003. Cell 112: 151–5 [DOI] [PubMed] [Google Scholar]
- 39.Luzwick JW, Nam EA, Zhao R, Cortez D. 2014. PLoS One 9: e99397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bosotti R, Isacchi A, Sonnhammer EL. 2000. Trends Biochem Sci 25: 225–7 [DOI] [PubMed] [Google Scholar]
- 41.Bakkenist CJ, Kastan MB. 2003. Nature 421: 499–506 [DOI] [PubMed] [Google Scholar]
- 42.Mordes DA, Glick GG, Zhao R, Cortez D. 2008. Genes Dev 22: 1478–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang X, Sun Y, Chen S, Roy K, Price BD. 2006. J Biol Chem 281: 15741–6 [DOI] [PubMed] [Google Scholar]
- 44.Ogi H, Goto GH, Ghosh A, Zencir S, Henry E, Sugimoto K. 2015. Mol Biol Cell 26: 3480–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baretic D, Pollard HK, Fisher DI, Johnson CM, Santhanam B, et al. 2017. Sci Adv 3: e1700933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Chu H, Lv M, Zhang Z, Qiu S, et al. 2016. Nat Commun 7: 11655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Ran T, Zhang X, Xin J, Zhang Z, et al. 2017. Science 358: 1206–9 [DOI] [PubMed] [Google Scholar]
- 48.Ma W, Westmoreland J, Nakai W, Malkova A, Resnick MA. 2011. Methods Mol Biol 745: 15–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kramer KM, Brock JA, Bloom K, Moore JK, Haber JE. 1994. Mol Cell Biol 14: 1293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oh J, Al-Zain A, Cannavo E, Cejka P, Symington LS. 2016. Mol Cell 64: 405–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andres SN, Williams RS. 2017. DNA Repair (Amst) 56: 109–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cassani C, Gobbini E, Vertemara J, Wang W, Marsella A, et al. 2018. Nucleic Acids Res 46: 2990–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lisby M, Mortensen UH, Rothstein R. 2003. Nat Cell Biol 5: 572–7 [DOI] [PubMed] [Google Scholar]
- 54.Waterman DP, Zhou F, Li K, Lee CS, Tsabar M, et al. 2019. PLoS Genet 15: e1008001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lobachev K, Vitriol E, Stemple J, Resnick MA, Bloom K. 2004. Curr Biol 14: 2107–12 [DOI] [PubMed] [Google Scholar]
- 56.Mimitou EP, Symington LS. 2008. Nature 455: 770–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shim EY, Chung WH, Nicolette ML, Zhang Y, Davis M, et al. 2010. EMBO J 29: 3370–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mimitou EP, Symington LS. 2010. EMBO J 29: 3358–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lisby M, Barlow JH, Burgess RC, Rothstein R. 2004. Cell 118: 699–713 [DOI] [PubMed] [Google Scholar]
- 60.Langerak P, Mejia-Ramirez E, Limbo O, Russell P. 2011. PLoS Genet 7: e1002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huertas P, Jackson SP. 2009. J Biol Chem 284: 9558–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ivanov EL, Sugawara N, White CI, Fabre F, Haber JE. 1994. Mol Cell Biol 14: 3414–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsubouchi H, Ogawa H. 1998. Mol Cell Biol 18: 260–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. 2008. Cell 134: 981–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Diede SJ, Gottschling DE. 2001. Curr Biol 11: 1336–40 [DOI] [PubMed] [Google Scholar]
- 66.Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A, Jackson SP. 2008. Nature 455: 689–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fishman-Lobell J, Haber JE. 1992. Science 258: 480–4 [DOI] [PubMed] [Google Scholar]
- 68.Cruz-Garcia A, Lopez-Saavedra A, Huertas P. 2014. Cell Rep 9: 451–9 [DOI] [PubMed] [Google Scholar]
- 69.Bhargava R, Onyango DO, Stark JM. 2016. Trends Genet 32: 566–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bae SH, Seo YS. 2000. J Biol Chem 275: 38022–31 [DOI] [PubMed] [Google Scholar]
- 71.Budd ME, Campbell JL. 1995. Proc Natl Acad Sci U S A 92: 7642–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stewart JA, Campbell JL, Bambara RA. 2010. Nucleic Acids Res 38: 920–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lydeard JR, Lipkin-Moore Z, Jain S, Eapen VV, Haber JE. 2010. PLoS Genet 6: e1000973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, et al. 2011. Genes Dev 25: 350–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Soniat MM, Myler LR, Kuo HC, Paull TT, Finkelstein IJ. 2019. Mol Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen X, Niu H, Chung WH, Zhu Z, Papusha A, et al. 2011. Nat Struct Mol Biol 18: 1015–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Clerici M, Mantiero D, Guerini I, Lucchini G, Longhese MP. 2008. EMBO Rep 9: 810–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nakada D, Matsumoto K, Sugimoto K. 2003. Genes Dev 17: 1957–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim JH, Grosbart M, Anand R, Wyman C, Cejka P, Petrini JHJ. 2017. Cell Rep 18: 496–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, et al. 2004. Curr Biol 14: 1703–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, et al. 2004. Nature 431: 1011–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bonilla CY, Melo JA, Toczyski DP. 2008. Mol Cell 30: 267–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mantiero D, Clerici M, Lucchini G, Longhese MP. 2007. EMBO Rep 8: 380–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zou L, Elledge SJ. 2003. Science 300: 1542–8 [DOI] [PubMed] [Google Scholar]
- 85.Saldivar JC, Cortez D, Cimprich KA. 2017. Nat Rev Mol Cell Biol 18: 622–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wanrooij PH, Burgers PM. 2015. DNA Repair (Amst) 32: 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mordes DA, Nam EA, Cortez D. 2008. Proc Natl Acad Sci U S A 105: 18730–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Navadgi-Patil VM, Burgers PM. 2008. J Biol Chem 283: 35853–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kumar S, Burgers PM. 2013. Genes Dev 27: 313–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bass TE, Luzwick JW, Kavanaugh G, Carroll C, Dungrawala H, et al. 2016. Nat Cell Biol 18: 1185–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Haahr P, Hoffmann S, Tollenaere MA, Ho T, Toledo LI, et al. 2016. Nat Cell Biol 18: 1196–207 [DOI] [PubMed] [Google Scholar]
- 92.Lee YC, Zhou Q, Chen J, Yuan J. 2016. Curr Biol 26: 3257–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Usui T, Ogawa H, Petrini JH. 2001. Mol Cell 7: 1255–66 [DOI] [PubMed] [Google Scholar]
- 94.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. 1998. J Biol Chem 273: 5858–68 [DOI] [PubMed] [Google Scholar]
- 95.Savic V, Yin B, Maas NL, Bredemeyer AL, Carpenter AC, et al. 2009. Mol Cell 34: 298–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Solovjeva LV, Svetlova MP, Chagin VO, Tomilin NV. 2007. Chromosome Res 15: 787–97 [DOI] [PubMed] [Google Scholar]
- 97.Caron P, van der Linden J, van Attikum H. 2019. DNA Repair (Amst) 82: 102686. [DOI] [PubMed] [Google Scholar]
- 98.Lee SE, Pellicioli A, Demeter J, Vaze MP, Gasch AP, et al. 2000. Cold Spring Harb Symp Quant Biol 65: 303–14 [DOI] [PubMed] [Google Scholar]
- 99.Manfrini N, Clerici M, Wery M, Colombo CV, Descrimes M, et al. 2015. Elife 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim JA, Kruhlak M, Dotiwala F, Nussenzweig A, Haber JE. 2007. J Cell Biol 178: 209–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee CS, Lee K, Legube G, Haber JE. 2014. Nat Struct Mol Biol 21: 103–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li J, Coic E, Lee K, Lee CS, Kim JA, et al. 2012. PLoS Genet 8: e1002630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dotiwala F, Haase J, Arbel-Eden A, Bloom K, Haber JE. 2007. Proc Natl Acad Sci U S A 104: 11358–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dotiwala F, Harrison JC, Jain S, Sugawara N, Haber JE. 2010. Curr Biol 20: 328–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Grenon M, Costelloe T, Jimeno S, O’Shaughnessy A, Fitzgerald J, et al. 2007. Yeast 24: 105–19 [DOI] [PubMed] [Google Scholar]
- 106.Hammet A, Magill C, Heierhorst J, Jackson SP. 2007. EMBO Rep 8: 851–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Giannattasio M, Lazzaro F, Plevani P, Muzi-Falconi M. 2005. J Biol Chem 280: 9879–86 [DOI] [PubMed] [Google Scholar]
- 108.van Leeuwen F, Gafken PR, Gottschling DE. 2002. Cell 109: 745–56 [DOI] [PubMed] [Google Scholar]
- 109.Granata M, Lazzaro F, Novarina D, Panigada D, Puddu F, et al. 2010. PLoS Genet 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pfander B, Diffley JF. 2011. EMBO J 30: 4897–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gritenaite D, Princz LN, Szakal B, Bantele SC, Wendeler L, et al. 2014. Genes Dev 28: 1604–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Puddu F, Granata M, Di Nola L, Balestrini A, Piergiovanni G, et al. 2008. Mol Cell Biol 28: 4782–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.di Cicco G, Bantele SCS, Reusswig KU, Pfander B. 2017. Sci Rep 7: 11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sun Z, Hsiao J, Fay DS, Stern DF. 1998. Science 281: 272–4 [DOI] [PubMed] [Google Scholar]
- 115.Usui T, Foster SS, Petrini JH. 2009. Mol Cell 33: 147–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Goldberg M, Stucki M, Falck J, D’Amours D, Rahman D, et al. 2003. Nature 421: 952–6 [DOI] [PubMed] [Google Scholar]
- 117.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. 2003. Nature 421: 961–6 [DOI] [PubMed] [Google Scholar]
- 118.Wu L, Luo K, Lou Z, Chen J. 2008. Proc Natl Acad Sci U S A 105: 11200–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Peng A, Chen PL. 2003. J Biol Chem 278: 8873–6 [DOI] [PubMed] [Google Scholar]
- 120.Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 2002. Science 298: 1435–8 [DOI] [PubMed] [Google Scholar]
- 121.Her J, Ray C, Altshuler J, Zheng H, Bunting SF. 2018. Mol Cell Biol 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. 2005. Cell 123: 1213–26 [DOI] [PubMed] [Google Scholar]
- 123.Baldock RA, Day M, Wilkinson OJ, Cloney R, Jeggo PA, et al. 2015. Cell Rep 13: 2081–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kleiner RE, Verma P, Molloy KR, Chait BT, Kapoor TM. 2015. Nat Chem Biol 11: 807–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang J, Gong Z, Chen J. 2011. J Cell Biol 193: 267–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Leung CC, Sun L, Gong Z, Burkat M, Edwards R, et al. 2013. Structure 21: 1450–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cescutti R, Negrini S, Kohzaki M, Halazonetis TD. 2010. EMBO J 29: 3723–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu Y, Cussiol JR, Dibitetto D, Sims JR, Twayana S, et al. 2017. J Cell Biol 216: 623–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ferrari M, Dibitetto D, De Gregorio G, Eapen VV, Rawal CC, et al. 2015. PLoS Genet 11: e1004928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bunting SF, Callen E, Wong N, Chen HT, Polato F, et al. 2010. Cell 141: 243–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hustedt N, Durocher D. 2016. Nat Cell Biol 19: 1–9 [DOI] [PubMed] [Google Scholar]
- 132.Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, et al. 2001. Nat Cell Biol 3: 958–65 [DOI] [PubMed] [Google Scholar]
- 133.Kumagai A, Dunphy WG. 2000. Mol Cell 6: 839–49 [DOI] [PubMed] [Google Scholar]
- 134.Zhao X, Chabes A, Domkin V, Thelander L, Rothstein R. 2001. EMBO J 20: 3544–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Desany BA, Alcasabas AA, Bachant JB, Elledge SJ. 1998. Genes Dev 12: 2956–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhao X, Muller EG, Rothstein R. 1998. Mol Cell 2: 329–40 [DOI] [PubMed] [Google Scholar]
- 137.Huang M, Zhou Z, Elledge SJ. 1998. Cell 94: 595–605 [DOI] [PubMed] [Google Scholar]
- 138.Jaehnig EJ, Kuo D, Hombauer H, Ideker TG, Kolodner RD. 2013. Cell Rep 4: 174–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Travesa A, Kuo D, de Bruin RA, Kalashnikova TI, Guaderrama M, et al. 2012. EMBO J 31: 1811–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bastos de Oliveira FM, Harris MR, Brazauskas P, de Bruin RA, Smolka MB. 2012. EMBO J 31: 1798–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Edenberg ER, Vashisht A, Benanti JA, Wohlschlegel J, Toczyski DP. 2014. Mol Cell Biol 34: 725–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kim EM, Jang YK, Park SD. 2002. Nucleic Acids Res 30: 643–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Eapen VV, Waterman DP, Bernard A, Schiffmann N, Sayas E, et al. 2017. Proc Natl Acad Sci U S A 114: E1158–E67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Allen JB, Zhou Z, Siede W, Friedberg EC, Elledge SJ. 1994. Genes Dev 8: 2401–15 [DOI] [PubMed] [Google Scholar]
- 145.Game JC. 1975. Basic Life Sci 5B: 541–4 [DOI] [PubMed] [Google Scholar]
- 146.Schwartz MF, Lee SJ, Duong JK, Eminaga S, Stern DF. 2003. Cell Cycle 2: 384–96 [PubMed] [Google Scholar]
- 147.Sweeney FD, Yang F, Chi A, Shabanowitz J, Hunt DF, Durocher D. 2005. Curr Biol 15: 1364–75 [DOI] [PubMed] [Google Scholar]
- 148.Schwartz MF, Duong JK, Sun Z, Morrow JS, Pradhan D, Stern DF. 2002. Mol Cell 9: 1055–65 [DOI] [PubMed] [Google Scholar]
- 149.Chen ESW, Hoch NC, Wang SC, Pellicioli A, Heierhorst J, Tsai MD. 2014. In Mol Cell Proteomics, pp. 551–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jia-Lin Ma N, Stern DF. 2008. Cell Cycle 7: 808–17 [DOI] [PubMed] [Google Scholar]
- 151.Morrison AJ, Kim JA, Person MD, Highland J, Xiao J, et al. 2007. Cell 130: 499–511 [DOI] [PubMed] [Google Scholar]
- 152.Morrison AJ, Shen X. 2009. Nat Rev Mol Cell Biol 10: 373–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kapoor P, Bao Y, Xiao J, Espejo A, Yang L, et al. 2015. Mol Cell 58: 863–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sanchez Y, Bachant J, Wang H, Hu F, Liu D, et al. 1999. Science 286: 1166–71 [DOI] [PubMed] [Google Scholar]
- 155.Blankley RT, Lydall D. 2004. J Cell Sci 117: 601–8 [DOI] [PubMed] [Google Scholar]
- 156.Abreu CM, Kumar R, Hamilton D, Dawdy AW, Creavin K, et al. 2013. PLoS Genet 9: e1003310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Toczyski DP, Galgoczy DJ, Hartwell LH. 1997. Cell 90: 1097–106 [DOI] [PubMed] [Google Scholar]
- 158.Dewar JM, Lydall D. 2012. Chromosoma 121: 117–30 [DOI] [PubMed] [Google Scholar]
- 159.Harper JW, Elledge SJ. 2007. Mol Cell 28: 739–45 [DOI] [PubMed] [Google Scholar]
- 160.Cuadrado M, Martinez-Pastor B, Murga M, Toledo LI, Gutierrez-Martinez P, et al. 2006. J Exp Med 203: 297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shiotani B, Zou L. 2009. Mol Cell 33: 547–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bartek J, Lukas J. 2003. Cancer Cell 3: 421–9 [DOI] [PubMed] [Google Scholar]
- 163.Schwarz JK, Lovly CM, Piwnica-Worms H. 2003. Mol Cancer Res 1: 598–609 [PubMed] [Google Scholar]
- 164.Xu X, Tsvetkov LM, Stern DF. 2002. Mol Cell Biol 22: 4419–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ng CP, Lee HC, Ho CW, Arooz T, Siu WY, et al. 2004. J Biol Chem 279: 8808–19 [DOI] [PubMed] [Google Scholar]
- 166.Ahn JY, Schwarz JK, Piwnica-Worms H, Canman CE. 2000. Cancer Res 60: 5934–6 [PubMed] [Google Scholar]
- 167.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, et al. 2000. Genes Dev 14: 1448–59 [PMC free article] [PubMed] [Google Scholar]
- 168.Zhao H, Piwnica-Worms H. 2001. Mol Cell Biol 21: 4129–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Buisson R, Boisvert JL, Benes CH, Zou L. 2015. Mol Cell 59: 1011–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Giono LE, Manfredi JJ. 2006. J Cell Physiol 209: 13–20 [DOI] [PubMed] [Google Scholar]
- 171.Carvajal LA, Manfredi JJ. 2013. EMBO Rep 14: 414–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lukas J, Lukas C, Bartek J. 2004. DNA Repair (Amst) 3: 997–1007 [DOI] [PubMed] [Google Scholar]
- 173.Boehme KA, Blattner C. 2009. Crit Rev Biochem Mol Biol 44: 367–92 [DOI] [PubMed] [Google Scholar]
- 174.Boutros R, Dozier C, Ducommun B. 2006. Curr Opin Cell Biol 18: 185–91 [DOI] [PubMed] [Google Scholar]
- 175.Sorensen CS, Syljuasen RG. 2012. Nucleic Acids Res 40: 477–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Cohen-Fix O, Koshland D. 1997. Proc Natl Acad Sci U S A 94: 14361–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yamamoto A, Guacci V, Koshland D. 1996. J Cell Biol 133: 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ciosk R, Zachariae W, Michaelis C, Shevchenko A, Mann M, Nasmyth K. 1998. Cell 93: 1067–76 [DOI] [PubMed] [Google Scholar]
- 179.Shirayama M, Toth A, Galova M, Nasmyth K. 1999. Nature 402: 203–7 [DOI] [PubMed] [Google Scholar]
- 180.Agarwal R, Tang Z, Yu H, Cohen-Fix O. 2003. J Biol Chem 278: 45027–33 [DOI] [PubMed] [Google Scholar]
- 181.Gardner R, Putnam CW, Weinert T. 1999. EMBO J 18: 3173–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Dotiwala F, Eapen VV, Harrison JC, Arbel-Eden A, Ranade V, et al. 2013. Proc Natl Acad Sci U S A 110: E41–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bartek J, Falck J, Lukas J. 2001. Nat Rev Mol Cell Biol 2: 877–86 [DOI] [PubMed] [Google Scholar]
- 184.Bastos de Oliveira FM, Kim D, Cussiol JR, Das J, Jeong MC, et al. 2015. Mol Cell 57: 1124–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Gapud EJ, Sleckman BP. 2011. Cell Cycle 10: 1928–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zha S, Jiang W, Fujiwara Y, Patel H, Goff PH, et al. 2011. Proc Natl Acad Sci U S A 108: 2028–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gapud EJ, Dorsett Y, Yin B, Callen E, Bredemeyer A, et al. 2011. Proc Natl Acad Sci U S A 108: 2022–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Jette N, Lees-Miller SP. 2015. Prog Biophys Mol Biol 117: 194–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Setiaputra D, Durocher D. 2019. EMBO Rep 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Jachimowicz RD, Beleggia F, Isensee J, Velpula BB, Goergens J, et al. 2019. Cell 176: 505–19 e22 [DOI] [PubMed] [Google Scholar]
- 191.Shibata A, Conrad S, Birraux J, Geuting V, Barton O, et al. 2011. EMBO J 30: 1079–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Vriend LE, Prakash R, Chen CC, Vanoli F, Cavallo F, et al. 2016. Nucleic Acids Res 44: 5204–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kass EM, Helgadottir HR, Chen CC, Barbera M, Wang R, et al. 2013. Proc Natl Acad Sci U S A 110: 5564–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Pierce AJ, Johnson RD, Thompson LH, Jasin M. 1999. Genes Dev 13: 2633–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Buisson R, Niraj J, Rodrigue A, Ho CK, Kreuzer J, et al. 2017. Mol Cell 65: 336–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kim D, Liu Y, Oberly S, Freire R, Smolka MB. 2018. Nucleic Acids Res 46: 8311–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lanz MC, Dibitetto D, Smolka MB. 2019. EMBO J 38: e101801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Szakal B, Branzei D. 2013. EMBO J 32: 1155–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. 2001. Mol Cell 7: 293–300 [DOI] [PubMed] [Google Scholar]
- 200.Palou R, Palou G, Quintana DG. 2017. Curr Genet 63: 275–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kim JA, Hicks WM, Li J, Tay SY, Haber JE. 2011. Mol Cell Biol 31: 507–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gobbini E, Villa M, Gnugnoli M, Menin L, Clerici M, Longhese MP. 2015. PLoS Genet 11: e1005685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ohouo PY, Bastos de Oliveira FM, Liu Y, Ma CJ, Smolka MB. 2013. Nature 493: 120–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ramos F, Villoria MT, Alonso-Rodriguez E, Clemente-Blanco A. 2019. Cell Stress 3: 70–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Dibitetto D, Ferrari M, Rawal CC, Balint A, Kim T, et al. 2016. Nucleic Acids Res 44: 669–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Millan-Zambrano G, Santos-Rosa H, Puddu F, Robson SC, Jackson SP, Kouzarides T. 2018. Mol Cell 72: 625–35 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. 1998. Cell 94: 399–409 [DOI] [PubMed] [Google Scholar]
- 208.Blank HM, Sheltzer JM, Meehl CM, Amon A. 2015. Mol Biol Cell 26: 1440–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Syljuasen RG, Jensen S, Bartek J, Lukas J. 2006. Cancer Res 66: 10253–7 [DOI] [PubMed] [Google Scholar]
- 210.Memisoglu G, Lanz MC, Eapen VV, Jordan JM, Lee K, et al. 2019. Cell Rep 28: 1090–102 e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Leroy C, Lee SE, Vaze MB, Ochsenbein F, Guerois R, et al. 2003. Mol Cell 11: 827–35 [DOI] [PubMed] [Google Scholar]
- 212.Keogh MC, Kim JA, Downey M, Fillingham J, Chowdhury D, et al. 2006. Nature 439: 497–501 [DOI] [PubMed] [Google Scholar]
- 213.Lee SE, Pellicioli A, Vaze MB, Sugawara N, Malkova A, et al. 2003. Mol Cell Biol 23: 8913–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Rawal CC, Riccardo S, Pesenti C, Ferrari M, Marini F, Pellicioli A. 2016. Cell Cycle 15: 2906–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Jazayeri A, Balestrini A, Garner E, Haber JE, Costanzo V. 2008. EMBO J 27: 1953–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Clerici M, Mantiero D, Lucchini G, Longhese MP. 2006. EMBO Rep 7: 212–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Eapen VV, Sugawara N, Tsabar M, Wu WH, Haber JE. 2012. Mol Cell Biol 32: 4727–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kotsantis P, Petermann E, Boulton SJ. 2018. Cancer Discov 8: 537–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Hanahan D, Weinberg RA. 2011. Cell 144: 646–74 [DOI] [PubMed] [Google Scholar]
- 220.Negrini S, Gorgoulis VG, Halazonetis TD. 2010. Nat Rev Mol Cell Biol 11: 220–8 [DOI] [PubMed] [Google Scholar]
- 221.Choi M, Kipps T, Kurzrock R. 2016. Mol Cancer Ther 15: 1781–91 [DOI] [PubMed] [Google Scholar]
- 222.Olivier M, Hollstein M, Hainaut P. 2010. Cold Spring Harb Perspect Biol 2: a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, et al. 2018. Cell 173: 371–85 e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lecona E, Fernandez-Capetillo O. 2018. Nat Rev Cancer 18: 586–95 [DOI] [PubMed] [Google Scholar]
- 225.Fishler T, Li YY, Wang RH, Kim HS, Sengupta K, et al. 2010. Oncogene 29: 4007–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Tho LM, Libertini S, Rampling R, Sansom O, Gillespie DA. 2012. Oncogene 31: 1366–75 [DOI] [PubMed] [Google Scholar]
- 227.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, et al. 2004. Cancer Res 64: 9152–9 [DOI] [PubMed] [Google Scholar]
- 228.Charrier JD, Durrant SJ, Golec JM, Kay DP, Knegtel RM, et al. 2011. J Med Chem 54: 2320–30 [DOI] [PubMed] [Google Scholar]
- 229.Weber AM, Ryan AJ. 2015. Pharmacol Ther 149: 124–38 [DOI] [PubMed] [Google Scholar]
- 230.Pilie PG, Tang C, Mills GB, Yap TA. 2019. Nat Rev Clin Oncol 16: 81–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, et al. 2018. Nat Med 24: 939–46 [DOI] [PubMed] [Google Scholar]
- 232.Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. 2018. Nat Med 24: 927–30 [DOI] [PubMed] [Google Scholar]
- 233.Niu D, Wei HJ, Lin L, George H, Wang T, et al. 2017. Science 357: 1303–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Brown KR, Mair B, Soste M, Moffat J. 2019. Mol Syst Biol 15: e8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Schiroli G, Conti A, Ferrari S, Della Volpe L, Jacob A, et al. 2019. Cell Stem Cell 24: 551–65 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Ho B, Baryshnikova A, Brown GW. 2018. Cell Syst 6: 192–205 e3 [DOI] [PubMed] [Google Scholar]
- 237.Hustedt N, Seeber A, Sack R, Tsai-Pflugfelder M, Bhullar B, et al. 2015. Mol Cell 57: 273–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Yu TY, Kimble MT, Symington LS. 2018. Proc Natl Acad Sci U S A 115: E11961–E9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Colombo CV, Menin L, Ranieri R, Bonetti D, Clerici M, Longhese MP. 2019. Genetics 211: 515–30 [DOI] [PMC free article] [PubMed] [Google Scholar]