


Abstract
Multi-drug resistant pathogens have become a major threat to human health and new antibiotics are urgently needed. Most antibiotics are derived from secondary metabolites produced by bacteria. In order to avoid suicide, these bacteria usually encode resistance genes, in some cases within the biosynthetic gene cluster (BGC) of the respective antibiotic compound. Modern genome mining tools enable researchers to computationally detect and predict BGCs that encode the biosynthesis of secondary metabolites. The major challenge now is the prioritization of the most promising BGCs encoding antibiotics with novel modes of action. A recently developed target-directed genome mining approach allows researchers to predict the mode of action of the encoded compound of an uncharacterized BGC based on the presence of resistant target genes. In 2017, we introduced the ‘Antibiotic Resistant Target Seeker’ (ARTS). ARTS allows for specific and efficient genome mining for antibiotics with interesting and novel targets by rapidly linking housekeeping and known resistance genes to BGC proximity, duplication and horizontal gene transfer (HGT) events. Here, we present ARTS 2.0 available at http://arts.ziemertlab.com. ARTS 2.0 now includes options for automated target directed genome mining in all bacterial taxa as well as metagenomic data. Furthermore, it enables comparison of similar BGCs from different genomes and their putative resistance genes.
INTRODUCTION
Due to the continuous increase of drug-resistant bacteria, antibiotic resistance is regarded as a global public health threat (1). The lack of new antibiotics with novel modes of action in the current drug development pipeline, makes finding new compounds to fight off resistant pathogens a critical task (2). Since the discovery of penicillin, secondary metabolites (SMs) produced by various living organisms have been foundational to the development of antimicrobial drugs (3). The majority of antibiotic compounds are isolated as natural products, from fungi and bacteria (4). For many decades, screening biological samples for desired bioactivity has been the traditional methodology for natural product discovery (5). Due to the high rediscovery rates and labor-intensive nature of the process, in silico methods have become a promising way to guide modern drug discovery efforts (6,7). Gene-centered methods, such as genome mining, enable researchers nowadays to computationally detect the biosynthetic gene clusters (BGCs) encoding enzymes necessary for the biosynthesis of antibiotics and predict encoded compounds (8). Over the last decade, greatly improved genome mining tools such as antiSMASH (9), EvoMining (10), PRISM (11) or DeepBGC (12) use methods like Hidden Markov Models, phylogeny or deep learning to highlight a variety of natural product classes. Combined with databases such as MIBiG (13), Natural Product Atlas (14) and the antiSMASH database (15), these tools allow for fast and efficient mining and dereplication of thousands of bacterial genomes and BGCs. According to the latest version of the Atlas of Biosynthetic Gene Clusters (IMG-ABC) (16) there currently are ∼400 000 predicted BGCs sequenced. Moreover, <1% of total clusters are experimentally verified, which leads to an important question: Which of these clusters should be further examined with wet lab experiments?
Recently, researchers adopted a prioritization approach for antibiotic discovery that is based on the observation that antibiotic producers have to be resistant against their own products to avoid suicide (17). This so called target-directed or self resistance based genome mining approach allows the prediction of the mode of action of the encoded compound of an uncharacterized BGC based on resistance genes, in some cases co-located within the antibiotic BGC (18). Multiple resistance mechanisms exist, such as inactivation and export of antibiotics as well as target modification. In the latter case, a duplicated and antibiotic-resistant homologue of an essential housekeeping gene is detectable within the antibiotic BGC and allows the prediction of the mode of action of the encoded compound even without knowing a chemical structure (19–21). Moore et al., for example, were able to identify a fatty acid synthase inhibiting antibiotic by screening for duplicated fatty acid synthase genes within orphan BGCs (22).
In 2017, we introduced the first version of the ‘Antibiotic Resistant Target Seeker’ (ARTS) (23), a user-friendly web server that automates target-directed genome mining to prioritize promising strains that produce antibiotics with new mode of actions. Since a resistant copy of the antibiotic target gene is typically detectable in the genome, can be observed within the BGC of the antibiotic and horizontally acquired with the BGC (23), ARTS automatically detects possible resistant housekeeping genes based on three criteria: duplication, localization within a biosynthetic gene cluster, and evidence of Horizontal Gene Transfer (HGT). One previous limitation of the ARTS pipeline was its focus on actinobacterial genomes. Although natural product discovery historically was highly focusing on the phylum Actinobacteria, prominent families from other phyla such as Proteobacteria or Firmicutes are known to have high natural product biosynthetic potential (24–26). Here, we introduce a greatly improved version 2 of the ARTS webserver, now allowing the analysis of the entire kingdom of bacteria, metagenomic data, and the comparison of multiple genomes. This update therefore will facilitate natural product prioritization and antibiotic discovery efforts beyond actinomycetes.
NEW FEATURES AND UPDATES
The workflow of the ARTS pipeline involves a few key steps: First, query genomes are screened for BGCs using antiSMASH (9). At the same time essential housekeeping (core) genes within the genome are determined using TIGRFAM models that have been identified by comparing a reference set of similar genomes (27) (Figure 1B). During the next steps the identified core and known resistance genes are screened for their location within BGCs. Duplication thresholds are determined for each core gene model, based on their respective frequencies among the reference set. Finally, possible HGT events are detected via phylogenetic screening with the help of constructed species trees and gene trees. All the results are summarized into interactive output tables.
Figure 1.
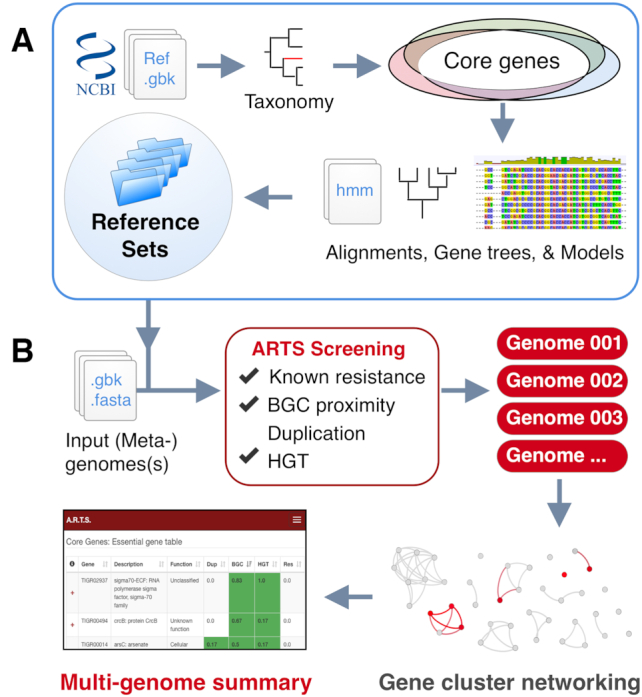
Outline representation of the ARTS pipeline. (A) Basic machinery of creating reference sets. Housekeeping core genes and duplication thresholds are detected per clade of organisms and gene alignments and trees are created for fast HGT detection. (B) Workflow with multi-genome comparative analysis. Input data is screened for ARTS selection criteria. All found BGCs are then subjected to BiG-SCAPE clustering algorithm. Finally, interactive output tables are presented for comparative analysis.
Reference sets of organisms and core genes
Since the determination of core gene content and the construction of phylogenetic trees is more specific and accurate when query genomes are compared with genomes from similar organisms, we aimed to generate phylum specific reference sets. However, since the number of genomes in the different phyla varied significantly, reference sets were sometimes also created by class or a group of closely related phyla (Supplementary Table S1).
In a first step, sequences of all classified bacteria were downloaded through NCBI’s RefSeq database (28) for further evaluation (Figure 1A). Redundant sequences were filtered with MASH (29) with a +95% similarity cut off. Where applicable, only complete genomes were used in a reference set. If the number and diversity of complete genomes within a phylum was not sufficient (distributed among a genus or two with <100 sequences), contig-level assemblies were also taken into consideration to expand the particular reference. Around 330 genome sequences were used for the creation of each individual reference set, which sum up to 4936 genomes in total.
Based on the number of genomes for each reference set, different boundaries were then selected for phyla with different levels of diversity. Given the diversity and large number of proteobacterial genomes deposited in Refseq (30), four different reference sets were created for proteobacterial genomes (Alpha, Beta, Gamma, Delta-Epsilon). In cases where a phylum does not comprise sufficient sequenced genome sequences (less than 100 genomes), multiple phyla were grouped into one reference set. In that way, 22 phyla were grouped into three reference sets. Groupings were based on phylogenetic distances in the tree of life (31) and the NCBI Lifemap (32). Another feature of the grouped sets is the high coverage of bacteria from harsh environments, allowing the analysis of extremophiles. For example, group 2, which was created from 214 organisms, is mainly comprised of the phyla Thermotogae and Chloroflexi (Supplementary Table S1), which are known to be mostly thermophilic (33,34).
Reference set and core gene analysis
Determination of core genes
Core genes were determined for each reference set using the method developed for the previous version of ARTS (23). Subsequently, the core genes from each set were compared with sequences from the Database of Essential Genes (DEG)v 1.5 (46). On average, 85% of genes had a match to one or more records (Supplementary Table S2). The majority of the genes that are not found in DEG belong to the gene categories ’unclassified’, ’unknown function’ or ’energy metabolism’. Furthermore, functional classification of each reference set revealed that, on average, genes with functions such as protein and amino acid synthesis, energy and metabolism were the most abundant as would be expected from essential genes (Supplementary Figure S1). The importance of individual reference sets is highlighted by the fact that one set only accounts for ∼40% of the total unique core genes from all sets (Supplementary Table S4).
Additionally, the reliability of the generated gene trees for each reference set were estimated by branch support (Supplementary Figure S2) and comparison to taxonomically correct species trees generated by the Accurate Species TRee ALgorithm (ASTRAL) (47) (Supplementary data).
Positive controls and detection frequencies
In order to test ARTS’ ability to detect resistant targets in non-actinobacterial genomes using the new reference sets, we analyzed known examples of self-resistance mechanisms. We identified several known non-actinobacterial examples as positive controls (Table 1). Out of 11 antibiotic natural products with identified resistance mechanisms, five of them had available genome sequences regarding specific isolates that contained respective BGCs. All of these cases showed at least two ARTS hits when run in normal mode with default cutoffs. To detect the accA gene, a known transferase, exploration mode had to be used. Otherwise, ARTS 2.0 predicted resistance genes in almost all control BGCs except one. The CoA reductase resistant gene was not detected because specific CoA reductase models were missing in both the core and known resistance set. We also analyzed ∼5000 genomes belonging to all reference sets for statistical evaluation (Supplementary Table S3). On average, only one gene model shows positive hits for three or more ARTS criteria. Also, most of the core genes from the respective sets are found in each analyzed genome. Around 2–5% of core genes are highlighted for each criterion. The percent of core genes that went through HGT is in conformity with the HGT estimate levels in the literature (48,49).
Table 1.
Default ARTS analysis for positive examples of genomes and BGCs with known self-resistance mechanisms
| Product | Resistance gene | Organism | ARTS hits | Criteria hits (>2, >3) | Genes (core, total) |
|---|---|---|---|---|---|
| Thiocillin | ribosomal protein L11(35) | Bacillus cereus ATCC 14579 | D,B,P | 9,1 | 472, 5231 |
| Myxovirescin | lspa: signal peptidase II(36) | Myxococcus xanthus DK 1622 | D,B,P | 15,2 | 372, 7267 |
| Thailandamide | accA: acetyl-CoA carboxylase(37) | Burkholderia thailandensis E264 | D,B,P,R* | 42, 5 | 838, 6347 |
| Indolmycin | trypS: tryptophan-tRNA synthetase(38) | Pseudoalteromonas luteoviolacea | D,B | 13, 2 | 540, 4963 |
| Agrocin 84 | leu tRNA synthase(39) | Agrobacterium radiobacter K84 | D,P | 41, 2 | 470, 6876 |
| Bengamide | methionine aminopeptidase(40) | Myxococcus virescens DSM 15898 | Core | N/A | 1, 18 |
| Mupirocin | Ile-tRNA synthetase(41) | Pseudomonas fluorescens NCIMB 10586 | Core | N/A | 1, 36 |
| Andrimid | accD: acetyl-CoA carboxylase(42) | Pantoea agglomerans Eh335 | Core | N/A | 1, 18 |
| Cystobactamid | Pentapeptide repeat protein(43) | Cystobacter sp. Cbv34 | R* | N/A | 0, 24 |
| Phaseolotoxin | ornithine carbamoyltransferase(44) | Pseudomonas savastanoi pv. phaseolicola | Core, R* | N/A | 3, 26 |
| Kalimantacin | fabl: enoyl reductase(45) | Pseudomonas fluorescens BCCM ID9359 | No hits | N/A | 3, 29 |
Hits to ARTS criteria are shown as; D: duplication, B: BGC proximity, P: phylogeny, R: resistance model. Rows in gray indicate only complete gene cluster as input rather than whole genome. Stars indicate exploration mode.
Reference sets for metagenomic data
Since metagenomic approaches are becoming increasingly popular in natural product research (50,51), submissions of whole metagenomes to the ARTS webserver are also showing a significant increase. Therefore, we have built an additional reference set available for metagenome analysis, which does not include phylogeny and duplications. Given that metagenomes are usually quite diverse and comprise more than one single phylum, core genes are defined as genes belonging to the Database of Essential Genes (DEG) (Supplementary Table S3).
Comparative analysis
ARTS 2.0 now makes it easier for users to analyze multiple genomes and applies a comparative analysis of provided organisms (Figure 2). Throughout the analysis, individual ARTS results are accessible upon completion of each run. Once all the sequences of interest are analyzed, an interactive summary table representing all genomes with each resulting criterion is provided. In addition, shared core genes with their respective hits and their observed frequencies among all genomes can be inspected via dynamic output tables. This aids in further prioritizing ARTS hits for those that are detected in multiple contexts or related BGCs and therefore are more likely to be involved in resistance. For example, users can now narrow HGT hits by inspecting those that are shared across multiple organisms. In addition to these data, the BiG-SCAPE algorithm (52) is applied on all detected BGCs, allowing users to investigate similar BGCs from multiple sources by constructing gene cluster sequence similarity networks and identifying gene cluster families inside these networks. Furthermore, each of the BGCs in a gene cluster family can be examined in order to assess whether they have core or resistance models as shared hits, as well as whether a cluster stands out with unique hits compared to its relatives from other species.
Figure 2.

Example output of multi-genome ARTS analysis. Top part of the page represents the summaries of individual arts runs and shared core genes throughout the whole analysis with respective ARTS hits. At the bottom, shared BGCs and resistance models can easily be navigated and an interactive BiG-SCAPE graph output can also be found via ‘Open BiG-SCAPE overview” option.
Server-side updates and speed up
In order to keep the ARTS pipeline at high standards, third party tools used in the workflow were updated. ARTS 2.0 now uses antiSMASH v5 and is able to analyze antiSMASH results from their newest JSON format. The most time consuming part of the ARTS pipeline is the creation of species and gene trees for phylogenetic analysis via ASTRAL. By updating antiSMASH and ASTRAL, the average runtime of the whole pipeline could now be cut down to half. Also, in order to satisfy the increasing demand, ARTS 2.0 is now hosted at the highly scalable de.NBI cloud system with seven times the computational power. With these hardware and software updates, the ARTS 2.0 webserver is now capable of analyzing multiple inputs up to 100MB and depending on the genomes and selected parameters, 3-8 times faster than the previous version.
CONCLUSIONS AND FUTURE PERSPECTIVES
Currently, ARTS is the only platform to automate resistance and putative drug-target based genome mining in bacteria via a user-friendly webserver. By design, ARTS aims to survey a wide scope of potential genes as drug targets while minimizing manual inspection by using the dynamic output and multiple screening criteria for more confident target predictions. Thus it is incumbent on the user to examine potential hits with provided metadata and contextual framing. Some of the ARTS hits might be more likely involved in biosynthesis and not associated with resistance. Although we removed common biosynthesis genes from the core gene sets to avoid false positives (23), it is currently not possible to automatically distinguish if genes are more likely involved in biosynthesis or resistance, for example fatty acid synthases are involved in both (22). The occasional high counts of positive hits in exploration mode, largely due to undefined cluster boundaries, can be easily and rapidly filtered in the interactive output page. As shown previously, this inspection can even serve to help define the true boundaries of clusters, which remains a largely unresolved challenge when dealing with bacterial BGCs (23). Newly introduced features now make ARTS 2.0 a fast and comprehensive pipeline allowing users to: analyze sequences from all bacterial genomes as well as metagenomic samples, apply comparative analysis on multiple genomes, and interrogate similar BGCs for shared resistant genes. For future applications, we are working on increasing ARTS’ availability by making it directly accessible through other webservers such as antiSMASH. This will enable researchers to easily apply target-directed genome mining approaches on sequences from different databases as a plugin. Furthermore, we are currently in process of creating the ARTS database, which will contain preanalyzed ARTS results for all bacterial genomes within the Refseq database, and will allow global analysis and comparisons of resistant targets within BGC. We hope that with this update, ARTS 2.0 will now provide an even broader access to resistance based genome mining methods and facilitate the discovery of competitive antibiotics.
Supplementary Material
ACKNOWLEDGEMENTS
The authors acknowledge the use of de.NBI cloud and the support by the High Performance and Cloud Computing Group at the Zentrum für Datenverarbeitung of the University of Tübingen and the Federal Ministry of Education and Research (BMBF) through grant no 031 A535A. We also acknowledge all ARTS users for helpful comments and feedback.
Contributor Information
Mehmet Direnç Mungan, Interfaculty Institute of Microbiology and Infection Medicine, University of Tübingen, Auf der Morgenstelle 28, 72076 Tübingen, Germany; German Centre for Infection Research (DZIF), Partner Site Tübingen, Germany.
Mohammad Alanjary, Bioinformatics Group, Wageningen University, Droevendaalsesteeg 1, 6708PB Wageningen, the Netherlands.
Kai Blin, The Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Kemitorvet Bygning 220, 2800 Kgs. Lyngby, Denmark.
Tilmann Weber, The Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Kemitorvet Bygning 220, 2800 Kgs. Lyngby, Denmark.
Marnix H Medema, Bioinformatics Group, Wageningen University, Droevendaalsesteeg 1, 6708PB Wageningen, the Netherlands.
Nadine Ziemert, Interfaculty Institute of Microbiology and Infection Medicine, University of Tübingen, Auf der Morgenstelle 28, 72076 Tübingen, Germany; German Centre for Infection Research (DZIF), Partner Site Tübingen, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
High Performance and Cloud Computing Group at the Zentrum für Datenverarbeitung of the University of Tübingen; State of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) [INST 37/935-1 FUGG]; N.Z., M.A. and M.D.M. acknowledge the German Center for Infection Research [DZIF TTU09.704]; T.W. and K.B. were supported by grants from the Novo Nordisk Foundation [NNF10CC1016517, NNF16OC0021746]; M.A. and M.H.M. were supported by an ERA NET CoBiotech (’Bestbiosurf’) grant through the Netherlands Organization for Scientific Research (NWO) [053.80.739]. Funding for open access charge: BMBF [DZIF TTU09.704].
Conflict of interest statement. None declared.
REFERENCES
- 1. Michael C.A., Dominey-Howes D., Labbate M.. The antimicrobial resistance crisis: causes, consequences, and management. Front. Public Health. 2014; 2:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cragg G.M., Newman D.J.. Natural products: a continuing source of novel drug leads. Biochim. Biophys. Acta. 2013; 1830:3670–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harvey A.L., Edrada-Ebel R., Quinn R.J.. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug. Discov. 2015; 14:111–129. [DOI] [PubMed] [Google Scholar]
- 4. Newman D.J., Cragg G.M.. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012; 75:311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ziemert N., Alanjary M., Weber T.. The evolution of genome mining in microbes–a review. Nat. Prod. Rep. 2016; 33:988–1005. [DOI] [PubMed] [Google Scholar]
- 6. Stokes J.M., Yang K., Swanson K., Jin W., Cubillos-Ruiz A., Donghia N.M., MacNair C.R., French S., Carfrae L.A., Bloom-Ackerman Z. et al.. A deep learning approach to antibiotic discovery. Cell. 2020; 180:688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li Z., Zhu D., Shen Y.. Discovery of novel bioactive natural products driven by genome mining. Drug Discov. Ther. 2018; 12:318–328. [DOI] [PubMed] [Google Scholar]
- 8. Bachmann B.O., Van Lanen S.G., Baltz R.H.. Microbial genome mining for accelerated natural products discovery: is a renaissance in the making?. J. Ind. Microbiol. Biot. 2014; 41:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blin K., Shaw S., Steinke K., Villebro R., Ziemert N., Lee S.Y., Medema M.H., Weber T.. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019; 47:W81–W87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sélem-Mojica N., Aguilar C., Gutiérrez-García K., Martínez-Guerrero C.E., Barona-Gómez F.. EvoMining reveals the origin and fate of natural product biosynthetic enzymes. Microb. Genom. 2019; 5:e000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Skinnider M.A., Merwin N.J., Johnston C.W., Magarvey N.A.. PRISM 3: expanded prediction of natural product chemical structures from microbial genomes. Nucleic Acids Res. 2017; 45:W49–W54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hannigan G.D., Prihoda D., Palicka A., Soukup J., Klempir O., Rampula L., Durcak J., Wurst M., Kotowski J., Chang D. et al.. A deep learning genome-mining strategy for biosynthetic gene cluster prediction. Nucleic Acids Res. 2019; 47:e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kautsar S.A., Blin K., Shaw S., Navarro-Muñoz J.C., Terlouw B.R., van der Hooft J.J., Van Santen J.A., Tracanna V., Suarez Duran H.G., Pascal Andreu V. et al.. MIBiG 2.0: a repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2020; 48:D454–D458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Van Santen J.A., Jacob G., Singh A.L., Aniebok V., Balunas M.J., Bunsko D., Neto F.C., Castaño-Espriu L., Chang C., Clark T.N. et al.. The natural products atlas: an open access knowledge base for microbial natural products discovery. ACS Cent. Sci. 2019; 5:1824–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blin K., Pascal Andreu V., de los Santos E. L.C., Del Carratore F., Lee S.Y., Medema M.H., Weber T.. The antiSMASH database version 2: a comprehensive resource on secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2019; 47:D625–D630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palaniappan K., Chen I.-M.A., Chu K., Ratner A., Seshadri R., Kyrpides N.C., Ivanova N.N., Mouncey N.J.. IMG-ABC v. 5.0: an update to the IMG/Atlas of Biosynthetic Gene Clusters Knowledgebase. Nucleic Acids Res. 2020; 48:D422–D430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Almabruk K.H., Dinh L.K., Philmus B.. Self-resistance of natural product producers: Past, present, and future focusing on self-resistant protein variants. ACS Chem. Biol. 2018; 13:1426–1437. [DOI] [PubMed] [Google Scholar]
- 18. Yan Y., Liu Q., Zang X., Yuan S., Bat-Erdene U., Nguyen C., Gan J., Zhou J., Jacobsen S.E., Tang Y.. Resistance-gene-directed discovery of a natural-product herbicide with a new mode of action. Nature. 2018; 559:415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brochet M., Couvé E., Zouine M., Poyart C., Glaser P.. A naturally occurring gene amplification leading to sulfonamide and trimethoprim resistance in Streptococcus agalactiae. J. Bacteriol. 2008; 190:672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Freel K.C., Millán-Aguiñaga N., Jensen P.R.. Multilocus sequence typing reveals evidence of homologous recombination linked to antibiotic resistance in the genus Salinispora. Appl. Environ. Microbiol. 2013; 79:5997–6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thaker M.N., Wang W., Spanogiannopoulos P., Waglechner N., King A.M., Medina R., Wright G.D.. Identifying producers of antibacterial compounds by screening for antibiotic resistance. Nat. Biotechnol. 2013; 31:922. [DOI] [PubMed] [Google Scholar]
- 22. Tang X., Li J., Millán-Aguiñaga N., Zhang J.J., O’Neill E.C., Ugalde J.A., Jensen P.R., Mantovani S.M., Moore B.S.. Identification of thiotetronic acid antibiotic biosynthetic pathways by target-directed genome mining. ACS Chem. Biol. 2015; 10:2841–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alanjary M., Kronmiller B., Adamek M., Blin K., Weber T., Huson D., Philmus B., Ziemert N.. The Antibiotic Resistant Target Seeker (ARTS), an exploration engine for antibiotic cluster prioritization and novel drug target discovery. Nucleic Acids Res. 2017; 45:W42–W48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cimermancic P., Medema M.H., Claesen J., Kurita K., Brown L. C.W., Mavrommatis K., Pati A., Godfrey P.A., Koehrsen M., Clardy J. et al.. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell. 2014; 158:412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Y., Li Z., Yamanaka K., Xu Y., Zhang W., Vlamakis H., Kolter R., Moore B.S., Qian P.-Y.. Directed natural product biosynthesis gene cluster capture and expression in the model bacterium Bacillus subtilis. Sci. Rep.-UK. 2015; 5:9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weissman K.J., Müller R.. Myxobacterial secondary metabolites: bioactivities and modes-of-action. Nat. Prod. Rep. 2010; 27:1276–1295. [DOI] [PubMed] [Google Scholar]
- 27. Haft D.H., Selengut J.D., Richter R.A., Harkins D., Basu M.K., Beck E.. TIGRFAMs and genome properties in 2013. Nucleic Acids Res. 2012; 41:D387–D395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D. et al.. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016; 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ondov B.D., Treangen T.J., Melsted P., Mallonee A.B., Bergman N.H., Koren S., Phillippy A.M.. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016; 17:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gupta R.S. The phylogeny of proteobacteria: relationships to other eubacterial phyla and eukaryotes. FEMS Microbiol. Rev. 2000; 24:367–402. [DOI] [PubMed] [Google Scholar]
- 31. Hug L.A., Baker B.J., Anantharaman K., Brown C.T., Probst A.J., Castelle C.J., Butterfield C.N., Hernsdorf A.W., Amano Y., Ise K. et al.. A new view of the tree of life. Nat. Microbiol. 2016; 1:16048. [DOI] [PubMed] [Google Scholar]
- 32. de Vienne D.M. Lifemap: exploring the entire tree of life. PLoS Biol. 2016; 14:e2001624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gupta R.S., Bhandari V.. Phylogeny and molecular signatures for the phylum Thermotogae and its subgroups. Anton. Leeuw. 2011; 100:1. [DOI] [PubMed] [Google Scholar]
- 34. Gregoire P., Bohli M., Cayol J.-L., Joseph M., Guasco S., Dubourg K., Cambar J., Michotey V., Bonin P., Fardeau M.-L. et al.. Caldilinea tarbellica sp. nov., a filamentous, thermophilic, anaerobic bacterium isolated from a deep hot aquifer in the Aquitaine Basin. Int. J. Syst. Evol. Micr. 2011; 61:1436–1441. [DOI] [PubMed] [Google Scholar]
- 35. Brown L. C.W., Acker M.G., Clardy J., Walsh C.T., Fischbach M.A.. Thirteen posttranslational modifications convert a 14-residue peptide into the antibiotic thiocillin. Proc. Natl. Acad. Sci. 2009; 106:2549–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiao Y., Gerth K., Müller R., Wall D.. Myxobacterium-produced antibiotic TA (myxovirescin) inhibits type II signal peptidase. Antimicrob. Agents Chemother. 2012; 56:2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wozniak C.E., Lin Z., Schmidt E.W., Hughes K.T., Liou T.G.. Thailandamide, a fatty acid synthesis antibiotic that is coexpressed with a resistant target gene. Antimicrob. Agents Chemother. 2018; 62:e00463-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Du Y.-L., Alkhalaf L.M., Ryan K.S.. In vitro reconstitution of indolmycin biosynthesis reveals the molecular basis of oxazolinone assembly. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:2717–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ryder M., Slota J., Scarim A., Farrand S.. Genetic analysis of agrocin 84 production and immunity in Agrobacterium spp. J. Bacteriol. 1987; 169:4184–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wenzel S.C., Hoffmann H., Zhang J., Debussche L., Haag-Richter S., Kurz M., Nardi F., Lukat P., Kochems I., Tietgen H. et al.. Production of the bengamide class of marine natural products in myxobacteria: biosynthesis and structure–activity relationships. Angew. Chem. Int. Ed. 2015; 54:15560–15564. [DOI] [PubMed] [Google Scholar]
- 41. El-Sayed A.K., Hothersall J., Cooper S.M., Stephens E., Simpson T.J., Thomas C.M.. Characterization of the mupirocin biosynthesis gene cluster from Pseudomonas fluorescens NCIMB 10586. Chem. Biol. 2003; 10:419–430. [DOI] [PubMed] [Google Scholar]
- 42. Liu X., Fortin P.D., Walsh C.T.. Andrimid producers encode an acetyl-CoA carboxyltransferase subunit resistant to the action of the antibiotic. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:13321–13326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Baumann S., Herrmann J., Raju R., Steinmetz H., Mohr K.I., Hüttel S., Harmrolfs K., Stadler M., Müller R.. Cystobactamids: myxobacterial topoisomerase inhibitors exhibiting potent antibacterial activity. Angew. Chem. Int. Ed. 2014; 53:14605–14609. [DOI] [PubMed] [Google Scholar]
- 44. Chen L., Li P., Deng Z., Zhao C.. Ornithine transcarbamylase ArgK plays a dual role for the self-defense of phaseolotoxin producing Pseudomonas syringae pv. phaseolicola. Sci. Rep.-UK. 2015; 5:12892–12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mattheus W., Masschelein J., Gao L.-J., Herdewijn P., Landuyt B., Volckaert G., Lavigne R.. The kalimantacin/batumin biosynthesis operon encodes a self-resistance isoform of the FabI bacterial target. Chem. Biol. 2010; 17:1067–1071. [DOI] [PubMed] [Google Scholar]
- 46. Luo H., Lin Y., Gao F., Zhang C.-T., Zhang R.. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2014; 42:D574–D580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang C., Rabiee M., Sayyari E., Mirarab S.. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics. 2018; 19:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jeong H., Sung S., Kwon T., Seo M., Caetano-Anollés K., Choi S.H., Cho S., Nasir A., Kim H.. HGTree: database of horizontally transferred genes determined by tree reconciliation. Nucleic Acids Res. 2016; 44:D610–D619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nakamura Y. Prediction of horizontally and widely transferred genes in prokaryotes. Evol. Bioinform. 2018; 14:doi:10.1177/1176934318810785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Trindade M., van Zyl L.J., Navarro-Fernández J., Abd Elrazak A.. Targeted metagenomics as a tool to tap into marine natural product diversity for the discovery and production of drug candidates. Front. Microbiol. 2015; 6:890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Garcia R., La Clair J.J., Müller R.. Future directions of marine myxobacterial natural product discovery inferred from metagenomics. Mar. Drugs. 2018; 16:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Navarro-Muñoz J.C., Selem-Mojica N., Mullowney M.W., Kautsar S.A., Tryon J.H., Parkinson E.I., De Los Santos E.L., Yeong M., Cruz-Morales P., Abubucker S. et al.. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020; 16:60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.