
Figure 2.
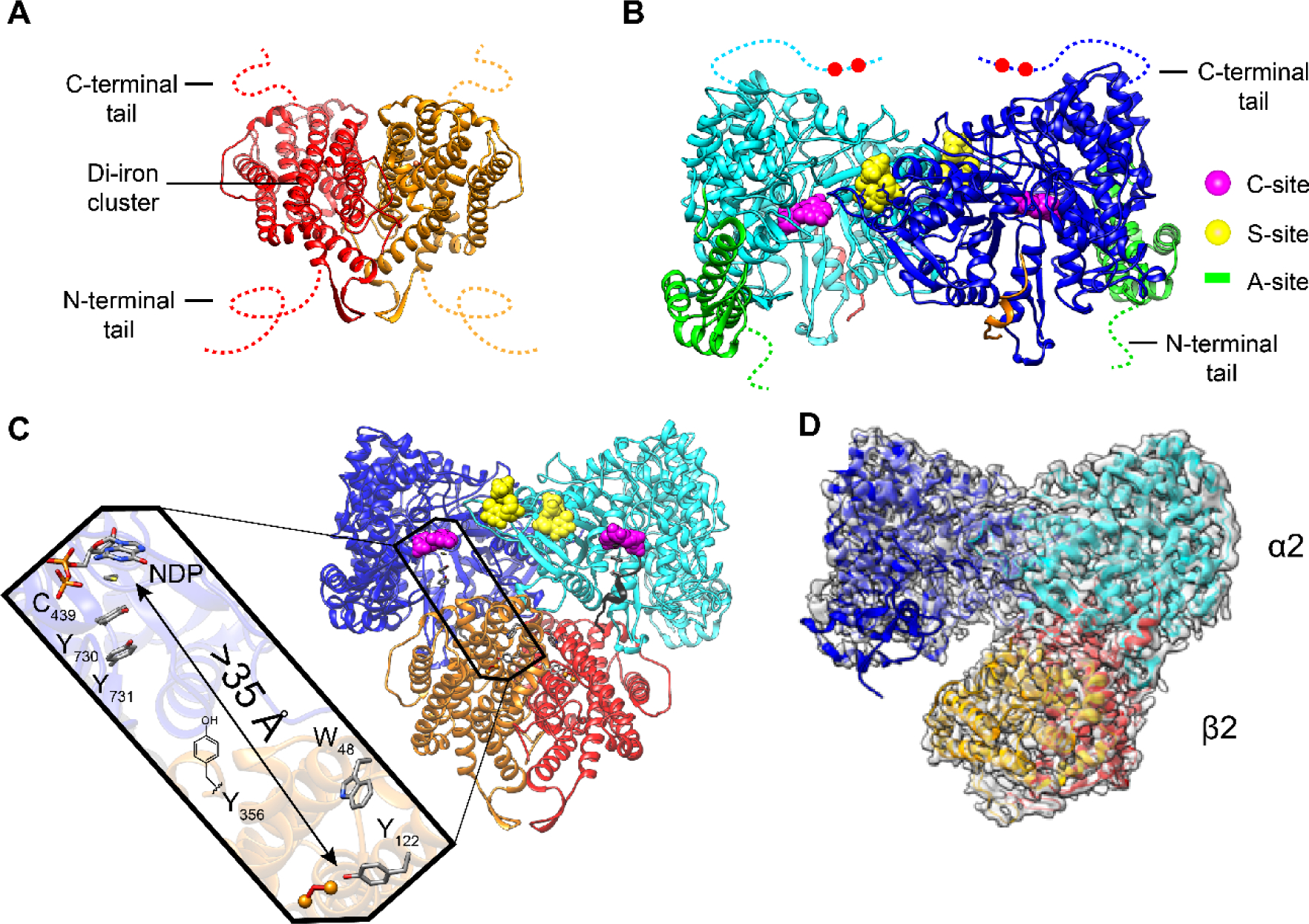
Structural models of class Ia RNR from E. coli. X-ray structures of A β2 (15), B α2 (13), C the Eklund docking model of α2β2 (13), and D a cryo-EM structure of an active α2β2 with two mutations in β2: F3Y122/E52Q (11). A β2, a homodimer in red/orange with disordered C-terminal tail residues (dashed lines 341–375 E. coli). B α2, a homodimer in light and dark blue with disordered C-terminal tail residues (dashed lines 737–761) that houses the two cysteines (red balls) which re-reduce the active site disulfide formed on NDP reduction (Figure 1A). α2 also houses the A-site (activity site or cone domain) that binds ATP (that activates RNR) or dATP (that inactivates RNR) in green; the C-site (catalytic site that binds CDP, UDP, GDP, and ADP) in magenta; the S-site (specificity site that binds the effectors dATP, ATP, TTP dGTP) in yellow. C Docking model of α2β2 with the long-range radical transfer pathway (left) (14). Shown also is a peptide in gray (residues 360–375 of β2) proposed to represent the tail of β2 responsible for α2 binding. D Asymmetric complex formed when F3Y122/E52Q-β2, interacts with α2, GDP, and TTP. 3.6-Å resolution cryo-EM density shown in transparent gray. This structure of the active α2β2 can be compared with the symmetric docking model in C.