
Extended Data Fig. 3. In silico confirmation of SVs in gnomAD-SV with long-read WGS.
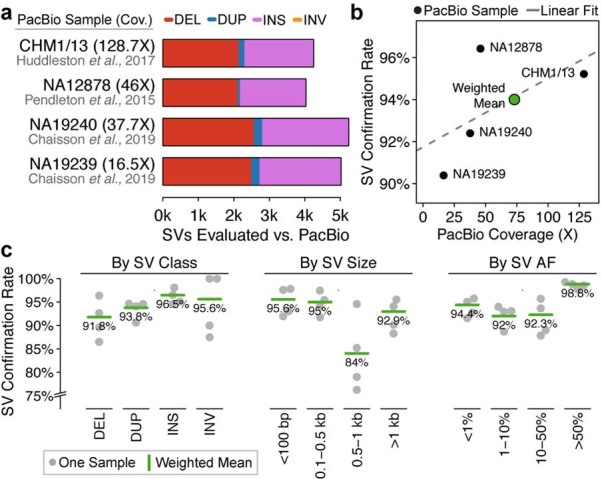
We used Pacific Biosciences (PacBio) long-read WGS data available for four samples in this study to perform in silico confirmation to estimate the positive predictive value and breakpoint accuracy for SVs in gnomAD-SV21,45,46 (Supplementary Fig. 10). a, Counts of SVs evaluated per sample in this analysis. SVs were restricted to those with breakpoint-level read support (that is, ‘split-read’ evidence, 92.8% of all SVs) and did not have breakpoints localized to annotated simple repeats or segmental duplications. b, An iterative local long-read WGS realignment algorithm, VaPoR47, was used to perform in silico confirmation of SVs predicted from short-read WGS in gnomAD-SV. As noted by the VaPoR developers47, the performance of this approach was sensitive to the sequencing depth of long-read WGS data. Therefore, the weighted mean of the four samples was used as a study-wide long-read WGS confirmation rate, weighting the confirmation rate of each sample based on the square root of its long-read WGS sequencing depth. c, Confirmation rates stratified by SV class, size and allele frequency. A mean of 4,829 SVs per sample were assessed. Horizontal green bars denote weighted means.