

Abstract
The COVID-19 caused by SARS-CoV-2 has spread globally and caused tremendous loss of lives and properties and it is of utmost emergency to understand its propagation process and find ways to slow down the epidemic. In this work we used a coarse grained model to calculate the binding free energy of SARS-CoV-2 or SARS-CoV to their human receptor ACE2. The investigation of the free energy contribution of the interacting residues indicates that the residues located outside the receptor binding domain are the source of the stronger binding of the novel virus. Thus the current results suggest that the essential evolution of SARS-CoV-2 happens remotely from the binding domain at the spike protein trimeric body. Such evolution may facilitate the conformational change and the infection process that occurs after the virus is bound to ACE2. By studying the binding pattern between SARS-CoV antibody m396 and SARS-CoV-2, it is found that the remote energetic contribution is missing, which might explain the absence of cross-reactivity of such antibodies.
Graphical Abstract
1. INTRODUCTION
The 2019 coronavirus disease (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been spread globally since its first outbroke in Wuhan, China in December 2019 1-3. Common symptoms of SARS-CoV-2 infected patients involves fever, cough, and fatigue with an estimated death rate of 3%-5% 4. It has already caused more than 7 million confirmed cases and more that 400,000 more deaths all over the world (at the time this work is written). In addition to health concerns, the disease also brought severe economic and social issues 5. While the situation of Wuhan has been stepped down, Europe and United State is experiencing major epidemic. Thus, it is crucial to understand the infection and spreading process and find measures to mitigate the epidemic situation.
The SARS-CoV-2 is a member of the beta-coronavirus genus 6 which also includes the acute respiratory syndrome coronavirus (SARS-CoV) and the middle-east respiratory syndrome virus (MERS). The SARS-CoV-2 virus appears to be optimized for binding to the human receptor ACE2 7,and the binding patterns between ACE2 and SARS-CoV-2 or SARS-CoV at the receptor binding domain (RBD) are thought to be almost identical 8. More specifically , SARS-CoV-2 shares 76%-78% sequence identity with SARS-CoV for the whole protein and 73%-76% for the RBD 9. The trimeric spike glycoprotein of SARS-CoV-2 is comprised of three S1/S2 units and the RBD locates at S1. One variation , the S1/S2 cleavage site of SARS-CoV-2, is a unique “RRAR” furin recognition site 10 ,while in SARS-CoV it is a single arginine 11. The three S1/S2 units undergoes a hinge-like conformational switch between “up” and “down” states. Only at the “up” state the RBD is exposed and is able to bind the receptor, while at the “down” state the RBD is hidden and is inaccessible by the receptor 12. For SARS-CoV, the spike trimer with two “down” and one “up” is the most populated state 13. This could very likely be the case for SARS-CoV-2, but to our knowledge no experimental statistical measurement has been reported yet. A recent study pointed out the possibility of two spike proteins binding with the same ACE214. After binding to the receptor, the following cascade of events is triggered: the spike protein undergoes a large conformational change, the S1 with the receptor is shed, S2 is transformed to a more stable post-fusion state, and finally the viral membrane is fused with the cell membrane15,16.
Despite the similarities in structures and binding patterns between the two viruses, SARS-CoV-2 spreads faster than SARS-CoV and this might be due to the stronger binding of the ACE2-SARS-CoV-2 complex 12. The range of experimental binding affinities of the two ACE2-virus is wide, with reports of 15 nM 12 and 150-185 nM 13 for the ACE2-SARS-CoV-2 and the ACE2-SARS-CoV complexes ,respectively, and also reports of 4.7 nM and 31 nM 8, and 1.2 nM and 5.0 nM 17 for both systems, respectively. In all cases, the ACE2-SARS-CoV-2 complex shows a larger binding affinity than ACE2-SARS-CoV. On the other hand, although the sequences and epitope has been studied extensively, it is still unclear what is the structural/energetic basis for the difference between the two complexes. Moreover, the receptor binding is a crucial step for drug and antibody interference with the infection process. Thus, this work will focus on understanding the detailed differences between the binding features of the coronavirus and the human receptor ACE2.
Recent works yilded high-resolution structure of SARS-CoV-2 at its pre-fusion state (14) , as well as the complex of its RBD domain and ACE2 8. These emerging structures provide an opportunity to use computational modeling to investigate the underlying mechanism behind the differences in binding strengths of the two ACE2-virus complexes.
However, such a task is very challenging. Recent theoretical work 18 analyzed the number of contacts, interface area, and fluctuations and concluded that different viruses has different strategy for binding. However, this work could not obtain the correct order of binding affinity between the ACE2-SARS-CoV-2 and ACE2-SARS-CoV complexes.
Obviously, the main issue is the differences in interaction free energies between the two types of viruses and the receptor, and this energy is essential for understanding the binding process. Evaluating the binding energy of a very large protein complexes is an enormous challenge for fully atomistic models and thus we chose here to use our coarse-grained (CG) model19-21 to study the energetics of the complexes. Our CG model has been consistently developed and systematically calibrated to account for proper evaluation of electrostatic free energies of proteins and membranes, including, of course, solvation and hydrophobic effects. The model was applied extensively to many systems, calculating protein folding free energies and related properties 22-24. Here we use the model to evaluate the binding of the virus to ACE2.
Our analysis of the binding pattern found that the substitutions of residues near the RBD of SARS-Cove in the conversion to SARS-CoV-2, is not the reason of increase in binding energy. It is found that the major contribution actually comes from the body of the spike protein that is away from the RBD. It is also found that the anti-body of SARS-CoV, that did not show cross-reactivity, might be partially due to the fact that the binding interface is partially covered, compare to the situation with ACE2.
2. METHODS
In this work, we used Modeller 25 to perform homology modeling in constructing the binding complexes of ACE2-SARS-CoV-2 and m396-SARS-CoV-2. The SARS-CoV-2 structure was taken from the recent cryo-EM study (PDB ID: 6VSB) 12 with an incomplete receptor binding domain. For the binding domain we used crystal structure of SARS-CoV-2 RBD that is bound to ACE2 (PDB ID: 6M0J) 8. The binding between m396 and SARS-CoV-2 was modeled using the m396-SARS-CoV structure as template (PDB ID: 2DD8) 26.
Subsequently, we utilized our CG model to calculate the free energy of each structure and the relevant binding energies. Our CG model is focused on the electrostatic free energy of the protein that involves the solvation energy and the interactions between charged and polar residues. The full CG treatment includes membrane terms (see SI) which are not included in the present case, since the membrane is out of the system studied. The total CG energy is defined as follows 20 (see also SI).
The terms on the right are: the main-chain solvation free energy, the electrostatic free energy, the hydrophobic solvation energy, the hydrophilic (polar) solvation energy, the effective van der Waals free energy, the effective hydrogen bond free energy, and the energy under external potential, respectively.
Before the evaluation of the free energy , we used a Monte Carlo Proton Transfer (MCPT) method 20 to determine the charge configuration of all ionizable residues in the system. In the MCPT approach, the MC procedures controls proton transfer moves between ionizable residues or between one ionizable residue and the bulk. The acceptance possibility of the move is determined by standard Metropolis criteria (see SI). By such calculations we are able to get the CG free energy of each protein configuration and also the electrostatic contribution of each residue when they are either in the ACE2-virus complex or the unbound state. Note that the CG already represents the free energy of the system and not the potential energy. All calculations and simulations were carried out using the MOLARIS-XG package 27,28. For more details see the SI
3. RESULTS AND DISCUSSION
We started by trying to evaluate the binding energy differences between the two complexes (ACE2-SARS-CoV-2 and ACE2-SARS-CoV) and to determine where the difference is coming from. This study utilized the recently published cryo-EM structure of SARS-CoV-2 (PDB ID: 6VSB)12 and the crystal structure of its RBD and ACE2 (PDB ID: 6M0J)8 , and performed homology-modeling in order to obtain the structure of the ACE2-SARS-CoV-2 complex. The structure of ACE2-SARS-CoV has been taken from a previous work (PDB ID: 6CS2) 13. After getting the structures we performed energy minimization and molecular dynamics for structural relaxation. This procedure is followed by the MCPT algorithm 20 (that determine the optimal charge distribution of the ionizable residues) to obtain the CG energies of the ACE2-SARS-CoV-2 and ACE2-SARS-CoV complexes. With the same treatment we obtained the CG energies of the ACE2, SARS-CoV, and SARS-COV-2 monomers at infinite distance separation. The ACE2-virus binding free energy is then calculated by .
Even though the binding mode of the two ACE2-virus complexes (Figure 1B) were argued to be almost identical 8, we still obtained binding energy of −70.7 kcal/mol for ACE2-SARS-CoV-2 and −66.4 for ACE2-SARS-CoV, respectively. As expected, the major difference come from electrostatic contributions. At this point, the binding energy difference could either be an effect of the non-conserved residues or the change in structures of the two complexes near or outside the RBDs.
Fig. 1.

(A) Structure of the ACE2-SARS-CoV-2 complex. ACE2 is in blue and SARS-CoV-2 is in cyan. (B) Overlap of receptor binding domain of ACE2-SARS-CoV-2 and ACE2-SARS-CoV complex. ACE2 of SARS-CoV-2 complex is in blue and SARS-CoV-2 spike protein is in cyan. ACE2 of SARS-CoV complex is in red and SARS-CoV spike protein is in orange.
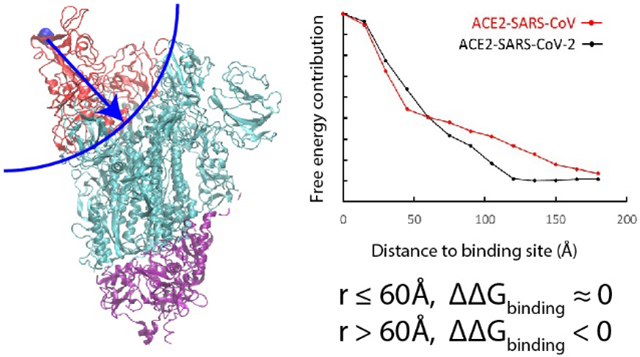
To understand this issue in a more quantitative way we evaluated the electrostatic contributions of each residues to the total binding free energy of the two complexes (see Figure 2A). It was found that some residues give positive contributions while others give negative contributions. This finding is consistent with the results that shows some interactions at the binding interface strengthen while others weaken the binding 14. To see whether the difference in binding comes from the RBD, we further classified the free energy contributions based on their distances from the binding site. Thus, we plotted in Fig.2B the contributions of the residues within given range to the total free energy (this was done according to the distances to the N501 residue of ACE2-SARS-CoV-2 or to T487 of ACE2-SARS-CoV). To our surprise, if we consider the residues within 60 Å of the binding site, it is found that the ACE2-SARS-CoV complex has a stronger binding affinity (its curve is below that of the ACE2-SARS-CoV-2 curve). Apparently the residues between 60 Å and 120 Å are the residues that switched the trend. This result indicates that the binding to the receptor in a remote position from the binding site possibly lead to a stronger binding in ACE2-SARS-CoV-2 than in ACE2-SARS-CoV. Interestingly, the three S1/S2 cleavage sites also locate within the 60 – 120 Å range.
Fig. 2.

(A) Electrostatic free energy changes of each residue before and after binding of ACE2-SARS-CoV-2 and ACE2-SARS-CoV complexes. (B) Binding energy contributions of residues depend on their distances to binding interface (N501 for SARS-CoV-2 and T487 for SARS-CoV). (C) Classification of charged residues based on their energy change of ACE2-SARS-CoV-2 complex and (D) of ACE2-SARS-CoV complex.
In view of the above conclusion we tried to examine whether the finding of long-range interactions is coincidental. Thus, we tested the effective dielectric constant in the CG model ( using a constant between 60 and 90) and also to use a function of the form: , where we used and . It was found that the trend stayed similar with the different dielectric constants. It is still possible that including the effect of the ionic strength would reduce the binding difference at the long distance, but the sign is very unlikely to change.
It should be noted that the value of the overall calculated binding energy is most probably an overestimate. One missing effect is the entropy contribution of the separate parts of the complex (which is equal for the two systems). Another missing effect is the above mentioned effect of the ionic strength that would reduce the electrostatic interaction.
To illustrate the contributions of each residue we plot their contributions in Figure 2C-2D. The figure assigns to each residue by its free energy contribution. Both complexes have residues with relatively large energy changes near the binding site (red and yellow colors) and according to Figure 2B these contributions are more negative for ACE2-SARS-CoV-2. However, when we move outside the RBD, more residues of this type appear in the SARS-CoV2’s thinner spike protein body. compared to the SARS-CoV’s fatter spike protein body. It suggests that for both ACE2-virus complexes, some residues at the RBD region strengthen the binding while others weaken it (Figure 2A). However, the interactions changes between 60 Å to 120 Å to the binding site make the binding affinity of SARS-CoV-2 stronger (Figure 2B), which may indicate an effective evolution of the spike protein body of the novel virus. This larger binding affinity might explain why SARS-CoV-2 spreads faster than SARS-CoV.
Because of the structural homology and similar binding patterns, several RBD-directed monoclonal anti-bodies (m396, S230, 80R) of SARS-CoV have been tested for SARS-CoV-2, but none of them could show cross-reactivity 8,12. Another antibody (CR3022) that was obtained from a convalescent SARS-CoV patient could bind to SARS-CoV-2 but still could not neutralize the virus even at concentration as high as 400 μg/mL and its cross-reactivity was attributed to the high percentage of targeted epitope residues (86%) 29.
In this work we tried to understand the absence of cross-reactivity by studying the binding pattern between SARS-CoV-2 with one of the SARS-CoV antibody m396. We used homology modeling to generate a structure of the m396-SARS-CoV-2 complex from SARS-CoV-2 (PDB ID: 6VSB) 12 and m396-SARS-CoV (RBD) (PDB ID: 2DD8) 26 structures. For the antibody-virus complexes we use . Figure 3A shows the overlapped structures of ACE2-SARS-CoV-2 and m396-SARS-CoV-2 complexes that are aligned toward the virus body. Visually the m396 antibody only covers a part of the ACE2-SARS-CoV-2 binding interface. As before, we analyzed the distance dependent electrostatic energy contributions of the m396-virus complex. As seen from figure 3B the binding of m396 and SARS-CoV-2 does not result in structural/energetic differences that can lead to increase of interactions in the range between 60 Å and 120 Å, which was observed in the ACE2-SARS-CoV-2 complex. The energy contribution near the RBD (< 60 Å ) is also weaker in comparison to the corresponding contribution in the ACE2-virus complex. Overall, m396 shows an ineffective binding pattern that missed part of epitopes of ACE2 that could trigger the following structural changes and this might also be the case for other antibodies that did not show cross-reactivity. To mimic the ACE2-SARS-CoV-2 binding pattern, Zhang et al. synthesized a 23-mer peptide fragment of the ACE2 peptidase domain α1 helix 30. However, the binding affinity was not strong. This might be an effect of the instability of a small helix fragment.
Fig. 3.

(A) Overlap of m396-SARS-CoV-2 and m396-SARS-CoV complex. m396 of SARS-CoV-2 complex is in blue and SARS-CoV-2 spike protein is in cyan. m396 of SARS-CoV complex is in red and SARS-CoV spike protein is in orange. (B) Binding energy contributions of residues depend on their distances to binding domain
Structural analysis show that there are 14 key residues that participate in the binding between ACE2 and SARS-CoV 17. Among them 8 are conserved in SARS-CoV-2 and the other 6 are mutated. The 6 mutated residues are: N439/R426 (SARS-CoV-2/SARS-CoV), L455/Y442, F486/L472, Q493/N478, Q498/Y484, and N501/T487 (Figure 4A). To understand how the substituted residues would affect binding energy, we performed mutation calculations for the two ACE2-virus complexes. The residues of ACE2-SARS-CoV-2 were mutated to the one in the correspond position in ACE2-SARS-CoV and vice versa. After introducing the mutation, we performed another relaxation run before the CG MCPT free energy evaluation. As shown in Table 1, all mutated ACE2-SARS-CoV-2 constructs are less stable compare to the wild-type system, while the mutated ACE2-SARS-CoV shows opposite results. This is consistent with our previous distance dependent binding energy contributions near the RBD , where we find that ACE2 and SARS-CoV gives a more favorable binding pattern than the ACE2 and SARS-CoV-2 (Figure 2B). The mutation calculation results suggest the effective evolvement of the novel virus might happens as remote protein body.
Fig. 4.

Positions of key residues at ACE2-SARS-CoV-2 binding domain that is substituted compare to SARS-CoV.
Table 1.
CG MCPT results after mutating residues at key positions between ACE2-SARS-CoV-2 (N439, L455, F486, Q493, Q498, N501) and ACE2-SARS-CoV (R426, Y442, L472, N479, Y484, T487) complexes. Energy unit in kcal/mol.
| Residue # | ACE2-SARS-CoV-2 mutation | ACE2-SARS-CoV mutation |
|---|---|---|
| Wild-type | −70.71 | −66.45 |
| N439/R426 | −70.79 | −61.98 |
| L455/Y442 | −75.61 | −62.20 |
| F486/L472 | −72.39 | −53.23 |
| Q493/N479 | −75.14 | −63.79 |
| Q498/Y484 | −73.90 | −57.10 |
| N501/T487 | −73.80 | −58.08 |
| All 6 | −73.05 | −58.81 |
The current work explored the structural/energetic basis of the difference in binding energy between the SARS-CoV-2/ACE2 and the SARS-CoV/ACE2 complexes. It is found that the SARS-CoV-2’s binding is more favorable , not because its RBD has been optimized, but because the SARS-CoV-2 glycoprotein trimer body has been evolved to bind stronger at a distanced sites (in fact, if we just consider the RBD then SARS-CoV is more favorable. It is not very clear if this stronger binding is converted to conformational changes, cleavage, and subsequent fusion events. However, if this is the case, we have an interesting explanation of the reasons why the novel coronavirus spreads faster and easier. The results also suggest to use the novel virus as template during drug/antibody design with the whole spike protein as binding template instead of a fraction of the RBD that might neglect the essential changes in the virus body and size effect.
Supplementary Material
ACKNOWLEDGEMENT
This work was supported by the National Institute of Health R35 GM122472 and the National Science Foundation Grant MCB 1707167. We thank Dr. Veselin Kolev for discussion and manuscript preparation. We thank the University of Southern California High Performance Computing and Communication Center for computational resources.
REFERRENCES
- 1.Zhou P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273, doi: 10.1038/s41586-020-2012-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269, doi: 10.1038/s41586-020-2008-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu N. et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. New England Journal of Medicine 382, 727–733, doi: 10.1056/NEJMoa2001017 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang D. et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA 323, 1061–1069, doi: 10.1001/jama.2020.1585 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKibbin WJ a. Roshen F,. The Global Macroeconomic Impacts of COVID-19: Seven Scenarios. CAMA 19, doi: 10.2139/ssrn.3547729 (2020). [DOI] [Google Scholar]
- 6.Lu R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet 395, 565–574, doi: 10.1016/S0140-6736(20)30251-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersen KG, Rambaut A, Lipkin WI, Holmes EC & Garry RF The proximal origin of SARS-CoV-2. Nature Medicine 26, 450–452, doi: 10.1038/s41591-020-0820-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lan J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature, doi: 10.1038/s41586-020-2180-5 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Wan Y, Shang J, Graham R, Baric RS & Li F Receptor Recognition by the Novel Coronavirus from Wuhan: an Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. Journal of Virology 94, e00127–00120, doi: 10.1128/jvi.00127-20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coutard B. et al. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Research 176, 104742, doi: 10.1016/j.antiviral.2020.104742 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bosch BJ, Bartelink W & Rottier PJM Cathepsin L Functionally Cleaves the Severe Acute Respiratory Syndrome Coronavirus Class I Fusion Protein Upstream of Rather than Adjacent to the Fusion Peptide. Journal of Virology 82, 8887–8890, doi: 10.1128/jvi.00415-08 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wrapp D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263, doi: 10.1126/science.abb2507 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirchdoerfer RN et al. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Scientific Reports 8, 15701, doi: 10.1038/s41598-018-34171-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan R. et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448, doi: 10.1126/science.abb2762 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annual Review of Virology 3, 237–261, doi: 10.1146/annurev-virology-110615-042301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song W, Gui M, Wang X & Xiang Y Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLOS Pathogens 14, e1007236, doi: 10.1371/journal.ppat.1007236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walls AC et al. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 181, 281–292.e286, doi: 10.1016/j.cell.2020.02.058 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brielle ES, Schneidman-Duhovny D & Linial M The SARS-CoV-2 exerts a distinctive strategy for interacting with the ACE2 human receptor. bioRxiv, 2020.2003.2010.986398, doi: 10.1101/2020.03.10.986398 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee M, Kolev V & Warshel A Validating a Coarse-Grained Voltage Activation Model by Comparing Its Performance to the Results of Monte Carlo Simulations. The Journal of Physical Chemistry B 121, 11284–11291, doi: 10.1021/acs.jpcb.7b09530 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vorobyov I, Kim I, Chu ZT & Warshel A Refining the treatment of membrane proteins by coarse-grained models. Proteins: Structure, Function, and Bioinformatics 84, 92–117, doi: 10.1002/prot.24958 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vicatos S, Rychkova A, Mukherjee S & Warshel A An effective coarse-grained model for biological simulations: recent refinements and validations. Proteins 82, 1168–1185, doi: 10.1002/prot.24482 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bai C & Warshel A Revisiting the protomotive vectorial motion of F0-ATPase. Proceedings of the National Academy of Sciences 116, 19484–19489, doi: 10.1073/pnas.1909032116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alhadeff R & Warshel A A free-energy landscape for the glucagon-like peptide 1 receptor GLP1R. Proteins: Structure, Function, and Bioinformatics 88, 127–134, doi: 10.1002/prot.25777 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee M, Bai C, Feliks M, Alhadeff R & Warshel A On the control of the proton current in the voltage-gated proton channel Hv1. Proceedings of the National Academy of Sciences 115, 10321–10326, doi: 10.1073/pnas.1809766115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Šali A & Blundell TL Comparative Protein Modelling by Satisfaction of Spatial Restraints. Journal of Molecular Biology 234, 779–815, doi: 10.1006/jmbi.1993.1626 (1993). [DOI] [PubMed] [Google Scholar]
- 26.Prabakaran P et al. Structure of Severe Acute Respiratory Syndrome Coronavirus Receptor-binding Domain Complexed with Neutralizing Antibody. Journal of Biological Chemistry 281, 15829–15836, doi: 10.1074/jbc.M600697200 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamerlin SCL, Vicatos S, Dryga A & Warshel A Coarse-Grained (Multiscale) Simulations in Studies of Biophysical and Chemical Systems. Annual Review of Physical Chemistry 62, 41–64, doi: 10.1146/annurev-physchem-032210-103335 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Lee FS, Chu ZT & Warshel A Microscopic and semimicroscopic calculations of electrostatic energies in proteins by the POLARIS and ENZYMIX programs. Journal of Computational Chemistry 14, 161–185, doi: 10.1002/jcc.540140205 (1993). [DOI] [Google Scholar]
- 29.Yuan M. et al. A highly conserved cryptic epitope in the receptor-binding domains of SARS-CoV-2 and SARS-CoV. Science, eabb7269, doi: 10.1126/science.abb7269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang G, Pomplun S, Loftis AR, Loas A & Pentelute BL The first-in-class peptide binder to the SARS-CoV-2 spike protein. bioRxiv, 2020.2003.2019.999318, doi: 10.1101/2020.03.19.999318 (2020). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.