
Figure 1: The mitochondrial apoptosis pathway.
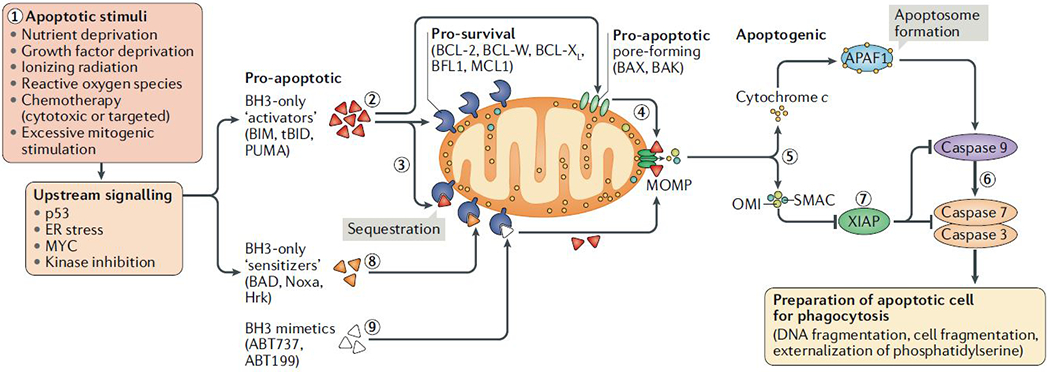
To initiate apoptosis, cellular stress or damage signals [1] unleash pro-apoptotic proteins (BH3-only ‘activators’ of apoptosis) via their upregulation (BIM or PUMA) or cleavage (BID cleaved to form truncated tBID) [2], which can either be bound and sequestered by pro-survival proteins such as BCL-2, BCL-XL or MCL1 [3] or, when these pro-survival proteins are saturated or absent, can activate BAX and/or BAK [4]. Activated BAX or BAK oligomerize and form pores to cause mitochondrial outer membrane permeabilization (MOMP), resulting in the release of apoptogenic molecules including SMAC, OMI and cytochrome c from the mitochondrial intermembrane space. Cytochrome c binds APAF1 in the cytosol to form the apoptosome (5), which serves as a platform for the activation of caspase 9, which then goes on to activate the effector caspases 3 and 7 (6) to dismantle the cell and prepare it for phagocytosis. Caspase activation can be blocked by XIAP (7), which in turn is inhibited by the released SMAC and OMI proteins from mitochondria (7). Upstream damage or stress signalling can also activate BH3-only ‘sensitizer’ proteins that don’t efficiently activate BAX and BAK but inhibit the activity of pro-survival BCL-2 family proteins to release any sequestered BH3-only activators, which trigger MOMP (8). BH3 mimetics are a novel class of agents that are able to sensitize cells to apoptosis by blocking the activity of pro-survival BCL-2 family proteins (9).