

Over fifty diseases called “proteinopathies” are caused by self-association of protein molecules to each other as these proteins misfold, become “sticky”, and aggregate. Protein aggregation also is involved in certain infectious diseases and central nervous system injuries (1). In some cases, a particular protein causes a certain disease, whereas in others, aggregation of the same protein underlies a family of diseases distinguished by different (though often overlapping) symptoms and by the fine details of the pathological aggregates in the affected tissue. One such family is synucleinopathies (2), in which the protein α-synuclein misfolds and self-assembles into neurotoxic oligomers and aggregates in the brain of affected people. The most common synucleinopathy is Parkinson’s disease (PD). The family also contains the rare diseases dementia with Lewy bodies, pure autonomic failure, and multiple system atrophy (MSA) (3).
MSA is a particularly aggressive disease, causing rapid deterioration of motor, autonomic, and cognitive functions and leading to death within 3–10 years from symptom onset. At early stages of disease, patients present predominantly either parkinsonian symptoms, including tremor, rigidity, and postural instability, or cerebellar ataxia-like symptoms characterized by difficulty with balance and gait (4,5). There are no disease-modifying therapies for synucleinopathies and unlike PD, for which dopamine-replacement therapy can offer relief for several years, even symptomatic treatment for MSA is very limited (6). Thus, new therapy for MSA is an urgent, unmet medical need.
Development of disease-modifying therapy for proteinopathies has been dominated in the dementia field by immunotherapeutic approaches targeting amyloid β-protein (Aβ) in Alzheimer’s disease (7), whereas in the case of synucleinopathies, the majority of clinical trials testing potential therapeutic agents used various neuroprotective drug candidates in patients with PD (8,9). Alongside these efforts, small-molecule inhibitors of abnormal protein self-assembly long have been considered a promising strategy for developing therapy against proteinopathies (10,11). Typically, such compounds are not specific to one protein but have the ability to modulate the oligomerization and aggregation of multiple amyloidogenic proteins.
Several strategies have been used to identify small-molecule inhibitors, including compound screening and rational design (11), yet probably the most common approach has been testing of nutraceuticals—compounds isolated from various foods that have been shown, or claimed, to have therapeutic benefits (12). Among those, one of the most frequently used family of compounds has been polyphenols (13). The allure of these compounds is clear: if they have been consumed by humans for centuries, the must be safe, lowering dramatically the bar for obtaining regulatory approval for clinical trials, e.g., Investigational New Drug designation by the American Food and Drug Administration, which can be costly and time consuming. As with anti-amyloid antibodies, however, none of these compounds has been successful in clinical trials for any proteinopathy to date.
There may be multiple reasons for the lack of success, perhaps primarily the fact that we do not understand how or why polyphenols inhibit the aggregation and toxicity of amyloidogenic proteins. Most compounds are tested initially for inhibition of aggregation against a target protein by using the common thioflavin T (ThT)-fluorescence assay, which is prone to artifacts (14,15), yet even if the compound indeed inhibits the aggregation of the target protein, it may do so simply because in the initial stages of the aggregation process, the interactions among the misfolded polypeptide chains are weak and easily modulated by non-specific binding.
The next step often is testing the compound’s effect in cell culture where the offending protein oligomers/aggregates are expressed or added exogenously. If the compound seems promising, it is further tested in animal models, such as worms, flies, fish, and rodents. However, polyphenols and many other small molecules tested in such assays are antioxidants, anti-inflammatory, and/or chelators, which may contribute to their apparently beneficial effects in these systems, regardless of their effect on the offending protein itself. Such multi-modal activity often is touted as promising for therapy development, yet in reality it may mask the actual mechanism of action and make optimizing the compounds more difficult because different activities require different effective concentrations and potentially different pharmacokinetics. The latter consideration is particularly important for compounds targeting the central nervous system due to the need to pass through the blood-brain barrier (BBB) and achieve therapeutic concentrations simultaneously at cellular (or extracellular) compartments where amyloidogenic proteins concentrate, where oxidative stress is rampant, at the active sites of receptors and enzymes mediating inflammatory responses, and/or where specific metal ions need to be chelated.
Epigallocatechin-3-gallate (EGCG, Figure 1), a polyphenol found in large quantities in green tea, likely is the most studied of the anti-amyloid polyphenols, though most of the literature about this compound actually comes from different fields, including cancer, heart disease, and other uses (16-21). In the amyloid field, it has been shown to inhibit the aggregation and toxicity of multiple proteins, including α-synuclein (10). Multiple studies using ThT fluorescence and in many cases morphological examination by electron microscopy have suggested that EGCG was a potent inhibitor of protein aggregation and amyloid fibril formation. However, several years ago, an investigation of EGCG’s interaction with Aβ40 and peptides derived from islet amyloid polypeptide or from the prion protein Sup35 showed that EGCG competes with ThT and can inhibit its binding to the cross-β structure of amyloid fibrils, possibly leading to a false positive aggregation inhibition signal (22). The same study also showed that the catechol moieties in EGCG (Figure 1) can undergo a self-redox reaction leading to formation of reactive carbonyl groups capable for forming Schiff bases with amino groups in the fibrils and cross-linking them. Similar chemistry was demonstrated for other catechol-containing polyphenolic compounds with anti-amyloid activity raising concerns regarding the safety of such compounds (23). Additional mechanistic studies of the protective effect of EGCG against amyloidogenic proteins have not delineated a specific binding site or a clear mechanism (24,25). However, positive therapeutic effects found in multiple pre-clinical models, including rodent (26) and non-human primate (27) models of synucleinopathy have built the case for the recent PROMESA clinical trial of EGCG in patients with MSA, which unfortunately failed to show a protective effect (28).
Figure 1.
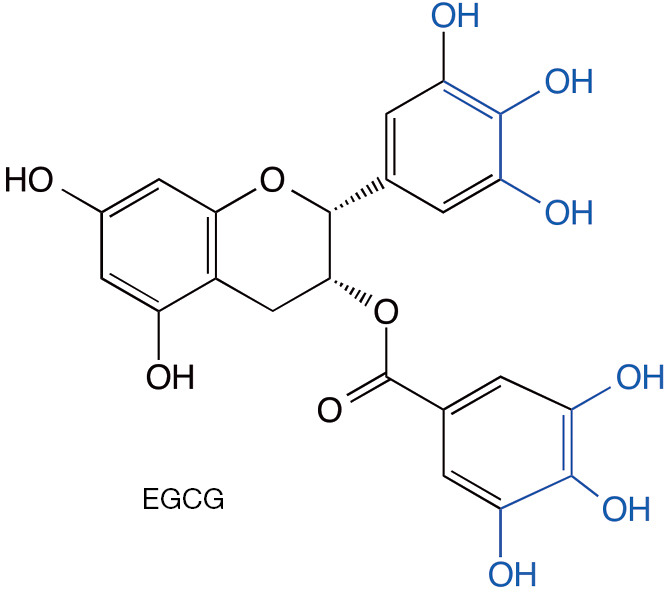
Schematic structure of EGCG. The catechol moieties that can undergo oxidation to form a reactive carbonyl and subsequent covalent attachment to amines are highlighted in blue. EGCG, epigallocatechin-3-gallate.
Additional support for initiating the PROMESA trial was based on the existence of a reasonable safety margin for EGCG in humans. The compound has been consumed in green, white, and black tea by humans and is approved in its purified form as a dietary supplement in many countries. However, EGCG may cause hepatotoxicity at daily doses of 600 mg or higher (29). Earlier pre-clinical studies found that at 100 mg/kg EGCG was lethal in mice and the doses needed for efficacy could be dangerously close to this lethal dose (30). The BBB penetration of EGCG has been known to be relatively low (31,32), yet was considered sufficient for previous clinical trials as well as for the PROMESA trial. The pros and cons of EGCG as a therapeutic drug for MSA are summarized in Table 1.
Table 1. Pros and cons of testing EGCG as therapy for MSA.
| Consideration | Pros | Cons |
|---|---|---|
| Pre-clinical efficacy | • Demonstrated efficacy in many in vivo models • Demonstrated efficacy in mouse and monkey models of synucleinopathy |
• Unclear mechanism of action |
| Clinical efficacy | • Cognitive improvement has been shown in Down syndrome (33) | • Unclear mechanism of action • Multiple clinical trials in proteinopathies have concluded without reporting data |
| Pharmacokinetics | • 2–3% penetration through the BBB | • 2–3% penetration through the BBB • May bind covalently to amino groups |
| Safety | • Centuries of consumption of green tea • Approved as a supplement in many countries • Did not cause toxicity in many pre-clinical and clinical trials |
• Studies suggesting relatively small safety window in mice (30) • Hepatotoxicity in humans at ≥600 mg per day |
| Trial design | • Relatively large number of completers • Well randomized and controlled • Careful and thoughtful statistical analysis |
• Most patients were at relatively late disease stages, perhaps too late for benefiting from the treatment |
In view of the issues listed above, it is perhaps not surprising that EGCG failed to show a therapeutic effect in patients with MSA. It is possible that insufficient amounts of the compound have reached their target—misfolded, oligomeric/aggregated α-synuclein—due to the limited BBB penetration of EGCG. The lack of a clear understanding of the mechanism by which the compound works makes interpreting the data difficult because it is not known specifically where in or outside cells, and in which cells—neurons, glia, or both—the most critical targets actually are. Interestingly, in a sub-group of patients who participated in a MRI sub-study, substantially lower striatal volume loss was observed compared to the placebo, suggesting that EGCG treatment exerted a neuroprotective effect and/or modulated neuroinflammation in these patients, which may or may not be related to its direct interaction with α-synuclein.
Sub-acute toxicity also might have contributed to the failure of the trial. Clear hepatotoxicity was observed in two patients who had to drop out of the trial whereas other adverse effects were not significantly different between the EGCG and placebo groups, yet sub-acute toxicity due to covalent modification of amino groups might have interfered with brain processes without being detected as overt adverse effects and led to a lack of improvement of disease symptoms. An interesting possibility is that mannitol, which was used as a placebo, did not work as a real placebo and inhibited α-synuclein aggregation, as has been reported recently in vitro (34), masking the effect of EGCG.
It is also possible that target engagement was achieved yet the treatment was applied too late in the disease to be effective. Notably, this is a particularly difficult problem in the case of MSA because diagnosis of the disease is challenging. To address this issue, 89 of the 92 patients enrolled in the trial had a probable-MSA diagnosis, which typically is given at a relatively advanced disease stage. Indeed, the diagnosis has been confirmed in all the five patients who passed away and their brains were donated for pathological analysis. Unfortunately, at an advanced disease stage a putative neurodegenerative cascade, involving many deleterious mechanisms, such as inflammation, apoptosis, and oxidative damage may have gone beyond the point of benefiting from an anti-α-synuclein aggregation therapy or any therapy. Another potential reason for the failure of the trial is the relatively low sensitivity of the clinical evaluation by the Unified Multiple System Atrophy Rating Scale (UMSARS) to detect subtle changes. Although the UMSARS currently is the gold standard for MSA evaluation, and although the evaluation was done by movement-disorders experts and every effort was made to standardize the evaluation across the twelve participating sites, clinical evaluation is intrinsically prone to the unintentional bias of the individual physicians, lowering the signal-to-noise ratio.
Both of these issues, the difficulty with early diagnosis of MSA and the relatively low sensitivity of the clinical assessment for measuring changes precisely, highlight a key issue in current MSA clinical practice and research—the absence of unbiased, sensitive, and specific biomarkers for the disease. If such biomarkers can be developed, they will assist with early diagnosis, measurement of disease progression, and evaluation of treatment effects in future clinical trials. In particular biomarkers measuring the levels of α-synuclein, aggregated α-synuclein, and/or specific disease-associated forms of the protein, such as phosphorylation at serine 129, will be useful for assessment of potential treatments targeting the toxic forms of α-synuclein in the brain. Encouragingly, major efforts currently are being dedicated to developing such biomarkers, raising hopes that they will become clinically available in the near future.
The failure of the PROMESA trial calls into question the strategy of using polyphenols as therapeutic agents for MSA in particular and proteinopathies in general. Despite promising initial data in test-tube, cell-culture, and animal models, polyphenols, including EGCG, have not fulfilled their promise in clinical trials testing them in various proteinopathies. Until and unless we have a detailed mechanistic understanding of the way these compounds affect living organisms, especially humans, testing them in additional clinical trials may be a drain of resources and time that could have been better spent on other therapeutic strategies.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: The author is thankful for support from National Institutes of Health/National Institute on Aging grants R01 AG050721 and RF1 AG054000, National Institutes of Health/National Institute of Neurological Disease and Stroke grant R01 NS107596 (PI: H. Kaufmann), Team Parkinson/Parkinson Alliance, Multiple System Atrophy Coalition grants 20170367 and 2017-10-007, California Department of Public Health grant 18-10926, The Alzheimer’s Association, The Michael J. Fox Foundation, Weston Brain Institute, and Alzheimer’s Research UK Biomarkers Across Neurodegenerative Diseases (BAND 3) grant 17990, CurePSP grant 665-2019-07, and The Michael J. Fox Foundation grant 18303.
Ethical Statement: The author is accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Provenance and Peer Review: This article was commissioned and reviewed by the Academic Editor Dr. Zhenxiang Zhao (Department of Neurology, Henan Provincial People’s Hospital, People’s Hospital of Zhengzhou University, People’s Hospital of Henan University, Zhengzhou, China).
Conflicts of Interest: The author has completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm.2020.01.117). GB serves as an unpaid editorial board member of Annals of Translational Medicine from Mar 2020 to Feb 2022.
References
- 1.Rahimi F, Bitan G. The structure and function of fibrillar and oligomeric assemblies of amyloidogenic proteins. In: Rahimi F, Bitan G, editors. Non-fibrillar amyloidogenic protein assemblies—common cytotoxins underlying degenerative diseases. Dordrecht: Springer; 2012:1-36. [Google Scholar]
- 2.Jellinger KA. Neuropathological spectrum of synucleinopathies. Mov Disord 2003;18 Suppl 6:S2-12. 10.1002/mds.10557 [DOI] [PubMed] [Google Scholar]
- 3.Marti MJ, Tolosa E, Campdelacreu J. Clinical overview of the synucleinopathies. Mov Disord 2003;18 Suppl 6:S21-7. 10.1002/mds.10559 [DOI] [PubMed] [Google Scholar]
- 4.Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670-6. 10.1212/01.wnl.0000324625.00404.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh RR, Krismer F, Galpern WR, et al. Recommendations of the Global Multiple System Atrophy Research Roadmap Meeting. Neurology 2018;90:74-82. 10.1212/WNL.0000000000004798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stefanova N, Wenning GK. Review: Multiple system atrophy: emerging targets for interventional therapies. Neuropathol Appl Neurobiol 2016;42:20-32. 10.1111/nan.12304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cummings J, Ritter A, Zhong K. Clinical Trials for Disease-Modifying Therapies in Alzheimer's Disease: A Primer, Lessons Learned, and a Blueprint for the Future. J Alzheimers Dis 2018;64:S3-22. 10.3233/JAD-179901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oertel WH. Recent advances in treating Parkinson's disease. F1000Res 2017;6:260. 10.12688/f1000research.10100.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayes MW, Fung VS, Kimber TE, et al. Updates and advances in the treatment of Parkinson disease. Med J Aust 2019;211:277-83. 10.5694/mja2.50224 [DOI] [PubMed] [Google Scholar]
- 10.Liu T, Bitan G. Modulating self-assembly of amyloidogenic proteins as a therapeutic approach for neurodegenerative diseases: strategies and mechanisms. ChemMedChem 2012;7:359-74. 10.1002/cmdc.201100585 [DOI] [PubMed] [Google Scholar]
- 11.Rahimi F, Li H, Sinha S, et al. Modulators of Amyloid β-Protein (Aβ) Self-Assembly. In: Wolfe MS, editor. Developing Therapeutics for Alzheimer's Disease. Boston: Academic Press, 2016:97-191. [Google Scholar]
- 12.Sadhukhan P, Saha S, Dutta S, et al. Nutraceuticals: An emerging therapeutic approach against the pathogenesis of Alzheimer's disease. Pharmacol Res 2018;129:100-14. 10.1016/j.phrs.2017.11.028 [DOI] [PubMed] [Google Scholar]
- 13.Rigacci S, Stefani M. Nutraceuticals and amyloid neurodegenerative diseases: a focus on natural phenols. Expert Rev Neurother 2015;15:41-52. 10.1586/14737175.2015.986101 [DOI] [PubMed] [Google Scholar]
- 14.Coelho-Cerqueira E, Pinheiro AS, Follmer C. Pitfalls associated with the use of Thioflavin-T to monitor anti-fibrillogenic activity. Bioorg Med Chem Lett 2014;24:3194-8. 10.1016/j.bmcl.2014.04.072 [DOI] [PubMed] [Google Scholar]
- 15.Gade Malmos K, Blancas-Mejia LM, Weber B, et al. ThT 101: a primer on the use of thioflavin T to investigate amyloid formation. Amyloid 2017;24:1-16. 10.1080/13506129.2017.1304905 [DOI] [PubMed] [Google Scholar]
- 16.Chakrawarti L, Agrawal R, Dang S, et al. Therapeutic effects of EGCG: a patent review. Expert Opin Ther Pat 2016;26:907-16. 10.1080/13543776.2016.1203419 [DOI] [PubMed] [Google Scholar]
- 17.Dhatwalia SK, Kumar M, Dhawan DK. Role of EGCG in Containing the Progression of Lung Tumorigenesis - A Multistage Targeting Approach. Nutr Cancer 2018;70:334-49. 10.1080/01635581.2018.1445762 [DOI] [PubMed] [Google Scholar]
- 18.Eng QY, Thanikachalam PV, Ramamurthy S. Molecular understanding of Epigallocatechin gallate (EGCG) in cardiovascular and metabolic diseases. J Ethnopharmacol 2018;210:296-310. 10.1016/j.jep.2017.08.035 [DOI] [PubMed] [Google Scholar]
- 19.Fujiki H, Watanabe T, Sueoka E, et al. Cancer Prevention with Green Tea and Its Principal Constituent, EGCG: from Early Investigations to Current Focus on Human Cancer Stem Cells. Mol Cells 2018;41:73-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mehta M, Ahmed S, Dryden G. Refractory pouchitis improves after administration of the green tea polyphenol EGCG: a retrospective review. Int J Colorectal Dis 2018;33:83-6. 10.1007/s00384-017-2906-5 [DOI] [PubMed] [Google Scholar]
- 21.Negri A, Naponelli V, Rizzi F, et al. Molecular Targets of Epigallocatechin-Gallate (EGCG): A Special Focus on Signal Transduction and Cancer. Nutrients 2018. doi: . 10.3390/nu10121936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palhano FL, Lee J, Grimster NP, Kelly JW. Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils. J Am Chem Soc 2013;135:7503-10. 10.1021/ja3115696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato M, Murakami K, Uno M, et al. Site-specific inhibitory mechanism for amyloid β42 aggregation by catechol-type flavonoids targeting the Lys residues. J Biol Chem 2013;288:23212-24. 10.1074/jbc.M113.464222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorenzen N, Nielsen SB, Yoshimura Y, et al. How epigallocatechin gallate can inhibit α-synuclein oligomer toxicity in vitro. J Biol Chem 2014;289:21299-310. 10.1074/jbc.M114.554667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang T, Zhang J, Derreumaux P, et al. Molecular mechanism of the inhibition of EGCG on the Alzheimer Aβ(1-42) dimer. J Phys Chem B 2013;117:3993-4002. 10.1021/jp312573y [DOI] [PubMed] [Google Scholar]
- 26.Xu Q, Langley M, Kanthasamy AG, et al. Epigallocatechin Gallate Has a Neurorescue Effect in a Mouse Model of Parkinson Disease. J Nutr 2017;147:1926-31. 10.3945/jn.117.255034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen M, Wang T, Yue F, et al. Tea polyphenols alleviate motor impairments, dopaminergic neuronal injury, and cerebral α-synuclein aggregation in MPTP-intoxicated parkinsonian monkeys. Neuroscience 2015;286:383-92. 10.1016/j.neuroscience.2014.12.003 [DOI] [PubMed] [Google Scholar]
- 28.Levin J, Maass S, Schuberth M, et al. Safety and efficacy of epigallocatechin gallate in multiple system atrophy (PROMESA): a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2019;18:724-35. 10.1016/S1474-4422(19)30141-3 [DOI] [PubMed] [Google Scholar]
- 29.Dekant W, Fujii K, Shibata E, et al. Safety assessment of green tea based beverages and dried green tea extracts as nutritional supplements. Toxicol Lett 2017;277:104-8. 10.1016/j.toxlet.2017.06.008 [DOI] [PubMed] [Google Scholar]
- 30.Yagiz K, Morre DJ, Morre DM. Transgenic mouse line overexpressing the cancer-specific tNOX protein has an enhanced growth and acquired drug-response phenotype. J Nutr Biochem 2006;17:750-9. 10.1016/j.jnutbio.2005.12.006 [DOI] [PubMed] [Google Scholar]
- 31.Mereles D, Hunstein W. Epigallocatechin-3-gallate (EGCG) for Clinical Trials: More Pitfalls than Promises? Int J Mol Sci 2011;12:5592-603. 10.3390/ijms12095592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pervin M, Unno K, Nakagawa A, et al. Blood brain barrier permeability of (-)-epigallocatechin gallate, its proliferation-enhancing activity of human neuroblastoma SH-SY5Y cells, and its preventive effect on age-related cognitive dysfunction in mice. Biochem Biophys Rep 2017;9:180-6. 10.1016/j.bbrep.2016.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de la Torre R, de Sola S, Hernandez G, et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down's syndrome (TESDAD): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol 2016;15:801-10. 10.1016/S1474-4422(16)30034-5 [DOI] [PubMed] [Google Scholar]
- 34.Paul A, Zhang BD, Mohapatra S, et al. Novel Mannitol-Based Small Molecules for Inhibiting Aggregation of α-Synuclein Amyloids in Parkinson's Disease. Front Mol Biosci 2019;6:16. 10.3389/fmolb.2019.00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as