

Assembly factors play a critical role in the biogenesis of mitochondrial respiratory chain complexes, some of which are present in large structures known as supercomplexes. Various assembly factors have been proposed as required for supercomplex assembly, including HIGD2A. We used quantitative proteomics and gene editing to clarify the function of human HIGD2A, revealing it to be a classical assembly factor required for biogenesis of mitochondrial DNA encoded COX3, a subunit of complex IV.
Keywords: Mitochondria function or biology, mitochondrial disease, energy metabolism, knockouts, translation, cytochrome c oxidase, mitochondria, mtDNA translation, OXPHOS, respirasome
Graphical Abstract

Highlights
Quantitative proteomics reveals HIGD2A is required for assembly of the COX3 module.
Pulse-SILAC demonstrates that HIGD2A is involved in COX3 biogenesis.
Supercomplexes in HIGD2A knockout cells are depleted of COX3.
HIGD2A is the first assembly factor identified for the COX3 module of Complex IV.
Abstract
Assembly factors play a critical role in the biogenesis of mitochondrial respiratory chain complexes I-IV where they assist in the membrane insertion of subunits, attachment of co-factors, and stabilization of assembly intermediates. The major fraction of complexes I, III and IV are present together in large molecular structures known as respiratory chain supercomplexes. Several assembly factors have been proposed as required for supercomplex assembly, including the hypoxia inducible gene 1 domain family member HIGD2A. Using gene-edited human cell lines and extensive steady state, translation and affinity enrichment proteomics techniques we show that loss of HIGD2A leads to defects in the de novo biogenesis of mtDNA-encoded COX3, subsequent accumulation of complex IV intermediates and turnover of COX3 partner proteins. Deletion of HIGD2A also leads to defective complex IV activity. The impact of HIGD2A loss on complex IV was not altered by growth under hypoxic conditions, consistent with its role being in basal complex IV assembly. Although in the absence of HIGD2A we show that mitochondria do contain an altered supercomplex assembly, we demonstrate it to harbor a crippled complex IV lacking COX3. Our results redefine HIGD2A as a classical assembly factor required for building the COX3 module of complex IV.
The electron transport chain (ETC) of human mitochondria is comprised of four multiprotein complexes (Complex I-IV) embedded in the inner mitochondrial membrane (IMM). In a process known as oxidative phosphorylation the ETC takes electrons from metabolites and uses their energy to build a proton gradient across the IMM, which in turn powers production of ATP through complex V (F1Fo ATP Synthase). Complex IV (CIV; CcO; cytochrome c oxidase) is the fourth and last enzyme of the ETC and catalyzes the transfer of electrons from cytochrome c to molecular oxygen. Complexes I, III, and IV have been shown to exist in several higher order structures known as respiratory chain supercomplexes or respirasomes (1–4). These structures have been suggested to reduce production of reactive oxygen species (ROS), improve complex stability and/or distribution throughout the protein dense environment of the IMM, and allow efficient channeling of substrates (3–8).
Complex IV is composed of 14 different subunits of dual genetic origin, the core membrane embedded subunits, COX1, COX2, and COX3 are encoded by mitochondria DNA (mtDNA) genes MT-CO1, MT-CO2, and MT-CO3, that are translated on mitochondrial ribosomes in the matrix, whereas the remaining 11 subunits are encoded by nuclear DNA, translated in the cytosol and imported into the mitochondria where they assemble with mtDNA encoded subunits. Nuclear encoded subunits can therefore be grouped into assembly modules seeded by each of the mtDNA encoded subunits (Fig. 1) (9, 10). During complex IV biogenesis, redox active heme and copper cofactors are incorporated into COX1 and COX2, which together with COX3 form the conserved core of the enzyme. Although COX3 does not have a direct role in electron transport, it is thought to play a regulatory role in modulating complex IV activity (11, 12). Homologs of COX3 are present in complex IV throughout the tree of life (13) and mutations within MT-CO3 lead to complex IV deficiency and mitochondrial encephalopathy or myopathy (MIM 516050) (14, 15) implicating the importance of COX3 in complex IV and mitochondrial function.
Fig. 1.

Human complex IV colored by subunit module (PDB 5Z62). Core mtDNA-encoded subunits (COX1 = light pink, COX2 = magenta, COX3 = blue) are rendered as surface representation whereas nuclear DNA-encoded subunits are presented as ribbons.
Biogenesis of complex IV occurs through the coordinated effort of more than 30 known assembly factors with roles including control of mitochondrial RNA stability and translation, membrane insertion of mtDNA encoded subunits, copper and heme biosynthesis and loading and stabilization of subunit modules during assembly (9, 10, 16). Although several assembly factors are known to be essential for stabilizing the modules containing COX1 and COX2, to date no assembly factors have been associated with the COX3 module (containing COX3, COX6A, COX6B, and COX7A) in humans. Two assembly factors have also been suggested to support complex IV-containing supercomplex assembly, COX7A2L (SCAFI) and hypoxia inducible gene 1 domain family member HIGD2A (17). COX7A2L was originally shown to be present within the supercomplex but not in the holo form of complex IV (6). A mouse strain harboring a truncated form of the protein lacked some forms of the supercomplexes and exhibited altered mitochondrial respiration, leading the authors to suggest the protein is required for supercomplex stability (6). Several studies have disputed this finding (18–23) and it is now understood that COX7A2L plays a role as a checkpoint for assembly of CIII2 into the supercomplex (17), a result that explains the differences in supercomplex distribution observed in mitochondria lacking full length COX7A2L. Yeast (S. cerevisiae) homologs of HIGD2A (Rcf1 and Rcf2) belong to the hypoxia inducible gene 1 domain containing (HIGD) family and were originally suggested to be required for assembly and stability of the yeast CIII2/IV supercomplex (24–26). Deletion of Rcf1 leads to impaired assembly of Cox12 and Cox13 (COX6B and COX6A in humans) into complex IV, reduced association with Rcf2, and a reduction in the levels CIII2/IV supercomplex (24–26). Recent studies have suggested a regulatory role for Rcf1 and Rcf2 in complex IV activity (27) through association with complex IV intermediates (28). Investigation into the function of mammalian homologs HIGD1A and HIGD2A have been limited to only a few studies. Both HIGD1A and HIGD2A were originally identified in a screen for genes up-regulated during HIF1α dependent hypoxia (29, 30). Following discovery of Rcf1 and Rcf2 in yeast supercomplex assembly, Chen et al. (26) showed that knockdown of Higd1a in mouse myoblasts had no impact on respiratory chain supercomplexes whereas, knockdown of Higd2a led to reduced amounts of CIII2/CIV and a higher-order assembly now known to be the respiratory chain megacomplex (CI2/III2/IV2) (2). No impact was observed on the levels of holo complex IV, leading the authors to suggest Higd2a is a supercomplex assembly factor, consistent with the studies in yeast (26). In contrast, HIGD1A has recently been suggested to associate non-stoichiometrically with an early assembly intermediate consisting of COX1 and associated subunits (10) and induce structural changes around the heme a active center altering complex IV activity during hypoxic stress (31).
We identified HIGD2A as a protein strongly up-regulated in lymphoblasts from a patient with ETC complex IV deficiency. Given that the yeast homolog of HIGD2A has been previously associated with either complex IV or respiratory chain supercomplex assembly, we aimed to clarify the function of human HIGD2A as well as its ortholog HIGD1A. Using CRISPR/Cas9 generated knockout cell lines we show that loss of HIGD1A leads to a modest defect in mitochondrial oxygen consumption and stability of subunits present within the COX1 module but does not affect complex assembly. Although we find HIGD1A is indeed induced during hypoxia, this results in no apparent changes to complex IV or supercomplex assembly. Conversely, we demonstrate that loss of HIGD2A leads to a strong defect in mitochondrial respiration, complex IV assembly and activity because of impaired biogenesis of newly translated COX3, and subsequent degradation of nuclear encoded subunits present within the COX3 module. We conclude that HIGD2A is a classical assembly factor involved in the early steps of COX3 biogenesis.
EXPERIMENTAL PROCEDURES
Tissue Culture and Generation of Cell Lines
HEK293T cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM High Glucose) supplemented with 10% (v/v) fetal calf serum (FCS; CellSera, Rutherford, NSW, Australia), penicillin/streptomycin (Thermo Fisher Scientific, Scoresby, VIC, Australia) and 50 μg ml−1 uridine (Sigma-Aldrich, North Ryde, NSW, Australia) at 37 °C under an atmosphere of 5% CO2. Patient lymphoblasts were obtained with familial informed consent as part of diagnostic investigations, and associated studies were approved by the Royal Children's Hospital Institutional Review Board (HREC reference number 34228). Early passage subclones of a single lymphoblast clone derived from whole patient and control blood were cultured under the same conditions, except that the medium was RPMI 1640 and the amount of FCS was 20% (v/v). Gene editing was performed in HEK293T (originally purchased from ATCC) and the NDUFV3KONDUFV3FLAG cell line published previously (32). Constructs for CRISPR-Cas9 genome editing were designed using the CHOPCHOP software (33) and oligonucleotides encoding gRNA sequences cloned into the pSpCas9(BB)-2A-GFP (PX458) plasmid (a gift from F. Zhang; Addgene, plasmid 48138; (34)) as previously described (32). The gRNA sequences used were 5′-ATGTCAGTCAAACTTACCAA for HIGD1A and 5′-ACTTACCTATGGGTACCACC for HIGD2A. Constructs were transfected using Lipofectamine LTX (Thermo Fisher Scientific) according to manufacturer's instructions. Single GFP positive cells were sorted into 96 well plates and clonal populations expanded for screening by SDS-PAGE and immunoblotting as described below. To verify the CRISPR/Cas9 induced insertions and deletions (indels) genomic DNA was isolated from candidate clones using the Quick-DNA kit (Zymo Research, Irvine, CA) according to manufacturer's specifications. Oligonucleotides provided by the CHOPCHOP software (33) were used for amplification of target regions and cloning into pGEM4Z (35) for M13 primed Sanger sequencing of individual alleles as we have done previously (32). HIGD1AKO contained the following mutations c.[97 + 2G_97 + 6Gdel];[97 + 2G_97 + 14Gdel];[97_97 + 3Tdel] predicted to impact splicing. HIGD2AKO contained c.[145del]; [131_150del];[133_149del];[138_158del];[143_153 + 49Gdel] predicted to alter the frame and introduce premature stop codons. NDUFV3KO NDUFV3FLAGHIGD2AKO contained the mutations c.[127_128insA;136_145del];[142_151del];[141_150del];[144_146del];[144_150del]; [143_147delinsGTGGAACGCCCGGCCTCAGTGAGCGAGCGAGCGCGCAGAGAGGGAGTGGCCAACTCCATCACTAGGGGTTCCTGCGGCCGCTCCCCAGCATGCCTGCTATTCTCTTCCCAATCCT] predicted to alter the reading frame and introduce premature stop codons. For hypoxia treatment, cells were cultured inside a hypoxia incubator chamber (STEMCELL Technologies, Vancouver, BC, Canada) pre-purged with 1% O2, 5% CO2, 94% N2 for 10 min at 25 liters/min. The sealed chamber was incubated at 37 °C for 3 or 24 h as indicated. For pulse-SILAC analysis of mitochondrial translation, cells were incubated in DMEM as above with the overnight addition of 50 μg ml−1 chloramphenicol (CAP; Sigma-Aldrich) prior to the start of the experiment. Media was replaced with neat DMEM for SILAC (Thermo Fisher Scientific) for 15 min following which the media was supplemented with 10% dialyzed FCS (Thermo Fisher Scientific), Sodium Pyruvate (Thermo Fisher Scientific), Glutamax (Thermo Fisher Scientific), penicillin/streptomycin (Thermo Fisher Scientific), 3.5 g/L glucose and 0.1 mg/ml cycloheximide (Sigma-Aldrich). Following an incubation for 30 min at 37 °C under an atmosphere of 5% CO2, media was further supplemented 1.2 g/ml l-proline, 285 mg/ml -13C615N2-l-lysine-HCl and 85 mg/L-13C615N2-l-arginine-HCl (Silantes, Munich, Germany). Time points were collected at 0, 1, 2, and 4 h post addition of SILAC amino acids.
Patient Sequencing and Enzymology Assays
The AARS2 patient was a boy with intrauterine growth retardation who had failure to thrive, hypotonia and developmental delay noted in the first months of life. He had metabolic acidosis with plasma lactate levels up to 12 mmol/L (normal range <2.5 mmol/L) and developed cortical blindness and temperature instability by 6 months of age with cerebral atrophy. From about 8 months of age he suffered a rapidly progressive neurodegenerative course with marked spasticity, decorticate posturing and was in a vegetative state prior to his death at 16 months of age. DNA was extracted from skin fibroblasts and whole exome sequencing was performed as published (per subject 1 in reference (36)). Our filtering strategy identified 20 rare, heterozygous, high quality, moderate-high impact variants in this patient. Most pediatric mitochondrial disorders are autosomal recessive in origin, AARS2 was the only gene in which two such variants were identified. Electron transport chain enzyme activities in skeletal muscle biopsies (post-660g supernatants) and mitochondrial preparations from Epstein-Barr Virus (EBV) transformed lymphocytes (lymphoblasts) were determined spectrophotometrically as previously described (37). Enzymes assayed were complex I (NADH-coenzyme Q1 oxidoreductase), complex II (succinate-coenzyme Q1 oxidoreductase), complex III (decylbenzylquinol-cytochrome c oxidoreductase), complex IV (cytochrome c oxidase) plus the mitochondrial marker enzyme citrate synthase.
Retroviral Transduction
To rescue HIGD2A expression in HIGD2AKO a construct encoding HIGD2A with a C-terminal FLAG tag and compatible overhangs for Gibson assembly was commercially synthesized (IDT technologies, Singapore). The construct was combined with pBABE-puro plasmid (Addgene, 1764) cut with BamHI-HF and HindIII-HF restriction enzymes (NEB, Notting Hill, VIC, Australia) and Gibson assembled using the NEBuilder HiFi DNA Assembly System (NEB) as per manufacturer's directions to yield pBabe-puro-HIGD2AFLAG. The construct was used to generate viral particles in HEK293T cells as previously described (32). Briefly, pBabe-puro-HIGD2AFLAG was co-transfected alongside pUMVC3 and pCMV-VSV-G (Addgene, 8449 and 8454) using Lipofectamine LTX (Thermo Fischer Scientific) according to manufacturer's instructions. Viral supernatant was collected at 48 h post-transfection, filtered with 0.45 μm PVDF membrane (Merck, Bayswater, VIC, Australia) and used to infect HIGD2AKO cells in the presence of 8 μg ml−1 polybrene. Cells were selected in puromycin at 2 μg/ml for 10 days. Transduction was verified by SDS-PAGE and immunoblotting.
SDS-PAGE, BN-PAGE and Western Blotting
SDS-PAGE was performed using samples solubilized in LDS sample buffer (Thermo Fisher Scientific) with presence of 50 mm DTT and separated on Invitrogen Bolt Bis-Tris protein gels (10% or 4–12%) using the MES running buffer according to manufacturer's instructions (Thermo Fischer Scientific). For assessing the effect of hypoxia treatment, cells were solubilized in RIPA buffer (25 mm Tris-HCl pH7.4, 150 mm NaCl, 0.1% (w/v) SDS, 1% (v/v) Triton X-100) supplemented with 1 mm PMSF and protease inhibitor mixture (cOmplete™, Merck) on ice. BN-PAGE was performed using mitochondrial protein solubilized in 1% digitonin (w/v) based solubilization buffer as described previously (38, 39). Mitochondria were isolated as previously described (40) with protein concentration determined using the Pierce Protein Assay Kit (Thermo Fisher Scientific). Detergent solubilized complexes were separated on Invitrogen NativePAGE Bis-Tris gels (3–12%) as per manufacturer's instructions. Western blotting of SDS-PAGE gels was undertaken using the Invitrogen iBlot2 Dry Blotting System using PVDF based stacks. The Invitrogen Mini Blot Module transfer system was used to transfer BN-PAGE gels onto PVDF (Merck) as per manufactures recommendations. Immunoblots were developed using horseradish peroxidase coupled mouse and rabbit secondary antibodies (Cell Signaling Technology, Danvers, MA) and ECL chemiluminescent substrates (Bio-Rad) and visualized on a ChemiDoc XRS+ gel documentation system (Bio-Rad, Gladesville, NSW, Australia). Commercial antibodies were obtained for COX1 (Abcam, Melbourne, VIC, Australia; ab14705), COX2 (Abcam, ab110258), COX4 (Abcam, ab110261), COX6A1 (Proteintech, Rosemont, IL, 11460-1-AP), FLAG (Sigma-Aldrich, F1804), GAPDH (Proteintech, HRP-60004), HIF-1α (Santa Cruz, Dallas, TX, SC-53546), HIGD1A (Proteintech, 21749-1-AP), HIGD2A (Proteintech, 21415-1-AP), SDHA (Abcam, Ab14715), UQCRC1 (Abcam, AB110252), β-actin (Proteintech, HRP-60008) and used at the recommended dilutions. In-house polyclonal rabbit antibodies for NDUFA9 were previously described (39). In-gel activity assays were performed as described previously (41).
Oxygen Consumption Measurements
Mitochondrial oxygen consumption rates were measured in live cells using a Seahorse Bioscience XFe-96 Analyzer (Agilent Mulgrave, VIC, Australia). Briefly, 25,000 cells were plated per well in culture plates treated with 50 μg/ml poly-d-lysine (Sigma-Aldrich). For each assay cycle, there were 4 measurements of 2 min followed by mixing, 2 min wait, and 5 min measure. The following inhibitor concentrations were used: 1 μm oligomycin, 1 μm carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), 0.5 μm rotenone and 0.5 μm antimycin A. Data were normalized using the Pierce Protein Assay Kit (Thermo Fisher Scientific) and analyzed using Wave (version 2.6.0, Agilent) and Prism (version 8.0.2, GraphPad) software packages. For substrate driven oxygen consumption, cells were permeabilized in 0.025% (w/v) digitonin as described (42). For each assay cycle, there were 4 measurements of 2 min mix, 2 min wait and 5 min measure. The following inhibitor concentrations and combinations were used. Port A: TMPD/ascorbate 0.5 mm/2 mm, 1 μm FCCP, ADP 1 mm; Port B: 1 μm oligomycin; Port C: Potassium azide 20 mm. Data were normalized and analyzed as above.
Acquisition and Analysis of Steady State Mass Spectrometry Data from Patient Lymphoblasts
Primary lymphoblasts (two subcultures from the same individual) and controls (from three separate individuals) were pre-normalized based on protein concentration using the Pierce Protein Assay Kit (Thermo Fisher Scientific). Pellets were solubilized in 1% (w/v) SDC, 100 mm Tris pH 8.1, 40 mm chloroacetamide (Sigma-Aldrich) and 10 mm tris(2-carboxyethyl)phosphine hydrochloride (TCEP; BondBreaker, Thermo Fisher Scientific) for 5 min at 99°C with 1500 rpm shaking followed by 15 min sonication in a waterbath sonicator. Protein digestion was performed with trypsin (Thermo Fisher Scientific) at a 1:50 trypsin:protein ratio at 37°C overnight. The supernatant was transferred to stagetips containing 3 × 14G plugs of 3M™ Empore™ SDB-RPS substrate (Sigma-Aldrich) as described previously (32, 43). Ethyl acetate (99% v/v) and 1% (v/v) TFA was added to the tip before centrifugation at 3000 g at room temperature. Stagetips were washed first with the same ethyl acetate containing solution and then subjected to an additional wash with 0.2% (v/v) TFA. Peptides were eluted in 80% (v/v) acetonitrile (ACN) and 1% (w/v) NH4OH, and then acidified to a final concentration of 1% TFA prior to drying in a CentriVap Benchtop Vacuum Concentrator (Labconco, Kansas City, MO) and reconstituted in 0.1% TFA and 2% ACN for analysis by Liquid chromatography (LC) - MS/MS on an Orbitrap QExactive Plus (Thermo Fisher Scientific) coupled with an Ultimate 3000 HPLC (Thermo Fisher Scientific) and NanoESI interface. Peptides were injected onto the trap column (Acclaim C18 PepMap nano Trap x 2 cm, 100 μm I.D, 5 μm particle size and 300 Å pore size; Thermo Fisher Scientific) at a flow rate of 15 μl/min before switching the trap in-line with the analytical column (Acclaim RSLC C18 PepMap Acclaim RSLC nanocolumn 75 μm × 50 cm, PepMap100 C18, 3 μm particle size 100 Å pore size; Thermo Fisher Scientific). The peptides were eluted using a 250 nL/min non-linear ACN gradient of buffer A (0.1% FA, 2% ACN) buffer B (0.1% FA, 80% ACN) increasing from 2.5% to 35.4% followed by a ramp to 99% over 278 min. Data were acquired in positive mode using data dependent acquisition 375–1800 m/z as a scan range, HCD for MS/MS of the top 12 intense ions with charges ≥ 2. MS1 scans were acquired at 70,000 resolution (at 200 m/z), MS max. injection time of 54 ms, AGC target 3e6, NCE at 27%, isolation window of 1.8 Da, MS/MS resolution of 17,500, MS/MS AGC target of 2e5. Raw files were processed using the MaxQuant platform (version 1.6.0.16) (44) and searched against UniProt human database (93,588 entries, Aug 2017) and a list of common contaminants using default settings for a label free quantification (LFQ) experiment with “LFQ,” “re-quant” and “Match between runs” enabled. For this search Trypsin/P cleavage specificity (cleaves after lysine or arginine, even when proline is present) was used with a maximum of 2 missed cleavages. Oxidation of methionine and N-terminal acetylation were specified as variable modifications. Carbamidomethylation of cysteine was set as a fixed modification. A search tolerance of 4.5 ppm was used for MS1 and 20 ppm for MS2 matching. False discovery rates (FDR) were determined through the target-decoy approach set to 1% for both peptides and proteins. The proteinGroups.txt output from the search was processed in Perseus (version 1.6.2.2) (45). Briefly, Log2-transformed LFQ Intensities were grouped into patient or control groups consisting of three controls and two replicates of the patient lymphoblasts. Identifications filtered to include only those that were quantified in at least two samples from each group. Annotations for proteins present in the Mitocarta2.0 dataset (46) were added through matching by gene name. A two-sided t-test with significance determined by permutation-based FDR statistics (FDR 5%, S0 = 0.1) was performed with results expressed as a volcano plot.
Acquisition and Analysis of Steady State Mass Spectrometry Data from HEK293T Cells
Crude mitochondrial protein from HEK293T, HIGD2AKO, and HIGD1AKO cells and whole cell material from hypoxia treated HEK293T cells (derived from two independent subcultures of each) were pre-normalized using the Pierce Protein Assay Kit (Thermo Fisher Scientific). Protein pellets were solubilized and digested into tryptic peptides for mass spectrometry using the iST-NHS kit (PreOmics GmbH, Planegg, Germany) as per manufacturer instructions. Peptides were labeled with 6plex Tandem Mass Tags (TMT) (Thermo Fisher Scientific) in 8:1 label:protein ratio as per manufacturer instructions. Pooled samples were fractionated using the Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo Fisher Scientific) as per manufacturer's instructions. Individual fractions were dried using a CentriVap Benchtop Vacuum Concentrator (Labconco) and reconstituted in 2% (v/v) acetonitrile (ACN) and 0.1% (v/v) trifluoroacetic acid (TFA). Liquid chromatography (LC) coupled MS/MS was carried out on an Orbitrap Lumos mass spectrometer (Thermo Fisher Scientific) with a nanoESI interface in conjunction with an Ultimate 3000 RSLC nanoHPLC (Dionex Ultimate 3000). The LC system was equipped with an Acclaim Pepmap nano-trap column (Dionex-C18, 100 Å, 75 μm × 2 cm) and an Acclaim Pepmap RSLC analytical column (Dionex-C18, 100 Å, 75 μm × 50 cm). The tryptic peptides were injected to the trap column at an isocratic flow of 5 μl/min of 2% (v/v) CH3CN containing 0.1% (v/v) formic acid for 5 min applied before the trap column was switched in-line with the analytical column. The eluents were 5% DMSO in 0.1% v/v formic acid (solvent A) and 5% DMSO in 100% v/v CH3CN and 0.1% v/v formic acid (solvent B). The flow gradient was (1) 0–6min at 3% B, (2) 6–95 min, 3–22% B (3) 95–105 min 22–40% B (4) 105–110 min, 40–80% B (5) 110–115 min, 80–80% B (6) 115–117 min, 80–3% and equilibrated at 3% B for 10 min before the next sample injection. The Synchronous Precursor Selection (SPS)–MS3 based TMT method was used. In brief, a full MS1 spectra was acquired in positive mode at 120,000 resolution scanning from 380–1500 m/z. AGC target was at 4e5 and maximum injection time of 50ms. Precursors for MS2/MS3 analysis were selected based on a Top 3 s method. MS2 analysis consists of collision induced dissociation (CID) with isolation window of 0.7 in the quadrupole. CID was carried out with normalized collision energy of 35 and activation time of 10 ms with detection in the ion trap. Following acquisition of each MS2 spectrum, multiple MS2 fragment ions were captured in the MS3 precursor population using isolation waveforms frequency notches. The MS3 precursors were fragmented by high energy collision-induced dissociation (HCD) and analyzed using the Orbitrap with scan range of 100–500 m/z, 60,000 resolution, normalized collision energy of 60%, AGC target of 1e5 and maximum injection time of 120ms. Raw files were processed using the MaxQuant platform (version 1.6.3.3) (44) and searched against UniProt human database (42,434 entries, June 2019) using default settings for a TMT 6plex experiment with the following modifications: deamination (NQ), oxidation of methionine and N-terminal acetylation were specified as variable modifications. Trypsin/P cleavage specificity (cleaves after lysine or arginine, even when proline is present) was used with a maximum of 2 missed cleavages. Carbamidomethylation of cysteine was set as a fixed modification. A search tolerance of 4.5 ppm was used for MS1 and 20 ppm for MS2 matching. False discovery rates (FDR) were determined through the target-decoy approach set to 1% for both peptides and proteins. The proteinGroups.txt output from the search was processed in Perseus (version 1.6.2.2) (45). Briefly, Log2-transformed TMT reporter intensity corrected values were grouped into wild-type, HIGD2A and HIGD1A groups consisting of two replicates each. Identifications filtered to include 100% valid values across all samples. Annotations for proteins present in the Mitocarta2.0 dataset (46) were added through matching by gene name and rows filtered to include only mitochondrial entries. A two-sided t test with significance determined by permutation-based FDR statistics (FDR 5%, S0 = 1) was performed with results expressed as a volcano plot. Ratios for individual subunits relative to controls were mapped onto the Cryo-EM PDB maps cited in the figure legends using a custom python script described previously (32).
Acquisition and Analysis of Pulse-SILAC Mass Spectrometry Data
Mitochondrial protein pellets (from three independent subcultures) were solubilized in 1% (w/v) SDC, 100 mm Tris pH 8.1, 40 mm chloroacetamide (Sigma) and 10 mm tris(2-carboxyethyl)phosphine hydrochloride (TCEP; BondBreaker, Thermo Fisher Scientific) for 5 min at 99°C with 1500 rpm shaking followed by 15 min sonication in a waterbath sonicator. Protein digestion was performed with trypsin (Thermo Fisher Scientific) at a 1:50 trypsin/protein ratio at 37°C overnight. The supernatant was transferred to stagetips containing 3 × 14G plugs of 3M™ Empore™ SDB-RPS substrate (Sigma-Aldrich) as described previously (32, 43) and above. Analysis of reconstituted peptides was performed by LC MS/MS on a QExactive Plus Orbitrap mass spectrometer (Thermo Fisher Scientific) in conjunction with an Ultimate 3000 RSLC nanoHPLC (Dionex Ultimate 3000). The LC system was as described above (Acquisition and Analysis of Steady State Mass Spectrometry Data from HEK293T Cells). For this experiment the mass spectrometer was operated in the data-dependent mode with a targeted inclusion list containing predicted peptides from the 13 mitochondrial DNA-encoded proteins. The inclusion list consists of mass/charge (m/z) and charge (z) of tryptic peptides (endogenous and SILAC labeled) predicted from in-silico digest of target proteins using the Skyline software (47). In addition, the inclusion list also contained peptides that have been previously observed in public data depositories through the PeptideAtlas site (48) and the present study. Full MS1 spectra were acquired in positive mode, 140,000 resolution, AGC target of 3e6 and maximum IT time of 50 ms. A loop count of 10 on the most intense targeted peptide were isolated for MS/MS. The isolation window was set at 1.2 m/z and precursors fragmented using stepped normalized collision energy of 28, 30 and 32. Resolution was at 35,000 resolution, AGC target at 2e5 and maximum IT time of 150 ms. Dynamic exclusion was set to be 30 s. Raw files were processed using the MaxQuant platform (version 1.6.3.3) (44) and searched against UniProt human database (20,336 entries, May 2018) using default settings for a SILAC experiment. For this search Trypsin/P cleavage specificity (cleaves after lysine or arginine, even when proline is present) was used with a maximum of 2 missed cleavages. Oxidation of methionine and N-terminal acetylation were specified as variable modifications. Carbamidomethylation of cysteine was set as a fixed modification. A search tolerance of 4.5 ppm was used for MS1 and 20 ppm for MS2 matching. False discovery rates (FDR) were determined through the target-decoy approach set to 1% for both peptides and proteins. The “Requant” option was set to “on.” The proteinGroups.txt output from the search was processed in Perseus (version 1.6.2.2) (45). Briefly, heavy intensity values for the detected mtDNA encoded proteins were Log2 transformed and values were normalized to the maximum value detected in the control 4 h condition. The means from three experiments were plotted over time using Prism (version 8.1.2, GraphPad) along with the standard deviation from the mean of replicates.
Acquisition and Analysis of Affinity Enrichment Mass Spectrometry Data
Affinity enrichment experiments were performed on complemented HIGD2AFLAG cells. In brief, 1 mg of whole-cell protein pellets from HEK293T and HIGD2AFLAG (derived from three subcultures of each) were harvested in triplicate and solubilized in 20 mm Tris-Cl pH 7.4, 50 mm NaCl, 10% (v/v) glycerol, 0.1 mm EDTA, 1% (w/v) digitonin and 125 units of benzonase (Merck). Pierce™ Spin Columns (Thermo Fisher Scientific) were loaded with 40 μl anti-FLAG M2 affinity gel (Sigma-Aldrich) and equilibrated with wash buffer (20 mm Tris-Cl pH 7.4, 60 mm NaCl, 10% v/v glycerol, 0.5 mm EDTA, 0.1% w/v digitonin). Clarified (20,000 × g, 5 mins at 4°C) lysate was added to spin columns containing FLAG beads and incubated at 4 °C for 2 h in rotating mixer. Columns were attached to a vacuum manifold and washed 30 times with 500 μl of wash buffer. For elution, 100 μl of wash buffer containing 100 μg/ml of FLAG peptide; Sigma-Aldrich) for 30 min at 4 °C prior to low speed centrifuge and collection into a microcentrifuge tube. Another 100 μl of wash buffer (not containing FLAG peptide) was added and both elutions were pooled into the same tube. Eluted proteins were precipitated with 5× volume of ice-cold acetone overnight at −20 °C. On the next day, samples were centrifuges at max speed for 10 min at room temperature and the precipitated protein pellet was solubilized with 8 m urea in 50 mm ammonium bicarbonate (ABC) followed by sonication for 15 min in a sonicator water bath. To reduce and alkylate proteins tris(2-carboxyethyl)phosphine hydrochloride (TCEP; Bondbreaker, Thermo Fisher Scientific) and chloroacetamide (Sigma-Aldrich) were added to a final concentration of 10 mm and 50 mm respectively and incubated at 37 °C for 30 min while shaking. Samples were diluted to 2 m urea using 50 mm ABC prior to digestion with 1 μg of trypsin (Thermo Fisher Scientific) at 37 °C overnight. Peptides were acidified to 1% TFA and desalted using stagetips (43) containing 2 × 14G plugs of 3M™ Empore™ SDB-XC Extraction Disks (Sigma-Aldrich) which were pre-activated with 100% acetonitrile (ACN) and equilibrated with 0.1% TFA, 2% ACN prior to sample loading. Column was washed with 0.1% TFA, 2% ACN and samples were eluted with 80% ACN, 0.1% TFA. All centrifugation steps were performed at 1,800 g. Elutions were dried using CentriVap concentrator (Labconco) and samples were reconstituted in 0.1% TFA, 2% ACN. LC-MS/MS was carried out on a LTQ Orbitrap Elite (Thermo Scientific) in conjunction with an Ultimate 3000 RSLC nanoHPLC (Dionex Ultimate 3000). The LC system was as described above (Acquisition and Analysis of Steady State Mass Spectrometry Data from HEK293T Cells). The LTQ Orbitrap Elite was operated in the data-dependent mode with nanoESI spray voltage of 1.8kV, capillary temperature of 250 °C and S-lens RF value of 55%. All spectra were acquired in positive mode with full scan MS spectra from m/z 300–1650 in the FT mode at 120,000 resolution. Automated gain control was set to a target value of 1.0e6. Lock mass of 445.120025 was used. The top 20 most intense precursors were subjected to rapid collision induced dissociation (rCID) with normalized collision energy of 30 and activation q of 0.25. Dynamic exclusion with of 30 s was applied for repeated precursors. Raw files were analyzed using MaxQuant platform (44) (version 1.6.5.0) and searched against UniProt human database (42,410 entries, January 2019) using default LFQ search parameters with the following modifications: LFQ min. ratio count and label min. ratio count = 1. For this search Trypsin/P cleavage specificity (cleaves after lysine or arginine, even when proline is present) was used with a maximum of 2 missed cleavages. Oxidation of methionine and N-terminal acetylation were specified as variable modifications. Carbamidomethylation of cysteine was set as a fixed modification. A search tolerance of 4.5 ppm was used for MS1 and 20 ppm for MS2 matching. False discovery rates (FDR) were determined through the target-decoy approach set to 1% for both peptides and proteins. Data analysis was performed using Perseus framework (45) (version 1.6.2.2). Briefly, LFQ intensities imported from the proteinGroups.txt output were Log2 transformed. Only proteins quantified across all three IP samples (with no restrictions on control identifications) were included and the missing control values imputed at the limit of detection based on normal distribution. Experimental groups were assigned to each set of triplicates and a two-sided t test performed with significance determined by permutation-based FDR statistics (FDR 5%, S0 = 1) to exclude all identifications enriched in control samples.
Experimental Design and Statistical Rationale
For label-free and TMT-labeled analyses of whole cells or mitochondria, the statistical approaches used to analyze the data were consistent with published analyses from ours (32, 49) and other labs employing similar instrumentation and methods. The log2 ratio values were normally distributed. The fold change threshold we used for significance was determined through a modified two-sided t test based on permutation-based FDR statistics between the two groups, with an FDR of 5% and the s0 value determined by the main distribution of quantified proteins. AEMS experiments were performed in triplicate using LFQ and compared with control cells as we have done previously (32, 49). Imputation was applied only to controls and random values drawn from a distribution equivalent to the limit of detection in each experiment. The log2 intensity values were normally distributed. Significantly enriched proteins were determined through a modified two-sided t test based on permutation-based FDR statistics (45) between the two groups, with an FDR of 5% and the s0 value set to exclude enriched proteins specific to the control group. For the pulse-chase SILAC experiment rows for mtDNA encoded proteins were isolated and log2 heavy proteinGroup intensities scaled to the maximum intensity observed for the proteinGroup in control cells.
RESULTS
HIGD2A is Up-regulated in Patient Lymphoblasts with an Isolated Complex IV Deficiency
We identified a patient presenting with intrauterine growth retardation, developmental delay, cortical blindness, possible deafness, lactic acidosis, and cardiomyopathy. The patient died at 16 months of age. Enzymology revealed an apparently isolated complex IV defect in muscle and lymphoblasts (Table I). Whole exome sequencing revealed two novel compound heterozygous missense mutations (c.1874G>A, p.Arg625His; c.665C>T p.Thr222Ile) in AARS2, which encodes for mitochondrial alanine-tRNA synthetase (MIM 612035). Both mutations are classified as class 4 or 'likely pathogenic' (PM2, PP3, PS3) using the ACMG standards and guidelines for the interpretation of sequence variants (50). It is not uncommon to find an apparently isolated ETC enzyme defect in genetic defects that would be expected to affect multiple ETC enzymes (51). Given that AARS2 lymphoblasts harbor an isolated complex IV defect, we utilized them to search for novel proteins involved in assembly of complex IV. We performed quantitative mass spectrometry on the patient lymphoblasts and on control lymphoblasts from three unaffected individuals, as we have done previously for patient fibroblasts (36). We quantified the levels of 3877 cellular proteins detected in AARS2 lymphoblasts and in at least two out of three control cell lines (supplemental Table S1), revealing significant reduction in the levels of complex IV subunits but not subunits of complexes I, III, or V (Fig. 2) consistent with enzymology data. HIGD2A was the most up-regulated mitochondrial protein in AARS2 lymphoblasts relative to control cell lines.
Table I. Primary human lymphoblasts with mutated AARS2 have an isolated complex IV defect. Electron transport chain enzyme activities in skeletal muscle homogenate and EBV-Lymphoblast mitochondria are expressed as %CS ratio, which represents % of the normal control mean value when expressed relative to the mitochondrial marker enzyme Citrate Synthase. Bold characters indicate clinically significant abnormal values. n.t.; not tested.
| Enzyme | Skeletal Muscle (%CS Ratio) |
Lymphoblasts (%CS Ratio) |
||
|---|---|---|---|---|
| Patient | Ref. Range (n = 9) | Patient | Ref. Range (n = 6) | |
| Complex I | 52 | 36–167 | 75 | 78–116 |
| Complex II | 129 | 52–156 | 92 | 47–142 |
| Complex III | n.t. | 62–185 | 155 | 64–132 |
| Complex IV | 12 | 36–192 | 38 | 76–126 |
Fig. 2.

HIGD2A is up-regulated in lymphoblasts with a specific defect in the translation of mtDNA-encoded complex IV subunits. Volcano plot comparing mitochondrial protein abundances for proteins quantified in AARS2 lymphoblasts relative to control lymphoblasts. Subunits of complexes I, III and IV are colored as indicated. The curved line indicates significantly altered protein abundance determined through a false discovery rate-based approach (5% FDR, s0 = 0.1). n = 2 subcultures for the patient cells and n = 3 independent controls.
Loss of HIGD2A Leads to Defective Assembly of Complex IV
To clarify the function of HIGD2A we used CRISPR/Cas9 to generate human embryonic kidney (HEK293T) cell lines lacking its expression (supplemental Fig. S1). We also generated cell lines lacking the HIGD2A ortholog HIGD1A. Loss of HIGD1A (HIGD1AKO) led to no changes in the steady state level of complex IV subunits (supplemental Fig. S1A), whereas loss of HIGD2A (HIGD2AKO) led to a strong reduction in the levels of COX2 (supplemental Fig. S1B), the core subunit of the COX2 module, and the total loss of COX6A, a nuclear encoded subunit present within the COX3 module. To assess the assembly of complex IV and supercomplexes we performed Blue Native (BN)-PAGE. When using the weak non-ionic detergent digitonin, BN-PAGE can resolve stalled complex IV assembly intermediates, holo complex IV and multiple high-order supercomplex structures (10, 17, 20, 52, 53). As can be seen in Fig. 3A, HIGD2AKO mitochondria contain reduced levels of the primary supercomplex assembly consisting of CI/III2/IV (compare lanes 1,2, and 2,5). Additionally, HIGD2AKO mitochondria accumulate both the S2 intermediate containing COX1 and COX4 (Fig. 3A compare lanes 7,8 and 10,11) and S3 intermediate containing COX2, consistent with defective assembly of complex IV (9, 16, 53). Strikingly, no signal was observed for COX3 module subunit COX6A1, either in accumulated intermediates or mature complex IV assemblies (Fig. 3A compare lanes 16,17). In contrast to HIGD2AKO, mitochondria lacking HIGD1A contained normal levels and distribution of supercomplex assemblies and accumulated complex IV intermediates were not detected (Fig. 3A).
Fig. 3.
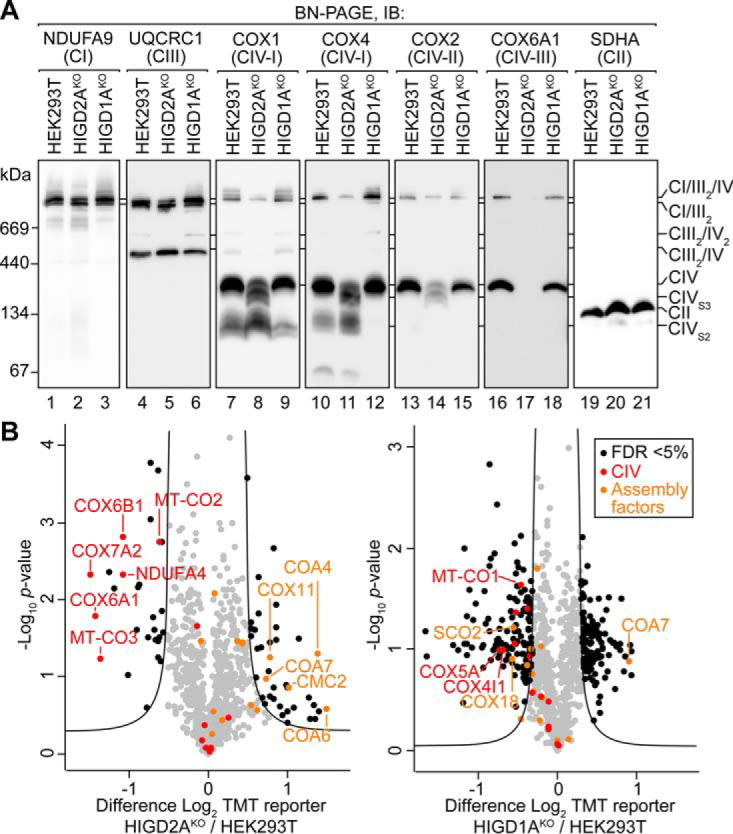
Loss of HIGD2A leads to defects in assembly of complex IV. A, Mitochondria isolated from the indicated cell lines were solubilized in 1% digitonin, analyzed by BN-PAGE and immunoblotted (IB) using antibodies specific for the indicated antibodies. CIVS2, complex IV stage II intermediate; CIVS3, complex IV stage 3 intermediate. B, Volcano plots depicting relative levels of mitochondrial proteins in HIGD2AKO and HIGD1AKO cell lines compared with HEK293T cells. Black dots and the curved line represent significantly altered protein abundance determined through a false discovery rate-based approach (5% FDR, s0 = 1). n = 2 independent subcultures.
HIGD2A is Required for the Stability of COX3 Module of Complex IV
To confirm the steady levels of complex IV subunits in HIGD2AKO mitochondria we performed quantitative mass-spectrometry. Mitochondrial proteins from HIGD2AKO, HIGD1AKO and control cells were digested, and tryptic peptides labeled with tandem mass tags (TMT) prior to pooled analysis and synchronous precursor selection (SPS)–MS3 based acquisition. We quantified the levels of 892 proteins present within the Mitocarta2.0 (46) inventory of mitochondrial proteins across all samples (supplemental Table S2). As can be seen in Fig. 3B (left panel) HIGD2AKO mitochondria contained a significant reduction in the levels of complex IV subunits COX3 (MT-CO3), COX6A1, COX6B1, COX7A2, as well as COX2 (MT-CO2) and NDUFA4, a subunit originally ascribed to complex I but now known to be present within complex IV (52). Other complex IV subunits were detected but found not to be significantly altered relative to control cells. HIGD2AKO mitochondria also contained significantly increased levels of complex IV assembly factors COA4, COA6, COA7, CMC2, and COX11 (9, 16). In contrast our, analysis of HIGD1AKO mitochondria revealed a significant reduction in the levels of a different cohort of complex IV subunits; COX1 (MT-CO1), COX4, and COX5A, as well as assembly factors SCO2 and COX18 (Fig. 3B, right panel). COA7 was also found in HIGD1AKO mitochondria at increased abundance relative to controls. Notably, the fold change in abundance of complex IV subunits was less in HIGD1AKO (∼1–1.5 fold change) than for HIGD2AKO (∼2–3 fold change) consistent with the strong defect in assembly we observed for the latter (Fig. 3A).
We used topographical heatmaps (32) to interpret our mass-spectrometry data in the context of the intact 14-subunit human holo complex IV (54) and CI/III2/IV respiratory chain supercomplex (2) structures recently determined by single particle cryo-electron microscopy. Both structures were determined using complexes isolated from the same HEK293T derived human cell line used in our study. Unsupervised hierarchical clustering of knockout to control protein ratios from both cell lines revealed several clusters of co-dependent subunits from complexes I, III, and IV (Fig. 4A). The most strongly clustered cohort of proteins in HIGD2AKO belonged to the COX3 module (Figs. 4A, 4B). We found NDUFA4 to be strongly correlated with these proteins, suggestive of it being a subunit of the COX3 module. Mapping of our data to the supercomplex (Fig. 4C) revealed the affected complex I and III subunits to be predominantly located at contact sites with complex IV. In contrast, loss of HIGD1A led to the comparatively weak clustering of COX1 and COX2 module subunits (Figs. 4A, 4D). Taken together, our data suggest that loss of HIGD2A strongly affects the COX3 module of complex IV, leads to accumulation of a crippled complex IV and its defective association with complexes I and III in the respiratory chain supercomplex.
Fig. 4.

Loss of HIGD2A affects the COX3 module of complex IV. A, Two-dimensional heatmap showing hierarchically clustered protein ratios for subunits of complex I, III and IV. Modules of complex IV are indicated with the colored squares. B, Topographical heatmap showing protein ratios for subunits of complex IV in HIGD2AKO cells (PDB 5Z62) C, Topographical heatmap showing protein ratios for subunits of the human supercomplex in HIGD2AKO cells (PDB 5XTH). The insets show the orientation of the complex with complexes I and III colored in orange and green respectively. D, Topographical heatmap showing protein ratios for subunits of complex IV in HIGD1AKO cells (PDB 5Z62).
Loss of HIGD2A Leads to Impaired Complex IV Activity
HIGD2AKO cells die in media where glucose has been replaced with galactose (data not shown), indicative of severely impaired mitochondrial respiration (32, 53). To investigate the impact of HIGD2A and HIGD1A on mitochondrial energy generation we measured the oxygen consumption rates in our HIGD2AKO and HIGD1AKO cell lines. Cells lacking HIGD2A were significantly impaired in basal and maximal respiration (Fig. 5A). In contrast, HIGD1AKO had impaired basal respiration but similar maximal respiration compared with control cells leading to an increase in spare respiratory capacity. To investigate the source of the respiration defects we performed an in-gel complex IV activity assay (41). As can be seen in Fig. 5B, the remnant complex IV found within HIGD2AKO mitochondria showed only background levels of activity. Although activity was detected in the HIGD2AKO CI/III2/IV supercomplex it was reduced relative to control cells. In contrast, complex IV activity in holo complex IV as well as the supercomplex was unaltered in HIGD1AKO mitochondria. Finally, to confirm the impact of HIGD2A loss on complex IV activity in vivo we measured complex IV-linked respiration (42). Briefly, the electron donor N,N,N′,N′-Tetramethyl-p-phenylenediamine (TMPD) is provided in the presence of ascorbate to semi-permeabilized cells where it donates electrons to cytochrome c thereby bypassing complex III (Fig. 5C). When TMPD was added to control HEK293T cells in the presence of ADP and uncoupler FCCP, we observed maximal mitochondrial respiration that could be partially dissipated with oligomycin. Inhibition of complex IV activity with azide reveals complex IV dependent oxygen consumption. In contrast to control cells, HIGD2AKO cells had significantly impaired complex IV dependent respiration (Fig. 5C). Taken together, we conclude that loss of HIGD2A leads to a specific defect in complex IV enzymatic activity leading to impaired mitochondrial respiration.
Fig. 5.

Biochemical characterization of mitochondrial respiration and complex IV activity in HIGD2AKO cells. A, Oxygen consumption rates (OCR) of HIGD2AKO, HIGD1AKO and control cell lines. Data reported as mean ± S.D. n = 5. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. B, Mitochondrial complexes solubilized in 1% digitonin were separated by BN-PAGE and the activity of complex IV determined through an in-gel assay. The gel was subsequently stained with coomassie (right panel). C, Flux analysis of complex IV activity in HIGD2AKO. OCR was determined in semi-permeabilized cells with the addition of electron donor N,N,N′,N′-Tetramethyl-p-phenylenediamine (TMPD) and ascorbate that donate electrons to cytochrome c bypassing complex III (see schematic). The presence of ADP and FCCP permit maximal respiration. Complex V is inhibited by the addition of oligomycin and complex IV through azide revealing complex IV activity (top right). Data reported as mean ± S.D. n = 2 and 3, *, p < 0.05.
HIGD2A Acts on Complex IV Assembly Independent of Oxygen Levels
Mammalian HIGD1A and HIGD2A were originally discovered as proteins induced following hypoxic stress (29, 30), an insult that is known to ultimately lead to turnover of OXPHOS subunits, a reduction in oxidative capacity and an increase in glycolysis. As a way of compensation to these changes in conditions, during the initial stages of hypoxia HIGD1A has been proposed to bind complex IV and promote its activity (31). To address whether the presence or absence of HIGD2A impacts complex IV under hypoxia we cultured HEK293T cells under an atmosphere of 1% oxygen for 0, 3 or 24 h. Growth under these conditions led to a robust hypoxic response that we measured through TMT-based quantitative mass-spectrometry (supplemental Table S3) and Western blotting (supplemental Fig. S2). Following 24 h under hypoxic conditions, proteins involved in glycolysis including SLC2A1, the primary plasma membrane glucose transporter, were significantly up-regulated (supplemental Fig. S2A). In contrast, subunits of the OXPHOS machinery and the mitochondrial ribosome were significantly decreased, as expected (55). Stabilization of HIF1α was observed within 3 h (supplemental Fig. S2B), as expected. Although we found HIGD1A and complex IV subunits were indeed increased in abundance at the early stages of hypoxia (3 h; supplemental Figs. S2B, S2C), the steady state level of HIGD2A was unaffected (supplemental Fig. S2B, compare lanes 1, 2). Finally, we investigated the effects of hypoxia on assembly of complex IV and the supercomplex. Loss of neither HIGD2A nor HIGD1A altered the effects of hypoxia on assembly of the supercomplex (supplemental Fig. S2B, lower panels). Moreover, following chronic hypoxia (24 h) we observed accumulation of the S2 intermediate in both cell lines consistent with the complexes being increasingly sensitive to destabilization in the absence of HIGD2A and HIGD1A. Taken together we conclude that HIGD2A acts on complex IV assembly independent of oxygen levels.
HIGD2A is Required for the Biogenesis of COX3
Because our results for HIGD1A function were broadly consistent with previous studies, suggesting its involvement in regulating the COX1 module following hypoxia (31), we focused our efforts on clarifying the function of HIGD2A under normoxic conditions. To identify the interaction partners of HIGD2A we used a retroviral system to express FLAG-tagged HIGD2A (HIGD2AFLAG) in our HIGD2AKO cell line. Ectopic expression of HIGD2AFLAG led to rescue of supercomplex distribution and an increase in the levels of fully assembled holo complex IV (Fig. 6A). HIGD2AFLAG cells were then solubilized in digitonin and complexes bound to FLAG-affinity gel, following which significantly enriched interactors were identified by affinity enrichment mass-spectrometry (AEMS). As can be seen in Fig. 6B, HIGD2AFLAG associates with subunits of complex IV with mtDNA encoded COX3 (MT-CO3) being the most strongly enriched (supplemental Table S4). Interestingly, we observed significant enrichment of complex I subunits, subunits of the mitochondrial ribosome (MRPL23, MRPL27 and MRPL50), HIGD1A, complex IV assembly factors COA3 (56–59) and SCO1 (60, 61), and complex I assembly factors NDUFAF2 (39, 62, 63) and NDUFAF4 (64) which is suggestive of the enriched cohort of proteins representing an assembly intermediate rather than the mature complex.
Fig. 6.
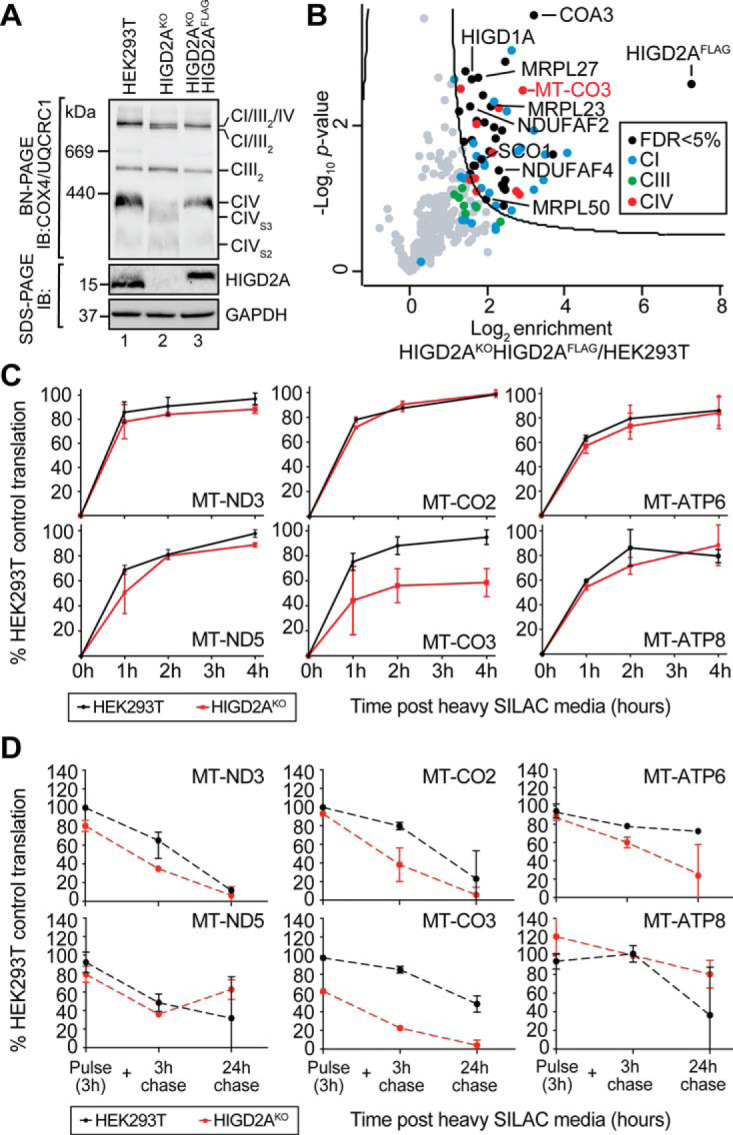
HIGD2A is required for the stability of newly translated COX3. A, The HIGD2AKO cell line was complemented with a retroviral HIGD2AFLAG construct and mitochondria analyzed by BN and SDS-PAGE and immunoblotting with the indicated antibodies. B, Affinity-enrichment mass spectrometry (AE-MS) was performed on the HIGD2AKOHIGD2AFLAG cell line and significant interaction partners visualized through a volcano plot. The curved line indicates significance determined through an FDR-based approach (FDR<5%, S0 = 1). n = 3. C, Pulse SILAC analysis of newly translated mtDNA encoded OXPHOS subunits in HIGD2AKO cells. SILAC media was added following overnight treatment with chloramphenicol (CAP) and analysis was performed at 0, 1, 2- and 4-hours post media addition. Log2 transformed heavy peptide-derived intensities were plotted relative to controls. Data reported as mean ± S.D. n = 3. D, As for C but heavy media exchanged with light media after a 3h pulse and cells cultured for the indicated times. n = 2.
Given the enrichment of the mitochondrial ribosome and the strong association of HIGD2AFLAG with mtDNA-encoded COX3, we asked if mtDNA-encoded subunit translation was affected upon loss of HIGD2A. Typically, translation and stability of mtDNA-encoded subunits is assessed using a radioactive pulse-chase assay and analysis by SDS-PAGE and autoradiography (65). Because COX2 and COX3 have a similar migration on SDS-PAGE (53) we sought to develop a mass-spectrometry based approach capable of clear differentiation between these two proteins. HIGD2AKO and control cells conditioned by overnight treatment with mtDNA translation inhibitor chloramphenicol were pulsed in media containing SILAC (stable isotope labeling by amino acids in cell culture; (66)) amino acids 13C615N4-arginine and 13C615N2-lysine for 1, 2 and 4 h prior to analysis by targeted mass-spectrometry (described in the methods). Using this technique, we could monitor the translation of 10 out of 13 mtDNA encoded subunits (supplemental Table S5) including COX2 and COX3. As can be seen in Fig. 6C we observed a significant defect in accumulation of newly translated COX3 (MT-CO3) whereas translation of other mtDNA-encoded subunits was comparable to control cells. This result led us to ask if the defect in biogenesis of newly translated COX3 occurs because of rapid turnover of the subunit. To address this question, HIGD2AKO and control cells treated as above were pulsed with SILAC media for 3 h to accumulate newly translated COX3. At the conclusion of the pulse, the amount of newly translated COX3 in HIGD2AKO was ∼60% of that observed in control cells (Fig. 6D). The accumulation of other newly translated subunits in HIGD2AKO was like control cells, as expected. SILAC media was exchanged with media containing normal amino acids and the stability of SILAC labeled proteins chased for 3 and 24 h. We observed time dependent turnover of most subunits as is observed in the classic radiolabeled translation assay (67). The turnover rate of newly translated COX3 was similar in both HIGD2AKO and control cells, suggesting the defect in COX3 occurs prior to the chase. Moreover, COX3 turnover in HIGD2AKO was like the rates observed for other mtDNA encoded proteins observed in both cell lines. Taken together we conclude that the defect in COX3 biogenesis observed in HIGD2AKO occurs early during subunit translation and maturation.
HIGD2AKO Mitochondria Contain Supercomplexes Lacking COX3
Given the apparent retention of the CI/III2/IV supercomplex in HIGD2AKO we sought an alternate approach to assess supercomplex content. We utilized a cell line we developed previously that stably expresses the FLAG-tagged complex I subunit NDUFV3 in the corresponding NDUFV3KO cell line (32). NDUFV3 is suggested to be the final subunit added to complex I during its biogenesis and we previously used the resulting NDUFV3FLAG cell line to enrich all detectable subunits of complexes I, III and IV (32) leading us to hypothesize that the cell line can be used to isolate the intact supercomplex. To this end we used CRISPR/Cas9 to generate HIGD2A knockout mutations in the NDUFV3FLAG cell line. HIGD2AKO and NDUFV3FLAGHIGD2AKO have comparable defects in complex IV assembly and supercomplex distribution (Fig. 7A). Next, we performed affinity enrichment and mass-spectrometry on both the original NDUFV3FLAG and NDUFV3FLAGHIGD2AKO cell lines. Although NDUFV3FLAG cells co-isolated complex I, III, and IV subunits as expected (Fig. 7B), we also detected HIGD2A, the known complex IV assembly factor COA3 and a number of other mitochondrial housekeeping proteins also detected in our original experiment (supplemental Table S6 and (32)). We conclude that although the NDUFV3FLAG eluate does not represent the purified supercomplex seen in the recent structures it likely represents a late stage assembly intermediate with a full complement of complex I, III, and IV subunits and selected assembly factors. Finally, to assess the impact of HIGD2A loss on the supercomplex we performed the affinity enrichment experiment from NDUFV3FLAGHIGD2AKO cells using NDUFV3FLAG as the control cell line. As can be seen in Fig. 7C, although the majority of complex I, III, and IV subunits were equally enriched from both cell lines, significantly lower amounts of COX3 were enriched from the pool of supercomplex intermediates isolated from cells lacking HIGD2A.
Fig. 7.

The supercomplex in HIGD2AKO cells lacks COX3. A, The indicated cell lines were analyzed by BN and SDS-PAGE and immunoblotting with the indicated antibodies. B, Affinity-enrichment mass spectrometry (AE-MS) was performed on the NDUFV3FLAG and control cell lines and significant FLAG interaction partners visualized through a volcano plot. The curved line indicates significance determined through an FDR-based approach (FDR<5%, S0 = 1). n = 3 independent subcultures. C, as for B however, the enrichment was performed on NDUFV3FLAG/HIGD2AKO relative to the NDUFV3FLAG cell line.
DISCUSSION
In this study we aimed to clarify the roles of hypoxia inducible gene 1 domain family members HIGD2A and HIGD1A. To achieve this, we utilized CRISPR/Cas9 gene editing to generate cell lines lacking detectable protein, mitochondrial biochemistry including BN-PAGE, as well as existing and novel mass-spectrometry based techniques. Although the mammalian proteins were originally identified in a screen for genes up-regulated upon hypoxic stress (29, 30) the majority of work has been focused on their yeast homologs Rcf1 and Rcf2 (named for Respiratory complex factors). Three independent groups reported the functions of Rcf1 and Rcf2 as required for the formation of the respiratory chain supercomplexes, which in the yeast S. cerevisiae are comprised of complexes III and IV (24–26). HIGD2A is thought to be the mammalian ortholog of Rcf1 and knockdown experiments in mouse myoblasts were shown to alter the distribution of complex IV-containing supercomplexes, suggesting the function of HIGD2A in supercomplex assembly is conserved from yeast to man (26). Until now this was the only study to investigate the impact of HIGD2A loss on mammalian complex IV and supercomplex assembly, and as a result the function of HIGD2A has largely been extrapolated from yeast studies. The solution structure of yeast Rcf1 was recently solved by NMR (68) revealing two charged transmembrane helices that form salt bridges leading to formation of a homodimer. Mammalian HIGD2A lacks the charged membrane helices responsible for Rcf1 dimerization, suggesting the functions of Rcf1 and HIGD2A may differ. Despite this point of difference, the results from yeast studies are generally in agreement with our observations. For example, as we found in HIGD2AKO cells, yeast mitochondria lacking Rcf1 still contain the CIII2/IV supercomplex observed using BN-PAGE (25, 26, 69) (compare with Fig. 3A). Secondly, using mass-spectrometry we observed turnover of COX6A and COX6B (Fig. 3B) in HIGD2AKO cells, consistent with the dissociation of their homologs (Cox13 and Cox12 in yeast) in yeast Rcf1 knockouts (24, 25). This suggests that like in yeast, the remaining assembly is still capable of association with complex III. Finally, we developed a novel pulse-SILAC approach to assess COX3 biogenesis, finding that accumulation of newly translated COX3 is reduced in HIGD2AKO cells (Fig. 6C). When we followed the fate of newly translated COX3 in a chase experiment (Fig. 6D) we found no difference in the rate of COX3 turnover between HIGD2AKO and control cells. Taken together, we concluded that HIGD2A is involved in the early stages of COX3 translation and/or maturation. Our SILAC based approach has a number of advantages over the established approach that uses radioactive methionine (65), including the use of non-radioactive isotopes, the ease of quantification, and the ability to determine the identity of the labeled protein directly from peptide sequences rather than protein migration on SDS-PAGE. On the other hand, disadvantages include our inability to quantify the levels of some SILAC labeled mtDNA encoded proteins from more than a single peptide, and the technique not being readily adaptable for monitoring complex assembly via BN-PAGE. Importantly, our results are consistent with the reduction in steady state levels of COX3 observed upon deletion of Rcf1 in yeast (25, 28, 69). Indeed, in their original study Strogolova and colleagues (25) suggest that Rcf1 may interact with newly translated COX3 to regulate its association with Cox12 (COX6B) and complex IV as a means of modulating complex IV activity. Although our data demonstrates a specific defect in the biogenesis of COX3 (Fig. 6C and 6D) it remains unclear if HIGD2A regulates translation of the subunit or stabilizes its early stages of biogenesis. The interaction between HIGD2A, COX3 and other complex IV and respiratory chain subunits detected in our HIGD2AFLAG affinity enrichment experiment (Fig. 6B) argues for HIGD2A being directly involved in adding COX3 to the partly assembled complex, whereas the up-regulation of HIGD2A in patient lymphoblasts with an isolated complex IV defect arising from impaired translation of mtDNA-encoded subunits could be taken as an argument for transcriptional regulation (Fig. 2B). Our previous analysis of fibroblasts from a patient with a mutation in a subunit of the mitoribosome and therefore defective translation of all mtDNA-encoded subunits, also showed up-regulation of complex IV assembly factors COX15, SCO2, SURF1 (36) suggesting their up-regulation is a general response to complex IV dysfunction. To resolve this argument, future studies could investigate the effect of HIGD2A loss on COX3 mRNA levels and processing and assess if inhibiting or ablating the function of matrix proteases can rescue the levels of COX3, and indeed restore Complex IV function in the HIGD2AKO. Finally, another possibility that warrants investigation is that HIGD2A may work with mitochondrial inner membrane insertase OXA1 (70) to promote membrane insertion of COX3. Although OXA1 was not quantified in our HIGD2AFLAG affinity enrichment, we previously showed that OXA1 interacts with HIGD2A as well as several other proteins detected in our experiment (70).
Although we cannot completely exclude the possibility that HIGD2A can directly regulate complex IV activity, we did not observe changes in either the levels of HIGD2A or the impact of its loss on complex IV or supercomplex assembly during acute or chronic hypoxia as has been suggested for HIGD1A (supplemental Fig. S2). Our conclusion is that the impact of HIGD2A on the supercomplex is a pleotropic effect because of impaired COX3 biogenesis and subsequently defective complex IV assembly. The intersection of the latter assembly steps of complexes I, III, and IV with supercomplex formation (8, 16–18, 20) is also a major factor to be considered in interpretation of the results from ours and other studies. Our use of a range of mass-spectrometry based techniques led us this conclusion. For example, we detected subunits of complexes I, III, and IV as well as assembly factors for complexes I and IV in our HIGD2AFLAG affinity enrichment and mass-spectrometry (AE-MS) experiment (Fig. 6B), suggesting that HIGD2A acts at the stage of both COX3 module integration into complex IV, and complex IV integration into the supercomplex. Moreover, in our HIGD2AKO cells we observed up-regulation of complex I and III subunits (Fig. 4A) and used topographic heatmapping (32, 49) to localize these to the interface between complexes I and IV (Fig. 4C). We hypothesize this is a mechanism to compensate for their destabilization following disruption of their interactions with complex IV subunits, in agreement with our previous results demonstrating that loss of the affected complex I subunits leads to total disassembly of the supercomplex and loss of mitochondrial respiration (32). Finally, our experiments employing the FLAG tagged complex I subunit NDUFV3 in the background of HIGD2AKO demonstrated the presence of a supercomplex containing subunits of complexes I, III, and IV, but depleted of COX3 (Fig. 7C). A caveat of this experiment is the equal enrichment of the other COX3 module subunits in eluates from both NDUFV3FLAGHIGD2AKO and NDUFV3FLAG cell lines. One possible explanation for this is that HIGD2AKO cells contain two populations of supercomplex, one lacking COX3 and one containing a fully assembled supercomplex, which is consistent with reduced but not absent COX3 biogenesis. Another possible explanation is the apparent stabilization of complex IV containing supercomplexes in the HIGD2AKO (e.g. Fig. 6A) in preference to holo-complex IV, a phenotype we have previously observed in HEK293T knockouts and patient cell lines harboring mutations in the complex IV assembly factor COA6 (53). HIGD2A would not the first assembly factor suggested to impact supercomplex assembly indirectly (8). The most well-known example is COX7A2L (SCAFI), which was originally found within the supercomplex but not holo complex IV (6). A role of COX7A2L in supercomplex assembly has been disputed by multiple groups (17–22) and the protein is now understood to act as a checkpoint for assembly of complex III into the supercomplex (17). Another example is UQCC3 (C11orf83), which is an assembly factor suggested to stabilize the CIII2/IV form of the supercomplex (71). UQCC3 has also been shown to be involved in early complex III assembly and is found mutated in patients with isolated complex III deficiency (MIM 616097; 72). Importantly, density for neither HIGD2A, COX7A2L, nor UQCC3 has been identified in the multiple recent structures representing several different forms of the mammalian supercomplex assembly (1, 2, 4, 7).
As part of this study we also attempted to validate the role of HIGD1A in respiratory chain assembly. The function of mammalian HIGD1A has been studied in more detail than HIGD2A and the protein has been shown to bind subunits of the COX1 module (specifically COX4, COX5A) (31). It has been proposed that during acute hypoxia, the increased amounts of HIGD1A bind complex IV and induce structural changes around the heme a active center, increasing complex IV activity (31). In agreement, we also showed up-regulation of HIGD1A protein following 3 h of growth under hypoxic conditions (supplemental Fig. S2) as well as up-regulation of complex IV subunits, which was previously suggested to be a mechanism of compensation for the reduced oxygen environment (55). Consistent with a general function for HIGD1A in regulation of the respiratory chain during metabolic stress, Ameri and colleagues (73) showed HIGD1A to be up-regulated upon glucose deprivation, however under these conditions the authors could only show an interaction with subunits of complex III and not complex IV. In contrast to these studies, under normoxic conditions, HIGD1A was most recently suggested to associate with an early complex IV assembly intermediate representing the nascent COX1 module (10). In agreement with this, we detected HIGD1A along with other complex IV assembly factors and subunits in our HIGD2AFLAG affinity enrichment experiment (Fig. 6B), suggestive of it being associated with complex IV during assembly. However, despite small changes in the levels of COX1 module subunits upon loss of HIGD1A (Fig. 4A, 4D), the lack of a strong complex IV or supercomplex assembly defect in our HIGD1AKO (Fig. 3A) argues against its role as an assembly factor.
In conclusion, although the hypoxia induced domain family member HIGD2A has previously been implicated in the maintenance of respiratory chain supercomplexes, we find it to be involved in the early biogenesis of mtDNA-encoded COX3. Loss of HIGD2A leads to turnover of nuclear encoded COX3 partner subunits and accumulation of a supercomplex containing a crippled complex IV. In addition to clarifying previous reports suggesting HIGD2A involvement in supercomplex assembly, our study identifies HIGD2A as the first assembly factor required for the biogenesis of the COX3 module of human complex IV.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD016864.
Supplementary Material
Acknowledgments
We thank the Stroud lab for input into experimental design and interpretation of data, Giel van Dooren and Jenni Hayward for advice on flux analysis experiments, the Bio21 Mass Spectrometry and Proteomics Facility (MMSPF) and the Monash Biomedical Proteomics Facility for the provision of instrumentation, training, and technical support. We acknowledge Bice Dibley for assistance in writing scripts used for the annotation and mapping of proteomics data onto Cryo-EM structures.
This article contains supplemental data.
Funding and additional information—We acknowledge funding from the National Health and Medical Research Council (NHMRC Project Grants 1125390, 1140906 to D.A.S. and M.T.R. and 1068409 to D.R.T.; NHMRC Fellowships 1140851 to D.A.S. and 1155244 to D.R.T.). D.H.H. is supported by a Melbourne International Research Scholarship and Mito Foundation Top-up Scholarship, and H.S.M. by an NHMRC Biomedical Postgraduate Scholarship (1017174). We acknowledge support from the Victorian Government's Operational Infrastructure Support Program.
Author contributions—D.H.H. and D.A.S. designed research; D.H.H., B.R., L.M.-W., H.S.M., and A.G.C. performed research; D.H.H., B.R., C.-S.A., H.S.M., A.G.C., M.T.R., D.R.T., and D.A.S. analyzed data; D.H.H. and D.A.S. wrote the paper; C.-S.A., M.T.R., D.R.T., and D.A.S. contributed new reagents/analytic tools.
Conflict of interest—Authors declare no competing interests.
Abbreviations—The abbreviations used are:
- AEMS
- affinity enrichment and mass spectrometry
- BN-PAGE
- Blue Native Polyacrylamide gel electrophoresis
- COX
- Cytochrome c oxidase
- ETC
- electron transport chain
- HIF,1α
- Hypoxia-inducible factor 1-alpha
- HIGD
- Hypoxia inducible gene 1
- IMM
- inner mitochondrial membrane
- KO
- Knock out of protein expression
- MIM
- Mendelian Inheritance in Man
- mtDNA
- mitochondrial DNA
- MT
- encoded by mitochondrial DNA
- NDUF
- NADH dehydrogenase (ubiquinone) oxidoreductase
- NMR
- Nuclear magnetic resonance
- OXPHOS
- oxidative phosphorylation
- Rcf
- Respiratory supercomplex factor
- ROS
- reactive oxygen species
- SCAFI
- Supercomplex Assembly Factor I
- SILAC
- Stable Isotope Labeling with Amino acids in Cell culture
- SPS-MS3
- Synchronous precursor selection triple stage mass spectrometry
- TMT
- Tandem Mass Tags
- TMPD
- N,N,N′,N′-Tetramethyl-p-phenylenediamine.
REFERENCES
- 1. Gu J., Wu M., Guo R., Yan K., Lei J., Gao N., and Yang M. (2016) The architecture of the mammalian respirasome. Nature 537, 639–643 [DOI] [PubMed] [Google Scholar]
- 2. Guo R., Zong S., Wu M., Gu J., and Yang M. (2017) Architecture of Human Mitochondrial Respiratory Megacomplex I2III2IV2. Cell 170, 1247–1257.e1212 [DOI] [PubMed] [Google Scholar]
- 3. Schagger H., and Pfeiffer K. (2000) Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 19, 1777–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu M., Gu J., Guo R., Huang Y., and Yang M. (2016) Structure of Mammalian Respiratory Supercomplex I1III2IV1. Cell 167, 1598–1609.e1510 [DOI] [PubMed] [Google Scholar]
- 5. Fedor J. G., and Hirst J. (2018) Mitochondrial supercomplexes do not enhance catalysis by quinone channeling. Cell Metab 28, 525–531.e524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lapuente-Brun E., Moreno-Loshuertos R., Acin-Perez R., Latorre-Pellicer A., Colas C., Balsa E., Perales-Clemente E., Quiros P. M., Calvo E., Rodriguez-Hernandez M. A., Navas P., Cruz R., Carracedo A., Lopez-Otin C., Perez-Martos A., Fernandez-Silva P., Fernandez-Vizarra E., and Enriquez J. A. (2013) Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 340, 1567–1570 [DOI] [PubMed] [Google Scholar]
- 7. Letts J. A., Fiedorczuk K., and Sazanov L. A. (2016) The architecture of respiratory supercomplexes. Nature 537, 644–648 [DOI] [PubMed] [Google Scholar]
- 8. Milenkovic D., Blaza J. N., Larsson N. G., and Hirst J. (2017) The enigma of the respiratory chain supercomplex. Cell Metab. 25, 765–776 [DOI] [PubMed] [Google Scholar]
- 9. Timon-Gomez A., Nyvltova E., Abriata L. A., Vila A. J., Hosler J., and Barrientos A. (2018) Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol. 76, 163–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vidoni S., Harbour M. E., Guerrero-Castillo S., Signes A., Ding S., Fearnley I. M., Taylor R. W., Tiranti V., Arnold S., Fernandez-Vizarra E., and Zeviani M. (2017) MR-1S interacts with PET100 and PET117 in module-based assembly of human cytochrome c oxidase. Cell Rep. 18, 1727–1738 [DOI] [PubMed] [Google Scholar]
- 11. Tsukihara T., Aoyama H., Yamashita E., Tomizaki T., Yamaguchi H., Shinzawa-Itoh K., Nakashima R., Yaono R., and Yoshikawa S. (1996) The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science 272, 1136–1144 [DOI] [PubMed] [Google Scholar]
- 12. Ogunjimi E. O., Pokalsky C. N., Shroyer L. A., and Prochaska L. J. (2000) Evidence for a conformational change in subunit III of bovine heart mitochondrial cytochrome c oxidase. J. Bioenerg. Biomembr. 32, 617–626 [DOI] [PubMed] [Google Scholar]
- 13. Soto I. C., Fontanesi F., Liu J., and Barrientos A. (2012) Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim. Biophys. Acta 1817, 883–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Keightley J. A., Hoffbuhr K. C., Burton M. D., Salas V. M., Johnston W. S., Penn A. M., Buist N. R., and Kennaway N. G. (1996) A microdeletion in cytochrome c oxidase (COX) subunit III associated with COX deficiency and recurrent myoglobinuria. Nat. Genet. 12, 410–416 [DOI] [PubMed] [Google Scholar]
- 15. Hanna M. G., Nelson I. P., Rahman S., Lane R. J., Land J., Heales S., Cooper M. J., Schapira A. H., Morgan-Hughes J. A., and Wood N. W. (1998) Cytochrome c oxidase deficiency associated with the first stop-codon point mutation in human mtDNA. Am. J. Hum. Genet. 63, 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Signes A., and Fernandez-Vizarra E. (2018) Assembly of mammalian oxidative phosphorylation complexes I-V and supercomplexes. Essays Biochem. 62, 255–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lobo-Jarne T., and Ugalde C. (2018) Respiratory chain supercomplexes: Structures, function and biogenesis. Semin Cell Dev. Biol. 76, 179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Davoudi M., Kotarsky H., Hansson E., Kallijarvi J., and Fellman V. (2016) COX7A2L/SCAFI and Pre-Complex III Modify Respiratory Chain Supercomplex Formation in Different Mouse Strains with a Bcs1l Mutation. PLoS ONE 11, e0168774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ikeda K., Shiba S., Horie-Inoue K., Shimokata K., and Inoue S. (2013) A stabilizing factor for mitochondrial respiratory supercomplex assembly regulates energy metabolism in muscle. Nat. Commun. 4, 2147. [DOI] [PubMed] [Google Scholar]
- 20. Mourier A., Matic S., Ruzzenente B., Larsson N. G., and Milenkovic D. (2014) The respiratory chain supercomplex organization is independent of COX7a2l isoforms. Cell Metab. 20, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun D., Li B., Qiu R., Fang H., and Lyu J. (2016) Cell type-specific modulation of respiratory chain supercomplex organization. Int. J. Mol. Sci. 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Williams E. G., Wu Y., Jha P., Dubuis S., Blattmann P., Argmann C. A., Houten S. M., Amariuta T., Wolski W., Zamboni N., Aebersold R., and Auwerx J. (2016) Systems proteomics of liver mitochondria function. Science 352, aad0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lobo-Jarne T., Nyvltova E., Perez-Perez R., Timon-Gomez A., Molinie T., Choi A., Mourier A., Fontanesi F., Ugalde C., and Barrientos A. (2018) Human COX7A2L regulates complex III biogenesis and promotes supercomplex organization remodeling without affecting mitochondrial bioenergetics. Cell Rep. 25, 1786–1799.e1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vukotic M., Oeljeklaus S., Wiese S., Vogtle F. N., Meisinger C., Meyer H. E., Zieseniss A., Katschinski D. M., Jans D. C., Jakobs S., Warscheid B., Rehling P., and Deckers M. (2012) Rcf1 mediates cytochrome oxidase assembly and respirasome formation, revealing heterogeneity of the enzyme complex. Cell Metab. 15, 336–347 [DOI] [PubMed] [Google Scholar]
- 25. Strogolova V., Furness A., Robb-McGrath M., Garlich J., and Stuart R. A. (2012) Rcf1 and Rcf2, members of the hypoxia-induced gene 1 protein family, are critical components of the mitochondrial cytochrome bc1-cytochrome c oxidase supercomplex. Mol. Cell. Biol. 32, 1363–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Y. C., Taylor E. B., Dephoure N., Heo J. M., Tonhato A., Papandreou I., Nath N., Denko N. C., Gygi S. P., and Rutter J. (2012) Identification of a protein mediating respiratory supercomplex stability. Cell Metab. 15, 348–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rydstrom Lundin C., von Ballmoos C., Ott M., Adelroth P., and Brzezinski P. (2016) Regulatory role of the respiratory supercomplex factors in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 113, E4476–E4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garlich J., Strecker V., Wittig I., and Stuart R. A. (2017) Mutational analysis of the QRRQ motif in the yeast Hig1 Type 2 protein Rcf1 reveals a regulatory role for the cytochrome c oxidase complex. J. Biol. Chem. 292, 5216–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kasper L. H., and Brindle P. K. (2006) Mammalian gene expression program resiliency: the roles of multiple coactivator mechanisms in hypoxia-responsive transcription. Cell Cycle 5, 142–146 [DOI] [PubMed] [Google Scholar]
- 30. Denko N., Schindler C., Koong A., Laderoute K., Green C., and Giaccia A. (2000) Epigenetic regulation of gene expression in cervical cancer cells by the tumor microenvironment. Clin. Cancer Res. 6, 480–487 [PubMed] [Google Scholar]
- 31. Hayashi T., Asano Y., Shintani Y., Aoyama H., Kioka H., Tsukamoto O., Hikita M., Shinzawa-Itoh K., Takafuji K., Higo S., Kato H., Yamazaki S., Matsuoka K., Nakano A., Asanuma H., Asakura M., Minamino T., Goto Y., Ogura T., Kitakaze M., Komuro I., Sakata Y., Tsukihara T., Yoshikawa S., and Takashima S. (2015) Higd1a is a positive regulator of cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 112, 1553–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stroud D. A., Surgenor E. E., Formosa L. E., Reljic B., Frazier A. E., Dibley M. G., Osellame L. D., Stait T., Beilharz T. H., Thorburn D. R., Salim A., and Ryan M. T. (2016) Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 538, 123–126 [DOI] [PubMed] [Google Scholar]
- 33. Labun K., Montague T. G., Krause M., Torres Cleuren Y. N., Tjeldnes H., and Valen E. (2019) CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47, W171–W174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yanisch-Perron C., Vieira J., and Messing J. (1985) Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33, 103–119 [DOI] [PubMed] [Google Scholar]
- 36. Lake N. J., Webb B. D., Stroud D. A., Richman T. R., Ruzzenente B., Compton A. G., Mountford H. S., Pulman J., Zangarelli C., Rio M., Boddaert N., Assouline Z., Sherpa M. D., Schadt E. E., Houten S. M., Byrnes J., McCormick E. M., Zolkipli-Cunningham Z., Haude K., Zhang Z., Retterer K., Bai R., Calvo S. E., Mootha V. K., Christodoulou J., Rotig A., Filipovska A., Cristian I., Falk M. J., Metodiev M. D., and Thorburn D. R. (2017) Biallelic mutations in MRPS34 lead to instability of the small mitoribosomal subunit and Leigh Syndrome. Am. J. Hum. Genet. 101, 239–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kirby D. M., Thorburn D. R., Turnbull D. M., and Taylor R. W. (2007) Biochemical assays of respiratory chain complex activity. Methods Cell Biol. 80, 93–119 [DOI] [PubMed] [Google Scholar]
- 38. Wittig I., Braun H. P., and Schagger H. (2006) Blue native PAGE. Nat. Protoc. 1, 418–428 [DOI] [PubMed] [Google Scholar]
- 39. Lazarou M., McKenzie M., Ohtake A., Thorburn D. R., and Ryan M. T. (2007) Analysis of the assembly profiles for mitochondrial- and nuclear-DNA-encoded subunits into complex I. Mol. Cell. Biol. 27, 4228–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnston A. J., Hoogenraad J., Dougan D. A., Truscott K. N., Yano M., Mori M., Hoogenraad N. J., and Ryan M. T. (2002) Insertion and assembly of human tom7 into the preprotein translocase complex of the outer mitochondrial membrane. J. Biol. Chem. 277, 42197–42204 [DOI] [PubMed] [Google Scholar]
- 41. Zerbetto E., Vergani L., and Dabbeni-Sala F. (1997) Quantification of muscle mitochondrial oxidative phosphorylation enzymes via histochemical staining of blue native polyacrylamide gels. Electrophoresis 18, 2059–2064 [DOI] [PubMed] [Google Scholar]
- 42. Salabei J. K., Gibb A. A., and Hill B. G. (2014) Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 9, 421–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kulak N. A., Pichler G., Paron I., Nagaraj N., and Mann M. (2014) Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324 [DOI] [PubMed] [Google Scholar]
- 44. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 45. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., and Cox J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 [DOI] [PubMed] [Google Scholar]
- 46. Calvo S. E., Clauser K. R., and Mootha V. K. (2016) MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 44, D1251–D1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Deutsch E. W., Lam H., and Aebersold R. (2008) PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 9, 429–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dibley M. G., Formosa L. E., Lyu B., Reljic B., McGann D., Muellner-Wong L., Kraus F., Sharpe A. J., Stroud D. A., and Ryan M. T. (2019) The mitochondrial acyl-carrier protein interaction network highlights important roles for LYRM family members in complex I and mitoribosome assembly. Mol. Cell. Proteomics 19, 65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W. W., Hegde M., Lyon E., Spector E., Voelkerding K., Rehm H. L., and Committee A. L. Q. A. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Frazier A. E., Thorburn D. R., and Compton A. G. (2019) Mitochondrial energy generation disorders: genes, mechanisms, and clues to pathology. J. Biol. Chem. 294, 5386–5395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Balsa E., Marco R., Perales-Clemente E., Szklarczyk R., Calvo E., Landazuri M. O., and Enriquez J. A. (2012) NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 16, 378–386 [DOI] [PubMed] [Google Scholar]
- 53. Stroud D. A., Maher M. J., Lindau C., Vogtle F. N., Frazier A. E., Surgenor E., Mountford H., Singh A. P., Bonas M., Oeljeklaus S., Warscheid B., Meisinger C., Thorburn D. R., and Ryan M. T. (2015) COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of mtDNA-encoded COX2. Hum. Mol. Genet. 24, 5404–5415 [DOI] [PubMed] [Google Scholar]
- 54. Zong S., Wu M., Gu J., Liu T., Guo R., and Yang M. (2018) Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 28, 1026–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fukuda R., Zhang H., Kim J. W., Shimoda L., Dang C. V., and Semenza G. L. (2007) HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129, 111–122 [DOI] [PubMed] [Google Scholar]
- 56. Ostergaard E., Weraarpachai W., Ravn K., Born A. P., Jonson L., Duno M., Wibrand F., Shoubridge E. A., and Vissing J. (2015) Mutations in COA3 cause isolated complex IV deficiency associated with neuropathy, exercise intolerance, obesity, and short stature. J. Med. Genet. 52, 203–207 [DOI] [PubMed] [Google Scholar]
- 57. Mick D. U., Vukotic M., Piechura H., Meyer H. E., Warscheid B., Deckers M., and Rehling P. (2010) Coa3 and Cox14 are essential for negative feedback regulation of COX1 translation in mitochondria. J. Cell Biol. 191, 141–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Clemente P., Peralta S., Cruz-Bermudez A., Echevarria L., Fontanesi F., Barrientos A., Fernandez-Moreno M. A., and Garesse R. (2013) hCOA3 stabilizes cytochrome c oxidase 1 (COX1) and promotes cytochrome c oxidase assembly in human mitochondria. J. Biol. Chem. 288, 8321–8331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Box J. M., Kaur J., and Stuart R. A. (2017) MrpL35, a mitospecific component of mitoribosomes, plays a key role in cytochrome c oxidase assembly. Mol. Biol. Cell 28, 3489–3499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Glerum D. M., Shtanko A., and Tzagoloff A. (1996) SCO1 and SCO2 act as high copy suppressors of a mitochondrial copper recruitment defect in Saccharomyces cerevisiae. J. Biol. Chem. 271, 20531–20535 [DOI] [PubMed] [Google Scholar]
- 61. Carr H. S., George G. N., and Winge D. R. (2002) Yeast Cox11, a protein essential for cytochrome c oxidase assembly, is a Cu(I)-binding protein. J. Biol. Chem. 277, 31237–31242 [DOI] [PubMed] [Google Scholar]
- 62. Ogilvie I., Kennaway N. G., and Shoubridge E. A. (2005) A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J. Clin. Invest. 115, 2784–2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vogel R. O., van den Brand M. A., Rodenburg R. J., van den Heuvel L. P., Tsuneoka M., Smeitink J. A., and Nijtmans L. G. (2007) Investigation of the complex I assembly chaperones B17.2L and NDUFAF1 in a cohort of CI deficient patients. Mol. Genet. Metab. 91, 176–182 [DOI] [PubMed] [Google Scholar]
- 64. Saada A., Edvardson S., Rapoport M., Shaag A., Amry K., Miller C., Lorberboum-Galski H., and Elpeleg O. (2008) C6ORF66 is an assembly factor of mitochondrial complex I. Am. J. Hum. Genet. 82, 32–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McKenzie M., Lazarou M., Thorburn D. R., and Ryan M. T. (2007) Analysis of mitochondrial subunit assembly into respiratory chain complexes using Blue Native polyacrylamide gel electrophoresis. Anal. Biochem. 364, 128–137 [DOI] [PubMed] [Google Scholar]
- 66. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., and Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 67. Formosa L. E., Hofer A., Tischner C., Wenz T., and Ryan M. T. (2016) Translation and assembly of radiolabeled mitochondrial DNA-encoded protein subunits from cultured cells and isolated mitochondria. Methods Mol. Biol. 1351, 115–129 [DOI] [PubMed] [Google Scholar]
- 68. Zhou S., Pettersson P., Huang J., Sjoholm J., Sjostrand D., Pomes R., Hogbom M., Brzezinski P., Maler L., and Adelroth P. (2018) Solution NMR structure of yeast Rcf1, a protein involved in respiratory supercomplex formation. Proc. Natl. Acad. Sci. U.S.A. 115, 3048–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Strogolova V., Hoang N. H., Hosler J., and Stuart R. A. (2019) The yeast mitochondrial proteins Rcf1 and Rcf2 support the enzymology of the cytochrome c oxidase complex and generation of the proton motive force. J. Biol. Chem. 294, 4867–4877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Thompson K., Mai N., Olahova M., Scialo F., Formosa L. E., Stroud D. A., Garrett M., Lax N. Z., Robertson F. M., Jou C., Nascimento A., Ortez C., Jimenez-Mallebrera C., Hardy S. A., He L., Brown G. K., Marttinen P., McFarland R., Sanz A., Battersby B. J., Bonnen P. E., Ryan M. T., Chrzanowska-Lightowlers Z. M., Lightowlers R. N., and Taylor R. W. (2018) OXA1L mutations cause mitochondrial encephalopathy and a combined oxidative phosphorylation defect. EMBO Mol. Med. 10, pii: e9060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Desmurs M., Foti M., Raemy E., Vaz F. M., Martinou J. C., Bairoch A., and Lane L. (2015) C11orf83, a mitochondrial cardiolipin-binding protein involved in bc1 complex assembly and supercomplex stabilization. Mol. Cell. Biol. 35, 1139–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wanschers B. F., Szklarczyk R., van den Brand M. A., Jonckheere A., Suijskens J., Smeets R., Rodenburg R. J., Stephan K., Helland I. B., Elkamil A., Rootwelt T., Ott M., van den Heuvel L., Nijtmans L. G., and Huynen M. A. (2014) A mutation in the human CBP4 ortholog UQCC3 impairs complex III assembly, activity and cytochrome b stability. Hum. Mol. Genet 23, 6356–6365 [DOI] [PubMed] [Google Scholar]
- 73. Ameri K., Jahangiri A., Rajah A. M., Tormos K. V., Nagarajan R., Pekmezci M., Nguyen V., Wheeler M. L., Murphy M. P., Sanders T. A., Jeffrey S. S., Yeghiazarians Y., Rinaudo P. F., Costello J. F., Aghi M. K., and Maltepe E. (2015) HIGD1A regulates oxygen consumption, ROS production, and AMPK activity during glucose deprivation to modulate cell survival and tumor growth. Cell Rep. 10, 891–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD016864.