

Abstract
Statistics show alarming numbers of infected and killed in the world, caused by the Covid-19 pandemic, which still doesn’t have a specific treatment and effective in combating all efforts to seek treatments and medications against this disease. Natural products are of relevant interest in the search for new drugs. Thus, Buriti oil (Mauritia flexuosa L.) is a natural product extracted from the fruit of the palm and is quite common in the legal Amazon region, Brazil. In the present work, the anti-Covid-19 biological activity of some constituents of Buriti oil was investigated using in silico methods of Molecular Docking and Molecular Dynamics Simulations. The main results of Molecular Docking revealed favorable interaction energies in the formation of the 2GTB peptidase complex (main peptidase of SARS-CoV) with the 13-cis-β-carotene ligands (ΔGbind = −10.23Kcal mol−1), 9-cis -β-carotene (ΔGbind = −9.82Kcal mol−1), and α-carotene (ΔGbind = −8.34Kcal mol−1). Molecular Dynamics simulations demonstrated considerable interaction for these ligands with emphasis on α-carotene. Such theoretical results encourage and enable a direction for experimental studies in vitro and in vivo, essential in the development of new drugs with enzymatic inhibitory action for Covid-19.
Communicated by Ramaswamy H. Sarma.
Keywords: Covid-19, buriti oil, molecular docking, molecular dynamic
1. Introduction
The new coronavirus (Sars-CoV-2) responsible for Covid-19 disease was described in December 2019 in China (World Health Organization, 2020). At the beginning of the spread of the virus, the first cases were contracted at the seafood and animal market in Huanan in the city of Wuhan located in Hubei province, then causing community transmission, which caused an exponential growth in the number of cases (Lai et al., 2020). The main consequences of Covid-19 are Severe Acute Respiratory Syndrome (SARS), in addition to digestive and systemic problems (Fauci et al., 2020; Wong et al., 2020). Due to the high rate of contagion among humans, the World Health Organization (WHO) decreed on March 11, 2020, the infection of Covid-19 as a pandemic (The Lancet, 2020). On April 9, ere were more than 1,500,000 cases registered worldwide, with a fatality rate of around 5.9% (Zhang et al., 2020).
Thus, a range of efforts is needed in the most diverse areas of knowledge for the development of new drug candidates for the treatment of Covid-19 (Liu et al., 2020). In this context, research with medicinal plant extracts is very promising, because Brazil has a great diversity in biomass able to be investigated in the search for pharmacological activities not yet proven by science (Ferreira et al., 2019).
Mauritia flexuosa L. popularly known as Buriti is a classic example of a plant popularly called medicinal (Barros et al., 2015). Buriti is a palm of the Arecaceae family, found in central and northern South America (Carneiro & Carneiro, 2011). In Brazil there is a predominance of this species mainly in the region of the legal Amazon (Silva et al., 2009). The use of Buriti oil based on the knowledge of common sense by popular people for various medicinal purposes in this region caught the attention of the authors of this work (Kovalski & Obara, 2013). In 2005, studies carried out with Buriti oil identified the molecules trans-β-carotene, 13-cis-β-carotene, phytofluene, zeaxanthin, β-10-apo-carotene, α-carotene, mutachrome, ζ-carotene, β- zeacarotene and, γ-carotene δ-carotene (Albuquerque et al., 2005). Knowledge of the molecular structure of molecules provides an opportunity to investigate and identify possible pharmacological activity against Covid-19 (Sharma et al., 2020).
In this context, the planning and development of new drugs has evolved in the last two decades, from the application of new experimental techniques and complementary technologies (Atanasov et al., 2015). Computational chemistry can perfectly act to assist experimental investigations (Monego et al., 2017). Therefore, two computational techniques of great applicability in research centers and in the pharmaceutical industries can be highlighted, the Molecular Docking and Molecular Dynamics (Alonso et al., 2006).
The Molecular Docking protocols guide the realization of the ideal fit of the ligand in the binding site of a protein target, previously analyzed (Rocha et al., 2018). For this, generate a set of conformations of the ligand-receptor complex, based on the positions of the ligand (Totrov & Abagyan, 2008). In relation to Molecular Dynamics, this methodology has become an important technique for designing bioactive molecules and investigating the behavior of the system over time (Tautermann et al., 2015). This being one of the most used techniques to study the balance and interactions of protein-ligand systems (Kerrigan, 2013; Piccirillo & Amaral, 2018).
The main peptidase of SARS-CoV (PDB ID: 2GTB) plays an important biological role in the virus life cycle (Lee et al., 2007). This peptidase is 96% similar to the main Covid-19 (SARS-CoV-2) protease (Xu et al., 2020). Peptidase 2GTB is used by scientists in the development of enzyme inhibitors for SARS-CoV and, due to its similarity, can therefore be used as a base molecule in the development of antiviral against Covid-19 (Bouchentouf & Missoum, 2020).
This work carried out an important preliminary study, in silico, with the application of Molecular Docking and Molecular Dynamics to the crystal of the SARS-Coronavirus peptidase (PDB ID: 2GTB) against the molecules present in Buriti oil (Mauritia flexuosa L.), in order to identify enzyme inhibitors against the SARS-Coronavirus peptidase in order to contribute to the fight against Covid-19.
2. Materials and methods
2.1. 3D Structure of the ligands and receptor
The crystal structure of the SARS-Coronavirus peptidase (PDB ID: 2GTB) was obtained from the PDB (Protein Data Bank) (Berman et al., 2000). The 3D structures of the ligands trans-β-carotene, 13-cis-β-carotene, phytofluene, β-10-apo-carotene, α-carotene, β-zeacarotene, and γ-carotene δ-carotene were obtained from PubChem Open Chemistry Database (Kim et al., 2019). It was not found structures of the zeaxantine, mutachrome and ζ-carotene in PubChem.
2.2. Molecular docking (MD)
The AutoDock Tools (ADT) version 1.5.6 package was used for all Molecular Docking simulations (Goodsell et al., 1996; Huey et al., 2012). All coupling procedures used the Autodock 4.2 package (Goodsell, 2005; Morris et al., 2008). The receiver being considered as rigid and the binders as flexible (Ravindranath et al., 2015). The global search was doing Lamarckian genetic algorithm (Morris et al., 1998). Molecular Docking calculations with small molecules and chloroquine showed a standard of interaction with the Met49 residue, which makes it an important amino acid (Samant & Javle, 2020). This information was considered for the choice of the Grid Box which was centered on the Met49 with coordinates x = 26.972 y= −15.931 and z = 15.125, cubic box size was 80 × 80 × 80 points, spacing 0.375 Å and number step equal 100. Hydrophobic and hydrogen bonding interactions were analyzed with Ligplot + version 2.2 (Laskowski & Swindells, 2011).
2.3. Molecular dynamics simulation (MDS)
The coordinates of the complexes formed with the best results of Molecular Docking were used in the Molecular Dynamics Simulations. The H++ server was used to protonate the SARS-Coronavirus peptidase (PDB ID: 2GTB) (Gordon et al., 2005). The molecular dynamics simulations were performed using the GROMOS9653a646 force field implemented in the GROMACS package version 2018.1 (Abraham et al., 2018; Oostenbrink et al., 2004). The simulations were carried out using water molecules explicit by the model of single point charge (SPC) (Van der Spoel et al., 1998). The simulations were performed for 30 ns using a 2fs integration time step (Ramos et al., 2012). Each system was heated in gradual increments at the following temperatures: 100 K (10ps), 150 K (5ps), 200 K (5ps) and 250 K (5ps) and, after these steps, the temperature was adjusted to 310 K (Arcanjo et al., 2017). The interaction energies of Coulomb (Coul), Lennard-Jones (LJ) and the sum of Coul + LJ were calculated to analyze the interactions between protein-ligand (Lemkul, 2019). The production analysis of each Molecular Dynamics Simulation performed was considered from 6 ns to 30 ns. PyMOL version 2.1.1 was used for visualization of results (Schrödinger LLC, 2018).
3. Results and discussion
3.1. Molecular docking (MD)
Initially to perform the molecular fit, a prediction of the structure of the ligand-receptor complex must be provided using computational methods, through the sets of conformations found of the ligands in the active site of the protein, followed by their classification by a scoring function (Meng et al., 2011). The most stable conformations after MD in 2GTB peptidase complexes according to Table 1 was with molecules 13-cis-β-carotene (Ki = 31.52ηmol L−1 and ΔGbind = −10.23Kcal mol−1), 9-cis-β-carotene (Ki = 63.73ηmol L−1 and ΔGbind = −9.82Kcal mol−1), and α-carotene (Ki = 770.11ηmolL−1 and ΔGbind = −8.34Kcal mol−1). Salim and Noureddine also investigated molecular docking in 2GTB peptidase complexes and obtained the following results: Chloroquine (ΔGbind = −6.21Kcal mol−1); hydroxychloroquine (ΔGbind = −5.51Kcal mol−1) and favipiravir (ΔGbind= −4.12 Kcal mol−1) (Bouchentouf & Missoum, 2020). These results were considered satisfactory by the authors, but the results obtained in this work show better values for ΔGbind in complex systems with 13-cis-β-carotene, 9-cis-β-carotene and α-carotene as shown in Table 1.
Table 1.
Inhibition constant (Ki) and binding energy (ΔGbind) obtained in Molecular Docking for 2GTB peptidase complexes.
| Ligand | Pubchem CID | Ki (mol L-1) | ΔGbind (Kcal mol-1) |
|---|---|---|---|
| trans-β-carotene | 5280489 | 4.76 × 10-6 | −7.26 |
| 13-cis-β-carotene | 10256668 | 31.52 × 10-9 | −10.23 |
| 9-cis-β-carotene | 9828626 | 63.73 × 10-9 | −9.82 |
| phytofluene | 6436722 | 6.15 × 10-3 | −3.02 |
| β-10-apo-carotene | 6450190 | 2.78 × 10-6 | −7.58 |
| α-carotene | 6419725 | 770.11 × 10-9 | −8.34 |
| β-zeacarotene | 5280790 | 1.19 × 10-6 | −8.08 |
| γ-carotene | 5280791 | 25.61 × 10-6 | −6.26 |
| δ-carotene | 5281230 | 33.96 × 10-6 | −6.10 |
The Figure 1 show global result of the molecular docking. In surface showed the ligand 13-cis-β-carotene was showed only hydrophobic contacts with the 2GTB peptidase and the main residues of the binding site were Ile43, Lys61, Ala46, Thr24, Asn142, His41, Gln189, Arg188, Met165, Tyr54, Asp187, His164, Cys44, Thr45, Cys22, Val42, and Gly23.
Figure 1.

(A) 2GTB peptidase receptor (green surface), ligand 13-cis-β-carotene (blue stick) and residue Met49 (red); (B) Ribbon representation of the 2GTB peptidase receptor (green), ligand 13-cis-β-carotene (blue stick) and residue Met49 (red); (C) LigPlot + diagram of the hydrophobic interaction between 2GTB peptidase receptor and 13-cis- β-carotene ligand.
In the Figure 2, the 9-cis-β-carotene molecule showed hydrophobic contacts with the 2GTB peptidase and the main residues of the binding site were Asp48, Ala46, Gln189, His164, Glu166, Gly143, Phe140, Leu141, Asn142, Cys145, Met49, Glu47, and Leu50. The ligand performs hydrogen bond with the following amino acids: His163 (2.98 Å), Ser144 (2.84 Å) and Asn51 (2.70 Å).
Figure 2.

(A) 2GTB peptidase receptor (green surface), ligand 9-cis-β-carotene (blue stick) and residue Met49 (red); (B) Ribbon representation of the 2GTB peptidase receptor (green), ligand 9-cis-β-carotene (blue stick) and residue Met49 (red); (C) LigPlot + diagram of the hydrogen interaction between 2GTB peptidase receptor and 9-cis-β-carotene ligand through residues Ser144 (green), His163 (green) and Asn51(green).
In the Figure 3, the α-carotene molecule showed only hydrophobic contacts with the 2GTB peptidase and the main residues of the binding site were Thr25, Cys44, Thr45, Thr25, Gly23, Ile43, Cys22, Lys61, Val42, Ser65, Met49, Asn142, Leu141, Glu166, and Phe140.
Figure 3.

(A) 2GTB peptidase receptor (green surface), ligand α-carotene (blue stick) and residue Met49 (red); (B) Ribbon representation of the 2GTB peptidase receptor (green), ligand α-carotene (blue stick) and residue Met49 (red); (C) LigPlot + diagram of the hydrophobic interaction between 2GTB peptidase receptor and ligand α-carotene.
The hydrophobic effect occurs due to the interaction between the nonpolar regions of the ligand and the active site with the solvent, being that, these are solvated by more organized layers of water molecules. During the ligand-receptor interaction, the presence of these nonpolar regions, cause the release and disorganization of water molecules causing an entropic effect that contributes to the minimization of Gibbs energy in the system. The reduction in Gibbs energy favors the formation of the ligand-receptor complex, thereby highlighting the important role of the aqueous solvent in the molecular recognition process (Guedes et al., 2014).
In addition, it was observed that the ligands (13-cis-β-carotene, 9-cis-β-carotene and, α-carotene) when interacting with the SARS-Coronavirus peptidase, had hydrophobic bonds in common with the amino acid Asn142 at the active site of the protein. However, the 13-cis-β-carotene and α-carotene ligands have a greater similarity in relation to their interactions with the peptidase, because have a greater amount of hydrophobic interactions with the same amino acid residues (Cys44, Thr45, Gly23, Ile43, Cys22, Lys61, Val42 and Asn142), moreover, do not have hydrogen bonds.
3.2. Molecular dynamics simulation (MDS)
In Figure 4, the molecular dynamics simulation demonstrated an interaction between the 13-cis-β-carotene ligand and 2GTB peptidase, furthermore, the ligand remained close to the amino acid Met49, that is part of the binding site. The interval of this interaction occurred between 6 ns-9ns. Subsequently, in the interval between 10 ns-13ns the ligand shifted and remained outside of the binding site of the 2GTB peptidase. However, between 14 ns-16ns the ligand returns to interaction with the same active site (close to the amino acid Met49). This periodic behavior is repeated for the intervals of 17 ns-24ns, 25 ns-29ns, and 30 ns.
Figure 4.

Analyse of the molecular dynamic simulation. 2GTB peptidase receptor (green surface), ligand 13-cis-β-carotene (blue stick), and residue Met49 (red).
Molecular dynamics simulations involving the formation of complex systems with the 9-cis-β-carotene and α-carotene ligands versus 2GTB peptidase were also investigated (as shown in Figures 5 and 6). For both systems investigated, behaviors similar to that described above were obtained.
Figure 5.

Analyse of the molecular dynamic simulation. 2GTB peptidase receptor (green surface), ligand 9-cis-β-carotene (blue stick), and residue Met49 (red).
Figure 6.

Analyse of the molecular dynamic simulation. 2GTB peptidase receptor (green surface), ligand α-carotene (blue stick) and residue Met49 (red).
The three molecular dynamics simulations showed a high degree of flexibility of the 2GBT peptidase in its conformation during interactions with the 13-cis-β-carotene, 9-cis-β-carotene, and α-carotene molecules, however, it can be seen a very important peculiarity. When the ligand interacts with the 2GTB peptidase, a new structural conformation must occur in both. Subsequently, due to the dynamic process of 2GTB peptidase, this equilibrium condition is affected and, consequently, there is destabilization of the system and subsequent expulsion of the ligand. After a new conformational stabilization of the 2GTB peptidase, the energy of the system reestablishes a new condition favorable to a new interaction with the ligand, which is why it returns to the same region close to the Met49 group. This leads us to believe that the region close to the Met49 group is, in fact, an active site of 2GTB peptidase thermodynamically favorable.
The processes described so far are dynamic and reversible processes, but that differ from each other in the time of interaction. Table 2 summarizes the interaction intervals for the three investigated ligands.
Table 2.
Ligand interaction intervals in relation to 2GTB peptidase receptor close to the Met49 residue.
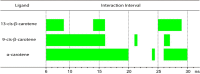 |
Thus, it is observed that for the three investigated ligands the greatest interval of interaction with the 2GTB peptidase occurred for α-carotene (between 6 ns-20ns), therefore, this is the most effective ligand for the inhibitory effect of 2GTB peptidase.
Table 3 shows the interaction energies of the 2GTB peptidase with the cis-β-carotene, 9-cis-β-carotene and α-carotene ligands over the analyzed range of molecular dynamics simulations.
Table 3.
Interaction energy of Coulomb (Coul), Lennard-Jones (LJ) and sum Coul + LJ for the 2GTB-13-cis-β-carotene, 2GTB-9-cis-β-carotene and 2GTB-α- carotene. Unit kJ mol−1.
| Complex (Protein-Ligand) | Coul | LJ | Coul + LJ |
|---|---|---|---|
| 2GTB-13-cis-β-carotene | 0.0 | −161.02 ± 6.10 | −161.02 ± 6.10 |
| 2GTB-9-cis-β-carotene | −28.95 ± 3.00 | −155.40 ± 4.70 | −184.35 ± 5.60 |
| 2GTB-α-carotene | 0.0 | −146.19 ± 7.90 | −146.19 ± 7.90 |
The energetic potentials of Coul and LJ are related to the interaction between atoms not covalently linked. In the Coul model, these interactions are mainly related to the effects of electrostatic poles, such as hydrogen bonds, whereas for LJ the interactions are related to less effective forces such as the van der Waals force (Namba et al., 2008). Table 3 illustrates the values of the Coul and LJ energy interactions for the complex systems 2GTB-13-cis-β-carotene, 2GTB-9-cis-β-carotene, and 2GTB-α-carotene. Only 2GTB-9-cis-β-carotene complex showed Coul energetic interaction (−28.95 ± 3.00 kJ mol−1), this value is consistent with the results of MD that suggest formation of hydrogen bonds for this complex. Moreover, all complexes showed interactions according to LJ (−161.02 ± 6.10 kJ mol−1 (2GTB-13-cis-β-carotene); −155.40 ± 4.70 kJ mol−1 (2GTB-9-cis-β-carotene); −146.19 ± 7.90 kJ mol−1(2GTB-α-carotene)). Thus, it is noted that the most stable interaction occurred for the 2GTB-9-cis-β-carotene complex (Coul + LJ = −184.35 ± 5.60 kJ mol−1). Although this complex is thermodynamically more stable, the α-carotene ligand showed greater efficiency in the interaction with 2GTB peptidase (2GTB-α-carotene complex). This shows that the energy factor is not the only determining factor in the ligand interaction process in relation to the active peptidase site. Probably the dynamic effect of the peptidase, as well as the ligand itself, affects thermodynamic equilibrium most effectively in 2GTB-9-cis-β-carotene in relation to the 2GTB-α-carotene complex in order to justify a longer enzymatic interaction time for this complex compared to the previous one. Thus, in inhibitory terms, the α-carotene ligand showed better results due to their interaction range being greater in relation to the 9-cis-β-carotene and 13-cis-β-carotene ligands.
4. Conclusion
The analysis of the interactions found in both Molecular Docking and Molecular Dynamics and, consequently, the values of energies free of favorable interactions for compounds 13-cis-β-carotene, 9-cis-β-carotene, and α-carotene against 2GTB peptidase demonstrate that these molecules are promising candidates for planning new drugs to combat Covid-19. The raw material for obtaining these compounds is available on a large scale and with low cost of production, obtaining and processing. As there are no experimental results with these molecules for the prevention and control of the disease, these theoretical results are of great importance, because allow a direction for studies in the mode in vitro and in vivo.
Disclosure statement
There are no conflicts to declare.
References
- Abraham M. J., Van Der Spoel D., Lindahl E., Hess B., & the GROMACS development team (2018). GROMACS user manual version 2018.1 www.gromacs.org
- Albuquerque M. L. S., Guedes I., Alcantara P. Jr., Moreira S. G. C., Barbosa Neto N. M., Correa D. S., & Zilio S. C. (2005). Characterization of buriti (Mauritia flexuosa L.) oil by absorption and emission spectroscopies. Journal of the Brazilian Chemical Society, 16(6a), 1113–1117. 10.1590/S0103-50532005000700004 [DOI] [Google Scholar]
- Alonso H., Bliznyuk A. A., & Gready J. E. (2006). Combining Docking and molecular dynamic simulations in drug design. Medicinal Research Reviews, 26(5), 531–568. 10.1002/med.20067 [DOI] [PubMed] [Google Scholar]
- Arcanjo D. D. R., Vasconcelos A. G., Nascimento L. A., Mafud A. C., Plácido A., Alves M. M. M., Delerue-Matos C., Bemquerer M. P., Vale N., Gomes P., Oliveira E. B., Lima F. C. A., Mascarenhas Y. P., Carvalho F. A. A., Simonsen U., Ramos R. M., & Leite J. R. S. A. (2017). Structure-function studies of BPP-BrachyNH 2 and synthetic analogues thereof with Angiotensin I-converting enzyme. European Journal of Medicinal Chemistry, 139, 401–411. 10.1016/j.ejmech.2017.08.019 [DOI] [PubMed] [Google Scholar]
- Atanasov A. G., Waltenberger B., Pferschy-Wenzig E.-M., Linder T., Wawrosch C., Uhrin P., Temml V., Wang L., Schwaiger S., Heiss E. H., Rollinger J. M., Schuster D., Breuss J. M., Bochkov V., Mihovilovic M. D., Kopp B., Bauer R., Dirsc V. M., & Stuppner H. (2015). Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnology Advances, 33(8), 1582–1614. 10.1016/j.biotechadv.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros E. M. L., Lira S. R. d S., Lemos S. I. A., Barros T. L. e., & Rizo M. D. S. (2015). Study of buriti (Mauritia flexuosa L.) cream in the healing process. ConScientiae Saúde, 13(4), 503–610. 10.5585/conssaude.v13n4.5175 [DOI] [Google Scholar]
- Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., & Bourne P. E. (2000). The protein data bank. Nucleic Acids Research, 28(1), 235–242. 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchentouf S., & Missoum N. (2020). Identification of compounds from Nigella sativa as new potential inhibitors of 2019 novel Coronasvirus (COVID-19): Molecular docking study. ChemRxiv, 1–14. [Google Scholar]
- Carneiro B. T., & Carneiro J. G. M. (2011). Frutos e polpa desidratada buriti (Mauritia flexuosa L.): Aspectos físicos, químicos e tecnológicos. Revista Verde de Agroecologia e Desenvolvimento Sustentável, 6, 105–111. [Google Scholar]
- Fauci A. S., Lane H. C., & Redfield R. R. (2020). Covid-19- navigating the uncharted. The New England Journal of Medicine, 382(13), 1268–1269. 10.1056/NEJMe2002387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira E. T., Santos E. S., Monteiro J. S., Gomes M. S. M., Menezes R. A. O., & Sousa M. J. C. (2019). The use of medicinal and phytotherapy plants: An integrational review on the nurses performance. Brazilian Journal of Health Review, 2, 1511–1523. [Google Scholar]
- Guedes I. A., Magalhães C. S., & Dardenne L. E. (2014). Capítulo 9: Atracamento molecular In VERLI, H. Bioinformática da biologia à flexibilidade molecular (1a ed., pp. 189–208). SBBq. [Google Scholar]
- Gordon J. C., Myers J. B., Folta T., Shoja V., Heath L. S., & Onufriev A. (2005). H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Research, 33(Web Server issue), w368–w371. 10.1093/nar/gki464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodsell D. S. (2005). Computational docking of biomolecular complexes with Auto-Dock In Golemis EA, Adams PD (Eds.), Protein-protein interactions: A molecular cloning mannual (2nd ed). Cold Spring Harbor Laboratory Press. [Google Scholar]
- Goodsell D. S., Morris G., & Olson A. J. (1996). Automated docking of flexible ligands: Applications of AutoDock. Journal of Molecular Recognition, 9(1), 1–5. 10.1002/(SICI)1099-1352(199601)9:1<1::AID-JMR241>3.0.CO;2-6 [DOI] [PubMed] [Google Scholar]
- Kerrigan J. E. (2013). Molecular dynamics simulations in drug design In In silico models for drug discovery (pp. 95–113). Humana Press. [Google Scholar]
- Huey R., Morris G. M., & Forli S. (2012). Using AutoDock4 and AutoDock Vina with AutoDockTools: A tutorial. The Scripps Research Institute. [Google Scholar]
- Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B. A., Thiessen P. A., Yu B., Zaslavsky L., Zhang J., & Bolton E. E. (2019). PubChem 2019 update: Improved access to chemical data. Nucleic Acids Research, 47(D1), D1102–D1109. 10.1093/nar/gky1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalski L. M., & Obara A. T. (2013). The ethno-botanical study of medicinal plants at school. Ciência & Educação (Bauru), 19(4), 911–927. 10.1590/S1516-73132013000400009 [DOI] [Google Scholar]
- Lai C.-C., Shih T.-P., Ko W.-C., Tang H.-J., & Hsueh P.-R. (2020). Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. International Journal of Antimicrobial Agents, 55(3), 105924. 10.1016/j.ijantimicag.2020.105924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A., & Swindells M. B. (2011). LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. Journal of Chemical Information and Modeling, 51(10), 2778–2786. 10.1021/ci200227u [DOI] [PubMed] [Google Scholar]
- Lee T.-W., Cherney M. M., Liu J., James K. E., Powers J. C., Eltis L. D., & James M. N. G. (2007). Crystal structures reveal an induced-fit binding of a substrate-like aza-peptide epoxide to SARS Coronavirus main peptidase. Journal of Molecular Biology, 366(3), 916–932. 10.1016/j.jmb.2006.11.078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemkul J. A. (2019). From proteins to perturbed hamiltonians: A suite of tutorials for the GROMACS-2018 molecular simulation package [Article v1.0]. Living Journal of Computational Molecular Science, 1(1), 1–53. 10.33011/livecoms.1.1.5068 [DOI] [Google Scholar]
- Liu C., Zhou Q., Li Y., Garner L. V., Watkins S. P., Carter L. J., Smoot J., Gregg A. C., Daniels A. D., Jervey S., & Albaiu D. (2020). Research and development on therapeutic agents and vaccines for COVID-19 and related human Coronavirus diseases. ACS Central Science, 6(3), 315–331. 10.1021/acscentsci.0c00272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X.-Y., Zhang H.-X., Mezei M., & Cui M. (2011). Molecular docking: A powerful approach for structure-based drug discovery. Current Computer-Aided Drug Design, 7(2), 146–157. 10.2174/157340911795677602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monego D. L., Rosa M. B., & Nascimento P. C. (2017). Applications of computational chemistry to the study of the antiradical activity of carotenoids: A review. Food Chemistry, 217, 37–44. 10.1016/j.foodchem.2016.08.073 [DOI] [PubMed] [Google Scholar]
- Morris G. M., Huey R., & Olson A. J. (2008). Using AutoDock for ligand-receptor docking. Current Protocols in Bioinformatics, 24(1), 8–14. 10.1002/0471250953.bi0814s24 [DOI] [PubMed] [Google Scholar]
- Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R. K., & Olson A. J. (1998). Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry, 19(14), 1639–1662. [DOI] [Google Scholar]
- Namba A. M., Silva V. B., & Silva C. H. T. P. (2008). Dinâmica molecular: teoria e aplicações de planejamento de fármacos. Eclética Química, 33(4), 13–24. 10.1590/S0100-46702008000400002 [DOI] [Google Scholar]
- Oostenbrink C., Villa A., Mark A. E., & Van Gunsteren W. F. (2004). A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. Journal of Computational Chemistry, 25(13), 1656–1676. 10.1002/jcc.20090 [DOI] [PubMed] [Google Scholar]
- Piccirillo E., & Amaral A. T. (2018). Busca virtual de compostos bioativos: Conceitos e aplicações. Química Nova, 41, 662–677. 10.21577/0100-4042.20170210 [DOI] [Google Scholar]
- Ramos R. M., Perez J. M., Baptista L. A., & de Amorim H. L. N. (2012). Interaction of wild type, G68R and L125M isoforms of the arylamine-N-acetyltransferase from Mycobacterium tuberculosis with isoniazid: a computational study on a new possible mechanism of resistance. Journal of Molecular Modeling, 18(9), 4013–4024. 10.1007/s00894-012-1383-6 [DOI] [PubMed] [Google Scholar]
- Ravindranath P. A., Forli S., Goodsell D. S., Olson A. J., & Sanner M. F. (2015). AutoDockFR: Advances in protein-ligand docking with explicitly specified binding site flexibility. PLOS Computational Biology, 11(12), e1004586 10.1371/journal.pcbi.1004586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha J. A., Rego N. C. S., Carvalho B. T. S., Silva F. I., Sousa J. A., Ramos R. M., Passos I. N. G., de Moraes J., Leite J. R. S. A., & Lima F. C. A. (2018). Computational quantum chemistry, molecular docking, and ADMET predictions of imidazole alkaloids of Pilocarpus microphyllus with schistosomicidal properties. PLOS One, 13(6), e0198476 10.1371/journal.pone.0198476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samant L. R., & Javle V. R. K. (2020). Comparative docking analysis of rational drugs against COVID-19 main protease. ChemRxiv, 1–21. [Google Scholar]
- Schrödinger LLC (2018).The PyMOL molecular graphics system (Version 1.2r3pre). Schrödinger, LLC.
- Sharma A., Tiwari V., & Sowdhamini R. (2020). Computational search for potential COVID-19 drugs from FDA-approved drugs and small molecules of natural origin identifies several anti-virals and plant products. ChemRxiv, 1–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva S. M., Sampaio K. A., Taham T., Rocco S. A., Ceriani R., & Meirelles A. J. A. (2009). Characterization of oil extracted from buriti fruit (Mauritia flexuosa) grown in the Brazilian Amazon region. Journal of the American Oil Chemists' Society, 86(7), 611–616. 10.1007/s11746-009-1400-9 [DOI] [Google Scholar]
- Tautermann C. S., Seeliger D., & Kriegl J. M. (2015). What can we learn from molecular dynamics simulations for GPCR drug design? Computational and Structural Biotechnology Journal, 13, 111–121. 10.1016/j.csbj.2014.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Lancet (2020). Global coalition to accelerate COVID-19 clinical research in resource-limited settings. The Lancet, 395(10233), 1322–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totrov M., & Abagyan R. (2008). Flexible ligand docking to multiple receptor conformations: a practical alternative. Current Opinion in Structural Biology, 18(2), 178–184. 10.1016/j.sbi.2008.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Spoel D., Van Maaren P. J., & Berendsen H. J. C. (1998). A systematic study of water models for molecular simulation: Derivation of water models optimized for use with a reaction field. The Journal of Chemical Physics, 108(24), 10220–10230. 10.1063/1.476482 [DOI] [Google Scholar]
- Wong S. H., Lui R. N., & Sung J. J. (2020). Covid-19 and the digestive system. Journal of Gastroenterology and Hepatology, 35(5), 744–748. 10.1111/jgh.15047 [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO). (2020). Retrieved April 14, 2020, from https://www.who.int/emergencies/diseases/novel-coronavirus-2019/global-research-on-novel-coronavirus-2019-ncov.
- Xu Z., Peng C., Shi Y., Zhu Z., Mu K., Wang X., & Zhu W. (2020). Nelfinavir was predicted to be a potential inhibitor of 2019 nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. BioRxiv, 1–20. [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., & Hilgenfeld R. (2020). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science, 368(6489), 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]