

Abstract
The most severe and fatal infections with SARS-CoV-2 result in the acute respiratory distress syndrome, a clinical phenotype of coronavirus disease 2019 (COVID-19) that is associated with virions targeting the epithelium of the distal lung, particularly the facultative progenitors of this tissue, alveolar epithelial type 2 cells (AT2s). Little is known about the initial responses of human lung alveoli to SARS-CoV-2 infection due in part to inability to access these cells from patients, particularly at early stages of disease. Here we present an in vitro human model that simulates the initial apical infection of the distal lung epithelium with SARS-CoV-2, using AT2s that have been adapted to air-liquid interface culture after their derivation from induced pluripotent stem cells (iAT2s). We find that SARS-CoV-2 induces a rapid global transcriptomic change in infected iAT2s characterized by a shift to an inflammatory phenotype predominated by the secretion of cytokines encoded by NF-kB target genes, delayed epithelial interferon responses, and rapid loss of the mature lung alveolar epithelial program. Over time, infected iAT2s exhibit cellular toxicity that can result in the death of these key alveolar facultative progenitors, as is observed in vivo in COVID-19 lung autopsies. Importantly, drug testing using iAT2s confirmed the efficacy of TMPRSS2 protease inhibition, validating putative mechanisms used for viral entry in human alveolar cells. Our model system reveals the cell-intrinsic responses of a key lung target cell to infection, providing a platform for further drug development and facilitating a deeper understanding of COVID-19 pathogenesis.
INTRODUCTION
Responding to the COVID-19 pandemic caused by the novel coronavirus, SARS-CoV-2, requires access to human model systems that can recapitulate disease pathogenesis, identify potential targets, and enable drug testing. Access to primary human lung in vitro model systems is a particular priority since a variety of respiratory epithelial cells are the proposed targets of viral entry1–3. A rapidly emerging literature now indicates that a diversity of epithelial cells of the respiratory tract from the nasal sinuses and proximal conducting airways through the distal lung alveoli express the cell surface receptor for SARS-CoV-2, angiotensin-converting enzyme 2 (ACE2), and appear permissive to infection with SARS-CoV-2 in vivo, and in some cases in vitro3. The most severe infections with SARS-CoV-2 result in acute respiratory distress syndrome (ARDS), a clinical phenotype that is thought to arise in the setting of COVID-19 pneumonia as the virus progressively targets the epithelium of the distal lung, particularly the facultative progenitors of this region, alveolar epithelial type 2 cells (AT2s)3. Little is known about the initial responses of human lung alveoli to SARS-CoV-2 infection due in part to the inability to access these cells from patients, particularly at early stages of disease.
To date, primary human AT2s that are harnessed from explanted lung tissues require 3D co-culture with supporting fibroblasts, cannot be maintained in culture for more than 3 passages, and tend to rapidly lose their AT2 phenotype ex vivo4. Thus, SARS-CoV-2 infection modeling has to this point been predominantly performed using either human airway (non-alveolar) cells, non-human cell lines that naturally express the ACE2 viral receptor and propagate the virus, such as the African Green Monkey Vero E6 cell line5, or transformed human cell lines with or without forced over-expression of ACE2. Although some of these transformed cell lines, such as A549 and Calu-3 cells, were originally generated from transformed cancerous lung epithelial cells, they no longer express NKX2–1, the master transcriptional regulator required for differentiated lung epithelial gene expression6, and thus are limited in their capacity to simulate an accurate lung cellular response to most perturbations, including viral infections.
To provide alternative sources of self-renewing human lung epithelial lineages, our group and others have recently developed human lung epithelial organoids and 2D air-liquid interface (ALI) lung cultures through the directed differentiation of induced pluripotent stem cells (iPSCs) in vitro6–16. Here we report the successful infection of a pure population of human iPSC-derived AT2-like cells (iAT2s) with SARS-CoV-2, providing a reductionist model that reveals the cell-intrinsic distal lung epithelial global transcriptomic responses to infection. By 1 day post-infection (dpi), SARS-CoV-2 induced a rapid global transcriptomic change in infected iAT2s characterized by a shift to an inflammatory phenotype associated with the secretion of cytokines encoded by NF-kB target genes. By 4 dpi, there were time-dependent epithelial interferon responses and progressive loss of the mature lung alveolar epithelial program. Our model system thus reveals the cell-intrinsic responses of a key lung target cell to infection, providing a novel platform for further drug development and facilitating a deeper understanding of COVID-19 pathogenesis.
RESULTS
In order to develop a human model system, we used the technique of directed differentiation4,10 to generate iAT2s from either human embryonic stem cells or iPSCs engineered to carry a tdTomato reporter targeted to the endogenous SFTPC locus9,10. In 3D Matrigel cultures, we employed our published method (4) to establish self-renewing epithelial spheres composed of purified iAT2s, >90% of which expressed surfactant protein-C (SFTPC), the canonical AT2 marker, as monitored by flow cytometry assessment of the SFTPCtdTomato reporter at each passage in culture (Fig. 1A, B). Serially passaging these epithelial spheres generated >1030 iAT2s per starting sorted tdTomato+ cell over 225 days in culture9, generating a stable source of human primary-like AT2 cells for viral infection disease modeling.
Figure 1. iPSC-derived alveolar epithelial type 2 cells (iAT2s) express functional SARS-CoV-2 entry factors ACE2 and TMPRSS2.
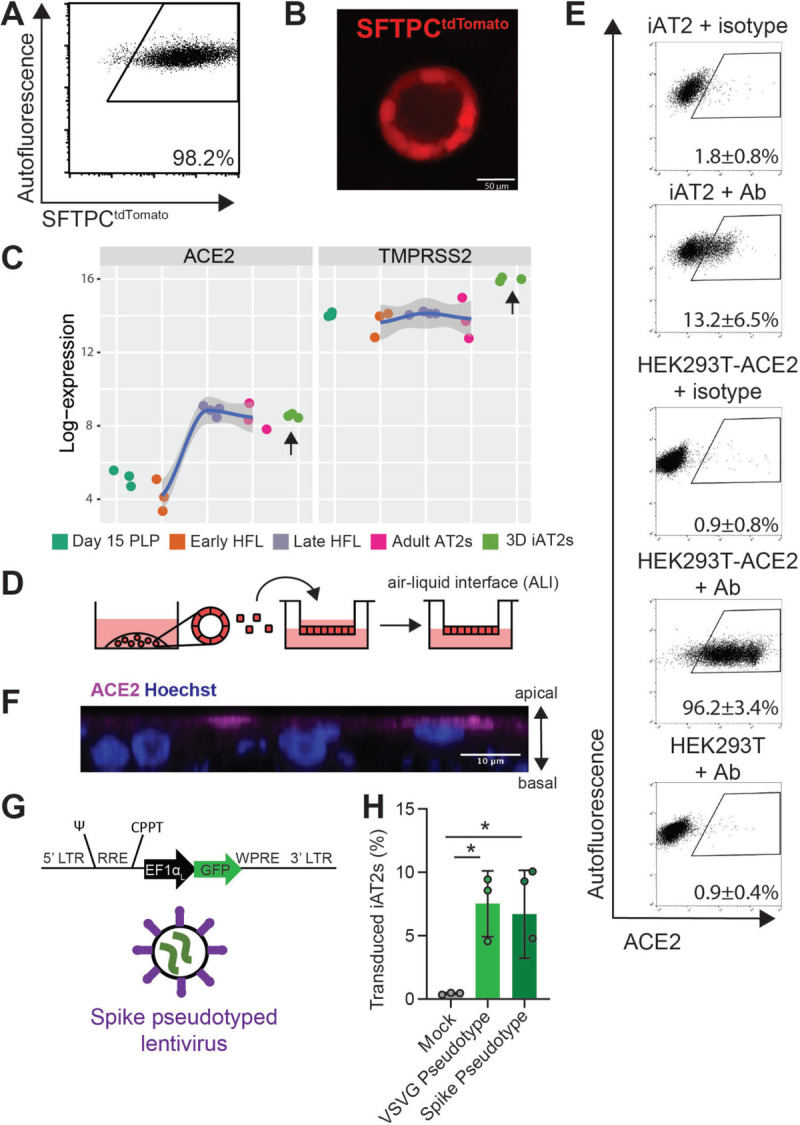
(A-B) iAT2s, carrying a tdTomato reporter targeted to the endogenous SFTPC locus by gene editing (SPC2 line), can be serially passaged, according to the method of Jacob et al (see Reference #4) while maintaining >90% SFTPCtdTomato+ expression in 3D sphere cultures (Day 160 of differentiation, passage 8 shown). (C) iAT2s (RUES2 line) express ACE2 and TMPRSS2 transcripts at comparable levels to purified primary adult human lung AT2s (adult AT2s; primary sample adult and fetal procurement described in detail in9). iAT2s (SPC2 line) adapted to 2D air-liquid interface (ALI) culture (D) express ACE2 protein, as observed by flow cytometry (E), n=9, which is apically localized, as observed by immunofluorescence staining (scale bar = 10 μm) (F). (G-H) iAT2s infected with a GFP-expressing lentivirus pseudotyped with either VSVG or SARS-CoV-2 Spike envelopes, n=3. Day 15 PLP=primordial lung progenitors derived from pluripotent stem cells at day 15 of differentiation, Early HFL=primary early human fetal lung alveolar epithelium at 16–17.5 weeks gestation; late HFL=alveolar epithelium at 20–21 weeks gestation, and adult AT2s=adult alveolar epithelial type 2 cells from 3 different individuals freshly sorted using the antibody HTII-280. *p<0.05, one-way ANOVA, all bars represent mean +/− standard deviation.
Since the directed differentiation of human iPSCs in vitro is designed to recapitulate the sequence of developmental milestones that accompanies in vivo human fetal organogenesis, we first sought to understand the ontogeny of expression of the coronavirus receptor, ACE2, during human fetal development from early endodermal progenitors through increasingly more mature stages of alveolar development. By analyzing our previously published transcriptomic time series profiles of developing human fetal and adult primary AT2s9, we found that ACE2 expression increases from early to late canalicular stages of distal human lung development, with expression levels similar to adult AT2 levels present by week 21 of gestation in developing alveolar epithelial cells (Fig. 1C). We found the directed differentiation of human pluripotent stem cells (RUES2) in vitro into purified distal lung AT2-like cells resulted in cells expressing similar levels of ACE2 to adult primary cell controls in head to head comparisons (Fig. 1C). We and others6,17 have recently profiled the frequency of ACE2 mRNA expressing primary adult and iPSC-derived AT2-like cells by single cell RNA sequencing (scRNA-Seq), finding that mRNA expression occurs in a minority of cells (1–3%) at any given time with similar frequencies observed in primary AT2s compared to iAT2s. In contrast, the gene encoding the protease utilized for viral entry, TMPRSS2, is expressed more robustly in both AT2s and iAT2s6 and is less developmentally variable, being stably expressed by week 16 of distal fetal lung development (Fig. 1C).
Because lung epithelial infection by SARS-CoV-2 occurs at the apical membrane of cells facing the air-filled lumens of airways and alveoli, submerged cultures of 3D epithelial spheres with apical membranes oriented interiorly are unlikely to faithfully recapitulate infection physiology. Therefore, we adapted our SFTPCtdTomato+ iAT2s (SPC2-ST-B2 iPSC line) to 2D ALI cultures (Fig. 1D), generating monolayered epithelial cultures of pure iAT2s with apical-basal polarization and barrier integrity, while preserving or augmenting expression of AT2-specific genes (e.g. SFTPC, SFTPB, SFTPA1, SFTPA2, NAPSA, and PGC) as detailed in our recent preprint6 as well as Figures 1 and S1.
To quantify protein-level expression frequency of the viral ACE2 receptor in iAT2s at ALI, we employed flow cytometry, observing that 13.2 ± 6.5% of live iAT2s demonstrated cell surface expression of ACE2 (Fig. 1E) and indicating more frequent expression at the protein level than had been predicted from analysis of published primary AT2 or ALI cultured iAT2 scRNA-Seq profiles6. We validated the sensitivity and specificity of the ACE2 antibody by staining controls consisting of human 293T cells, which lack ACE2, and 293T cells lentivirally transduced to over-express ACE218 (Fig. 1E). Apical localization of ACE2 protein was confirmed by immunofluorescence staining (Fig. 1F, S1B). To test functionality of ACE2 as the viral receptor, we used a GFP-expressing lentivirus pseudotyped with either viral spike protein (S) or VSV-G envelope to infect iAT2s, finding that both pseudotypes infected iAT2s equally (Fig. 1G, H).
To test whether ALI cultures of iAT2s are permissive to SARS-CoV-2 infection, we employed escalating viral doses representing a broad range of multiplicities of infections (MOIs). iAT2s at ALI were apically exposed to SARS-CoV-2 (Fig. 2A), and viral infection was visualized by immunofluorescence analysis using an antibody against the viral nucleoprotein (N). The number of N-positive cells increased over time (Fig. 2B) and with increasing MOIs (Fig. 2C), indicative of viral replication and spread. Quantification of N-positive cells by flow cytometry revealed an infection level of about 20% at 1 dpi and 60% at 4 dpi (Fig. 2D). The time- and dose-dependent increases in infection efficiencies were observed at both low MOIs (0.0004–0.04; unpurified virus) and high MOIs (up to 140; purified virus), with MOI 140 saturating N transcript levels by 1 dpi (Fig. 2E–G). Infectious virus mainly was released from the apical side of iAT2 ALI cultures at increasing titers over time (4 dpi vs 1 dpi; Fig. 2H), providing further evidence of ongoing viral replication in iAT2s in this model system. The discrepancy between the number of ACE2+ iAT2s (13%) and N+ cells at 1 dpi as well 4 dpi may suggest alternative viral entry mechanisms or the possibility that infections with large numbers of virions per target cell (such as occurs at MOI of 140) may enhance viral entry into cells with low ACE2 density. Immunofluorescence analysis of infected iAT2s suggests time-dependent cytopathogenicity as indicated by increasing numbers of fragmented nuclei from 1 to 4 dpi (Fig. 2I). Transmission electron microscopy (TEM) confirmed the successful infection of mature, functional iAT2s. Viral particles were visible both intracellularly in lamellar body-containing iAT2s as well as in the apical extracellular space adjacent to tubular myelin, an ultrastructure specific for secreted surfactant (Fig. 2J–L, S2).
Figure 2. SARS-CoV-2 infects iAT2s in a dose- and time-dependent manner.

(A) Schematic of the iAT2 directed differentiation protocol, in which robustly self-renewing iAT2s can be plated at air-liquid interface (ALI) for SARS-CoV-2 infections. “CK+DCI”=distal lung medium components detailed in the Materials and Methods. (B) Immunofluorescence images of viral nucleoprotein (N, green) of iAT2s infected with SARS-CoV-2 (MOI=140) at 1 and 4 days post infection (dpi), or (C) with increasing MOIs (14, 70, 140) shown at 1 dpi (20x, scale bar = 50μm). (D) Efficiency of iAT2 infections scored by representative FACS plots of SARS-CoV-2 N at 1 and 4 dpi (MOI 140) compared to mock; mean gated percentages +/− standard deviation for n=3 replicates are shown. (E) RT-qPCR of viral N gene expression at 1 and 4 dpi using a range of low MOIs of an unpurified SARS-CoV-2 virus stock, n=3. (F, G) RT-qPCR of N gene expression at 1 and 4 dpi using a purified virus stock to infect with an MOI of 10 (F) or an MOI of 140 (G), n=3. Fold change expression compared to Mock [2−ΔΔCt] after 18S normalization is shown. (H) Viral titers were determined in apical washes and basolateral media at 1 and 4 dpi (n=5). (I) Mean percent fragmented nuclei in immunofluorescence images of infected iAT2s at 1 and 4 dpi (MOI 140; n=3). (J) Electron micrograph of infected iAT2s showing virions (arrow heads) intracellularly, including in a lamellar body and (K) extracellular virions around tubular myelin, the meshwork (L, arrow) that forms upon secretion of pulmonary surfactant (size bar=200 nm). All bars represent mean +/− standard deviation with biological replicates indicated for each panel. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, unpaired, two-tailed Student’s t-test (H) or one-way ANOVA with multiple comparisons (E, F, G, H) were performed.
Having established a putative human model system for SARS-CoV-2 infection of AT2-like cells, next we sought to define the global, time-dependent transcriptomic responses of cells infected at the MOI we had identified as achieving the highest infection efficiency of the viral doses we had tested. We performed bulk RNA sequencing of iAT2s at 1 dpi or 4 dpi, compared to mock-infected iAT2 controls (n=3 biological replicates at each time point, Fig. 3A). Profound and progressive changes in the global transcriptomes of infected iAT2s were observed based on principle components analyses and differential gene expression (Fig. 3B, Table S1). For example, 4519 genes were differentially expressed between mock and SARS-CoV-2-infected cells at 1 dpi, 10725 genes between SARS-CoV-2-infected cells at 1 dpi and 4 dpi, and 10462 between the infected samples as a whole and mock (FDR<0.05; empirical Bayes moderated t-test; Table S1). Viral transcripts, including the viral genome, were amongst the top differentially expressed transcripts at both 1 dpi and 4 dpi, with viral transcripts representing 28% to 33% of all reads mapped at 1 dpi (Fig. 3C). AT2-specific genes, such as SFTPC, were amongst the top downregulated genes by 1 dpi, and progressive loss of the AT2 program continued to 4 dpi with significant continued loss of SFTPC, SFTPA1, SFTPD, and SFTPCtdTomato encoding transcripts (FDR<0.05; Table S1; Fig. 3C). Gene set enrichment analysis (GSEA) revealed significant upregulation of inflammatory pathways both at 1 dpi (FDR<0.05; Fig. 3D) and 4 dpi, with TNF-NFkB mediated inflammatory signaling representing the first- and second- most upregulated pathways at 1 dpi and 4 dpi, respectively, and the most upregulated pathway over the entire time course (Fig. 3D–G). Interferon (IFN) signaling was in the top 10 pathways significantly upregulated at both time points, and pathways reported to be activated by interferon signaling were also significantly upregulated (FDR<0.5) including KRAS (MAPK) signaling, IL6-JAK-STAT3, and IL2-STAT5 signaling (Figure 3D). Despite the GSEA findings, the global transcriptomic analysis showed no significant induction of individual type I and III IFN genes (e.g. IFNB1, IFNL1, or IFNL2) at any time point p.i. and only moderate upregulation of multiple IFN signaling related genes and targets (IFNAR2, IRF 1/4/7/8/9, IFIT1, MX1, CXCL10, CXCL11, SOCS3, and ISG15) mainly at 4 dpi (Fig. 3F–H). These results were confirmed by RT-qPCR (Figure 4, Fig S3) and indicate that SARS-CoV-2 infection of iAT2s elicits a delayed, modest IFN response. Of note, this IFN response pattern was observed over a wide range of MOIs (0.0004 to 140; Fig. S3).
Figure 3. SARS-CoV-2 elicits transcriptomic changes in iAT2s that highlight epithelial-intrinsic inflammatory responses to infection.

(A) Schematic of the iAT2 ALI samples (starting with Day 208 iAT2s) infected with SARS-CoV-2 (MOI 140) and collected at 1 and 4 dpi (mock collected 1 dpi) for bulk RNA sequencing (RNA-seq). B) Principal component analysis (PCA) of iAT2 samples (n=3 biological replicates per condition) showing global transcriptomic variance (%) of PC1 and PC2 components. (C) Local regression (LOESS) plots of viral, AT2, NF-kB, and interferon (IFN) gene expression levels quantified by RNA-seq normalized expression (counts per million reads). (D) Gene set enrichment analysis (GSEA, Camera using Hallmark gene sets) of the top 10 upregulated gene sets in 1 dpi vs. mock or 4 dpi vs. 1 dpi conditions (black color indicates statistical significance; FDR<0.05). (E) Unsupervised hierarchical clustered heat maps of differentially expressed genes (DEGs; FDR<0.05) in the Hallmark gene sets “TNFA signaling in NFkB” and (F) “interferon gamma response”, as plotted with row normalized Z-score; a selected subset of these DEGs are highlighted with large font. (G) Volcano plots of differentially expressed genes in 1 dpi vs. mock and (H) 4 dpi vs. 1 dpi.
Figure 4. Infection of iAT2s with SARS-CoV-2 prompts the loss of the lung AT2 program, activation of the NF-kB pathway, and modest, delayed activation of IFN signaling.

(A) Immunofluorescence staining of pro-surfactant protein C (pro-SFTPC) in tissue sections of non-COVID-19 and COVID-19 lungs. Arrow indicates AT2 hyperplasia. (B) COVID-19 descendents’ autopsied lung tissue sections, stained with H&E and (C) cytokeratin AE1/AE3 (brown) showing early acute phase of diffuse alveolar damage with sloughed alveolar epithelium (200x magnification). (D) RT-qPCR of AT2 and (E) NFkB-related transcripts in iAT2s infected with SARS-CoV-2 (MOI 140; n=3; Fold change expression over “Mock”= 2−ΔΔCt) at 1 and 4 dpi. (F) Luminex analysis of apical washes and basolateral media collected from iAT2 ALI cultures (n=5). (G) RT-qPCR of interferon-stimulated genes (ISGs) in infected (MOI 140) iAT2s at 1 and 4 dpi (n=3). (H) RT-qPCR of N gene expression at 2 dpi (MOI 1) with vehicle control, camostat (TMPRSS2 inhibitor), and E-64D (cathepsin inhibitor) treatment for 30 min, n=3. All bars represent mean +/− standard deviation.*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, one-way ANOVA with multiple comparisons were performed.
Consistent with the cytopathogenicity suggested by our microscopy studies, apoptosis was significantly upregulated over the entire time course post infection, and stress related signaling was evident by 4 dpi as multiple heat shock proteins were in the top most upregulated transcripts, comparing 4 dpi to 1 dpi (Fig. 3H; e.g. HSPA1A, HSPA1B, HSPA6, HSP90AB1). Significant downregulation of proliferation markers (TOP2A and MKI67; Figure 3G, H) was evident by 4 dpi. Taken together, these results suggest that SARS-CoV-2 infection of human iAT2s results in a cell-intrinsic shift away from an AT2 program toward an inflammatory program with NF-kB mediated inflammatory and interferon signaling pathways significantly upregulated.
To compare our findings to changes in AT2s in vivo, we performed pro-SFTPC immunostaining of lung tissue sections from the autopsies of two individuals who died from SARS-CoV-2 induced respiratory failure (clinical information provided in Supplementary Materials). In contrast to the typical, frequent pattern of pro-SFTPC immunostaining in control lung sections, COVID-19 decedent lungs exhibited regions of reduced and sporadic pro-SFTPC staining interspersed with AT2 cell hyperplasia, with additional histopathologic findings of diffuse alveolar damage, such as hyaline membrane formation. Sloughing of cells that stained positively for cytokeratin AE1/AE3 further confirmed regional injury to the alveolar epithelium, consistent with injury observed in the iAT2 in vitro model. (Fig. 4A–C).
We validated downregulation of iAT2-specific genes by RT-qPCR and observed significantly diminished expression of SFTPC, SFTPA1 and LAMP3 at 4 dpi (Fig. 4D). Moreover, we demonstrated functional activation of NF-kB signaling in infected iAT2s, as predicted by our bioinformatics analysis, by quantifying expression of NF-kB modulated target mRNAs and proteins. Upregulation of NF-kB target transcripts IL6, TNF, CXCL8, CXCL2, CXCL3, CXCL10, and CXCL11, as well as NF-kB related mRNA NFKB1, NFKB2, RELA, and RELB, was validated by RT-qPCR (Fig. 4E). Secretion of NF-kB target proteins by infected iAT2s was determined by Luminex analysis of apical washes and basolateral supernatants. IL-6 and CXCL8 (IL-8) were increased both apically and basolaterally, while GM-CSF and VEGF were secreted into the basolateral media, as has been shown previously in other models of AT2 injury19 (Fig. 4F).
Finally, to assess the potential of iAT2s to screen for COVID-19 therapeutics that might target the alveolar epithelium, we tested the effect of a recently published TMPRSS2 inhibitor, camostat mesylate, that blocks SARS-CoV-2 infection in Vero cells, Calu-3 cells, and human airway epithelial cells1, but has not been tested previously in human alveolar cells. We found camostat significantly reduced the levels of detectable viral N transcript at 2 dpi (Fig. 4H) indicating its potential as an anti-infective drug and suggesting that SARS-CoV-2 infection of iAT2s likely relies on priming by the protease, TMPRSS2, which is expressed at similar levels in both iAT2s and primary adult human AT2s (Fig. 1 and S1).
DISCUSSION
Taken together, our approach provides a new human model of SARS-CoV-2 infection of a key lung target cell which is otherwise difficult to access in vivo and hard to maintain in vitro. Since iAT2s can be propagated indefinitely in 3D culture in a form that is easily shareable between labs4, 9, our adaptation of these cells to 2D ALI cultures now allows straight-forward simulations of apical viral respiratory infections of a self-renewing cell type that can be scaled up, nearly inexhaustibly, and studied in highly pure form, thus simulating cell-autonomous or “epithelial-only” host responses to pathogens. Our results implicate AT2s as inflammatory signaling centers that respond to SARS-CoV-2 infection within 24 hours with TNF-NFkB signaling predominating this response. The significant loss of surfactant gene expression and cellular stress, toxicity, and death of iAT2s observed in our model are likely to be clinically relevant as similar observations were made in vivo in the lung autopsies of multiple COVID-19 decedents.
Importantly, IFN responses were found to be milder than TNF-NFkB signaling in our model. SARS-CoV-2 has been shown to be sensitive to IFNλ and IFNβ treatment, so the absence of a robust IFN response in AT2s, if verified in vivo, would have significant clinical implications and might suggest pathways to augment therapeutically in COVID-19 patients before they progress to ARDS20–22. In prior studies of cell lines that lacked AT2-specific gene expression, the absence of a robust IFN response was overcome by using a high MOI for infection21. This is in contrast to our results which indicate a moderate and delayed IFN response across a broad range of MOIs. Interestingly, a delayed IFN response was also observed in SARS-CoV- and MERS-CoV-infected human airway epithelial cells and is a determinant of SARS disease severity23,24.
An important caveat of our study is the well-published observation that most human lineages derived in vitro from iPSCs are immature or fetal in phenotype, possibly confounding disease modeling. However, our adaptation of iAT2s to ALI culture significantly augmented expression of maturation genes, including surfactant proteins (Fig. S1,6). This, together with the observation of SARS-CoV-2 virions intracellularly in lamellar bodies and extracellularly in the vicinity of tubular myelin confirms that surfactant-secreting, functionally mature AT2-like cells were the targets of infection in our studies. The presence of virions within lamellar bodies also implies that this surfactant-packaging organelle, specific to mature AT2 cells and absent in lung cell lines, may be a site directly utilized for and potentially dysregulated by SARS-CoV-2 infection. Thus, our model system reveals the cell-intrinsic responses of a key lung target cell to infection, facilitating a deeper understanding of COVID-19 pathogenesis and providing a platform for drug discovery.
Supplementary Material
Acknowledgments:
The authors wish to thank all members of the Boston University COVID-19 affinity research collaborative (ARC), as well as the Wilson, Hawkins, Mühlberger, Saeed, and Kotton Labs for helpful discussions. We thank Olivia Hix, Michael Herriges, Andrea Alber, Nora Lee, Marally Vedaie, Mitchell White, and Baylee Heiden for excellent technical assistance and histologic scoring; George Murphy for donation of the N RT-qPCR probe, Alejandro Balazs for donation of the spike pseudotype plasmid, 293T-ACE2 cell line, and guidance; Suryaram Gummuluru for donation of camostat and E-64D; Vickery Trinkaus-Randall for help with confocal microscopy, Yuriy Aleksyeyev for RNA sequencing, Brian Tilton for cell sorting, and Ronald Corley, Katya Ravid, and Jay Mizgerd for critical review of results.
Funding: This work was supported by Evergrande MassCPR awards to DNK, EM, and MS; NIH grant F30HL147426 to KMA; a CJ Martin Early Career Fellowship from the Australian National Health and Medical Research Council to RBW; the I.M. Rosenzweig Junior Investigator Award from the Pulmonary Fibrosis Foundation to KDA; a Harry Shwachman Cystic Fibrosis Clinical Investigator Award, Gilead Research Scholars, Gilda and Alfred Slifka, and Gail and Adam Slifka funds, and CFMS fund to RW; a Cystic Fibrosis Foundation (CFF) grant HAWKIN19XX0, Fast Grants award to EM, and NIH grants U01HL148692 and R01HL139799 to FH; NIH grants U01HL134745, U01HL134766 and R01HL095993 to DNK; NIH grants U01TR001810, UL1TR001430, R01DK101501, and R01DK117940 to AAW; R21AI135912 to EM, iPSC distribution and disease modeling is supported by NIH grants U01TR001810 and N01 75N92020C00005.
REFERENCES
- 1.Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Muller MA, Drosten C, Pohlmann S. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W, China Novel Coronavirus I, Research T. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med 2020;382:727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hou YJ, Okuda K, Edwards CE, et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacob A, Vedaie M, Roberts DA, Thomas DC, Villacorta-Martin C, Alysandratos KD, Hawkins F, Kotton DN. Derivation of self-renewing lung alveolar epithelial type II cells from human pluripotent stem cells. Nat Protoc 2019;14:3303–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harcourt J, Tamin A, Lu X, Kamili S, Sakthivel SK, Murray J, Queen K. Severe Acute Respiratory Syndrome Coronavirus 2 from Patient with Coronavirus Disease, United States. Emerging Infectious Diseases 2020;26:1266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abo KM, Ma L, Matte T, Huang J, Alysandratos KD, Werder RB, Mithal A, Beermann ML, Lindstrom-Vautrin J, Mostoslavsky G, Ikonomou L, Kotton DN, Hawkins F, Wilson A, Villacorta-Martin C. Human iPSC-derived alveolar and airway epithelial cells can be cultured at air-liquid interface and express SARS-CoV-2 host factors. bioRxiv 2020:2020.06.03.132639. [Google Scholar]
- 7.Hawkins F, Kramer P, Jacob A, Driver I, Thomas DC, McCauley KB, Skvir N, Crane AM, Kurmann AA, Hollenberg AN, Nguyen S, Wong BG, Khalil AS, Huang SX, Guttentag S, Rock JR, Shannon JM, Davis BR, Kotton DN. Prospective isolation of NKX2–1-expressing human lung progenitors derived from pluripotent stem cells. J Clin Invest 2017;127:2277–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang SX, Islam MN, O’Neill J, Hu Z, Yang YG, Chen YW, Mumau M, Green MD, Vunjak-Novakovic G, Bhattacharya J, Snoeck HW. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat Biotechnol 2014;32:84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hurley K, Ding J, Villacorta-Martin C, Herriges MJ, Jacob A, Vedaie M, Alysandratos KD, Sun YL, Lin C, Werder RB, Huang J, Wilson AA, Mithal A, Mostoslavsky G, Oglesby I, Caballero IS, Guttentag SH, Ahangari F, Kaminski N, Rodriguez-Fraticelli A, Camargo F, Bar-Joseph Z, Kotton DN. Reconstructed Single-Cell Fate Trajectories Define Lineage Plasticity Windows during Differentiation of Human PSC-Derived Distal Lung Progenitors. Cell Stem Cell 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacob A, Morley M, Hawkins F, McCauley KB, Jean JC, Heins H, Na CL, Weaver TE, Vedaie M, Hurley K, Hinds A, Russo SJ, Kook S, Zacharias W, Ochs M, Traber K, Quinton LJ, Crane A, Davis BR, White FV, Wambach J, Whitsett JA, Cole FS, Morrisey EE, Guttentag SH, Beers MF, Kotton DN. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell 2017;21:472–88 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Longmire TA, Ikonomou L, Hawkins F, Christodoulou C, Cao Y, Jean JC, Kwok LW, Mou H, Rajagopal J, Shen SS, Dowton AA, Serra M, Weiss DJ, Green MD, Snoeck HW, Ramirez MI, Kotton DN. Efficient derivation of purified lung and thyroid progenitors from embryonic stem cells. Cell Stem Cell 2012;10:398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCauley KB, Alysandratos KD, Jacob A, Hawkins F, Caballero IS, Vedaie M, Yang W, Slovik KJ, Morley M, Carraro G, Kook S, Guttentag SH, Stripp BR, Morrisey EE, Kotton DN. Single-Cell Transcriptomic Profiling of Pluripotent Stem Cell-Derived SCGB3A2+ Airway Epithelium. Stem cell reports 2018;10:1579–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCauley KB, Hawkins F, Kotton DN. Derivation of Epithelial-Only Airway Organoids from Human Pluripotent Stem Cells. Curr Protoc Stem Cell Biol 2018;45:e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCauley KB, Hawkins F, Serra M, Thomas DC, Jacob A, Kotton DN. Efficient Derivation of Functional Human Airway Epithelium from Pluripotent Stem Cells via Temporal Regulation of Wnt Signaling. Cell Stem Cell 2017;20:844–57 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serra M, Alysandratos KD, Hawkins F, McCauley KB, Jacob A, Choi J, Caballero IS, Vedaie M, Kurmann AA, Ikonomou L, Hollenberg AN, Shannon JM, Kotton DN. Pluripotent stem cell differentiation reveals distinct developmental pathways regulating lung- versus thyroid lineage specification. Development 2017;144:3879–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamamoto Y, Gotoh S, Korogi Y, Seki M, Konishi S, Ikeo S, Sone N, Nagasaki T, Matsumoto H, Muro S, Ito I, Hirai T, Kohno T, Suzuki Y, Mishima M. Long-term expansion of alveolar stem cells derived from human iPS cells in organoids. Nat Methods 2017;14:1097–106. [DOI] [PubMed] [Google Scholar]
- 17.Ziegler CGK, Allon SJ, Nyquist SK, et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020;181:1016–35.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crawford KHD, Eguia R, Dingens AS, Loes AN, Malone KD, Wolf CR, Chu HY, Tortorici MA, Veesler D, Murphy M, Pettie D, King NP, Balazs AB, Bloom JD. Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pham I, Uchida T, Planes C, Ware LB, Kaner R, Matthay MA, Clerici C. Hypoxia upregulates VEGF expression in alveolar epithelial cells in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol 2002;283:L1133–42. [DOI] [PubMed] [Google Scholar]
- 20.Broggi A, Ghosh S, Sposito B, Spreafico R, Balzarini F, Lo Cascio A, Clementi N, De Santis M, Mancini N, Granucci F, Zanoni I. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D, Wang TT, Schwartz RE, Lim JK, Albrecht RA, tenOever BR. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020;181:1036–45.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clementi N, Ferrarese R, Criscuolo E, Diotti RA, Castelli M, Scagnolari C, Burioni R, Antonelli G, Clementi M, Mancini N. Interferon-β 1a inhibits SARS-CoV-2 in vitro when administered after virus infection. J Infect Dis 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, Perlman S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016;19:181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menachery VD, Eisfeld AJ, Schäfer A, Josset L, Sims AC, Proll S, Fan S, Li C, Neumann G, Tilton SC, Chang J, Gralinski LE, Long C, Green R, Williams CM, Weiss J, Matzke MM, Webb-Robertson BJ, Schepmoes AA, Shukla AK, Metz TO, Smith RD, Waters KM, Katze MG, Kawaoka Y, Baric RS. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. mBio 2014;5:e01174–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olejnik J, Forero A, Deflubé LR, Hume AJ, Manhart WA, Nishida A, Marzi A, Katze MG, Ebihara H, Rasmussen AL, Mühlberger E. Ebolaviruses Associated with Differential Pathogenicity Induce Distinct Host Responses in Human Macrophages. J Virol 2017;91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thi Nhu Thao T, Labroussaa F, Ebert N, et al. Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform. Nature 2020;582:561–5. [DOI] [PubMed] [Google Scholar]
- 27.Wilson AA, Murphy GJ, Hamakawa H, Kwok LW, Srinivasan S, Hovav AH, Mulligan RC, Amar S, Suki B, Kotton DN. Amelioration of emphysema in mice through lentiviral transduction of long-lived pulmonary alveolar macrophages. J Clin Invest 2010;120:379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Law CW, Alhamdoosh M, Su S, Dong X, Tian L, Smyth GK, Ritchie ME. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Res 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu D, Smyth GK. Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res 2012;40:e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Habermann AC, Gutierrez AJ, Bui LT, et al. Single-cell RNA-sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. bioRxiv 2019:753806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.