

Abstract
The anti-CD19 chimeric antigen receptor (CAR) T cells showed excellent effect against acute lymphoblastic leukemia (ALL) in bone marrow (BM) in clinical trials. However, it remains to be elucidated whether the CD19 CAR T cell therapy is effective for ALL cells in central nervous system (CNS) because the patients with isolated or advanced CNS disease were excluded from clinical trials of systemic intravenous (i.v.) delivery of CAR T cells. Therefore, the preclinical evaluation for the efficacy of CAR T cell therapy against ALL cells in CNS is essential for clinical application. We evaluated the effect and adverse reaction of CD19 CAR T cells against ALL in CNS using a xenograft mouse model by i.v. or intra-cerebroventricular (i.c.v.) delivery of CAR T cells. Injection of piggyBac CD19 CAR T cells by i.v. had partial effects, whereas all CAR T i.c.v.-delivered mice had eliminated ALL in CNS. Although some CAR T i.c.v.-delivered mice showed transient changes of clinical symptoms during the first few days after treatment, none of CAR T i.c.v.-delivered mice displayed fatal adverse events. In this study, we demonstrated that direct delivery into CNS of CAR T cells is a possible therapeutic approach with the xenograft mouse model.
Keywords: CNS-ALL, xenograft mouse model, piggyBac transposon, CD19 CAR T cells, intra thecal delivery, intra-cerebroventricular, preclinical study
Graphical Abstract
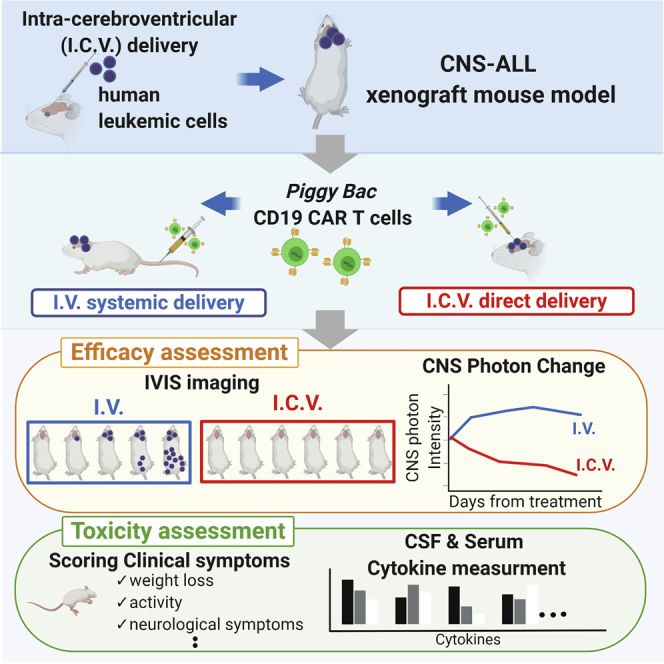
It remains to be elucidated whether the CD19 CAR T cell therapy is effective for ALL cells in the central nervous system (CNS). Tanaka et al. demonstrated that direct delivery of CAR T cells into CNS is an effective therapeutic approach with the preclinical xenograft model. Keywords: CNS-ALL, xenograft mouse model, piggyBac transposon, CD19 CAR T cells, intra thecal delivery, intra-cerebroventricular, preclinical study
Introduction
Recent clinical trials show that the CD19 chimeric antigen receptor (CAR) T cell therapy has an excellent effect against refractory and relapsed pediatric acute lymphoblastic leukemia (ALL) in bone marrow (BM).1, 2, 3, 4, 5, 6 Some reports showed the effects of systemic intravenous (i.v.) delivery of CAR T cells against ALL cells in the central nervous system (ALL in CNS),4,5 which led us to the therapeutic strategy against ALL in CNS using CAR T cells. However, because the patients with isolated or advanced CNS disease have been excluded,1, 2, 3, 4 there are no detailed data about the efficacy of CAR T cell therapy for ALL in CNS even with the animal model. In addition, blood-brain-barrier restricts the diffusion of cells in the blood into the brain7 and there are a variety of differences in microenvironmental conditions between BM and CNS.8 Therefore, it is required to clarify whether the CD19 CAR T cell therapy is effective for ALL in CNS in the preclinical model. Recently, the direct delivery of CAR T cells into solid tumors was reported to be superior to other approaches,9,10 and we and others have shown that intrathecal (IT) donor lymphocyte infusion can be used to treat ALL in CNS without severe adverse events.11 These reports inspired us to apply direct CAR T cell infusion into CNS as a new therapeutic approach against ALL in CNS.
In this study, we utilize an ALL in CNS xenograft mouse model by intra-cerebroventricular (i.c.v.) injection of ALL cells and demonstrated that i.c.v.-delivery of CAR T cells could be an encouraging therapeutic approach for ALL in CNS.
Results
A Xenograft Model by i.c.v. Delivery Is a Useful Model to Estimate the Effect against ALL in CNS
In our study, we used a human Philadelphia chromosome-positive pre-B ALL cell line, SU/SR,12 which was confirmed CD19 expression by flow cytometry (Figure S1A). SU/SR was stably transduced with a GFP-luciferase lentiviral construct (SU/SRGFP/luc; Figures S1B and S1C).
We have previously reported ALL patient-derived xenograft model with CNS infiltration in non-obese diabetic/severe combined immunodeficiency/γc Null (NOD/SCID/γc Null, NOG) mouse by i.v. injection via tail vein.8 In this study, to assess clearly the effect against ALL in CNS, we made an ALL in CNS xenograft mouse model using the procedure of i.c.v. delivery of NOG mouse (Figures S2A and S2B). We demonstrated that SU/SRGFP/luc cells delivered by i.c.v. were engrafted into CNS and then spread systemically and few ALL cells are detected in systemic organs at 7 days after injection (Figures 1A and 1B). Histopathology showed infiltration around the leptomeninges (Figure 1C) and absence of gross parenchymal involvement, which closely resembled the pathology of CNS involvement ALL.13 These data mean that this xenograft mouse model reproduces isolated CNS relapse of ALL at early timing from tumor transplantation.14 This i.c.v. xenograft mouse model was a useful model for analyzing ALL in CNS.
Figure 1.

A CNS-ALL Xenograft Model Can Be Established by i.c.v. Delivery of Human ALL Cells
(A and B) Bioluminescent imaging (A) and flow cytometry analysis (B) showing that SU/SRGFP/luc cells injected by i.c.v. into NOG mice engrafted in the CNS and then spread systemically. (C) H&E staining and immunohistochemical staining of human CD19 in brain showing engraftment of human CD19-positive ALL cells in the CNS of a NOG mouse. Scale bars for images are 100 μm.
Injection of piggyBac CD19 CAR T Cells by i.c.v Had Eliminated ALL in CNS
We conducted an in vivo study using the xenograft model to determine whether i.v.-delivered CAR T cells can be efficacious against ALL in CNS and whether i.c.v.-delivery of CAR T cells has any influence on anti-leukemic effect.
PiggyBac CD19 CAR T cells were produced using a pIRII-CD19.CD28.z_CAR transposon plasmid without the immunoglobulin G1 (IgG1)-CH2CH3 spacer and a pCMV-piggyBac transposase plasmid (Figure S3A) as previously described.15,16 On day 14, cultured cells were harvested and used for further experiment. CAR expression and immunophenotypic composition of the product were analyzed using flow cytometry (Figures S3B and S3C). Non-transduced T cells from the same donor were also cultured and used as Mock T cells in this study.
Two hundred thousands SU/SRGFP/luc cells were injected by i.c.v. into NOG mice. Seven days later, 2 × 106 piggyBac CD19 CAR T cells manufactured simultaneously from the single donor were injected by i.v. via tail vein (CAR T i.v.; n = 7) or i.c.v. (CAR T i.c.v.; n = 8). As control, some mice received no treatment (n = 4). To evaluate the influence of the procedure of i.c.v. injection, we injected medium alone by i.c.v. in Medium i.c.v. group (n = 5). Besides, to evaluate the influence of nonspecific allogenic antitumor response via T cell receptor by injected T cells,17 we injected 2 × 106 Mock T cells by i.c.v. in Mock T i.c.v. group (n = 6). ALL invasion was followed by bioluminescent imaging. At day 4 and day 10, one mouse of each group was culled and sacrificed for further analysis (Figure 2A).
Figure 2.

Injection of piggyBac CD19 CAR T Cells by Intra-Cerebroventricular Had Eliminated ALL in CNS
(A) Experimental outline of the in vivo study. NOG mice received 2 × 105 SU/SRGFP/luc cells by intracerebroventricular (i.c.v.) injection on day -7, followed by piggyBac CD19 CAR T intravenous (i.v.) or i.c.v. injection on day 0. As control, non-transduced T cells or vehicle were injected by i.c.v. on day 0. Some mice received no treatment on day 0. Bioluminescent imaging and clinical symptoms were monitored. At day 4 and day 7, one mouse of human T cell injected groups was culled for further analysis. (B) Kaplan-Meier survival curve of mice in each group showing improvement in survival of piggyBac CD19 CAR T cell treated mice comparing with Medium i.c.v. group. p value was analyzed using a log-rank test with the Bonferroni correction. ∗p < 0.05. (C) Bioluminescent images of each group at time points post treatment. Red bold “X” indicates removal because of morbidity and the timing of each event was shown. The mice surrounded by the blue line showed residual disease in the Mock T i.c.v. or CAR T i.v. group. The mice surrounded by the red line showed no residual disease in the CAR T i.c.v. group. (D) The scheme of region of interest of CNS. The region of interest (ROI) was defined as the square depicted by the nose to the tip of auricle as length and right and left of auricle as width. (E) The line graph showing the changes of luminescence of CNS region among groups. The gray dash line shows average background photon flux (5.99 ± 0.99 × 105) of a group of mice (n = 6) that did not receive injections of tumor or T cells. Data are shown as mean ± standard error of the mean (SEM). The comparisons of photon intensity of bioluminescent imaging between CAR T i.v. and i.c.v. group after day 25 were statically analyzed using Wilcoxon rank sum test. ∗p < 0.05. ∗∗p < 0.01.
All mice in the no treatment group and the Medium i.c.v. group and two of five mice in the Mock T i.c.v. group died by day 45, whereas all mice treated by CAR T cells regardless of route of injection were alive at day 45. (Figure 2B). This indicated that i.v.-delivered CAR T cells have some effect against ALL in CNS as predicted from some clinical cases. However, bioluminescent imaging revealed that three out of five mice in the CAR T i.v. group had residual disease. On the other hand, i.c.v.-delivered CAR T cells eliminated ALL in CNS of all mice (Figure 2C). Likewise, the quantification of the luminescence of CNS region also revealed the reduction of tumor burden in CNS by i.c.v.-delivered CAR T cells (Figures 2D and 2E). Despite the improvement of leukemic burden, unexpected fetal events occurred possibly due to the development of graft-versus-host disease (x-GVHD) with the systemic invasion of human T cells (Figure S4). These events disabled to monitor the direct therapeutic benefit of CAR T cell after 45 days of treatment.
I.c.v.-Delivered CAR T Cells Showed Earlier Distribution than i.v.-Delivered CAR T Cells
On day 4 after treatment, we culled and analyzed one mouse of each group treated by human T cells whose luminescence of ALL cells in CNS region had the lowest progression rate (Figure 3A). Histopathological and flow cytometry analysis in CNS showed a decrease in ALL cells and expansion of human CD3-positive T cells in the CAR T i.c.v. mouse, whereas no human CD3-positive T cells were detected in the CAR T i.v. mouse (Figures 3B and 3C). The difference of distribution of human CD3-positive T cells between two groups was also observed in the flow cytometry analysis of cerebral spinal fluid (CSF) and peripheral blood (Figure S5).
Figure 3.

I.c.v.-Delivered CAR T Cells Showed Earlier Distribution than i.v.-Delivered CAR T Cells
(A) IVIS images of each mouse analyzed at day 4. (B) Representative H&E staining and immunohistochemical staining of human CD19 and human CD3 in brain tissue sections from Mock T i.c.v., CAR T i.v., and CAR T i.c.v. treated mice on day 4 post treatment. The area surrounded by dash lines indicates human CD19-positive ALL cells engrafted on the brain surface and red arrow heads indicate human CD3-positive cells. Scale bars for images are 100 μm. (C) Flow cytometry plots of human CD3-positive cells and GFP-positive ALL cells on day 4 in the CNS. Dead cells were excluded using 4’,6-diamidoino-2-phenylindole (DAPI).
On day 10 after treatment (Figure S6A), few mononuclear cells were isolated from CNS in the CAR T treated mice. Neither of ALL cells nor human CD3-positive cells were detected by histopathological and flow cytometry analysis in CNS (data not shown) and BM (Figure S6B). Quantitative real-time polymerase chain reaction analysis revealed the existence of CAR T cells in the BM of the CAR T i.v. mouse, but not in that of the CAR T i.c.v. mouse (Figure S6C).
I.c.v. Delivery of CD19 CAR T Cells Had No Neurological Symptoms and Fatal Adverse Effects
As lethal neurotoxicity (NT) has been reported in CAR T i.v. injection clinical trials,18,19 we estimated adverse effects of treatment in our xenograft mice model. We serially assigned clinical symptom scores using a modified scoring system previously described20 (Tables S1 and S2) and measured body weight and rectal temperature every morning after treatment. Regarding these clinical symptoms, some mice in the CAR T i.c.v. group showed transient changes during the first few days after treatment (Figure 4A). None of the mice displayed neurological symptoms and fatal adverse events (Table S2). Because a NOD/SCID/interleukin-2 Rgcnull (NOD/SCID/IL-2Rgcnull, NSG) xenograft model of glioma treated by anti-GD2 CAR T cells showed ventriculomegaly and fatal brain swelling,21 we analyzed histopathological changes of CNS on day 4. It revealed the existence of inflammation for leukemia clearance such as slight thickening of the choroid plexus epithelium and an edema localized around the lateral ventricle in the CAR T i.c.v. mouse. However, neither ventriculomegaly nor extensive brain edema was observed (Figure 4B). It is known that several cytokines are related to severe cytokine releasing syndrome (CRS) and NT18,19,22,23 and have the peak concentrations in serum within about 2 weeks post infusion of CAR T cells.19 To clarify whether the changes of clinical symptoms in our study were accompanied with severe cytokine production, we measured multiple human and mouse cytokine levels in serum and CSF on day 4 and day 10 after treatment when the luminescence of CNS region and clinical symptoms of mice in the CAR T treated groups had changed most significantly. At either timing, there were no apparent differences in serum levels of human and mouse cytokines between Mock T i.c.v., CAR T i.v., and CAR T i.c.v. groups. Likewise, in CSF, there were not severe multiple cytokine elevations indicating CRS in all groups (Figure 5; Figure S7).
Figure 4.

Mice Treated by CD19 CAR T i.c.v. Did Not Show Fatal Adverse Effects
(A) The changes in weight from baseline, rectal temperature, and clinical scores of each mouse are shown. (B) H&E staining and immunohistochemical staining for human CD19 around the ventricles of brain tissue sections from CAR T i.v. and i.c.v. injected mice analyzed on day 4. White arrowheads indicate ependymal multilayered cells with infiltration of human CD3 (yellow arrows). The area surrounded by dash lines indicates localized edema. Scale bars for images are 100 μm.
Figure 5.

There Was No Significant Difference of Cytokines in Serum and CSF between Mock T i.c.v., CAR T i.v., and CAR T i.c.v Group
(A–D) Human and mouse cytokine concentrations in (A) peripheral blood serum and (B) cerebrospinal fluid (CSF) on day 4 (n = 4 mice per group) and in (C) peripheral blood serum and (D) CSF on day 10 (n = 4 mice per group). CSF was gathered from each group and analyzed as one sample. Cytokine concentrations were measured using the Bio-Plex Cytokine assay system (Bio-Rad Laboratories). Data are shown as mean ± SEM. Data were analyzed by one-way ANOVA, and then differences among means were analyzed using Tukey-Kramer multiple comparison tests. n.s., no significant difference. ∗, not detected.
Discussion
Although treatment intensification adapted by risk stratification of childhood ALL have reduced CNS relapse,24, 25, 26 residual CNS disease predicts increased risk of CNS relapse despite hematopoietic stem cell transplantation27 and the prognosis of relapsed ALL with CNS involvement is still not satisfactory.27,28 Moreover, existing intensive CNS-directed treatments, namely multiple IT infusion of cytotoxic agents and pre-emptive cranial radiotherapy, can lead to acute and late adverse complications, such as endocrine disorders, neurocognitive impairments, and secondary neoplasms.25,29,30 These complications have been serious problems especially for survivors of pediatric ALL. Even today, a more effective and targeted treatment strategy for ALL in CNS is needed. Although the CAR T cell therapy is an expected strategy, it is necessary to directly determine the effect against ALL in CNS.
Consistent with previous clinical and experimental reports,4,5,31 our study demonstrated that injection of CD19 CAR T cells by i.v. had a partial effect with residual disease in some mice, whereas i.c.v.-delivered CAR T cells had a complete response against ALL in CNS of all mice. The quantitative analysis of photons in CNS regions also showed the prominent effect of i.c.v.-delivered CAR T cells. Moreover, i.c.v.-delivered T cells were detected earlier than i.v.-delivered T cells in the CNS at the same dose. The reason of this would be that i.c.v.-delivered cells were injected locally right to the tumor over the blood-brain barrier, rather than i.v.-delivered cells. Our investigation into the importance of the route of administration is timely given the result of recent reports showing the superior effect of direct CAR T cell injection for solid tumors.32,33 In our study, i.c.v.-transplanted ALL cells caused fatal systemic infiltration in no treatment and Medium i.c.v. groups. However, in CAR T i.c.v. group, systemic disease had not been detected. Our study suggests that, in clinical, elimination of ALL in CNS by direct administration of CAR T cells could lead to reduction of additional systemic therapy for patients with isolated CNS relapsed disease. In addition to that, it is known that submicroscopic BM involvement is detected in some patients diagnosed as isolated CNS relapses in childhood ALL.14 i.c.v.-delivered T cells were detected in peripheral blood at day 4 after i.c.v. delivery. Although we could not show directly whether i.c.v.-delivered CAR T cells eliminated a combined relapse of BM and CNS in our xenograft model, there is the possibility that early leakage of i.c.v.-delivered CAR T cells would also prevent the combined submicroscopic systemic disease from progressing.
We could not estimate the durability of CAR T cells in CNS after i.c.v. administration, because few mononuclear cells could be collected from CNS after the elimination of leukemic cells. The long-term disease control and the strategy of multiple administration remain to be elucidated by further analysis.
In our study, neither neurotoxic symptoms nor fatal adverse events were observed even in the CAR T i.c.v. group. Of course, there are limitations of using xenograft model as a preclinical tool; it is known that mice are less reactive to human cytokines than mouse cytokines, and a decreased number and impaired function of these microenvironmental components, which are known to release the cytokines, are found in immunodeficient mice,34 so there would be concern that CRS and NT induced by CD19 CAR T cell therapy could be not reproduced faithfully using xenograft mouse models. Within these limitations of xenograft model, there are reports that human cytokines derived from injected human T cells are detectable in x-GVHD mouse models35 and an orthotopic NSG xenograft model of glioma could reproduce the fatal brain edema after i.v. delivery of GD2 CAR T cells21 that were not observed in our study. One of the reasons of the minimal adverse effects of i.c.v.-delivered CAR T cells could be few systemic tumor burdens at the time of treatment in our xenograft model. It has been shown that systemic CRS and NT can be observed when high CAR T cell numbers rapidly engage high tumor burdens.19
Conclusions
In conclusion, we demonstrated that direct delivery into CNS of CAR T cells is a promising therapeutic approach within the xenograft mouse model. Although further consideration will be needed to yield toxicities in clinical situation, our study can be a rationale for translating this attractive therapy to patients with CNS involvement.
Materials and Methods
CD19 CAR T Cells and Pre-B ALL Cell Lines
SU/SR was human Philadelphia chromosomepositive pre-B ALL cell line derived from SU-Ph2 by culture with imatinib as described previously12 and maintained in suspension culture RPMI 1640 medium with 10% heat-in activated fetal bovine serum and 100 U/mL penicillin/streptomycin.
Lentivirus was generated by plating 293T cells transfected with packaging plasmids and the lentiviral reporter construct pHAGE PGK-GFP-IRES-Luciferase-w (Addgene). Supernatants were collected after 2 to 3 days and concentrated via high-speed centrifugation (20,000 × g) for 4 h at 4°C. Lentiviral pellets were then resuspended and stored at −80°C for later use. GFP-positive fraction of SU/SR transfected with this lentivirus were sorted on a BD fluorescence-activated cell sorting (FACS) Aria and then were expanded.
PiggyBac CD19 CAR T cells were produced as previously described.15,16 In brief, mononuclear cells were freshly isolated from peripheral blood of healthy donors and then immediately transfected by electroporation with a pIRII-CD19.CD28.z_CAR transposon plasmid without the IgG1-CH2CH3 spacer and a pCMV-piggyBac transposase plasmid (Figure S3A). Electroporated cells were co-cultured with irradiated autologous activated T cells (ATCs) pulsed with four viral peptide pools (ACE; AdV5 Hexon, CMV pp65, EBV EBNA-1, and BZLF1) in T cell culture medium supplemented with IL-7 (10 ng/mL)/IL-15 (5 ng/mL) on day 0. On day 7, cells were re-stimulated with ACE-pulsed irradiated ATCs. On day 14, cultured cells were harvested and used for further experiment.
Mice
Mice used in this study were 8- to 10-week-old-male NOG mice. All animal experiments were carried out under protocols approved by Institute of Laboratory Animals of Graduate School of Medicine, Kyoto University.
I.c.v. Injection into Mice and CSF Collection
The mice were initially anesthetized during the procedure and fastened using a stereotactic instrument (Narishige, Tokyo, Japan). The hair was removed, then an incision was made at midline of the head. A 1-mm burr hole was drilled in the skull at proper position (1.0 mm lateral, 0.5 mm posterior from bregma; Figure S2A). Through this hole, cells were suspended in 5 μL RPMI and injected into the lateral ventricle at 3 mm deep from the surface of skull over 5 min automatically using a 26-gauge needle 10 μL syringe (Hamilton Company, Boston, MA, USA) and IMS-20 micro injector (Narishige, Tokyo, Japan). The needle was left in place for 5 min to prevent the loss of cells through needle track. The incision was sutured. This procedure was validated with dye injection. When injection was performed at the correct site injected dye spread along the lateral ventricle (Figure S2B).
CSF collection was performed as previously reported.36 In brief, after drilling through the skull, a 5 μL syringe (Hamilton Company, Boston, MA, USA) was lowered in the lateral ventricle and CSF was withdrawn over 5 min. 5 μL of CSF was collected per mouse.
In Vivo Bioluminescence Imaging of Xenograft Model
Bioluminescence imaging was acquired using the IVIS Lumina LT In Vivo Imaging System (Perkin Elmer, Boston, MA, USA) with Living Image version 4.5.5 software (Perkin Elmer, Boston, MA, USA). Mice were infused by intraperitoneal injection with 150 mg/kg D-luciferin potassium salt (Wako, Osaka, Japan) suspended in 200 μL PBS. 5–15 minutes later, mice were imaged under 2% isoflurane anesthesia. Images were acquired on a 25-cm field of view at medium binning level for within 1 min exposure time.
Mononuclear Cells Isolation from Mice
Harvest of mononuclear cells from mice were performed according to previously reported methods.37 In brief, mice were euthanized, and then the organs were perfused with PBS before removal and mechanical dispersion. Mononuclear cells were isolated by Ficoll-Hypaque density gradient centrifugation.
Flow Cytometry
For analysis of leukemic cells and CAR T cells in organs, samples of each organs were stained with antibodies after isolation of mononuclear cells as described above. Dead cells were excluded by 4’,6-diamidino-2-phenylindole (DAPI) staining. Samples were analyzed using a BD FACS Verse and BD FACSuite software according to the manufacturer’s protocol. Antibodies used for flow cytometric analysis were anti-human CD3-allophycocyanin (APC; BD PharMingen, Franklin Lakes, NJ, USA), anti-human CD3 fluorescein isothiocyanate (FITC; BD PharMingen, Franklin Lakes, NJ, USA), anti-human CD4-APC (BD PharMingen, Franklin Lakes, NJ, USA), anti-human CD4- phycoerythrin (PE; Miltenyi Biotec, Bergisch Gladbach, Germany), anti-human CD8-PE (BD PharMingen, Franklin Lakes, NJ, USA), anti-human CD8-APC (Miltenyi Biotec, Bergisch Gladbach, Germany), anti-human CD56-PE (Miltenyi Biotec, Bergisch Gladbach, Germany), anti-human CD19-PE (eBiosciences, San Diego, CA, USA), anti-human CCR7-PE (Miltenyi Biotec, Bergisch Gladbach, Germany), anti-human CD62L-PE (Miltenyi Biotec, Bergisch Gladbach, Germany), anti-human CD45RO-APC (Miltenyi Biotec, Bergisch Gladbach, Germany), anti-human CD45RA-APC (Miltenyi Biotec, Bergisch Gladbach, Germany), and anti-mouse CD45-Brilliant Violet 421 (BD PharMingen, Franklin Lakes, NJ, USA). To evaluate CAR expression, we used a specific anti-idiotype single-chain variable fragment monoclonal antibody,38 which was kindly provided by Dr. Lawrence Cooper (MD Anderson Cancer Center) and anti-mouse Ig antibody (BD PharMingen, Franklin Lakes, NJ, USA). We use FITC-labeled CD19 protein tagged by histidine39 (Acrobiosystems, Newark, NJ, USA) in another method for CAR detection. The data were analyzed using FlowJo version 10 software (Tree Star, Ashland, OR, USA).
Histological Analysis of Mouse Tissue Sections
Morphological (using hematoxylin and eosin [H&E] staining) and immunohistochemical analysis were applied in the xenograft murine tissue sections of BM and brain. The organs were harvested, fixed with 4% paraformaldehyde, and embedded in paraffin. Each slide was deparaffinized, underwent citrate-based antigen retrieval, and stained with rabbit anti-human CD19 (Spring Bioscience, Pleasanton, CA, USA) or rabbit anti-human CD3 (Leica Biosystems, San Diego, CA, USA) followed by detection using VECTASTAIN Elite ABC Rabbit IgG Kit (Vector Laboratories, San Diego, CA, USA) with 50% hematoxylin counterstain. BZ-9000 (Keyence, Osaka, Japan) was used for microscopic observation.
Measurement of Cytokine/Chemokine in CSF and Serum
Cerebrospinal fluid (CSF) and serum samples were analyzed for cytokines/chemokines, namely, using the Bio-Plex human 27-plex panel, mouse 23-Plex Panel, and the Bio-Plex Cytokine assay system (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer’s instructions. Because the amount of collectable CSF is 5 μL per mouse, CSF samples of each treatment group were gathered and diluted 2-fold with standard dilution solution (Bio-Rad Laboratories, Hercules, CA, USA) and stored at −80°C for later analysis. Serum samples of each mice were diluted 2-fold with standard dilution solution and stored. Cytokine/chemokine concentrations were measured using Bio-Plex Array Reader (Bio-Rad Laboratories, Hercules, CA, USA) and calculated using a standard curve.
Quantitative Real-Time PCR
DNA was extracted from mononuclear cells of the brains and BM of mice using QIAamp DNA MINI kit (QIAGEN, Valencia, CA, USA). The quantitative real-time PCR targeting sequences specific for the promoter of CD19 CAR was performed according to our previous report.15 The copy number of CD19 CAR was calculated and expressed as the copy number per nanogram of DNA.
Statistical Analysis
Statistical tests were generated with using R (version 3.4.4; R Development Core Team) software. For survival analysis, a log-rank test was used with Bonferroni correction. For comparison of photon intensity between CAR T i.v. and i.c.v. group, statistical comparisons were analyzed using Wilcoxon rank sum test. For parametric data of cytokine production, statistical comparisons were analyzed by one-way ANOVA, and then differences among means were analyzed using Tukey-Kramer multiple comparison tests. In each analysis, p values of less than 0.05 were considered statistically significant.
Author Contributions
K.T. and I.K. designed and performed experiments and analyzed data. M.T., D.M., and Y.N. manufactured CD19 CAR T cells. K.M. and Y.N. performed quantitative real-time PCR analysis and discussion. Y.T., T.N., K.U, H.H., S.A., J.T., and Y.N. supervised the project and experimental design, and K.T. and I.T. wrote the paper with input from M.T., D.M., K.M., Y.T., T.N., K.U., H.H., S.A., J.T., and Y.N.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
I.K.’s work has been funded by JSPS KAKENHI (grant number: 17K16251), Japan Leukemia Research Fund, Novartis Research grants, and Kyoto Preventive Medical Center grant. D.M. has been funded by JSPS KAKENHI (grant number: 18K15311). Y.T. has been funded by Japan Agency for Medical Research and Development (AMED; grant number: 17824690). Y.N. has been funded by JSPS KAKENHI (grant number: 17K10106). The authors thank Yoshifumi Nakane and all other members of the Institute of Laboratory Animals at Kyoto University for support in animal experiments. The authors also thank Tatsuaki Tsuruyama and all other members of the Center for Anatomical, Pathological and Forensic Medical Research at Kyoto University for pathological analysis.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.05.013.
Supplemental Information
Reference
- 1.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park J.H., Rivière I., Gonen M., Wang X., Sénéchal B., Curran K.J., Sauter C., Wang Y., Santomasso B., Mead E. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018;378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davila M.L., Riviere I., Wang X., Bartido S., Park J., Curran K., Chung S.S., Stefanski J., Borquez-Ojeda O., Olszewska M. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee D.W., Kochenderfer J.N., Stetler-Stevenson M., Cui Y.K., Delbrook C., Feldman S.A., Fry T.J., Orentas R., Sabatino M., Shah N.N. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gardner R.A., Finney O., Annesley C., Brakke H., Summers C., Leger K., Bleakley M., Brown C., Mgebroff S., Kelly-Spratt K.S. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–3331. doi: 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bollard C.M., Barrett A.J. Cytotoxic T lymphocytes for leukemia and lymphoma. Hematology (Am. Soc. Hematol. Educ. Program) 2014;2014:565–569. doi: 10.1182/asheducation-2014.1.565. [DOI] [PubMed] [Google Scholar]
- 7.Abbott N.J., Patabendige A.A., Dolman D.E., Yusof S.R., Begley D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 8.Kato I., Nishinaka Y., Nakamura M., Akarca A.U., Niwa A., Ozawa H., Yoshida K., Mori M., Wang D., Morita M. Hypoxic adaptation of leukemic cells infiltrating the CNS affords a therapeutic strategy targeting VEGFA. Blood. 2017;129:3126–3129. doi: 10.1182/blood-2016-06-721712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Priceman S.J., Tilakawardane D., Jeang B., Aguilar B., Murad J.P., Park A.K., Chang W.C., Ostberg J.R., Neman J., Jandial R. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2+ Breast Cancer Metastasis to the Brain. Clin. Cancer Res. 2018;24:95–105. doi: 10.1158/1078-0432.CCR-17-2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nellan A., Rota C., Majzner R., Lester-McCully C.M., Griesinger A.M., Mulcahy Levy J.M., Foreman N.K., Warren K.E., Lee D.W. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J. Immunother. Cancer. 2018;6:30. doi: 10.1186/s40425-018-0340-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yanagisawa R., Nakazawa Y., Sakashita K., Saito S., Tanaka M., Shiohara M., Shimodaira S., Koike K. Intrathecal donor lymphocyte infusion for isolated leukemia relapse in the central nervous system following allogeneic stem cell transplantation: a case report and literature review. Int. J. Hematol. 2016;103:107–111. doi: 10.1007/s12185-015-1902-1. [DOI] [PubMed] [Google Scholar]
- 12.Hirase C., Maeda Y., Takai S., Kanamaru A. Hypersensitivity of Ph-positive lymphoid cell lines to rapamycin: Possible clinical application of mTOR inhibitor. Leuk. Res. 2009;33:450–459. doi: 10.1016/j.leukres.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 13.Price R.A., Johnson W.W. The central nervous system in childhood leukemia. I. The arachnoid. Cancer. 1973;31:520–533. doi: 10.1002/1097-0142(197303)31:3<520::aid-cncr2820310306>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 14.Hagedorn N., Acquaviva C., Fronkova E., von Stackelberg A., Barth A., zur Stadt U., Schrauder A., Trka J., Gaspar N., Seeger K., Resistant Disease Committee of the International BFM study group Submicroscopic bone marrow involvement in isolated extramedullary relapses in childhood acute lymphoblastic leukemia: a more precise definition of “isolated” and its possible clinical implications, a collaborative study of the Resistant Disease Committee of the International BFM study group. Blood. 2007;110:4022–4029. doi: 10.1182/blood-2007-04-082040. [DOI] [PubMed] [Google Scholar]
- 15.Saito S., Nakazawa Y., Sueki A., Matsuda K., Tanaka M., Yanagisawa R., Maeda Y., Sato Y., Okabe S., Inukai T. Anti-leukemic potency of piggyBac-mediated CD19-specific T cells against refractory Philadelphia chromosome-positive acute lymphoblastic leukemia. Cytotherapy. 2014;16:1257–1269. doi: 10.1016/j.jcyt.2014.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morita D., Nishio N., Saito S., Tanaka M., Kawashima N., Okuno Y., Suzuki S., Matsuda K., Maeda Y., Wilson M.H. Enhanced Expression of Anti-CD19 Chimeric Antigen Receptor in piggyBac Transposon-Engineered T Cells. Mol. Ther. Methods Clin. Dev. 2017;8:131–140. doi: 10.1016/j.omtm.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh A., Smith M., James S.E., Davila M.L., Velardi E., Argyropoulos K.V., Gunset G., Perna F., Kreines F.M., Levy E.R. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat. Med. 2017;23:242–249. doi: 10.1038/nm.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gust J., Hay K.A., Hanafi L.A., Li D., Myerson D., Gonzalez-Cuyar L.F., Yeung C., Liles W.C., Wurfel M., Lopez J.A. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017;7:1404–1419. doi: 10.1158/2159-8290.CD-17-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santomasso B.D., Park J.H., Salloum D., Riviere I., Flynn J., Mead E., Halton E., Wang X., Senechal B., Purdon T. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018;8:958–971. doi: 10.1158/2159-8290.CD-17-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mook-Kanamori B., Geldhoff M., Troost D., van der Poll T., van de Beek D. Characterization of a pneumococcal meningitis mouse model. BMC Infect. Dis. 2012;12:71. doi: 10.1186/1471-2334-12-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mount C.W., Majzner R.G., Sundaresh S., Arnold E.P., Kadapakkam M., Haile S., Labanieh L., Hulleman E., Woo P.J., Rietberg S.P. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat. Med. 2018;24:572–579. doi: 10.1038/s41591-018-0006-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teachey D.T., Lacey S.F., Shaw P.A., Melenhorst J.J., Maude S.L., Frey N., Pequignot E., Gonzalez V.E., Chen F., Finklestein J. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016;6:664–679. doi: 10.1158/2159-8290.CD-16-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hay K.A., Hanafi L.A., Li D., Gust J., Liles W.C., Wurfel M.M., López J.A., Chen J., Chung D., Harju-Baker S. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. 2017;130:2295–2306. doi: 10.1182/blood-2017-06-793141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen K., Devidas M., Cheng S.C., La M., Raetz E.A., Carroll W.L., Winick N.J., Hunger S.P., Gaynon P.S., Loh M.L., Children’s Oncology Group Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia. 2008;22:2142–2150. doi: 10.1038/leu.2008.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pui C.H., Cheng C., Leung W., Rai S.N., Rivera G.K., Sandlund J.T., Ribeiro R.C., Relling M.V., Kun L.E., Evans W.E., Hudson M.M. Extended follow-up of long-term survivors of childhood acute lymphoblastic leukemia. N. Engl. J. Med. 2003;349:640–649. doi: 10.1056/NEJMoa035091. [DOI] [PubMed] [Google Scholar]
- 26.Hunger S.P., Lu X., Devidas M., Camitta B.M., Gaynon P.S., Winick N.J., Reaman G.H., Carroll W.L. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J. Clin. Oncol. 2012;30:1663–1669. doi: 10.1200/JCO.2011.37.8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aldoss I., Al Malki M.M., Stiller T., Cao T., Sanchez J.F., Palmer J., Forman S.J., Pullarkat V. Implications and Management of Central Nervous System Involvement before Allogeneic Hematopoietic Cell Transplantation in Acute Lymphoblastic Leukemia. Biol. Blood Marrow Transplant. 2016;22:575–578. doi: 10.1016/j.bbmt.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 28.Hamidieh A.A., Monzavi S.M., Kaboutari M., Behfar M., Esfandbod M. Outcome Analysis of Pediatric Patients with Acute Lymphoblastic Leukemia Treated with Total Body Irradiation-Free Allogeneic Hematopoietic Stem Cell Transplantation: Comparison of Patients with and Without Central Nervous System Involvement. Biol. Blood Marrow Transplant. 2017;23:2110–2117. doi: 10.1016/j.bbmt.2017.08.036. [DOI] [PubMed] [Google Scholar]
- 29.Laningham F.H., Kun L.E., Reddick W.E., Ogg R.J., Morris E.B., Pui C.H. Childhood central nervous system leukemia: historical perspectives, current therapy, and acute neurological sequelae. Neuroradiology. 2007;49:873–888. doi: 10.1007/s00234-007-0300-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krull K.R., Brinkman T.M., Li C., Armstrong G.T., Ness K.K., Srivastava D.K., Gurney J.G., Kimberg C., Krasin M.J., Pui C.H. Neurocognitive outcomes decades after treatment for childhood acute lymphoblastic leukemia: a report from the St Jude lifetime cohort study. J. Clin. Oncol. 2013;31:4407–4415. doi: 10.1200/JCO.2012.48.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mamonkin M., Rouce R.H., Tashiro H., Brenner M.K. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126:983–992. doi: 10.1182/blood-2015-02-629527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown C.E., Alizadeh D., Starr R., Weng L., Wagner J.R., Naranjo A., Ostberg J.R., Blanchard M.S., Kilpatrick J., Simpson J. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahmed N., Brawley V., Hegde M., Bielamowicz K., Kalra M., Landi D., Robertson C., Gray T.L., Diouf O., Wakefield A. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017;3:1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kasahara K., Fukunaga Y., Igura S., Andoh R., Saito T., Suzuki I., Kanemitsu H., Suzuki D., Goto K., Nakamura D. Background data on NOD/Shi-scid IL-2Rγnull mice (NOG mice) J. Toxicol. Sci. 2017;42:689–705. doi: 10.2131/jts.42.689. [DOI] [PubMed] [Google Scholar]
- 35.van Rijn R.S., Simonetti E.R., Hagenbeek A., Hogenes M.C., de Weger R.A., Canninga-van Dijk M.R., Weijer K., Spits H., Storm G., van Bloois L. A new xenograft model for graft-versus-host disease by intravenous transfer of human peripheral blood mononuclear cells in RAG2-/- gammac-/- double-mutant mice. Blood. 2003;102:2522–2531. doi: 10.1182/blood-2002-10-3241. [DOI] [PubMed] [Google Scholar]
- 36.Smith J.S., Angel T.E., Chavkin C., Orton D.J., Moore R.J., Smith R.D. Characterization of individual mouse cerebrospinal fluid proteomes. Proteomics. 2014;14:1102–1106. doi: 10.1002/pmic.201300241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kato I., Niwa A., Heike T., Fujino H., Saito M.K., Umeda K., Hiramatsu H., Ito M., Morita M., Nishinaka Y. Identification of hepatic niche harboring human acute lymphoblastic leukemic cells via the SDF-1/CXCR4 axis. PLoS ONE. 2011;6:e27042. doi: 10.1371/journal.pone.0027042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jena B., Maiti S., Huls H., Singh H., Lee D.A., Champlin R.E., Cooper L.J. Chimeric antigen receptor (CAR)-specific monoclonal antibody to detect CD19-specific T cells in clinical trials. PLoS ONE. 2013;8:e57838. doi: 10.1371/journal.pone.0057838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li S., Zhang J., Wang M., Fu G., Li Y., Pei L., Xiong Z., Qin D., Zhang R., Tian X. Treatment of acute lymphoblastic leukaemia with the second generation of CD19 CAR-T containing either CD28 or 4-1BB. Br. J. Haematol. 2018;181:360–371. doi: 10.1111/bjh.15195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.