

Abstract
Background
Glycogen storage disease type IV (GSD IV; Andersen’s disease) is a rare autosomal recessive disease caused by mutation in the GBE1 gene. Presentation of GSD IV varies on a continuum of severity and symptomatology ranging from neonatal death to mild adult-onset disease with variable involvement of hepatic, muscular, neurologic, dermatologic, and cardiac systems. Cardiomyopathy seen in GSD IV is also heterogeneous and its appearance on cardiac magnetic resonance imaging (CMR) is rarely described.
Case summary
A 29-year-old man without previous medical history was admitted to our facility multiple times over 2 years for focal sensorimotor deficits, gout arthropathy, chronic hyperlactataemia and hyperuricaemia, and severe decompensated non-ischaemic cardiomyopathy complicated by episodes of thromboembolic organ infarction. Echocardiography and CMR showed severe biventricular failure with the presence of intraventricular thrombi with increased right ventricular trabeculation and absent late gadolinium enhancement. He underwent muscle biopsy which showed prominent glycogen in skeletal muscle followed by genetic testing showing a single heterozygous splicing mutation c.993-1G>T found at the junction of intron 7 and exon 8 of the GBE1 gene which had not previously been reported and was predicted to be pathologic. He was referred to a tertiary care centre with glycogen storage disease specialists but expired prior to establishing care at that facility.
Discussion
Discovery of GSD IV in our patient was unexpected due to a highly variant clinical presentation. Our case stresses the clinical heterogeneity of GSD IV and the importance of genetic sequencing studies in the evaluation of potential glycogen storage disease.
Keywords: Glycogen storage disease type IV, Cardiac MRI, Hypertrabeculation, Manifesting heterozygote, Case report, Dilated cardiomyopathy
Learning points
Clinical presentation of glycogen storage disease type IV (GSD IV) is highly variable and the diagnosis should be within the differential in patients with otherwise unexplained multi-organ disease. Genetic testing, including genetic sequencing, is essential in diagnosis.
Glycogen storage disease type IV associated cardiomyopathy is also highly variable and multiple morphologies have been reported.
The mechanism of cardiomyopathy associated with GSD IV may not necessarily be mediated by fibrosis detectable via gadolinium enhancement on cardiac magnetic resonance imaging. Native T1 mapping has not been reported in GSD IV-associated cardiomyopathy and attention should be paid to this assessment in future cases.
Introduction
Glycogen storage disease type IV (GSD IV; Andersen’s disease) is a rare autosomal recessive disease caused by mutation in the GBE1 gene which impairs glycogen branching enzyme (GBE). Reduced GBE activity causes formation of abnormally long glycogen chains called polyglucosans.1 Variable involvement of tissue-specific isozymes causing polyglucosan build-up in skin, liver, skeletal muscle, cardiac muscle, and central nervous system are thought to underlie the gamut of presentations seen.2
The clinical presentation of GSD IV is extremely heterogeneous ranging from death in utero to mild adult-onset disease.3,4 Cardiomyopathy in GSD IV is also heterogeneous and numerous morphologies have been reported, including hypertrophic cardiomyopathy during infancy,5,6 neonatal dilated cardiomyopathy,7 and adult-onset dilated cardiomyopathy.5,6 Within the last category is the only other case in which cardiac magnetic resonance imaging (CMR) findings were reported which demonstrated late gadolinium enhancement (LGE) in segments of the interventricular septum. We report a case of GSD IV-related dilated cardiomyopathy with absent LGE on CMR and increased right ventricular (RV) trabeculation.
Timeline
| Date | Event |
|---|---|
| January 2013 | First presented with upper extremity weakness and paresthesias along with diffuse polyarthritis. Workup non-diagnostic, treated as seronegative spondyloarthritis and neuropathy and discharged with plan for outpatient neurological workup but lost to follow-up |
| January 2014 | Presents with dyspnoea and bilateral knee arthritis, found to have non-ischaemic cardiomyopathy (NICM) with ejection fraction (EF) of 25%. Started on medical therapy and refused implantable cardioverter-defibrillator placement |
| December 2014 | Presents with haemoptysis found to be due to thromboembolic pulmonary infarcts. Interval decrease in EF to 18% with the presence of biventricular apical thrombi |
| January 2015 | Accepts joint aspiration which finds monosodium urate crystals |
| February 2015 | Left vastus lateralis muscle biopsy performed for muscle weakness finds vacuolar myopathy and prominent glycogen |
| March 2015 | Glycogen storage disease next-generation sequencing panel showed a heterozygous mutation in the GBE1 gene for sequence variant c.993-1G>T found at the junction of intron 7 and exon 8 |
| April 2015 | Referred to tertiary centre with glycogen storage disease specialists; however, expired prior to presentation there |
Case presentation
A 29-year-old man without previous medical history initially presented with chronic left upper extremity weakness, paresthesias, and diffuse polyarticular pain and swelling. He was found to have elevated erythrocyte sedimentation rate to 95 mm/h (0–13 mm/h) and C-reactive protein to 154.5 mg/L (0–5 mg/L) with erosive changes in multiple joint radiographs. He was treated with non-steroidal anti-inflammatory drugs, gabapentin, and low-dose oral prednisone with plan for further outpatient workup but did not follow-up.
He presented the next year with inability to ambulate due to severe dyspnoea and bilateral knee arthritis. Family history included a maternal grandmother with arthritis; however, there was no family history of cardiovascular disease. He was euvolemic, his bilateral knees were swollen and there was severe pes cavus deformity of the feet. Labs included chronic lactataemia of 4–6 mmol/L (0.5–2.2 mmol/L), hyperammonaemia to 177 µmol/dL (11–33 µmol/dL), and hyperuricaemia to 11.5 mg/dL (3.7–7 mg/dL). Autoimmune panel, anti-Trypanosoma cruzi antibodies, and Coxsackie virus antibodies were negative. There was radiographic enlargement of the cardiac silhouette with echocardiographic left ventricular dilation, ejection fraction (EF) of 25%, and diffuse hypokinesis (Figure 1). Electrocardiogram (EKG) showed left axis, left bundle branch block, and diffuse tall QRS complexes (Figure 2). No evidence of coronary disease was found during cardiac catheterization at an outside facility a month prior to presentation. The patient resumed treatment for arthritis with oral steroids and began medical therapy for heart failure with carvedilol, furosemide, and lisinopril. He refused implantable cardioverter-defibrillator (ICD) placement and was lost to follow-up upon discharge.
Figure 1.

Anteroposterior (AP) radiograph of the chest showing enlargement of the cardiac silhouette (A). Echocardiographic four-chamber view (B) reveals severe left ventricular dilation. M-mode of left ventricular mid-chamber diameter with minimal fractional shortening (C). LA, left atrial; LV, left ventricular; RA, right atrial; RV, right ventricular.
Figure 2.

Electrocardiogram showing sinus tachycardia with left axis deviation, left bundle branch block, and increased QRS amplitude. aVF, augmented vector foot; aVL, augmented vector left ; aVR, augmented vector right.
He presented later that year with shortness of breath and episodes of haemoptysis and was admitted to the medical intensive care unit due to elevated lactate to 18 mg/dL without acidosis (bicarbonate 27.3 mmol/L; 22–29 mmol/L). Vitals and physical exam were otherwise benign. Troponin-T peaked at 0.798 ng/mL (<0.01 ng/mL) but EKG was unchanged. Repeat echocardiography showed worsened EF to 18% with marked reduction of tricuspid annular plane systolic excursion, RV dilation, and presence of biventricular thrombi in the apical walls (Figure 3). Intravenous contrast-enhanced computed tomography of the abdomen and pelvis found wedge-shaped hypodensities in the lung parenchyma and kidney representing thrombotic infarctions (Figure 4). He was anticoagulated and stabilized. Biventricular ICD placement was considered but deferred in the setting of intracardiac thrombus. The patient was not a candidate for cardiac transplant due to continual medication non-compliance and social instability.
Figure 3.
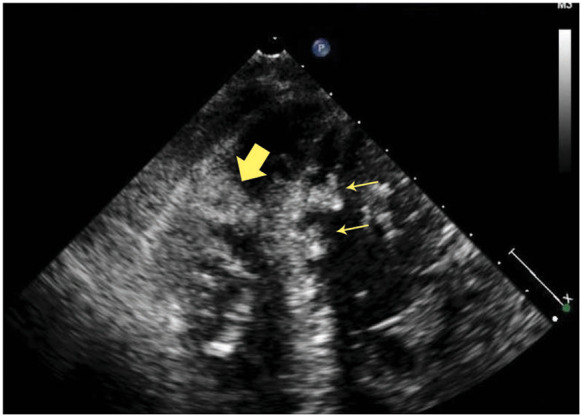
A four-chamber view showing right ventricular (thick arrow) and left ventricular (thin arrows) mural echogenic masses concerning for thrombi.
Figure 4.

Coronal images from contrast-enhanced computed tomography of the abdomen showing wedged shaped infarctions in the right lung (orange arrows) (A) and right kidney (yellow arrows) (B).
In the following year, he was admitted multiple times for polyarthritic flares and dyspnoea. Arthrocentesis found monosodium urate crystals and he was treated with intra-articular and systemic steroids and colchicine. He had multiple episodes of hypoglycaemia and concurrent evaluation showed non-insulin-mediated hypoglycaemia with decreased glucose, insulin, and c-peptide. Electromyography was performed to evaluate weakness and showed mild, diffuse myopathy affecting the lower extremities and distal hand muscles. Left vastus lateralis biopsy showed prominent glycogen in skeletal muscle on electron microscopy. Targeted next-generation sequencing of genomic deoxyribonucleic acid (DNA) for the full coding regions plus ∼20 bp of non-coding DNA flanking each exon of the 14 genes known to be involved in GSDs detected a novel heterozygous c.993-1G>T mutation at the junction of intron 7 and exon 8 of the GBE1 gene which was further confirmed by Sanger sequencing. A splicing prediction software, Human Splicing Finder, predicted that the patient’s novel variant alters GBE1 splicing resulting in pathogenicity.8
Follow-up echocardiogram with contrast showed worsening biventricular function with an RV thrombus within a hypertrabeculated RV. Cardiac magnetic resonance imaging found absent LGE with increased RV trabeculation and thrombus (Figures 4and5) . He was referred to a tertiary centre with glycogen storage disease specialists and was subsequently lost to follow-up from our facility. He was later found to have expired under unknown circumstances prior to presentation to that facility and no further information could be obtained.
Figure 5.

(A) Echocardiography showing densely trabeculated right ventricle (yellow arrows). (B and C) Cardiac magnetic resonance imaging showing increased right ventricular trabeculation in short-axis FIESTA view (B). Increased trabeculation of the right ventricle with thrombus in a trabecular recess (C, yellow arrow) is evident in the four-chamber horizontal long-axis FIESTA (C).
Discussion
Variant presentation
Diagnosis of GSD IV was unexpected due to our patient’s variant presentation. Although biopsy results limited differential to the glycogen storage diseases; presence of hyperlactataemia, hyperuricaemia, and gouty arthrosis led members of the team to expect confirmation of glycogen storage disease Type I.3 Given family history of arthropathy, these findings are thought to represent a synchronous disease process. Glycogen storage disease type IV was diagnosed based on involvement of the GBE1 gene and organ systems known to affected by GSD IV.
Manifesting heterozygosity
Confirmatory diagnosis of GSD IV is established via genetic testing showing homozygous or compound heterozygous pathologic mutations in the GBE1 gene;9,10 however, only a single mutation was identified in our patient indicating manifesting heterozygous disease. The phenomenon of manifesting heterozygosity has been investigated in adult body polyglucosan disease (ABPD)—a neurological disorder involving the GBE1 gene. In one retrospective study of 35 patients with ABPD, 16 were heterozygous for a c.986A>C mutation in GBE1 without a second mutation. Reverse transcription of the messenger ribonucleic acid for the GBE1 gene found a deep intronic mutation creating an ectopic last exon in all manifesting heterozygotes.11 It is possible that a similar process may be responsible for the manifesting heterozygous disease seen in our patient.
Heterogeneity of cardiomyopathy associated with glycogen storage disease type IV
Absent LGE and development of RV hypertrabeculation were unique findings on our patient’s CMR. Presence of LGE represents fibrosis and is an independent risk factor for adverse cardiac events.12 Although LGE was present in the only other reported case of GSD IV-associated cardiomyopathy evaluated with CMR;5 its absence in our patient suggests that the mechanism of GSD-associated cardiomyopathy involves a process which does not necessarily involve fibrosis detectable by LGE. Myocardial native T1 mapping is an emerging biomarker which has demonstrated reduction of myocardial T1 values in carriers of glycogen storage diseases. Unfortunately, native T1 mapping was not available on our magnetic resonance imaging unit at the time of this examination and was not reported in the only other case of GSD IV cardiomyopathy. Further CMR evaluation in patients with suspected GSD cardiomyopathy should include this technique.13
Imaging also showed interval increase in RV trabeculation which resembled an RV analogue of left ventricular non-compaction cardiomyopathy. Left ventricular non-compaction cardiomyopathy is a severe cardiomyopathy characterized by embryological endomyocardial arrest causing persistent spongy trabeculated myocardium. Non-compaction cardiomyopathy of the RV has previously been reported; however, its existence as a clinical entity is debated as the RV is normally trabeculated and normal vs. pathologic hypertrabeculation is difficult to distinguish.14 This finding in our patient may represent an adaptation to increased RV load.15
Conclusion
Our patient’s severe adult-onset, multisystemic presentation with a novel heterozygous mutation, and cardiomyopathy with novel findings on CMR exemplifies the extreme heterogeneity seen in GSD IV. These unique findings stress the importance of genetic sequencing studies in the diagnosis of Andersen’s disease.
Lead author biography

Dr Shawn Lyo completed his internship in Internal Medicine at Albert Einstein College of Medicine - Jacobi Medical Center where he was awarded a Leo M. Davidoff society award for excellence in teaching medical students. He began diagnostic radiology residency at SUNY Downstate Health Sciences University in 2019. Dr Lyo has a wide interest in all modalities of medical imaging, informatics, and image-guided interventions.
Supplementary material
Supplementary material is available at European Heart Journal - Case Reports online.
Slide sets: A fully edited slide set detailing this case and suitable for local presentation is available online as Supplementary data.
Consent: The author/s confirm that written consent for submission and publication of this case report including image(s) and associated text has been obtained from the patient in line with COPE guidance.
Conflict of interest: none declared.
Supplementary Material
References
- 1. Magoulas PL, El-Hattab AW.. Glycogen Storage Disease Type IV. Seattle, WA: University of Washington, Seattle; 1993–2019; 2013. https://www.ncbi.nlm.nih.gov/books/NBK115333/ (2 November 2019). [PubMed] [Google Scholar]
- 2. Moses SW, Parvari R.. The variable presentations of glycogen storage disease type IV: a review of clinical, enzymatic and molecular studies. Curr Mol Med 2002;2:177–188. [DOI] [PubMed] [Google Scholar]
- 3. Ozen H. Glycogen storage diseases: new perspectives. World J Gastroenterol 2007;13:2541–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mochel F, Schiffmann R, Steenweg ME, Akman HO, Wallace M, Sedel F, Laforêt P, Levy R, Powers JM, Demeret S, Maisonobe T, Froissart R, Da Nobrega BB, Fogel BL, Natowicz MR, Lubetzki C, Durr A, Brice A, Rosenmann H, Barash V, Kakhlon O, Gomori JM, van der Knaap MS, Lossos A.. Adult polyglucosan body disease: natural history and key magnetic resonance imaging findings. Ann Neurol 2012;72:433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Szymańska E, Szymańska S, Truszkowska G, Ciara E, Pronicki M, Shin YS, Podskarbi T, Kępka A, Śpiewak M, Płoski R, Bilińska ZT, Rokicki D.. Variable clinical presentation of glycogen storage disease type IV: from severe hepatosplenomegaly to cardiac insufficiency. Some discrepancies in genetic and biochemical abnormalities. Arch Med Sci 2018;14:237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aksu T, Colak A, Tufekcioglu O.. Cardiac involvement in glycogen storage disease type IV: two cases and the two ends of a spectrum. Case Rep Med 2012;2012:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tang TT, Segura AD, Chen Y-T, Ricci LM, Franciosi RA, Splaingard ML, Lubinsky MS.. Neonatal hypotonia and cardiomyopathy secondary to type IV glycogenosis. Acta Neuropathol 1994;87:531–536. [DOI] [PubMed] [Google Scholar]
- 8. Desmet F-O, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C.. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009;37:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bruno C, Cassandrini D, Assereto S, Akman HO, Minetti C, Di Mauro S.. Neuromuscular forms of glycogen branching enzyme deficiency. Acta Myol 2007;26:75–78. [PMC free article] [PubMed] [Google Scholar]
- 10. Schene IF, Korenke CG, Huidekoper HH, van der Pol L, Dooijes D, Breur JMPJ, Biskup S, Fuchs SA, Visser G.. Glycogen storage disease type IV: a rare cause for neuromuscular disorders or often missed? JIMD Rep 2019;45:99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Akman HO, Kakhlon O, Coku J, Peverelli L, Rosenmann H, Rozenstein-Tsalkovich L, Turnbull J, Meiner V, Chama L, Lerer I, Shpitzen S, Leitersdorf E, Paradas C, Wallace M, Schiffmann R, DiMauro S, Lossos A, Minassian BA.. Deep intronic GBE1 mutation in manifesting heterozygous patients with adult polyglucosan body disease. JAMA Neurol 2015;72:441–445. [DOI] [PubMed] [Google Scholar]
- 12. Wu KC, Weiss RG, Thiemann DR, Kitagawa K, Schmidt A, Dalal D, Lai S, Bluemke DA, Gerstenblith G, Marbán E, Lima JAC, Tomaselli GF.. Late gadolinium enhancement by cardiovascular magnetic resonance heralds an adverse prognosis in nonischemic cardiomyopathy. J Am Coll Cardiol 2008;51:2414–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Poyhonen P, Hiippala A, Ollila L, Kaasalainen T, Hanninen H, Helio T.. Cardiovascular magnetic resonance findings in patients with PRKAG2 gene mutations. J Cardiovasc Magn Reson 2015;17:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oechslin EN, Attenhofer Jost CH, Rojas JR, Kaufmann PA, Jenni R.. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol 2000;36:493–500. [DOI] [PubMed] [Google Scholar]
- 15. Arbustini E, Weidemann F, Hall JL.. Left ventricular noncompaction: a distinct cardiomyopathy or a trait shared by different cardiac diseases? J Am Coll Cardiol 2014;64:1840–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.