

Abstract
Background:
Small cell neuroendocrine carcinoma (SCNC) of the prostate is an aggressive subtype with frequent TP53 mutation and RB1 inactivation, however the molecular phenotype remains an area of investigation. Here, we compared telomere lengths in prostatic SCNC and usual-type prostatic adenocarcinoma (AdCa).
Methods:
We studied 32 cases of prostatic SCNC (including 11 cases with concurrent AdCa) and 347 cases of usual-type AdCa on tissue microarrays. Telomere lengths in tumor cells were qualitatively compared to that in normal cells using a telomere-specific fluorescence in situ hybridization assay. ERG, PTEN and TP53 status were assessed in a proportion of cases using genetically validated immunohistochemistry protocols. Clinical-pathologic and molecular characteristics of cases were compared between telomere groups using the chi-square test.
Results:
A significantly higher proportion of prostatic SCNC cases (50%, 16/32) showed normal/long telomeres compared to AdCa cases (11%, 39/347; p<0.0001). In 82% (9/11) of cases with concurrent SCNC and AdCa, the paired components were concordant for telomere length status. Among AdCa cases, the proportion of cases with normal/long telomeres significantly increased with increasing tumor Grade Group (p=0.01) and pathologic stage (p=0.02). Cases with normal/long telomeres were more likely to be ERG positive (p=0.04) and to have TP53 missense mutation (p=0.01) compared to cases with short telomeres.
Conclusions:
Normal or long telomere lengths are significantly more common in prostatic SCNC compared to AdCa and are similar between concurrent SCNC and AdCa tumors supporting a common origin. Among AdCa cases, longer telomere lengths are significantly associated with high risk pathologic and molecular features.
Keywords: Telomeres, prostate cancer, neuroendocrine, adenocarcinoma, small cell carcinoma
1. INTRODUCTION
Telomeres are repetitive DNA elements located at chromosomal ends that are essential for maintenance of genomic integrity (1, 2). With each cell division, telomeres shorten due to incomplete DNA replication on the lagging strand (3). Cancer cells often have critically shortened telomeres compared to surrounding normal cells, likely due to their high proliferative rate, and these critically short telomeres promote genomic instability (4, 5). Most tumors are able to activate telomerase to stabilize telomeric ends, enabling the capacity for unlimited replication (6), while others undergo telomerase-independent telomere lengthening by the recombination-based mechanism called alternative lengthening of telomeres (ALT) (7, 8). In prostate cancer, telomere shortening is common in precursor and invasive prostatic adenocarcinomas, and measures of both tumor cell and stromal cell telomere length are associated with risk of death from prostate cancer (9, 10). Using a robust telomere-specific fluorescence in situ hybridization (FISH) assay that provides relative telomere lengths on a per cell basis, we previously observed that men with more variable telomere lengths in their cancer cells (in addition to shorter stromal cell telomeres) had a significantly increased risk of prostate cancer death compared to men with less variable cancer telomeres (and longer stromal telomeres) (11). To date, however, the histologic and molecular correlates of telomere length or telomere length variability in prostate cancer remain unclear.
Small cell neuroendocrine prostate cancer (SCNC) is a rare but extremely aggressive histologic subtype in which the median survival is of around 6 months (12). SCNC is characterized by distinct morphologic features, an extremely high proliferative rate compared to usual type prostatic adenocarcinoma (AdCa), with increased expression of neuroendocrine and neural associated genes and loss of androgen receptor (AR) expression and downstream signaling in many cases (13, 14). Molecularly, prostatic SCNC is similar to lung SCNC, with frequent inactivation of the RB1 and TP53 tumor suppressors (15–18). In the prostate, SCNC occurs only rarely in the primary setting and more commonly arises due to lineage plasticity as a mechanism of resistance to androgen deprivation therapy (ADT) in metastatic disease (18, 19). Accordingly, SCNC may occur admixed with AdCa, and the two components are frequently clonally related (13, 20).
Given the unique clinical and molecular features of prostatic SCNC and its highly proliferative phenotype, we examined tumor cell telomere length in a cohort of SCNC cases, including a subset with concurrent adenocarcinoma. Next, we compared telomere lengths in SCNC to a large set of pure adenocarcinomas, enriched for high pathologic grade. We find that there is a significantly higher proportion of prostatic SCNC cases with normal or long telomeres compared to AdCa cases where telomeres are predominantly short, without evidence of ALT in any cases. Interestingly, among AdCa cases, the proportion of cases with normal or long telomeres increases with increasing Grade Group and frequency of TP53 missense mutation.
2. MATERIALS AND METHODS
2.1. Tissue samples
The study was approved by the Institutional Review Board at the Johns Hopkins University School of Medicine. Seven separate tissue microarray (TMA) sets were used, including one for SCNC (radical prostatectomy, needle biopsy and transurethral resection of the prostate [TURP] specimens) which has been previously described (n=32 with informative telomere data) (13, 15, 21), and six sets of surgically treated AdCa cases from radical prostatectomy specimens, also previously described. SCNC cases were retrieved from the surgical pathology and consultation files of the Johns Hopkins Hospitals as previously described in detail (15) and were generally diagnosed by established morphologic criteria (12): high nuclear-cytoplasmic ratios, with nuclear molding, indistinct nucleoli, nuclear crush artifact and visibly high proliferation/apoptotic rate. As previously described (15), the majority of these cases were transurethral resections of the prostate (TURP) samples, with a minority derived from contiguous site biopsies (bladder, rectum) or radical prostatectomy specimens; none were metastatic samples. Prostatic origin was documented for the majority of cases using the following criteria: a concurrent or prior history of prostatic acinar carcinoma by histologic diagnosis, or positive PSA (prostate specific antigen) immunostaining. Given that most cases were derived from consultations, it was not clear how many represented de novo SCNC vs. treatment-related SCNC (15). AdCa TMA sets were selected to increase the diversity of pathologic Grade Groups analyzed, with a particular focus on higher Grade Groups to enable better comparison between the AdCa and SCNC cases. AdCa TMA sets included a cohort of patients without germline mutations in HOXB13 (n=94) (22), a cohort of European-American patients with Grade Group 3 or higher (n=76) (23), a cohort of tumors enriched for cribriform pattern 4 cancer (n=13) (24), a cohort of patients with Grade Group 5 cancer (n=70) (25), a cohort of patients with metastatic disease who were subsequently treated with abiraterone and enzalutamide (n=50) (26), and cohort of patients with cancers ranging from Grade Groups 1–4 (n=72).
LuCaP xenografts (unrelated to cases described above) were grown as previously described (27), including 36 AdCa lines and 6 neuroendocrine prostate cancer lines (#49, 77CR, 93, 145.1, 145.2 and 173.1). Of the neuroendocrine lines, one (#49) was cultured from a patient with SCNC (28), while others were classified based in part on SCNC morphology and on gene expression/immunohistochemistry analyses (27).
2.2. Telomere-specific FISH
Telomere lengths were assessed by fluorescence staining for telomeric DNA. Briefly, deparaffinized slides were hybridized with a Cy3-labeled peptide nucleic acid (PNA) probe complementary to the mammalian telomere repeat sequence [(N-terminus to C-terminus) CCCTAACCCTAACCCTAA]. As a positive control for hybridization efficiency, an Alexa Fluor 488-labeled PNA probe with specificity for centromeric DNA repeats (CENP-B–binding sequence; ATTCGTTGGAAACGGGA) was included in the hybridization solution. Slides were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) for labeling total nuclear DNA as previously described (10, 11) and mounted with Prolong anti-fade mounting medium.
As previously described (29, 30), telomere lengths were qualitatively scored in cancer cells on TMA spots by direct visual assessment of the stained slides, comparing telomere signals from tumor cells to telomere signals from normal cells (normal epithelial cells and/or stromal cells) from the same case. In all cases, signals from normal cells were considered 3+. Telomeres in the tumor cells of different cases ranged from short (1+, 2+) to normal (3+) to long (4+, 5+).
2.3. Telomere Q-PCR
Extracted DNA from LuCaP xenografts was re-purified to remove potential residual PCR inhibitors using the DNeasy Blood and Tissue kit (Qiagen). Quantitative PCR was used to estimate the ratio of telomeric DNA to that of a single copy gene (β-globin) as previously described (31) with modification (32).
2.4. Assessment of TERT and TERC expression by RT-qPCR
Extracted RNA from LuCaP xenografts was reverse transcribed using 1μg of RNA using the iScript Advanced cDNA synthesis kit (Bio-Rad). As previously described (33), RT-qPCR was performed on the equivalent of 50 ng of cDNA using iTaq Universal SYBR Green Supermix (Bio-Rad). TERT and TERC expression was normalized to the expression of the reference gene, TBP.
2.5. ERG, PTEN and p53 immunohistochemistry
PTEN and ERG IHC were performed on all TMAs using automated assays as previously described (22–26, 34). We have previously published that these assays are highly sensitive and specific for the presence of underlying PTEN gene deletion and ERG gene rearrangement by gold-standard fluorescence in situ hybridization FISH (35, 36). Briefly, the PTEN protocol uses the Ventana automated staining platform (Ventana Discovery Ultra, Ventana Medical Systems, Tucson, AZ) and a rabbit anti-human PTEN antibody (Clone D4.3 XP; Cell Signaling Technologies, Danvers, MA). The assay was blindly scored by a urologic pathologist (TLL) using a genetically validated scoring system. A tumor biopsy was considered to have PTEN protein loss if the intensity of cytoplasmic and nuclear staining for PTEN was markedly decreased or entirely negative across >10% of tumor cells compared to surrounding benign glands and/or stroma, which provide internal positive controls for PTEN protein expression. If the tumor showed PTEN protein expressed in >90% of sampled tumor glands, the tumor was scored as PTEN intact. If PTEN was lost in <100% of the tumor cells sampled in a given core, the core was annotated as showing heterogeneous PTEN loss in some, but not all, cancer glands (focal loss). Alternatively, if the core showed PTEN loss in 100% of sampled tumor glands, the core was annotated as showing homogeneous PTEN loss. Finally, a small percentage of cores were scored as having ambiguous PTEN IHC results. This occurred when the intensity of the tumor cell staining was light or absent in the absence of evaluable internal benign glands or stromal staining.
ERG immunohistochemistry was performed on the Ventana Benchmark autostaining system using a rabbit monoclonal antibody (EPR 3864) after antigen retrieval in CC1 buffer followed by detection with the Optiview HRP system (Roche/Ventana Medical Systems, Tucson, AZ). Each TMA spot containing tumor was visually dichotomously scored for presence or absence of nuclear ERG signal by a urologic pathologist blinded to the gene expression data (TLL). A spot was considered to be ERG-positive if any tumor nuclei showed ERG positivity, utilizing endothelial cells as an internal positive control in all cases. A tumor was considered ERG positive if any sampled spots were scored as ERG positive, and as ERG negative if all sampled spots were scored as ERG negative.
p53 IHC was performed on the Ventana Benchmark autostaining system using a mouse monoclonal antibody (BP53–11) after antigen retrieval in CC1 buffer followed by detection with the iView HRP system (Roche/Ventana Medical Systems, Tucson, AZ). Each tissue microarray spot containing tumor cells was visually dichotomously scored for presence or absence of nuclear p53 accumulation by a urologic pathologist (TLL). A spot was considered to show p53 nuclear accumulation if >10% of tumor nuclei showed p53 positivity. A tumor was considered to show p53 nuclear accumulation if any sampled spot was scored as p53-positive, and as lacking p53 nuclear accumulation if all sampled spots were scored as p53 negative. Nuclear accumulation of p53 by this assay is more than 90% sensitive and specific for the presence of an underlying missense mutation in TP53, which generally stabilizes the protein (26, 37). Notably, this assay does not detect other loss-of-function alteration in TP53, such as frame shift mutations, splice site mutation or homozygous deletions.
2.6. Statistical analysis
For statistical analyses, the normal and long telomere groups were combined and compared to the short telomere group for a dichotomized scoring system. Medians and proportions for clinical-pathologic and molecular characteristics were calculated by sample set (AdCa vs. SCNC) stratified by dichotomized telomere status (short vs. normal/long). Chi-square test was used to compare proportions. Kaplan-Meier curves were used to visualize time-to-event data. All statistical tests were two-sided, with p<0.05 considered to be statistically significant.
3. RESULTS
Using a robust telomere FISH assay, telomere lengths in tumor cells were qualitatively compared to that in adjacent benign cells. Initially, we assessed 37 cases of prostatic SCNC. Telomere length was evaluable in 86% (32/37) of cases. Of these, 50% (16/32) harbored normal/long telomeres (Figure 1A,C). In contrast to a subset (3/13; 23%) of SCNC arising in the bladder (38), none of these prostatic SCNC cases displayed ultra-bright telomeric foci indicative of ALT. Additionally, there were no discernable associations with previously reported RB1, p53, PTEN, or ERG status in these tumors (Table 1) (15).
Figure 1. Telomere length analysis by FISH.
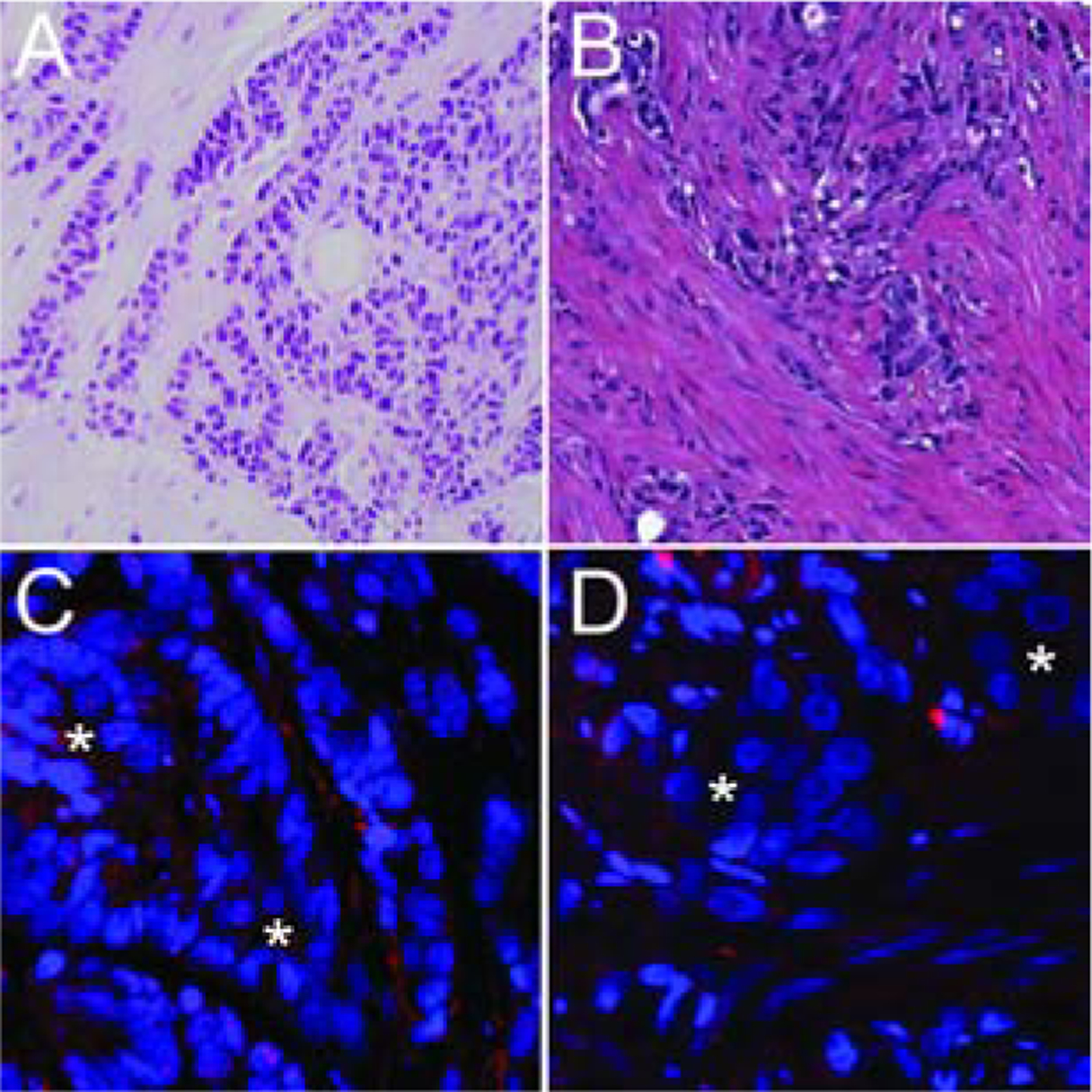
(A, C) A small cell carcinoma case shows extremely bright telomere signals (indicative of longer telomeres) in the cancer cells (*) when compared to the surrounding benign stroma. (B, D) In contrast, this representative adenocarcinoma case shows greatly reduced telomere signals in the cancer cells (*) as compared to the surrounding benign stroma. The centromere DNA, stained with the FITC-labeled centromere-specific peptide nucleic acid probe, has been omitted from the image to emphasize the differences in telomere lengths. Original magnification × 400.
Table 1.
Telomere lengths and molecular status in small cell neuroendocrine carcinomas of the prostate.
| Case # | Telomere Length | RB1 | P53 | PTEN | ERG |
|---|---|---|---|---|---|
| 1 | Long | Neg | Pos | Intact | Neg |
| 2 | Long | Neg | Pos | Intact | Neg |
| 3 | Long | Neg | Pos | Intact | Neg |
| 4 | Long | Neg | Neg | Lost | Neg |
| 5 | Long | Neg | Pos | Lost | Pos |
| 6 | Long | Neg | Pos | Lost | Neg |
| 7 | Long | Neg | Pos | Lost | Neg |
| 8 | Long | Neg | Pos | Lost | Neg |
| 9 | Long | Pos | N/A | Intact | N/A |
| 10 | Long | Pos | Pos | Intact | Neg |
| 11 | Normal | Neg | Neg | Intact | Neg |
| 12 | Normal | Neg | Neg | Intact | Neg |
| 13 | Normal | Neg | Pos | Intact | Pos |
| 14 | Normal | Neg | Neg | Lost | Neg |
| 15 | Normal | Neg | Pos | Lost | Neg |
| 16 | Normal | Pos | Neg | Lost | Pos |
| 17 | Short | Neg | Neg | Intact | N/A |
| 18 | Short | Neg | Neg | Intact | Neg |
| 19 | Short | Neg | Neg | Intact | Pos |
| 20 | Short | Neg | Neg | Intact | Neg |
| 21 | Short | Neg | Pos | Intact | Neg |
| 22 | Short | Neg | Pos | Intact | Neg |
| 23 | Short | Neg | Neg | Lost | Neg |
| 24 | Short | Neg | Neg | Lost | Neg |
| 25 | Short | Neg | Neg | Lost | Neg |
| 26 | Short | Neg | Neg | Lost | Neg |
| 27 | Short | Neg | Pos | Lost | Neg |
| 28 | Short | Neg | Pos | Lost | N/A |
| 29 | Short | Neg | Pos | Lost | N/A |
| 30 | Short | Neg | Pos | Lost | N/A |
| 31 | Short | Neg | Pos | Lost | Neg |
| 32 | Short | Pos | Neg | Intact | Pos |
In some cases of prostatic SCNC, the tumor likely arises from transdifferentiation of an adenocarcinoma (AdCa) occurring in the same patient (39). In many cases, both tumor components (SCNC and AdCa) are sampled and we and others have previously shown that they are frequently clonally related based on ERG rearrangement and other molecular markers (13, 20, 40). Thus, we examined telomere length in 11 evaluable cases of concurrent SCNC and AdCa, including 7 with normal or long telomere length, and comprising a subset of the total 32 SCNC cases. Strikingly, there was concordance of telomere length for the SCNC and AdCa components in 82% (9/11) of cases, supporting the notion that these two components share a common origin and molecular phenotype in many cases (Figure 2, Table 2).
Figure 2. Telomere-specific FISH in a case with small cell carcinoma and adenocarcinoma components.
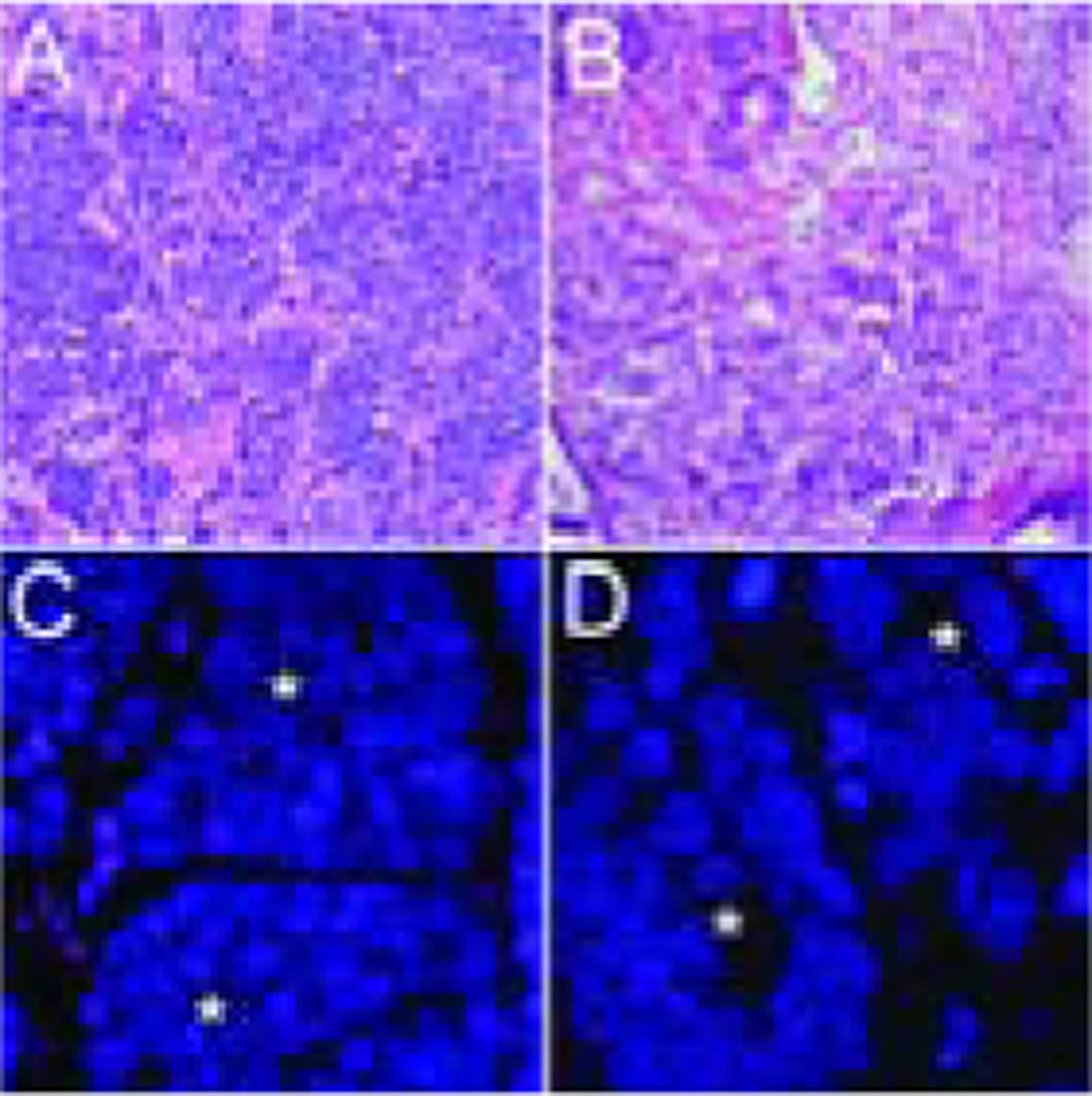
In a representative case, the telomere signals are robust in the cancer cells (*) from both the (A, C) small cell carcinoma and the (B, D) concurrent adenocarcinoma. In all images, the DNA is stained with DAPI (blue) and telomere DNA is stained with the Cy3-labeled telomere-specific PNA probe (red). The centromere DNA, stained with the FITC-labeled centromere-specific peptide nucleic acid probe, has been omitted from the image to emphasize the differences in telomere lengths. Original magnification × 400.
Table 2.
Telomere lengths in cases that contained small cell neuroendocrine carcinomas with concurrent adenocarcinoma.
| Telomere Lengths | ||||||
|---|---|---|---|---|---|---|
| Very Short | Short | Normal | Long Very | Long | ||
| Case 23 | SCNC | |||||
| Case 13 | SCNC | |||||
| Case 1 | SCNC | |||||
| Case 25 | SCNC | |||||
| Case 5 | SCNC | |||||
| Case 22 | SCNC | |||||
| Case 31 | SCNC | |||||
| Case 3 | SCNC | |||||
| Case 20 | SCNC | |||||
| Case 8 | SCNC | |||||
| Case 12 | SCNC | |||||
Next, for comparison to the prostatic SCNC cases, we studied cancer cell telomere lengths in prostatic AdCa cases at radical prostatectomy using previously constructed tissue microarray (TMA) sets, many of which were designed to enrich for cases with adverse pathologic features and outcomes. Of 361 unique cases present in these TMA sets, 96% (347/361) were evaluable for telomere lengths. A significantly lower proportion of AdCa cases (11%, 39/347; p<0.0001) displayed normal/long telomere lengths compared to SCNC cases (50%, 16/32) (Figure 1B, D). However, the prevalence of normal/long telomeres in AdCa cases varied significantly by Grade Group (GG), with only 4% (3/81) of GG1 tumors showing normal/long telomeres compared to 19% (20/106) of GG5 tumors (p=0.01) (Table 3, Figure 3). A similar association was seen for increasing pathologic stage among AdCa cases (p=0.02). Interestingly, we also observed an association of telomere length with self-identified race, with 24% (7/29) of non-white patients harboring tumors with normal/long telomeres, compared to 10% (32/313) of white patients (p=0.02).
Table 3.
Clinical-Pathologic and Molecular Characteristics of Adenocarcinoma Cases by Telomere Status
| Short telomeres | Normal/Long telomeres | p value | |
|---|---|---|---|
| Clinical characteristics | |||
| Median age, Years | 59 | 59 | |
| Race, N (%) | |||
| White | 281 (92.7) | 32 (82.1) | 0.02 |
| Non-White | 22 (7.3) | 7 (17.9) | |
| Gleason score, N (%) | |||
| 6 (Grade Group 1) | 78 (25.3) | 3 (7.7) | 0.01 |
| 3+4 (Grade Group 2) | 42 (13.6) | 2 (5.1) | |
| 4+3 (Grade Group 3) | 71 (23.1) | 9 (23.1) | |
| 8 (Grade Group 4) | 31 (10.1) | 5 (12.8) | |
| 9 and 10 (Grade Group 5) | 86 (27.9) | 20 (51.3) | |
| Stage, N (%) | |||
| T2 | 133 (44.2) | 8 (20.5) | 0.02 |
| T3a | 90 (29.9) | 14 (36.0) | |
| T3b | 51 (16.9) | 8 (20.5) | |
| N1 | 33 (11.0) | 9 (23.1) | |
| Molecular characteristics | |||
| ERG, N (%) | |||
| Negative | 157 (58.1) | 14 (40.0) | 0.04 |
| Positive | 113 (41.9) | 21 (60.0) | |
| PTEN, N (%) | |||
| Intact | 171 (63.3) | 19 (54.3) | 0.30 |
| Loss | 99 (36.7) | 16 (45.7) | |
| P53, N (%) | |||
| Nuclear accumulation absent | 161 (86.1) | 18 (66.7) | 0.01 |
| Nuclear accumulation present | 26 (13.9) | 9 (33.3) | |
Figure 3. Telomere length status by histologic diagnosis.
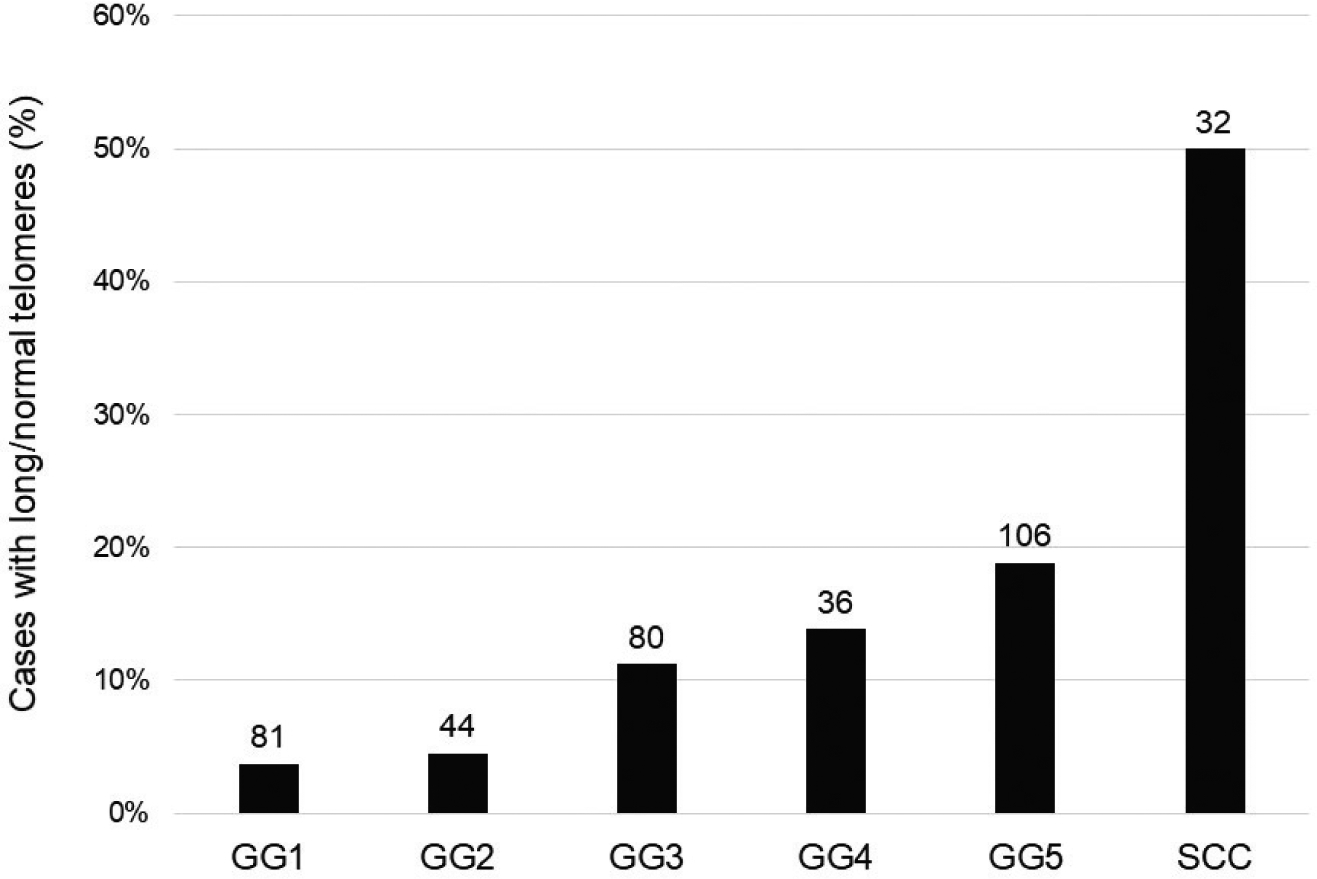
There is a step-wise increase in the prevalence of normal/long telomere lengths among adenocarcinoma cases with increasing Grade Group category. Prostatic small cell carcinoma cases show the highest prevalence of normal/long telomere lengths. The numbers over each bar represent the total number of cases examined for each category.
Next, we examined association of telomere length with other genomic alterations in the AdCa cohorts. Data for ERG, PTEN and p53 status, using genetically validated and previously reported immunohistochemistry (IHC) assays (22, 23, 25, 26, 34), were available for the majority of the AdCa cases with telomere length data (Table 3, Figure 4). Compared to cases with short telomeres, cases with normal/long telomeres were more likely to be ERG positive (60% or 21/35 vs 42% or 113/270; p=0.04) and to have p53 nuclear accumulation signifying underlying TP53 missense mutation (33% or 9/27 vs. 14% or 26/187; p=0.01). Of note, in a rare AdCa case where there was distinctive heterogeneity of telomere length within the sampled tumor, this heterogeneity seemed to correlate with apparent subclonal tumor gland populations based on PTEN status (Figure 4). In order to begin to assess whether telomere lengths were associated with telomerase levels, we also measured TERT and TERC gene expression in a set of previously described patient-derived xenografts (LuCaP xenografts), a small subset of which were classified as neuroendocrine prostate cancer (NEPC) based on morphology and immunohistochemical staining (41). However, we did not observe any associations between telomere content (as measured by telomere-specific Q-PCR) and TERT or TERC gene expression level (Supplemental Table S1). Taken together, these data suggest that cancer cell telomere lengths may be associated with underlying molecular phenotypes in prostatic AdCa, though there does not appear to be a clear association with telomerase gene expression in xenograft models.
Figure 4. Heterogeneity in telomere lengths and ERG/PTEN status in prostatic adenocarcinoma.
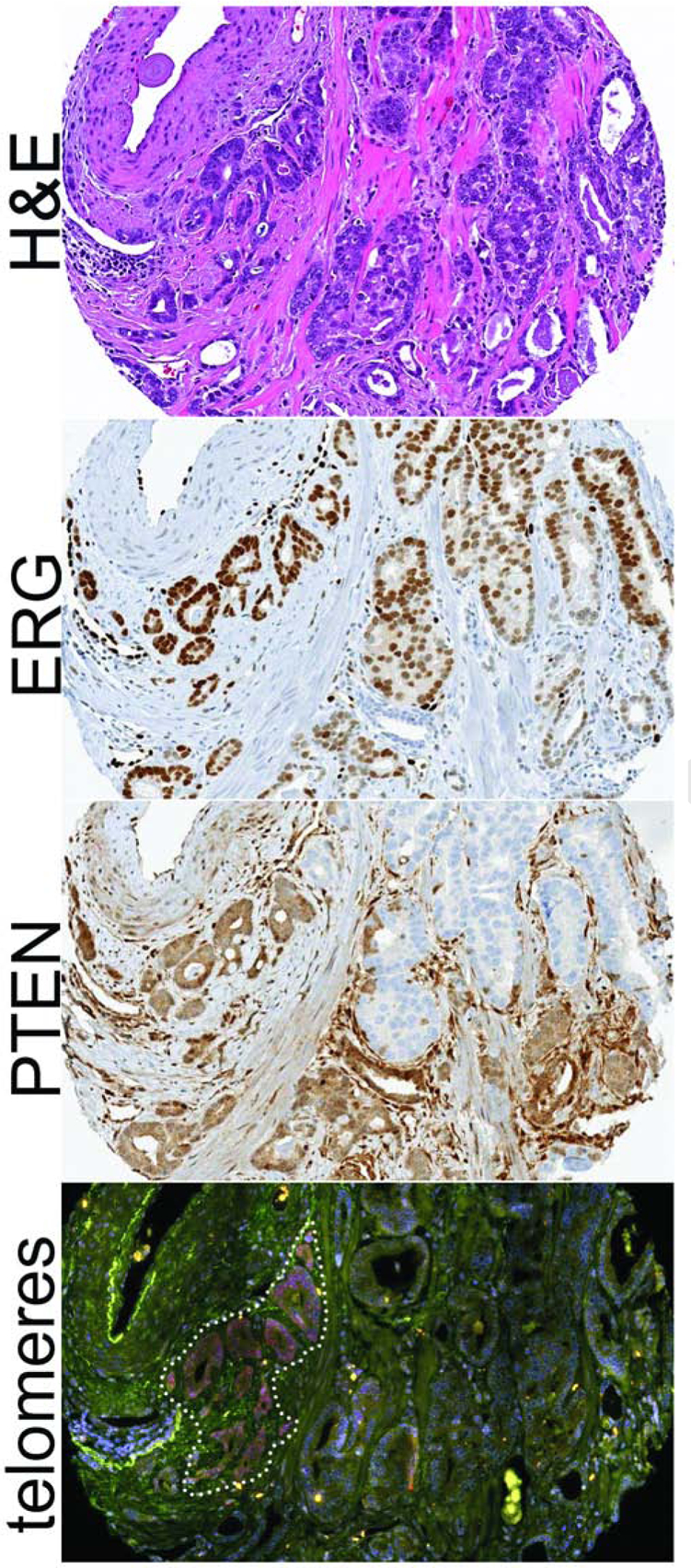
In this rare adenocarcinoma case with dramatic telomere length heterogeneity, the telomere signals are robust, thereby marking the relatively longer telomeres, in a distinct subset of tumor glands (as outlined on the bottom panel). While the entire tumor expresses ERG (signifying a clonal ERG gene rearrangement in all tumor cells), PTEN is lost in a subset of tumor cells (signifying subclonal underlying PTEN genomic inactivation). The subset of tumor cells with intact PTEN appears to exclusively overlap with the subclone showing extremely long telomeres. In the bottom panel, the DNA is stained with DAPI (blue), telomere DNA is stained with the Cy3-labeled telomere-specific PNA probe (red), and centromere DNA is stained with an Alexa 488-labeled centromere-specific PNA probe (green). Original magnification × 200.
Next, we assessed the association between cancer cell telomere length category and biochemical recurrence-free survival among a cohort of 54 surgically-treated and molecularly characterized GG5 AdCa cases with primary Gleason pattern 5 (Gleason Score 5+4=9 and 5+5=10) (42). While no apparent association was observed, because of the small cohort size and retrospective nature of these data, more definitive conclusions could not be derived (Supplemental Figure S1). Finally, since these cases had previously reported targeted sequencing data available, we also examined whether telomere length was associated with a specific mutational profile, however there was no readily apparent correlation between cancer cell telomere lengths and common genomic alterations in this cohort, such as DNA damage repair gene deficiency (Supplemental Table S2).
4. DISCUSSION
Telomeres consist of a highly conserved, repetitive hexanucleotide sequence (TTAGGGn) that are bound by the shelterin protein complex to stabilize eukaryotic chromosomal ends. Since telomeres shorten with each cell division, they play an integral role in ensuring finite cellular replicative capacity. In normal cells, with intact cell cycle checkpoint proteins such as p53 and RB1, progressive telomere shortening induces cellular senescence or apoptosis (2, 43). However, abrogation of these checkpoints – which frequently occurs in cancer cells – leads to aberrant cell proliferation and ultimately results in critically shortened telomeres. This, in turn, can compromise genomic integrity by driving additional somatic copy number alterations, aneuploidy, and DNA rearrangements. Along these lines, previous studies of prostate cancer have shown significant telomere shortening in cancer cells compared with normal epithelial cells (9, 44). This appears to be an early event, occurring even in prostatic intraepithelial neoplasia (PIN), the presumed precursor lesion to prostatic adenocarcinoma (9, 45). However, even in the absence of intact cell cycle checkpoints, critically short telomeres will ultimately limit the number of viable cell divisions leading to genomic catastrophe. Accordingly, cancer cells must activate a telomere maintenance mechanism, either through upregulation of telomerase or by the telomerase-independent mechanism, ALT. In mouse tumor models driven by transgenic oncogene expression, such as Myc or KrasG12D, it is notable that animals with longer telomeres at baseline develop more aggressive tumors (46, 47).
In this study, we found that small cell neuroendocrine prostate cancer – the most aggressive histologic variant of the disease – very commonly harbors normal or even long cancer cell telomeres compared to the surrounding stromal cells. While short telomeres in cancer cells are the norm in prostatic adenocarcinomas, occurring in nearly 90% of cases we studied, only half of small cell carcinomas (SCNC) had short telomeres. Because there is no evidence of ALT in prostatic SCNC (38), these data suggest that prostatic SCNC reactivates telomerase, perhaps early during its progression, thereby maintaining telomere length and supporting the high proliferation rate that is characteristic of these tumors. While we did not observe associations between TERT expression, telomere content, and cancer type (NEPC vs AdCa) in the patient-derived xenografts, Lung SCNC, which shares many genomic and clinical-pathologic features with prostatic SCNC, has been shown to have increased telomerase expression and activity when compared to non-SCNC lung cancers or lower grade neuroendocrine carcinomas of the lung (48–52). Notably, TERT promoter mutations are not seen in lung SCNC (53), thus the mechanism of this increase in telomerase expression and activity in lung SCNC remains to be elucidated.
In the current study, we found that telomere lengths are generally similar between SCNC and concurrently occurring AdCa tumors in the prostate, supporting the notion that they share a common origin and suggesting that there may be underlying clonal genomic alterations associated with the telomere length phenotype. Consistent with this hypothesis, we found that some aggressive subsets of adenocarcinoma – such as those with very high-grade or pathologic stage – are highly enriched for normal/long cancer cell telomere lengths. Tumors with ERG gene rearrangement or TP53 mutation also were more likely to have normal/long telomeres compared to those without these alterations. The association with ERG is puzzling, especially given that ERG expression is not associated with aggressive prostate cancer (54). Future studies are necessary to substantiate this association and investigate possible mechanisms for this effect. The association of TP53 mutations with telomere length has been previously reported in other tumor types (55) and may be due in part to a direct or indirect effect of TP53 inactivation on telomerase expression (56–58). This is a particularly intriguing hypothesis given that prostatic SCNC has extremely high rates of TP53 loss and future studies may examine this potential mechanism in more detail. However, in prostate cancer, since TP53 mutation prevalence also increases with tumor grade (37), it is possible that the association between TP53 mutations and telomere length may be confounded by the underlying correlation of telomere length with Grade Group. Despite the association of longer cancer cell telomere lengths in adenocarcinomas with TP53 mutation and higher grade, we did not see evidence for an association of long telomere length with clinical outcomes, such as metastasis, independent of grade. In a cohort of very high-grade adenocarcinomas with previously reported clinical follow-up, stratification by telomere length category was not significantly associated with time to metastasis. However, it must be noted that our previous large study linking telomere alterations with progression to metastasis and prostate cancer death (11) specifically used quantitative image analysis to measure telomere length variability (cell-to-cell) among prostate cancer cells, not overall length per se, in combination with telomere lengths in prostate cancer–associated stromal cells. Finally, ALT has not been reported in primary prostatic adenocarcinoma (38), now including in prostatic SCNC, although we have identified a lethal, metastatic case that was associated with a clear clonal evolution in which a late developing subclone showed a genomic ATRX inversion (59).
In summary, we investigated cancer cell telomere lengths in a cohort of SCNC, including a subset with concurrent AdCa, as well as AdCa cohorts; all of which had extensive molecular phenotyping data. The fact that telomere lengths are similar between concurrent SCNC and AdCa tumors supports a common histogenesis for these two components. The finding that telomere lengths are longer in prostatic SCNC relative to most AdCa cases is unexpected and suggests the potential for early reactivation of telomerase in these tumors. Finally, we demonstrate that longer telomere lengths are significantly associated with high risk pathologic and molecular features in AdCa, thereby warranting further prospective studies.
Supplementary Material
Grant support:
This work is supported by a PCF Young Investigator Award (C. Heaphy), as well as the NIH/NCI Prostate SPORE P50CA58236 and the NCI Cancer Center Support Grant 5P30CA006973-52.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: None
REFERENCES
- 1.Blackburn EH. Structure and function of telomeres. Nature 1991;350:569–73. [DOI] [PubMed] [Google Scholar]
- 2.O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol 2010;11:171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levy MZ, Allsopp RC, Futcher AB, Greider CW, Harley CB. Telomere end-replication problem and cell aging. J Mol Biol 1992;225:951–60. [DOI] [PubMed] [Google Scholar]
- 4.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer 1997;33:787–91. [DOI] [PubMed] [Google Scholar]
- 5.Hackett JA, Feldser DM, Greider CW. Telomere dysfunction increases mutation rate and genomic instability. Cell 2001;106:275–86. [DOI] [PubMed] [Google Scholar]
- 6.Counter CM, Avilion AA, LeFeuvre CE, et al. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J 1992;11:1921–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunham MA, Neumann AA, Fasching CL, Reddel RR. Telomere maintenance by recombination in human cells. Nat Genet 2000;26:447–50. [DOI] [PubMed] [Google Scholar]
- 8.Henson JD, Neumann AA, Yeager TR, Reddel RR. Alternative lengthening of telomeres in mammalian cells. Oncogene 2002;21:598–610. [DOI] [PubMed] [Google Scholar]
- 9.Meeker AK, Hicks JL, Platz EA, et al. Telomere shortening is an early somatic DNA alteration in human prostate tumorigenesis. Cancer Res 2002;62:6405–9. [PubMed] [Google Scholar]
- 10.Heaphy CM, Gaonkar G, Peskoe SB, et al. Prostate stromal cell telomere shortening is associated with risk of prostate cancer in the placebo arm of the Prostate Cancer Prevention Trial. Prostate 2015;75:1160–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heaphy CM, Yoon GS, Peskoe SB, et al. Prostate cancer cell telomere length variability and stromal cell telomere length as prognostic markers for metastasis and death. Cancer Discov 2013;3:1130–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Epstein JI, Amin MB, Beltran H, et al. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am J Surg Pathol 2014;38:756–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lotan TL, Gupta NS, Wang W, et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod Pathol 2011;24:820–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W, Epstein JI. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am J Surg Pathol 2008;32:65–71. [DOI] [PubMed] [Google Scholar]
- 15.Tan HL, Sood A, Rahimi HA, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res 2014;20:890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tzelepi V, Zhang J, Lu JF, et al. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clinical cancer research: an official journal of the American Association for Cancer Research 2012;18:666–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ku SY, Rosario S, Wang Y, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017;355:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mu P, Zhang Z, Benelli M, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355:84–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin D, Wyatt AW, Xue H, et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer research 2014;74:1272–83. [DOI] [PubMed] [Google Scholar]
- 20.Hansel DE, Nakayama M, Luo J, et al. Shared TP53 gene mutation in morphologically and phenotypically distinct concurrent primary small cell neuroendocrine carcinoma and adenocarcinoma of the prostate. Prostate 2009;69:603–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai HK, Lehrer J, Alshalalfa M, et al. Gene expression signatures of neuroendocrine prostate cancer and primary small cell prostatic carcinoma. BMC cancer 2017;17:759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lotan TL, Torres A, Zhang M, et al. Somatic molecular subtyping of prostate tumors from HOXB13 G84E carriers. Oncotarget 2017;8:22772–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaur HB, Lu J, Guedes LB, et al. TP53 missense mutation is associated with increased tumor-infiltrating T-cells in primary prostate Cancer. Hum Pathol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herawi M, Epstein JI. Immunohistochemical antibody cocktail staining (p63/HMWCK/AMACR) of ductal adenocarcinoma and Gleason pattern 4 cribriform and noncribriform acinar adenocarcinomas of the prostate. Am J Surg Pathol 2007;31:889–94. [DOI] [PubMed] [Google Scholar]
- 25.Isaacsson Velho P, Lim D, Wang H, et al. Clinical Outcomes and molecular characterization of clinically localized very high risk (primary Gleason score of 5) prostate cancer after radical prostatectomy. Journal of Clinical Oncology precision medicine 2019, in press;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maughan BL, Guedes LB, Boucher K, et al. p53 status in the primary tumor predicts efficacy of subsequent abiraterone and enzalutamide in castration-resistant prostate cancer. Prostate Cancer Prostatic Dis 2018;21:260–8. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen HM, Vessella RL, Morrissey C, et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease an--d Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017;77:654–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.True LD, Buhler K, Quinn J, et al. A neuroendocrine/small cell prostate carcinoma xenograft-LuCaP 49. Am J Pathol 2002;161:705–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heaphy CM, Subhawong AP, Gross AL, et al. Shorter telomeres in luminal B, HER-2 and triple-negative breast cancer subtypes. Mod Pathol 2011;24:194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heaphy CM, Asch-Kendrick R, Argani P, Meeker AK, Cimino-Mathews A. Telomere length alterations unique to invasive lobular carcinoma. Hum Pathol 2015;46:1197–203. [DOI] [PubMed] [Google Scholar]
- 31.Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res 2002;30:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurwitz LM, Heaphy CM, Joshu CE, et al. Telomere length as a risk factor for hereditary prostate cancer. Prostate 2014;74:359–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graham MK, Kim J, Da J, et al. Functional Loss of ATRX and TERC Activates Alternative Lengthening of Telomeres (ALT) in LAPC4 Prostate Cancer Cells. Mol Cancer Res 2019;17:2480–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morais CL, Herawi M, Toubaji A, et al. PTEN loss and ERG protein expression are infrequent in prostatic ductal adenocarcinomas and concurrent acinar carcinomas. Prostate 2015;75:1610–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lotan TL, Wei W, Ludkovski O, et al. Analytic validation of a clinical-grade PTEN immunohistochemistry assay in prostate cancer by comparison with PTEN FISH. Mod Pathol 2016;29:904–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaux A, Albadine R, Toubaji A, et al. Immunohistochemistry for ERG expression as a surrogate for TMPRSS2-ERG fusion detection in prostatic adenocarcinomas. Am J Surg Pathol 2011;35:1014–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guedes LB, Almutairi F, Haffner MC, et al. Analytic, Preanalytic, and Clinical Validation of p53 IHC for Detection of TP53 Missense Mutation in Prostate Cancer. Clin Cancer Res 2017;23:4693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heaphy CM, Subhawong AP, Hong SM, et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol 2011;179:1608–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin D, Wyatt AW, Xue H, et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res 2014;74:1272–83. [DOI] [PubMed] [Google Scholar]
- 40.Beltran H, Prandi D, Mosquera JM, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 2016;22:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Labrecque MP, Coleman IM, Brown LG, et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J Clin Invest 2019;130:4492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Velho PI, Lim D, Wang H, et al. Molecular Characterization and Clinical Outcomes of Primary Gleason Pattern 5 Prostate Cancer After Radical Prostatectomy. JCO Precis Oncol 2019;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vaziri H Critical telomere shortening regulated by the ataxia-telangiectasia gene acts as a DNA damage signal leading to activation of p53 protein and limited life-span of human diploid fibroblasts. A review. Biochemistry (Mosc) 1997;62:1306–10. [PubMed] [Google Scholar]
- 44.Graham MK, Meeker A. Telomeres and telomerase in prostate cancer development and therapy. Nat Rev Urol 2017;14:607–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vukovic B, Park PC, Al-Maghrabi J, et al. Evidence of multifocality of telomere erosion in high-grade prostatic intraepithelial neoplasia (HPIN) and concurrent carcinoma. Oncogene 2003;22:1978–87. [DOI] [PubMed] [Google Scholar]
- 46.Feldser DM, Greider CW. Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell 2007;11:461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perera SA, Maser RS, Xia H, et al. Telomere dysfunction promotes genome instability and metastatic potential in a K-ras p53 mouse model of lung cancer. Carcinogenesis 2008;29:747–53. [DOI] [PubMed] [Google Scholar]
- 48.Hiyama K, Hiyama E, Ishioka S, et al. Telomerase activity in small-cell and non-small-cell lung cancers. J Natl Cancer Inst 1995;87:895–902. [DOI] [PubMed] [Google Scholar]
- 49.Sarvesvaran J, Going JJ, Milroy R, Kaye SB, Keith WN. Is small cell lung cancer the perfect target for anti-telomerase treatment? Carcinogenesis 1999;20:1649–51. [DOI] [PubMed] [Google Scholar]
- 50.Zaffaroni N, De Polo D, Villa R, et al. Differential expression of telomerase activity in neuroendocrine lung tumours: correlation with gene product immunophenotyping. J Pathol 2003;201:127–33. [DOI] [PubMed] [Google Scholar]
- 51.Lantuejoul S, Soria JC, Moro-Sibilot D, et al. Differential expression of telomerase reverse transcriptase (hTERT) in lung tumours. Br J Cancer 2004;90:1222–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zaffaroni N, Villa R, Pastorino U, et al. Lack of telomerase activity in lung carcinoids is dependent on human telomerase reverse transcriptase transcription and alternative splicing and is associated with long telomeres. Clin Cancer Res 2005;11:2832–9. [DOI] [PubMed] [Google Scholar]
- 53.Zheng X, Zhuge J, Bezerra SM, et al. High frequency of TERT promoter mutation in small cell carcinoma of bladder, but not in small cell carcinoma of other origins. J Hematol Oncol 2014;7:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pettersson A, Graff RE, Bauer SR, et al. The TMPRSS2:ERG rearrangement, ERG expression, and prostate cancer outcomes: a cohort study and meta-analysis. Cancer Epidemiol Biomarkers Prev 2012;21:1497–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barthel FP, Wei W, Tang M, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet 2017;49:349–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanaya T, Kyo S, Hamada K, et al. Adenoviral expression of p53 represses telomerase activity through down-regulation of human telomerase reverse transcriptase transcription. Clin Cancer Res 2000;6:1239–47. [PubMed] [Google Scholar]
- 57.Xu D, Wang Q, Gruber A, et al. Downregulation of telomerase reverse transcriptase mRNA expression by wild type p53 in human tumor cells. Oncogene 2000;19:5123–33. [DOI] [PubMed] [Google Scholar]
- 58.Shats I, Milyavsky M, Tang X, et al. p53-dependent down-regulation of telomerase is mediated by p21waf1. J Biol Chem 2004;279:50976–85. [DOI] [PubMed] [Google Scholar]
- 59.Haffner MC, Mosbruger T, Esopi DM, et al. Tracking the clonal origin of lethal prostate cancer. J Clin Invest 2013;123:4918–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.