

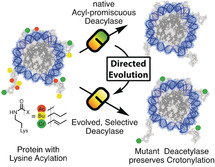
Abstract
Lysine acylations, a family of diverse protein modifications varying in acyl‐group length, charge, and saturation, are linked to many important physiological processes. Only a small set of substrate‐promiscuous lysine acetyltransferases and deacetylases (KDACs) install and remove this vast variety of modifications. Engineered KDACs that remove only one type of acylation would help to dissect the different contributions of distinct acylations. We developed a bacterial selection system for the directed evolution of KDACs and identified variants up to 400 times more selective for butyryl‐lysine compared to crotonyl‐lysine. Structural analyses revealed that the enzyme adopts different conformational states depending on the type of acylation of the bound peptide. We used the butyryl‐selective KDAC variant to shift the cellular acylation spectrum towards increased lysine crotonylation. These new enzymes will help in dissecting the roles of different lysine acylations in cell physiology.
Keywords: directed evolution, genetic code expansion, lysine acylation, post-translational modifications, sirtuins
Ch‐ch‐changes: Cellular proteins exhibit complex lysine acylation patterns, which are reversed by a small set of substrate‐promiscuous lysine deacetylases (KDACs). A strategy was developed to create KDAC variants with increased selectivity towards acetyl (Ac) over butyryl (Bu) and crotonyl (Cr) modifications through directed evolution by using a novel selection system. These engineered KDACs can be used to dissect the roles of different lysine acylations in cells.
Introduction
Lysine acetylation was first discovered more than fifty years ago as a posttranslational modification of histone proteins.1 The past two decades have revealed a wide variety of functional roles of this modification in almost every physiological process,2 such as transcription,3 chromatin structure,4 cytoskeleton organization,5 and energy metabolism.6 Unsurprisingly, defects in the enzymes governing lysine acetylation are linked to a variety of diseases such as diabetes,7 cancer,8 neurodegeneration,9 and aging.10
The acylation spectrum of lysine side chains is not restricted to acetylation (here abbreviated as ac), but ranges from short acyl chains, for example, butyryl (bu) or crotonyl (cr), to fatty acids and charged functional groups.11 All of these modifications are reversed by a comparably small set of lysine deacetylases (KDACs) grouped into four enzyme families. The related class 1, 2, and 4 enzymes12 are structurally and mechanistically distinct from class 3 KDACs.13 The former classes contain an active site zinc ion to orient a water molecule and to polarize the substrate. The latter class, the sirtuins, consist of a zinc‐binding and a Rossmann‐fold domain, which are responsible for acyl peptide binding and NAD+ binding, respectively, connected by a flexible cofactor‐binding loop.13b, 14 They couple the hydrolysis of the amide bond to the cleavage of NAD+,15 producing nicotinamide (NAM) and O‐acetyl‐ADP‐ribose.16
KDACs differ in their preference towards various acylations and peptide sequences, the biological relevance and purpose of which are still unclear.17 For example, class 1 KDACs (HDAC1, HDAC2, HDAC3, and HDAC8) and SirT1 exhibit significant decrotonylase activity in vivo.18 SirT1–3 are active towards various uncharged acylations in vitro, while SirT5 mainly removes negatively charged acylations and SirT6 and HDAC11 exhibit preference for long‐chain fatty acids.19
Despite extensive structural and mechanistic studies,13b, 20 how KDACs discriminate between different types of acylations remains poorly understood. In order to deconvolute the role of different lysine acylations, acyl‐type‐selective KDAC variants would be tremendously useful tools. However, their rational design is very challenging. For example, an attempt to create an acetyl‐specific HDAC1 variant by rational design resulted in an enzyme that lost deacetylation and retained decrotonylation activity.17, 18 The inability to predict the outcome of mutagenesis campaigns can be compensated by testing an increasingly large number of mutants. Ideally, the desired activity is coupled to a phenotypically selectable trait of a microorganism. Such selection systems allow the identification of desirable variants in large mutant pools (>109).
Herein, we report on the design of a selection system for KDACs that enables the directed evolution of variants with altered deacylation selectivity. The system relies on replacing an essential active‐site lysine residue of orotidine‐5′‐monophosphate (OMP) decarboxylase with lysine derivatives by genetic code expansion.21 In the absence of uracil, cells have to remove the acylation to turn on uracil biosynthesis, thereby coupling deacetylase activity to the survival of the host cell (Figure 1 A). This allows us to evolve KDACs for selectivity for particular lysine acylations. We show that these KDAC variants can be used to shift the cellular acylation pattern, which will help to reveal the physiological significance of alternative lysine acylations.
Figure 1.
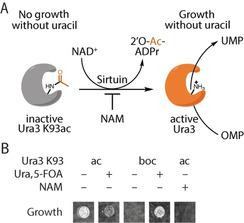
Design of the KDAC selection system. A) Deacetylation of OMP decarboxylase K93ac (Ura3 K93ac) by sirtuins enables growth of E. coli in the absence of uracil. B) E. coli producing the OMP decarboxylase Ura3 K93ac as the sole source for UMP depend on KDAC activity. Cell growth is shown for E. coli DB6656 (ΔpyrF) expressing plasmids to encode Ura3 K93ac or K93boc (not cleavable, tert‐butyloxycarbonyl‐lysine) on agar plates with or without uracil and 5‐FOA. Nicotinamide (NAM), which inhibits endogenous CobB, prevents growth.
Results and Discussion
Design of a Selection System for Acyl‐Type‐Specific Lysine Deacetylases
In order to develop a selection system for acyl‐type‐specific KDACs, we first searched for a selectable marker with an essential lysine residue that could be masked by acylation. Mutation of an active‐site lysine (K93) to alanine in budding yeast OMP decarboxylase (Ura3, which is required for the biosynthesis of uracil), is known to reduce its activity by more than five orders of magnitude.22 To test whether acetylation of K93 has a similar impact on catalysis, we complemented E. coli ΔpyrF (the homologue of Ura3) with Ura3 K93ac (Figure 1 B). Ura3 K93ac is produced by replacing the respective lysine codon in the Ura3 gene with an amber (UAG) codon and encoding the incorporation of N(ϵ)‐acetyl‐lysine (AcK) with M. barkeri AcKRS3/PylT (an AcK‐specific variant of pyrrolysyl‐tRNA synthetase (PylS) and its cognate amber suppressor tRNA23).
We tested the selection system by cultivating E. coli expressing Ura3 K93ac as the sole source of OMP decarboxylase on minimal media without uracil. The cells were able to grow in the absence of uracil, thus indicating that CobB (the major lysine deacetylase of E. coli) was able to remove the acetyl group from the active site lysine of Ura3 K93ac, but did not grow when CobB was inhibited with NAM. Growth of the same cells is inhibited when 5‐fluoro‐orotic acid (5‐FOA), a compound converted to a toxic metabolite by Ura3,24 is added to the medium. Incorporation of N(ϵ)‐tert‐butyl‐oxycarbonyl‐lysine (BocK) in place of AcK did not complement pyrF deficiency, thus indicating that this lysine modification is not a substrate of CobB.
Hence, this system is able to positively (auxotrophic selection) and negatively (5‐FOA sensitivity) select E. coli harboring an active KDAC. To demonstrate the applicability of our selection system to other KDACs, we used it to select for the activity of human SirT1–3 and HDAC8, a mammalian class I KDAC structurally and mechanistically distinct from the sirtuin family member CobB (Figure S1 in the Supporting Information).
Isolation of Acyl‐Type‐Specific Deacetylases
Next, we aimed to create acyl‐type‐selective variants of CobB. We constructed a mutant library by randomizing five active‐site residues (A76, Y92, R95, I131 and V187) of CobB to all possible combinations of natural amino acids, thereby creating 205 (3.2×106) different mutants (Figure 2 A). To identify CobB mutants that selectively remove acetyl but not crotonyl groups, we subjected the library to three rounds of selection (positive, negative, positive).
Figure 2.
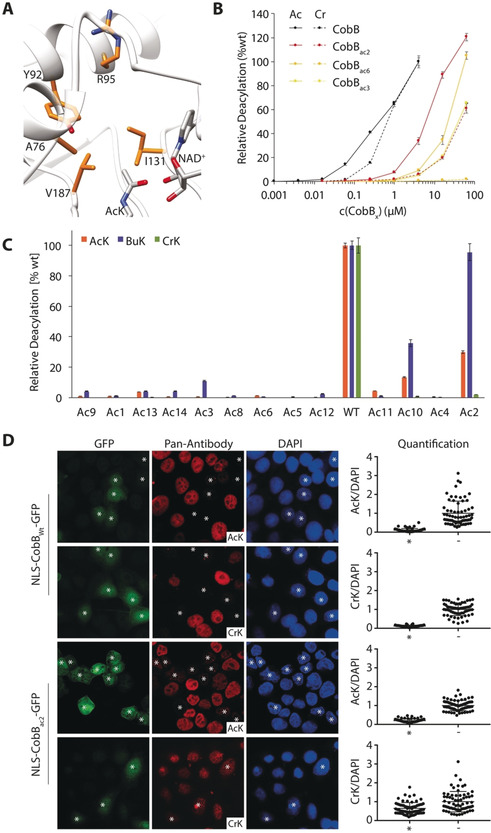
Creation of AcK‐selective CobB variants to modulate cellular acylation patterns. A) CobB library design. Highlighted amino acid residues were randomized to all possible combinations of natural amino acids to generate a library of thirty million mutants in the active site of CobB (image was created based on PDB IDs: 1S5P, 2H4F). B) Deacylation selectivity of Ni2+‐NTA purified CobB and selected variants (load control and CobBAc7 in Figure S3) using FLuc K529ac/bu/cr. C) Demodification of FLuc K529ac and FLuc K529cr by CobB and AcK‐selective variants. NLS‐CobBwt‐GFP and NLS‐CobBac2‐GFP were expressed in HeLa cells for 24 h and cells stained by immunofluorescence for lysine acetylation or crotonylation (treated with 2 mm sodium crotonate 2 h prior fixation, see Supplementary Figure 7). The change of the nuclear acylation level was quantified in transfected (*) and untransfected (−) cells (n≥50) as a ratio of the antibody signal divided by the DAPI fluorescence and normalized to the average ratio of the untransfected cells. Shown are all individual measurements with the standard deviation of the mean. Ac=acetyl, Bu=butyryl, Cr=crotonyl.
In the first round of positive selection for CobB variants able to remove acetyl groups, we grew E. coli ΔpyrF ΔcobB transformed with the CobB mutant library and expressing Ura3 K93ac in the presence of AcK on medium without uracil. The subsequent round of negative selection aimed to remove CobB variants active towards crotonyl groups from the remaining pool. Therefore, the library plasmids were isolated from the pool of surviving clones of the first round of selection and used to transform E. coli ΔpyrF ΔcobB producing Ura3 K93cr (encoded with wild‐type PylS25). Cells were grown on plates containing N(ϵ)‐crotonyl‐lysine (CrK) and 5‐FOA to select against CobB mutants capable of removing crotonyl groups from Ura3 K93cr. After a third positive round of selection for deacetylase activity, CobB library plasmids were isolated from individual clones and re‐tested for their ability to allow cells to survive on uracil‐free medium when expressing Ura3 K93ac.
We sequenced 60 isolates corresponding to 14 unique CobB mutants. Eight of the sequences dominated this set and accounted for 53 of the sequenced CobB isolates (Table S1). This shows that the selection system is capable of enriching individual sequences from a mean frequency of 3.125×10−7 (1/3.2×106) in the original library to 0.3 (18 out of 60) after three rounds of selection, thus suggesting amplification by a factor of almost one million.
To identify the most active KDAC variants among the isolates, we set out to modulate the stringency of our selection system. The reporter enzyme Ura3 is inhibited by 6‐azauridine (6‐AU). Addition of this compound to the medium should therefore raise the threshold level of this enzyme required for cell growth. We arrayed the 14 different clones isolated in the selection on plates containing increasing concentrations of 6‐AU and monitored growth at 37 °C (Figure S2). The ability of the clones to survive increasing 6‐AU concentrations varied between 0.16 to 2 mm.
Biochemical Characterization of Evolved CobB Variants
Next, we purified the mutant enzymes and analyzed their ability to reverse the modification of purified firefly luciferase (FLuc) K529ac, K529bu and K529cr26 (Figure 2 B). This in vitro assay uses the same principle described for Ura3 of capping an essential lysine residue with an acylation. If a KDAC deacylates FLuc, a luminescence signal can be measured. With 25 % activity against FLuc K529ac and almost 100 % against FLuc K529bu compared to wild‐type, CobBac2 turned out to be the most active variant, approximately fourfold more active than CobBac6 (Figure 2 C and Figure S3), which is consistent with providing the highest resistance to 6‐AU (Figure S2).
The diminished activity of CobBac3 and CobBac6 for AcK may be explained by an increased K M for NAD+ (Figure S4 A). These mutants retained similar sensitivity to inhibition by NAM. CobBac6 showed a decreasing deacylation activity with increasing acyl chain length (Figure S4 B). Interestingly, CobBac2 and CobBac3 preferred BuK by more than tenfold over AcK and 50‐ or 400‐fold over CrK, respectively, thus demonstrating remarkable selectivity of these variants (Figure 2 B). CobBac2 still showed measurable decrotonylation activity compared to CobBac3/6, however, when we tested this mutant using myoglobin with different acylations at residue four23b as substrates, it was able to remove acetylation and butyrylation but not crotonylation (Figure S5). The more selective variants, CobBac3 and CobBac6, were unable to remove the acetylation (or any other acylation tested) of this residue to a measurable extent (Figure S5). We attribute this lack of activity to the difference in the substrates and the differential sensitivity of the assays to detect the unmodified protein. Similarly, CobBac2 was capable of removing H4 K16ac from isolated HeLa histones, while its ability to reverse histone crotonylation was strongly reduced compared to wild‐type CobB (Figure S6).
To show that the unique deacylation activity of CobBac2 can be used to shift the cellular acylation spectrum, we expressed the mutant and wild‐type protein, fused to GFP and a nuclear localization sequence (NLS), in HeLa cells. Indeed, transfected cells (*) showed drastic changes in their acylation levels. Wild‐type CobB strongly reduced the nuclear levels of lysine acetylation, butyrylation, and crotonylation to the same extent, while CobBac2 only removed acetylation and butyrylation efficiently, with the signal for lysine crotonylation being largely maintained (Figure 2 D and Figure S7 A). In fact, the reduction in the signal for crotonylation by CobBac2 is probably the result of the removal of lysine butyrylation, which is recognized equally well by the pan‐crotonyl‐lysine antibody (Figure S7 B). To enhance the level of crotonylation, we grew the cells in the presence of crotonate, which increased the intensity of the persistent signal in CobBac2‐transfected cells (Figure 2 D).
In summary, our combined selection and screening system can be used to identify acyl‐type‐specific variants in a large pool of KDAC mutants. In particular, CobBac2 can be used to shift the lysine acylation pattern of the proteome towards crotonylation or to selectively deplete lysine butyrylation, and thus may help to uncover the specific role of these modifications in cell physiology.
Molecular Mechanism Responsible for the Loss of Decrotonylation Activity in CobBac2
The mutations A76G and I131C in CobBac2 selectively abolish decrotonylation activity, while the enzyme still removes acetyl groups with 25 % and butyryl groups with 100 % activity (Figure 2 C). To better understand the molecular mechanism, we crystalized CobB and CobBac2 in complex with histone H4 peptides acylated at lysine 16 with either acetyl, butyryl or crotonyl (see Table S2 for crystallographic data). The overall structure of CobBac2 is very similar to wild‐type CobB, with a rmsd of less than 0.5 Å (Figure 3). The most pronounced changes in the CobBac2 structures bound to different peptides concentrate on the cofactor‐binding loop connecting Helix α1 and Helix α2, whereas the wild‐type structures are little affected by the type of acylation of the bound peptide (Figure 3).
Figure 3.
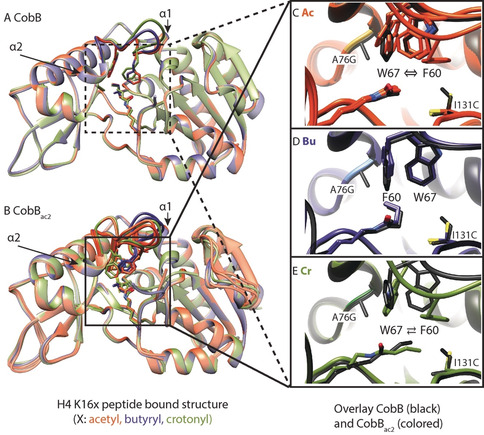
A, B) Crystal structures of wild‐type CobB (A) and the A76G, I131C mutant CobBac2 (B) in complex with K16‐modified H4 peptides. Red‐brown=acetyl, blue=butyryl, green=crotonyl. Overlay of different substrates shows high conformational variability of the cofactor binding loop connecting Helix α1 and Helix α2 (highlighted) of CobBac2 compared to wild‐type. C–E) Overlay of active‐site residues (black wild‐type, colored CobBac2) in the presence of a peptide substrate with Ac (C), Bu (D), or Cr (E) modifications. Depending on the type of modification, the substrates either induce the native conformation (Bu), a flipped conformation (Cr, ⇄), or a mixture of both (Ac, ⇔) in CobBac2. Several chains from an elementary cell are overlaid (for chains D and H in CobBac2:H4 K16ac and chain A in CobBac2:H4 K16cr see Figure S8).
The A76G and I131C mutations in CobBac2 allow the loop to adopt two alternative conformations, the choice of which is dictated by the substrate. The loop conformations differ mainly in the arrangement of W67 and F60. The butyryl group stabilizes the original arrangement of these residues through its kinked binding mode. A crotonyl group lacks this flexibility due to its conjugated bonds and, therefore, dislocates F60 from its original position. The vacated space is then occupied by W67, causing a distortion of the cofactor‐binding loop (Figure 3 E). Although the enzyme prefers to adopt this new conformation, it is still able to rearrange to its original conformation, since a second chain in the same unit cell shows a superposition of both structures (Figure S8).
The acetyl group does not stabilize either conformation, resulting in multiple arrangements of the loop observed for different chains within the same unit cell. Crotonyl‐lysine is likely to stabilize the loop in a new conformation through an induced‐fit mechanism.27 The dynamic nature of this loop becomes apparent in the acetyl‐lysine‐bound structures, which display several different loop conformations (Figure 3 C), thus indicating that the apo‐enzyme already exists in different conformational states.
We observed similar structural alterations for CobBac3 bound to the acetylated peptide (Figure S9 A,B), thus indicating that it acquires its selectivity by a similar mechanism. To test whether swapping of F60 and W67 is the main cause of the butyryl selectivity, we interchanged both residues by mutagenesis. CobB F60W W67F displayed a similar preference for butyryl lysine as CobBac2 (Figure S9 C). On condition that the cofactor‐binding loop of the mutated enzyme adopts the same conformation as in wild‐type CobB, it can be assumed that the apo‐enzyme is inactive. Only when a butyrylated peptide binds, the cofactor‐binding loop rearranges itself so that catalysis can take place. Hence, this structural alteration is the key feature responsible for butyryl selectivity. However, since we could not obtain crystals of CobBac3 bound to butyrylated or crotonylated peptides, we cannot exclude other explanations for the selectivity of CobBac3.
Since we expected the altered conformation to be inactive, we attempted to solve the structure of the complex of CobBac2 with NAD+ and crotonyl peptide (Figure 4). We obtained crystals of the complex by co‐crystallization after 16 h (Figure 4 A) and by soaking for 36 h (Figure 4 B). We observed a continuous density that linked the ADP‐ribose moiety to the crotonyl group via the 2′‐OH of the ribose (Figure 4 B), thus suggesting the presence of a reaction intermediate. This density is best explained by reaction intermediate III (Int. III) that was previously reported by Wang et al. for the SirT2 reaction cycle in the presence of a thiomyristoyl inhibitor.28 We modelled the analogous crotonyl‐Int. III with 60 % (Figure 4 A) and 100 % (Figure 4 B) occupancy for both states. Usually, this intermediate is hydrolyzed, but the repositioned W67 blocks the access of water to the active site (Figure 4 A). Indeed, water is only near Int. III after W67 has returned to its original position, as seen in the 36 h structure (Figure 4 B). Nevertheless, there is no clear indication in this structure for hydrolysis of the intermediate, thus suggesting that other factors may also be required.
Figure 4.
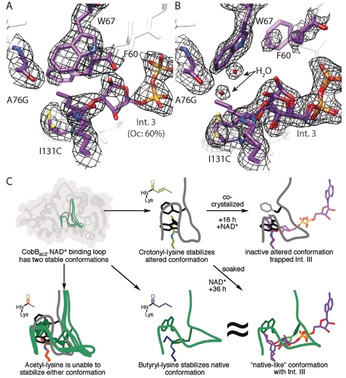
Demodification of crotonylated peptide stalls at intermediate III in the CobBac2 mutant. A, B) Active‐site views are shown for CobBac2/H4K16cr co‐crystallized with NAD+ for 16 h (A) and soaked with NAD+ for 36 h (B). The 2 F o−F c omit map (black, 1 σ) is shown for key residues. The density map shows a clear continuous density between the crotonyl group and the 2′‐OH group of the ADP‐ribose. Therefore, the intermediate was modelled analogously to the intermediate III described by Wang et al.28 C) A cartoon model highlights the critical changes in the cofactor binding loop of CobBac2 by which the enzyme recognizes and specifically stalls the decrotonylation reaction.
This conclusion is supported by the observation that the equivalent of F60 (F33) in Thermotoga maritima Sir2 shields the reaction intermediates from premature hydrolysis and ADP‐ribose formation,14 and that the equivalent of W67 (Y40) is predicted by MD simulations to control the access of water to the active site.29
Residue I131, which is mutated to Cys in CobBac2, is part of the conserved NID motif30 of sirtuins. Interestingly, its mutation to cysteine is naturally present in human SirT7. The A76G mutation, to our knowledge, does not exist in other proteins. However, at the equivalent position in SirT3, the mutation F180L was found to increase selectivity for acetyl by 20‐fold over crotonyl‐lysine, possibly by removing the π–π stacking interaction with the crotonyl group.19d
In summary, we have shown that CobBac2 acquires its selectivity for butyrylated peptides through a substrate‐induced mechanism in which the mutations A76G and I131C allow the cofactor‐binding loop to adopt two alternative conformations (Figure 4 C). The crotonyl‐induced conformation is inactive because access of water to the active site is blocked. This leads to an accumulation of Int. III, which is slowly hydrolyzed upon reversion of the loop conformation.
Creation of Acyl‐Type Selective SirT1 Variants
To test the generality of our selection approach, we created a library in the active site of SirT1 by randomizing five residues (A313, I316, I347, F366, I411) to all possible combinations of natural amino acids (Figure 5 A). We subjected this library to three rounds of selection (positive on Ura3 K93ac; negative on either Ura3 K93pr/bu/cr; positive on Ura3 K93ac) and sequenced in total 82 isolates after the third round (Figure 5 B).
Figure 5.
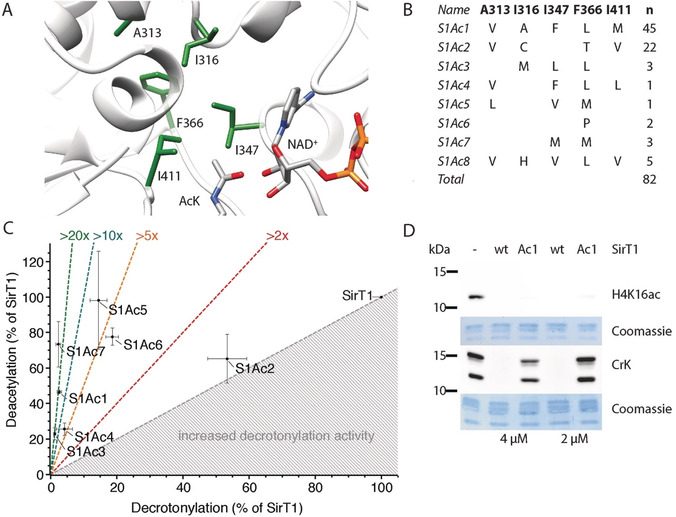
Creation of SirT1 variants with AcK preference. A) Structure of the SirT1 active site (PDB ID: 4KXQ) with ligands NAD+ and AcK imported from PDB IDs 2H4F and 1S5P, respectively, shown as stick models. Residues highlighted in green were randomized to all possible combinations of natural amino acids in the mutant library. B) SirT1 variants isolated after three rounds of selection from the library. C) Acyl‐type preference of SirT1 variants assayed on FLuc K529ac and K529cr. D) SirT1 variant S1Ac1 deacetylates H4 K16ac but is compromised in histone decrotonylation. Activity of SirT1 and SirT1Ac1 (2 μm or 4 μm) was measured by Western blot on isolated human histones using antibodies against H4 K16ac or pan‐anti‐CrK.
The spectrum of isolates retrieved from different negative selections was very similar and converged on eight different sequences, whereby two of these corresponded to more than 80 % of the sequenced clones. We compared the selectivity of the SirT1 mutants for their ability to deacylate FLuc K529ac and K529cr. All mutants were shifted in their substrate preference towards lysine deacetylation compared to SirT1 (Figure 5 C). The best mutants discriminated between acetylated and crotonylated FLuc more than 20 times better than SirT1, while maintaining close to wild‐type activity towards the acetylated form. This increased selectivity was maintained towards histones. SirT1Ac1 deacylated H4 K16ac with near wild‐type activity but was almost 20 times less efficient in removing lysine crotonylation (Figure 5 D). The activity of the SirT1 variants decreased with increasing acyl chain length, thus indicating that a reduction in the volume of the substrate binding site causes the preference for acetylated lysine residues (Figure S10). Similar rearrangements of hydrophobic pockets were reported for engineering of SH3 domains,31 ubiquitin,32 and human carbonic anhydrase II.33 This shows that the directed evolution approach is portable to other KDAC isoforms.
Conclusion
The substrate promiscuity of sirtuins20b impairs the clean delineation of individual functional roles of different lysine acylations in cellular processes. The KDAC selection system reported here facilitates the rapid modulation of their selectivity, thereby enabling us to identify acyl‐type‐specific variants of the naturally promiscuous sirtuins CobB and SirT1. We show that these acyl‐type‐specific KDAC variants can be used to shift the acylation pattern of the cellular proteome and, hence, will be useful to deconvolute the individual contributions of different types of lysine acylations to cell physiology. They can be employed to erase a particular acylation by overexpression in the wild‐type background or to partially complement knockout cell lines. Targeted to specific genomic regions, they can be used to locally deplete particular types of acylation and to investigate the impact of metabolites on gene expression.34
In comparison, the rational design of acyl‐type‐selective mutants is challenging and often results in partial success. CobB mutants with up to 43‐fold increased preference for AcK over N(ϵ)‐succinyl‐lysine have been created in this way.35 A crotonyl‐lysine selective HDAC1 was used to investigate the role of this modification in transcriptional regulation and self‐renewal of mouse embryonic stem cells.18 In these cases, selectivity was successfully engineered by removing acyl‐specific interactions. For sirtuins, however, there appears to be a more complex interaction between active‐site residues, thus making the rational design of selective variants very difficult.
Our selection system is a powerful tool to find acyl‐selective KDAC mutants for any desired lysine acylation through genetic code expansion, which will be tremendously useful for future mechanistic studies. Furthermore, the selection system could also be used to facilitate the creation of selective mutant/inhibitor pairs by a bump‐and‐hole strategy,36 drug discovery of plasmid encoded compound libraries,37 or the creation of enzymes with novel bioorthogonal reactivity.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Petra Geue for technical support and Sascha Gentz for peptide synthesis. Datasets were collected at PXII X10SA beamline at the Suisse Light Source, Villigen, Switzerland. We thank local contacts and beamline scientists for their support. This project was supported financially by the Heisenberg Program of the German Research Foundation (DFG) [NE1589/5‐1], the Human Frontier Science Program (HFSP, RPG0031/2017) and the Max‐Planck‐Institute of Molecular Physiology.
M. Spinck, P. Neumann-Staubitz, M. Ecke, R. Gasper, H. Neumann, Angew. Chem. Int. Ed. 2020, 59, 11142.
A previous version of this manuscript has been deposited on a preprint server (https://www.biorxiv.org/content/10.1101/723684v1).
References
- 1. Allfrey V. G., Faulkner R., Mirsky A. E., Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Verdin E., Ott M., Nat. Rev. Mol. Cell. Biol. 2015, 16, 258–264. [DOI] [PubMed] [Google Scholar]
- 3. Feige J. N., Auwerx J., Curr. Opin. Cell Biol. 2008, 20, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilkins B. J., Rall N. A., Ostwal Y., Kruitwagen T., Hiragami-Hamada K., Winkler M., Barral Y., Fischle W., Neumann H., Science 2014, 343, 77. [DOI] [PubMed] [Google Scholar]
- 5. Tas R. P., Chazeau A., Cloin B. M. C., Lambers M. L. A., Hoogenraad C. C., Kapitein L. C., Neuron 2017, 96, 1264–1271.e1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Houtkooper R. H., Pirinen E., Auwerx J., Nat. Rev. Mol. Cell Biol. 2012, 13, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jing E., Emanuelli B., Hirschey M. D., Boucher J., Lee K. Y., Lombard D., Verdin E. M., Kahn C. R., Proc. Natl. Acad. Sci. USA 2011, 108, 14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haberland M., Johnson A., Mokalled M. H., Montgomery R. L., Olson E. N., Proc. Natl. Acad. Sci. USA 2009, 106, 7751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stilling R. M., Fischer A., Neurobiol. Learn. Mem. 2011, 96, 19–26. [DOI] [PubMed] [Google Scholar]
- 10. Rogina B., Helfand S. L., Proc. Natl. Acad. Sci. USA 2004, 101, 15998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Choudhary C., Weinert B. T., Nishida Y., Verdin E., Mann M., Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [DOI] [PubMed] [Google Scholar]
- 12. Yang X. J., Seto E., Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Feldman J. L., Dittenhafer-Reed K. E., Denu J. M., J. Biol. Chem. 2012, 287, 42419–42427; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Sanders B. D., Jackson B., Marmorstein R., Biochim. Biophys. Acta Proteins Proteomics 2010, 1804, 1604–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hawse W. F., Hoff K. G., Fatkins D. G., Daines A., Zubkova O. V., Schramm V. L., Zheng W., Wolberger C., Structure 2008, 16, 1368–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Landry J., Sutton A., Tafrov S. T., Heller R. C., Stebbins J., Pillus L., Sternglanz R., Proc. Natl. Acad. Sci. USA 2000, 97, 5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tanner K. G., Landry J., Sternglanz R., Denu J. M., Proc. Natl. Acad. Sci. USA 2000, 97, 14178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao S., Zhang X., Li H., Curr. Opin. Struct. Biol. 2018, 53, 169–177. [DOI] [PubMed] [Google Scholar]
- 18. Wei W., Liu X., Chen J., Gao S., Lu L., Zhang H., Ding G., Wang Z., Chen Z., Shi T., Li J., Yu J., Wong J., Cell Res. 2017, 27, 898–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Feldman J. L., Baeza J., Denu J. M., J. Biol. Chem. 2013, 288, 31350–31356; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Tan M., Peng C., Anderson K. A., Chhoy P., Xie Z., Dai L., Park J., Chen Y., Huang H., Zhang Y., Ro J., Wagner G. R., Green M. F., Madsen A. S., Schmiesing J., Peterson B. S., Xu G., Ilkayeva O. R., Muehlbauer M. J., Braulke T., Muhlhausen C., Backos D. S., Olsen C. A., McGuire P. J., Pletcher S. D., Lombard D. B., Hirschey M. D., Zhao Y., Cell Metabolism 2014, 19, 605–617; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19c. Du J., Zhou Y., Su X., Yu J. J., Khan S., Jiang H., Kim J., Woo J., Kim J. H., Choi B. H., He B., Chen W., Zhang S., Cerione R. A., Auwerx J., Hao Q., Lin H., Science 2011, 334, 806–809; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19d. Bao X., Wang Y., Li X., Li X.-M., Liu Z., Yang T., Wong C. F., Zhang J., Hao Q., Li X. D., eLife 2014, 3, e02999; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19e. Cao J., Sun L., Aramsangtienchai P., Spiegelman N. A., Zhang X., Huang W., Seto E., Lin H., Proc. Natl. Acad. Sci. USA 2019, 116, 5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Sauve A. A., Biochim. Biophys. Acta Proteins Proteomics 2010, 1804, 1591–1603; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Bheda P., Jing H., Wolberger C., Lin H., Annu. Rev. Biochem. 2016, 85, 405–429; [DOI] [PubMed] [Google Scholar]
- 20c. Zhao K., Harshaw R., Chai X., Marmorstein R., Proc. Natl. Acad. Sci. USA 2004, 101, 8563–8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neumann-Staubitz P., Neumann H., Curr. Opin. Struct. Biol. 2016, 38, 119–128. [DOI] [PubMed] [Google Scholar]
- 22. Miller B. G., Snider M. J., Wolfenden R., Short S. A., J. Biol. Chem. 2001, 276, 15174–15176. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Neumann H., Hancock S. M., Buning R., Routh A., Chapman L., Somers J., Owen-Hughes T., van Noort J., Rhodes D., Chin J. W., Mol. Cell 2009, 36, 153–163; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Neumann H., Peak-Chew S. Y., Chin J. W., Nat. Chem. Biol. 2008, 4, 232–234. [DOI] [PubMed] [Google Scholar]
- 24. Meng X., Smith R. M., Giesecke A. V., Joung J. K., Wolfe S. A., Biotechniques 2006, 40, 179–184. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Gattner M. J., Vrabel M., Carell T., Chem. Commun. 2013, 49, 379–381; [DOI] [PubMed] [Google Scholar]
- 25b. Kim C. H., Kang M., Kim H. J., Chatterjee A., Schultz P. G., Angew. Chem. Int. Ed. 2012, 51, 7246–7249; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7358–7361; [Google Scholar]
- 25c. Wilkins B. J., Hahn L. E., Heitmuller S., Frauendorf H., Valerius O., Braus G. H., Neumann H., ACS Chem. Biol. 2015, 10, 939–944. [DOI] [PubMed] [Google Scholar]
- 26. Spinck M., Ecke M., Sievers S., Neumann H., Biochemistry 2018, 57, 3552–3555. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Bosshard H. R., News Physiol. Sci. 2001, 16, 171–173; [DOI] [PubMed] [Google Scholar]
- 27b. Koshland D. E., Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Y., Fung Y. M. E., Zhang W. Z., He B., Chung M. W. H., Jin J., Hu J., Lin H. N., Hao Q., Cell Chem. Biol. 2017, 24, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shi Y. W., Zhou Y. Z., Wang S. L., Zhang Y. K., J. Phys. Chem. Lett. 2013, 4, 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brachmann C. B., Sherman J. M., Devine S. E., Cameron E. E., Pillus L., Boeke J. D., Gene Dev. 1995, 9, 2888–2902. [DOI] [PubMed] [Google Scholar]
- 31. Ben-David M., Huang H. M., Sun M. G. F., Corbi-Verge C., Petsalaki E., Liu K., Gfeller D., Garg P., Tempel W., Sochirca I., Shifman J. M., Davidson A., Min J. R., Kim P. M., Sidhu S. S., J. Mol. Biol. 2019, 431, 336–350. [DOI] [PubMed] [Google Scholar]
- 32. Haririnia A., Verma R., Purohit N., Twarog M. Z., Deshaies R. J., Bolon D., Fushman D., J. Mol. Biol. 2008, 375, 979–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hunt J. A., Ahmed M., Fierke C. A., Biochemistry 1999, 38, 9054–9062. [DOI] [PubMed] [Google Scholar]
- 34. Heitmüller S., Neumann-Staubitz P., Herrfurth C., Feussner I., Neumann H., Metab. Eng. 2018, 47, 453–462. [DOI] [PubMed] [Google Scholar]
- 35. Colak G., Xie Z., Zhu A. Y., Dai L., Lu Z., Zhang Y., Wan X., Chen Y., Cha Y. H., Lin H., Zhao Y., Tan M., Mol. Cell. Proteomics 2013, 12, 3509–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shah K., Liu Y., Deirmengian C., Shokat K. M., Proc. Natl. Acad. Sci. USA 1997, 94, 3565–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tavassoli A., Curr. Opin. Chem. Biol. 2017, 38, 30–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary