


Abstract
Structural variants (SVs) that alter DNA sequence emerge as a driving force involved in the reorganisation of DNA spatial folding, thus affecting gene transcription. In this work, we describe an improved version of our integrated web service for structural modeling of three-dimensional genome (3D-GNOME), which now incorporates all types of SVs to model changes to the reference 3D conformation of chromatin. In 3D-GNOME 2.0, the default reference 3D genome structure is generated using ChIA-PET data from the GM12878 cell line and SVs data are sourced from the population-scale catalogue of SVs identified by the 1000 Genomes Consortium. However, users may also submit their own structural data to set a customized reference genome structure, and/or a custom input list of SVs. 3D-GNOME 2.0 provides novel tools to inspect, visualize and compare 3D models for regions that differ in terms of their linear genomic sequence. Contact diagrams are displayed to compare the reference 3D structure with the one altered by SVs. In our opinion, 3D-GNOME 2.0 is a unique online tool for modeling and analyzing conformational changes to the human genome induced by SVs across populations. It can be freely accessed at https://3dgnome.cent.uw.edu.pl/.
INTRODUCTION
There is a plethora of evidence to be found in the literature for the significant role of genome spatial organization in gene regulation for both health and disease (1–3). Structural variation (SV), encompassing deletions, duplications, insertions and inversions, is the main source of genetic variation in humans and it is shown to have a critical impact on higher-order chromatin conformation and gene functioning (1,4). Deletions, duplications, insertions and their combination may reorganize chromatin interactions by altering the DNA segments that are involved in the establishment of three-dimensional (3D) contacts. Inversions, on the other hand, may alter the directionality of the binding motifs of the CTCF proteins that bring the mentioned above segments together. SVs that do not overlap any interacting sites do not affect the resulting 3D structure (the number and relative arrangement of loops in the model), but they still contribute to the shortening or extending of chromatin loops.
By the effort of the 1000 Genomes Consortium, a catalogue of SVs identified in over 2500 human samples from 26 populations was created (5). Several examples of SVs present in non-coding regions were already reported to disrupt local 3D genome organization leading to an altered gene transcription (6,7) and in some cases causing disease (8–12). Nevertheless, this area of research is dominated by studies identifying spatial DNA structure for a selected and limited number of human cell lines (13,14). Thus, it lacks the broader perspective acquired by investigating the processes that shape genomic diversity in the human population as a whole - a focus of international studies such as the 1000 Genomes Project (1kGP) (5) or the Simons Genome Diversity Project (15). In this regard, the population analysis of SVs in the context of 3D genome structure can provide unique insights into biophysical mechanisms regulating chromatin organisation and gene transcription at the population scale (4).
To address this issue, we have implemented our recently published SV-based modifying algorithm (4), adopted to generate altered chromatin interaction patterns, to our previously developed 3D genome modeling engine 1.0 (3D-GENOME) web server (16). This web service generates chromatin 3D structures using a Monte Carlo approach based on Chromatin Conformation Capture (3C) data, providing tools for their visualization and analysis. Users can generate 3D structures of a genomic region of interest by simply specifying its coordinates. Here we present the 2.0 version of 3D-GNOME. With this update, users gain the ability to predict SVs-driven changes in 3D conformations of specific loci in human genomes. In the default setting, 3D-GNOME 2.0 employs chromatin interaction paired-end tag sequencing (ChIA-PET) data for the GM12878 cell line (6) as the reference chromatin interaction map and sets of SVs emerging across human populations constructed by 1kGP as the source of genetic variation (18). When relying on our precomputed reference 3D model, users may also provide a particular individual's SVs. 3D-GNOME 2.0 returns both the reference 3D structure and the structure altered by SVs. Alternatively, users can submit their list of loops obtained from 3C based methods such as Hi-C or ChIA-PET and a custom SV list and generate 3D structures of their loci and genomes of choice (Figure 1).
Figure 1.

A schematic representation of the workflow of 3D-GNOME 2.0.
3D-GNOME 2.0 provides novel tools to inspect, visualize and analyze 3D models of chromatin regions modified by SVs in the samples of interests at different levels of spatial chromatin organisation, starting with individual loops, genomic domains (chromatin contact domains-CCDs) and continuing to full chromosomes. Differences between the reference and altered structure can be analyzed with diagrams of contacts, statistical plots, and three-dimensional models. According to our knowledge, this is the first easily accessible online tool for modeling conformational changes in the human genome induced by SVs in different populations. A schematic representation of the workflow of 3D-GNOME 2.0 is presented in Figure 1.
NEW FEATURES AND UPDATES
Basic web server architecture with extensions
New features introduced to 3D-GNOME consist of tools for both the processing and analysis of SVs associated data. The overall architecture of the web server is maintained. In detail, 3D-GNOME 2.0 has been implemented by using the WSGI application Flask framework (https://palletsprojects.com/p/flask/), and upgraded to be compatible with Python 3.6+. A validated request, potentially including SVs information, is saved to the MySQL database. The data processing pipeline is written in Python, together with external scripts written in C++, PHP and R. The web server generates contact diagrams, plots, statistics and 3D models. To view the models, we maintain an interactive viewer implemented in WebGL (https://www.khronos.org/webgl/), developed as part of the previous version of 3D-GNOME.
An important change in version 2.0 is the implementation of a method for recovering individualized genomic interaction patterns based on reference interaction patterns and an individual set of SVs published previously (4). It is also available as a separate python script at https://bitbucket.org/4dnucleome/spatial_chromatin_architecture/.
Modeling changes of spatial chromatin architecture induced by structural variants
We developed an algorithm for modeling changes of the genome topology by removing and creating contacts between chromatin interaction anchors based on genetic alterations introduced by SVs. The algorithm uses high-quality CTCF or RNA Polymerase II (RNAPII) ChIA-PET data collected for the GM12878 cell line as a reference chromatin interaction pattern (17). When given a set of SVs present in other lymphoblastoid genomes, it applies a series of simple rules to recover their individualized chromatin interaction patterns from the reference data. The resulting genomic interactions are then passed to the 3D-GNOME modeling engine to build 3D models of individualized chromatin structures. Of note, while GM12878 ChIA-PET data is set as the reference for modeling 3D genomes of the samples genotyped by the 1000 Genomes Consortium, in principle any genomic interaction data can be used as the reference. The method acts on the following types of SVs: deletions (DEL), duplications (DUP), insertions (INS) and inversions (INV). The algorithm's behavior as a function of the input SV type is described below and represented schematically in Figure 2. The method was described in detail previously (4).
Figure 2.
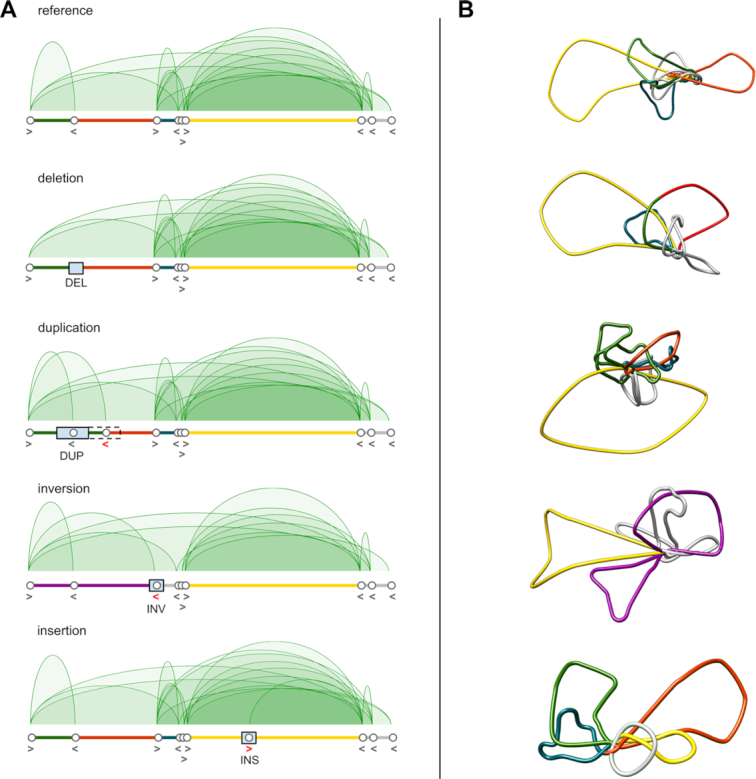
Schemes representing the behaviour of the computational algorithm implemented in 3D-GNOME 2.0 for prediction of changes in 3D chromatin contacts induced by SVs. (A) CTCF ChIA-PET contact diagrams for exemplary region chr1:47656996-48192898 containing TAL1 locus for the reference genome (GM12878) and upon introduction of SVs. Alteration of CTCF-mediated contact patterns upon addition of DUP, DEL, INV or INS to the genomic sequence is shown. SVs are marked as blue rectangles. CTCF anchors and their directionality are represented as white circles and arrows, correspondingly. Red arrows represent CTCF anchors’ alterations induced by SVs. (B) 3D models of CTCF mediated chromatin structures corresponding to genomic regions shown in (A). Loops are coloured as genomic regions represented below CTCF contact diagrams depicted in (A).
Deletions
Deletion removes all anchors within its scope, and therefore all the interactions they form with other anchors. Loops directly adjacent to the introduced deletion are elongated or shortened depending on the interaction pattern and size of the deletion. A deletion, which partially or completely overlaps the outermost anchor of a particular CCD, removes its boundary and merges it with the genomic region on the other side of the boundary.
Duplications and multiallelic copy number variants
Interactions that reside entirely in the duplicated region are repeated as a whole and introduced adjacently downstream in the genomic sequence. Similarly, a new copy of an anchor is created downstream from the original anchor. The original anchor maintains only the upstream interactions it formed before being duplicated. The downstream interactions are in turn connected to the duplicated anchor instead of the original one. Regarding CTCF mediated interactions, if the original anchor is lacking downstream contacts, naturally there are no interactions that the duplicated anchor could take over. In such cases, the algorithm finds the closest CTCF anchor with which it can create a convergent loop and connects them. This strategy reflects the currently assumed mechanism of CTCF-mediated chromatin loop formation, known as extrusion (19). According to this model, chromatin is pulled through the ring-shaped cohesin complex until the cohesin ring stops at an obstacle larger than the ring lumen. In this model, these obstacles correspond to CTCF proteins bound to the DNA motifs on the opposite DNA strands. Furthermore, if a duplication expands over a CCD boundary, parts of the CCDs, placed at the breakpoint after duplication, are fused. On top of having a direct impact on interacting loci, duplications can also increase the lengths of chromatin-loop spanning the duplicated regions. The effects of Multiallelic Copy Number Variants (mCNVs) are introduced in the 3D genome by performing multiple duplications or deletions.
Insertions
Inserted sequences are scanned for CTCF motifs using the PFM matrix. If a motif is found, the algorithm treats it as a new CTCF anchor and finds the closest anchor with which it can create a convergent loop.
Inversion
The algorithm reverses the directionality of the anchors affected by the inversion and removes all original contacts they formed. Next, it matches each of these anchors with the closest anchor of opposite orientation to create a convergent loop. If the considered anchors correspond to protein factors that do not bind any specific DNA motifs or if the orientation of the motifs they bind is irrelevant for the loop formation, the algorithm links such anchors with the closest ones, regardless of their directionality. If as a result of inversion, the algorithm links anchors from adjacent CCDs, the boundary between them is removed and the CCDs are merged.
To summarize, if as a result of the introduction of a given SV, an anchor loses all its interactions, it is joined with the nearest anchor of the opposite orientation (in the case of CTCF-mediated interactions), or to the nearest anchor without additional criteria. If an SV does not intersect any CTCF binding sites (or correspondingly, any other protein binding site, like RNAPII), no changes will be introduced to the chromatin interaction pattern, except for the change in length of certain loops.
The algorithm introduces changes in PET clusters, leaving singletons unchanged, thus, singleton data in sample variants will be equivalent to their reference singleton sets.
Input
The 3D-GNOME 2.0 web server is able to model 3D structures across individuals. In particular, one can model conformation of the genomes genotyped by the 1000 Genomes Consortium, and study topological genome variability in the human population. Users can choose to model chromatin structures using CTCF interactions only or CTCF and RNPII interactions at the same time. The only input information that needs to be provided is the location of the genomic region of interest and the list of SVs identified in a given genome. The former could be solely specified by chromosome number and coordinates, for example, chr14:35138000-36160000 (GM12878 ChIA-PET data will be used as the reference in this case), or the user can upload a BEDPE-format file with their own 3D chromatin interaction map data containing locations and frequencies of long-range contacts (obtained from 3C based methods such as Hi-C or ChiA-PET). The latter can be provided either as a custom list of SVs in the VCF format or by choosing SVs of interest from the predefined list of IDs identical to the ones from the 1kGP. The VCF file can be uploaded using the Upload VCF file option in the field ‘List of structural variants’. If more than one sample is to be analysed, sample IDs should also be entered, separated by commas into the text box ‘IDs of selected samples’. Alternatively, users can select IDs of 1kGP samples by choosing the ‘Select Sample IDs’ option in the ‘List of structural variants’ field.
To reduce calculation time, we cached interaction data for 2,504 genomes in both allelic variants. The ‘Submit’ button starts a simulation and a URL, pointing to the results page, appears. During the computations, the results page is constantly refreshed until the task is completed, after which the results are displayed.
Output
In 3D-GNOME 2.0 we extended the user interface by adding a menu on the left side contains a list of checkboxes that allow users to show or hide selected results such as contact diagrams, singleton heatmaps, data statistics, and plots. The desired result details may be displayed for chosen genomic samples, for both allelic variants separately. The selected elements are shown in the centre of the screen.
The main improvement in the output content includes contact diagrams that allow users to compare changes in interaction patterns introduced by SVs of interest. SVs emerging in a selected region are annotated on a reference contact diagram by labels and lines colored according to the SV type. Two separate diagrams of chromatin contacts are displayed for each reference and variant case, one representing genomic interactions mediated by CTCF, another showing RNAPII-mediated contacts (Figure 3A). Next, all statistics, such as Number of Interactions, Minimal/Maximal/Average Length of interactions and Maximal Count Frequency of the CTCF, RNAPII and singleton interactions are calculated and presented in separate plots for each variant. Additionally, a plot showing the distribution of interaction lengths is generated for each sample (Figure 3B). This enables a convenient comparison between the reference and the variant. Interaction sets displayed on the charts may be downloaded in the BEDPE format.
Figure 3.

Output of 3D-GNOME 2.0. for an exemplary region (chr14:35138000-36160000) affected by two deletions: (chr14:35401403-35401724 and chr14:35605439-35615196). (A) Screenshot of the result page with diagrams of chromatin contacts mediated by CTCF (green) and RNAPII (purple) for both the reference genome (based on GM12878 data) and the variant genome (based on SVs from HG00099); the deletion chr14:35605439-35615196 (right DEL) disrupts CTCF and RNAPII interactions. (B) Clusters length distribution for CTCF and RNAPII protein factors for reference genome (top) and HG00099 (bottom). (C, D) Representation of 3D models in the 3D viewer, proposed using only CTCF interactions (panel C) or both CTCF and RNAPII data (panel D) from the reference genome (left) and that affected by SVs (right).
The menu contains URLs that link to the page containing an interactive 3D viewer with the pre-loaded chromatin model of the selected structure. 3D-GNOME 2.0 extends the possibilities of the 3D viewer with the ability to display both 3D models of the reference structure and the variant side by side (Figure 3C, D). Additionally, the 3D-Viewer Control Menu has the option to save the 3D model locally in PDB (Protein Data Bank) or XYZ format. This allows local examination of the model with a molecular visualization software such as UCSF Chimera (https://www.cgl.ucsf.edu/chimera/). It should be highlighted that taking into consideration the nature of 3C experiments, which are performed using millions of cells, these models represent the average of structures existing in the population of the cells rather than structures from individual cells. Several successful analyses have been, however, already published using the former kind of data. For example, as described in (4), analyzing the distribution of distances between specific genomic elements, like enhancers and promoters, and gene expression profiles in a given region can lead to uncovering meaningful associations.
Computational time of modeling depends on different parameters, such as: the size of the region, the number of interactions-mediated proteins (CTCF only or CTCF and RNAPII together) or whether the user selects/provides the list of SVs or not. For example, for a region chr14:35138000-36160000 computations time is (I) ∼30 s if only CTCF interactions are considered for modeling, (II) ∼2 min if both CTCF and RNAPII interactions are used, and (III) ∼8 min. if both CTCF and RNAPII interactions are used and the sample is modified by SVs from the HG00099 cell line.
CONCLUSIONS AND FUTURE PLANS
3D-GNOME 2.0 provides users with a new capacity to process structural variation data. Novel features enable users to model changes in chromatin contacts caused by SVs and allow to compare these changes using diagrams of contacts, statistical plots and 3D models. We believe that 3D-GNOME 2.0 is an easily accessible tool, valuable to scientists who wish to study processes that shape 3D chromatin architecture in the nucleus, specifically from a population perspective. In the future, we plan to implement a GPU-accelerated version of our modeling algorithm. This will help reduce the computational time and therefore provide a more effective way to analyse large population datasets. Furthermore, we also aim to improve 3D visualization and model analysis.
ACKNOWLEDGEMENTS
We thank Veronika Mancikova for language editing and proofreading of the manuscript and Agnieszka Bucka and Agnes Alcantara Paculdar for critical reading of the text.
Contributor Information
Michal Wlasnowolski, Centre of New Technologies, University of Warsaw, Warsaw 02-097, Poland; Faculty of Mathematics and Information Science, Warsaw University of Technology, Warsaw 00-662, Poland.
Michal Sadowski, Centre of New Technologies, University of Warsaw, Warsaw 02-097, Poland.
Tymon Czarnota, Faculty of Mathematics and Information Science, Warsaw University of Technology, Warsaw 00-662, Poland.
Karolina Jodkowska, Centre of New Technologies, University of Warsaw, Warsaw 02-097, Poland.
Przemyslaw Szalaj, Centre of New Technologies, University of Warsaw, Warsaw 02-097, Poland; Centre for Bioinformatics and Data Analysis, Medical University of Bialystok, Bialystok 15-089, Poland; I-BioStat, Hasselt University, 3500 Hasselt, Belgium.
Zhonghui Tang, The Jackson Laboratory for Genomic Medicine, Farmington, CT 06032, USA.
Yijun Ruan, The Jackson Laboratory for Genomic Medicine, Farmington, CT 06032, USA; Department of Genetics and Genome Sciences, UConn Health, Farmington, CT 06030-6403, USA.
Dariusz Plewczynski, Centre of New Technologies, University of Warsaw, Warsaw 02-097, Poland; Faculty of Mathematics and Information Science, Warsaw University of Technology, Warsaw 00-662, Poland; The Jackson Laboratory for Genomic Medicine, Farmington, CT 06032, USA.
FUNDING
Polish National Science Centre [2014/15/B/ST6/05082]; Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund (TEAM to D.P.); ‘Nucleome Positioning System for Spatiotemporal Genome Organization and Regulation’ within the 4DNucleome National Institute of Health program, and by the European Commission as European Cooperation in Science and Technology COST actions: CA18127 ‘International Nucleome Consortium’ (INC) [1U54DK107967-01]; 'Impact of Nuclear Domains On Gene Expression and Plant Traits’ [CA16212]; Horizon 2020 Marie Skłodowska-Curie ITN Enhpathy grant 'Molecular Basis of Human enhanceropathies'; D.P. and M.W. were supported by the RENOIR Project from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie [691152]; Ministry of Science and Higher Education (Poland) [W34/H2020/2016, 329025/PnH/2016]. Funding for open access charge: Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund (TEAM to DP).
Conflict of interest statement. None declared.
REFERENCES
- 1. Spielmann M., Lupianez D.G., Mundlos S.. Structural variation in the 3D genome. Nat. Rev. Genet. 2018; 19:453–467. [DOI] [PubMed] [Google Scholar]
- 2. Hnisz D., Weintraub A.S., Day D.S., Valton A.L., Bak R.O., Li C.H., Goldmann J., Lajoie B.R., Fan Z.P., Sigova A.A. et al.. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science (New York, N.Y.). 2016; 351:1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schoenfelder S., Fraser P.. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 2019; 20:437–455. [DOI] [PubMed] [Google Scholar]
- 4. Sadowski M., Kraft A., Szalaj P., Wlasnowolski M., Tang Z., Ruan Y., Plewczynski D.. Spatial chromatin architecture alteration by structural variations in human genomes at the population scale. Genome Biol. 2019; 20:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., Abecasis G.R.. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tang Z., Luo O.J., Li X., Zheng M., Zhu J.J., Szalaj P., Trzaskoma P., Magalska A., Wlodarczyk J., Ruszczycki B. et al.. CTCF-Mediated human 3D genome architecture reveals chromatin topology for transcription. Cell. 2015; 163:1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heinz S., Texari L., Hayes M.G.B., Urbanowski M., Chang M.W., Givarkes N., Rialdi A., White K.M., Albrecht R.A., Pache L. et al.. Transcription elongation can affect genome 3D structure. Cell. 2018; 174:1522–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lupianez D.G., Kraft K., Heinrich V., Krawitz P., Brancati F., Klopocki E., Horn D., Kayserili H., Opitz J.M., Laxova R. et al.. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015; 161:1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weischenfeldt J., Dubash T., Drainas A.P., Mardin B.R., Chen Y., Stutz A.M., Waszak S.M., Bosco G., Halvorsen A.R., Raeder B. et al.. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat. Genet. 2017; 49:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kantidze O.L., Gurova K.V., Studitsky V.M., Razin S.V.. The 3D genome as a target for anticancer therapy. Trends Mol. Med. 2020; 26:141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J. et al.. Systematic localization of common disease-associated variation in regulatory DNA. Science (New York, N.Y.). 2012; 337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Talkowski M.E., Mullegama S.V., Rosenfeld J.A., van Bon B.W., Shen Y., Repnikova E.A., Gastier-Foster J., Thrush D.L., Kathiresan S., Ruderfer D.M. et al.. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am. J. Hum. Genet. 2011; 89:551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. et al.. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Szalaj P., Tang Z., Michalski P., Pietal M.J., Luo O.J., Sadowski M., Li X., Radew K., Ruan Y., Plewczynski D.. An integrated 3-dimensional genome modeling engine for data-driven simulation of spatial genome organization. Genome Res. 2016; 26:1697–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mallick S., Li H., Lipson M., Mathieson I., Gymrek M., Racimo F., Zhao M., Chennagiri N., Nordenfelt S., Tandon A. et al.. The simons genome diversity Project: 300 genomes from 142 diverse populations. Nature. 2016; 538:201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szalaj P., Michalski P.J., Wroblewski P., Tang Z., Kadlof M., Mazzocco G., Ruan Y., Plewczynski D.. 3D-GNOME: an integrated web service for structural modeling of the 3D genome. Nucleic Acids Res. 2016; 44:W288–W293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fullwood M.J., Ruan Y.. ChIP-based methods for the identification of long-range chromatin interactions. J. Cell. Biochem. 2009; 107:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chaisson M.J.P., Sanders A.D., Zhao X., Malhotra A., Porubsky D., Rausch T., Gardner E.J., Rodriguez O.L., Guo L., Collins R.L. et al.. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019; 10:1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. et al.. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (New York, N.Y.). 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]