

Abstract
目的
探索伊布替尼治疗复发/难治原发性自身免疫性溶血性贫血(AIHA)的疗效及安全性。
方法
2例原发性AIHA患者多次复发,1例有脾切除史,经糖皮质激素、利妥昔单抗及多种免疫抑制药物治疗无效,诊断为复发/难治原发性AIHA。给予伊布替尼起始剂量280 mg/d(例1第3~8周加量到420 mg/d)治疗,观察疗效及安全性。
结果
例1治疗2周后脱离输血,HGB增长>20 g/L,获得部分疗效;10周后HGB 113 g/L伴有代偿性溶血实验室改变,获得完全缓解伴代偿性溶血状态(CRi)。例2治疗2周后脱离输血,HGB增长>20 g/L,获得部分疗效;6周后HGB 118 g/L伴有代偿性溶血实验室改变,获得CRi。2例患者分别随访16周和12周,已维持疗效14周和10周,未复发。伊布替尼治疗期间2例患者血液学不良反应轻,非血液学不良反应轻微。
结论
伊布替尼单药治疗复发/难治原发性AIHA有初步疗效,尚需规范的临床研究试验进一步验证其疗效及安全性。
Keywords: 自身免疫性溶血性贫血, 复发/难治, 伊布替尼
Abstract
Objective
To explore the efficacy and safety of ibrutinib treatment for relapsed/refractory(R/R)primary autoimmune hemolytic anemia(AIHA).
Methods
Two cases of primary AIHA with relapse events were refractory to glucocorticoid, anti-CD20 monoclonal antibody, immunosuppressive drugs, and splenectomy(case 1 only). Ibrutinib treatment was administered at an initial dose of 280 mg/d(420 mg/d for case 1 from the 3rd to 8th week).
Results
Both patients achieved transfusion independence and HGB>20 g/L above baseline after 2 weeks(partial response). For case 1, HGB concentration restored to 113 g/L but with incomplete hemolysis recovery after 10 weeks;HGB reached the level of 118 g/L, also with incomplete hemolysis recovery, after 6 weeks in case 2. They all acquired complete response with incomplete hemolysis recovery(CRi). The responses sustained 14 weeks and 10 weeks after follow-up at 16 weeks and 12 weeks, respectively. During the treatment, hematologic and nonhematologic toxicity is mild and acceptable.
Conclusion
Ibrutinib alone is effective for the 2 R/R primary AIHA cases. We need further clinical trial to identify its efficacy and safety.
Keywords: Autoimmune hemolytic anemia, Relapsed/refractory, Ibrutinib
自身免疫性溶血性贫血(AIHA)是一类自身抗体介导的溶血性疾病,B淋巴细胞在自身抗体产生中发挥了重要作用[1]–[3]。糖皮质激素一线治疗有效率高达80%,约三分之一的患者可以获得长期无治疗缓解,而糖皮质激素治疗无效、治疗依赖或多次复发患者二线治疗应优先选择利妥昔单抗[4]–[6]。目前国际诊疗共识将脾切除术及其他免疫抑制剂如硫唑嘌呤、环孢素A、霉酚酸酯作为三线治疗,治疗失败者可尝试应用环磷酰胺、抗CD52单克隆抗体或蛋白酶体抑制剂等药物治疗[4]–[5]。但目前复发/难治AIHA尚无有效治疗方案,患者生存质量差,病死率为5%~10%[7]–[10]。如何提高该类患者的临床疗效,改善生存质量,减少治疗药物相关并发症是亟待解决的临床难题。
利妥昔单抗治疗AIHA重要机制是清除B淋巴细胞,减少自身抗体产生[7],[10]。而B细胞信号通路关键分子Bruton酪氨酸激酶(BTK)抑制剂伊布替尼有效治疗惰性小B细胞淋巴瘤的主要分子机制是靶向抑制B淋巴细胞活化、增殖及功能[11]–[13]。利妥昔单抗已经成功用于治疗AIHA,提示我们能靶向抑制B淋巴细胞的伊布替尼是否也具有良好的治疗疗效,这是我们开展这项研究的科学假设。已有文献报道伊布替尼成功治疗继发于惰性小B细胞淋巴瘤AIHA[14]–[17],但BTK抑制剂治疗原发性AIHA患者国际国内尚未有报道。我们受此启发,经我院伦理委员会批准(NI2020001-EC-1),收治了2例原发性、复发/难治AIHA患者,给予伊布替尼探索性治疗,2周时获得疗效并已持续缓解10~14周,现报道如下。
病例资料
例1,女性,17岁,“诊断AIHA 8年余,4个月前溶血再次发作,激素治疗无效”于2019年8月21日入我院。4岁时诊断“原发免疫性血小板减少症(ITP)”并行脾切除术。9岁时外院确诊AIHA,糖皮质激素治疗有效,HGB正常并可停药,但溶血每年发作1次。2018年始联合服用霉酚酸酯治疗至本次入院。入院HGB 42 g/L,网织红细胞(Ret)比例16.11%、绝对值177.2×109/L;直接Coombs试验IgG+C3阳性,冷凝集素试验阳性(效价1∶128,积分46);骨髓检查流式细胞免疫分型、免疫组化未见淋巴瘤证据;IgH、Igκ、TCR βγδ重排均阴性,游离轻链比值正常,单克隆免疫球蛋白电泳阴性;EB病毒、巨细胞病毒、微小病毒B19、乙型肝炎病毒均阴性;风湿免疫全套抗体阴性。予利妥昔单抗100 mg每周1次,连续4周治疗并逐渐减停甲泼尼龙。利妥昔单抗治疗后第9周时溶血急性加重,HGB 19 g/L,Ret低于检测下限,中性粒细胞及血小板正常,诊断为复发/难治原发性AIHA(温冷双抗体型)。给予紧急分次输血5.5 U,甲泼尼龙40 mg/d联合丙种球蛋白急救治疗3 d。征得患者与家属知情同意并签署知情同意书后,于10月30日开始口服伊布替尼单药治疗。治疗前血常规:WBC 7.03×109/L,PLT 666×109/L,HGB 47 g/L,Ret绝对值84.5×109/L,淋巴细胞绝对值2.41×109/L;生化:总胆红素199.1 µmol/L,LDH 680 U/L;结合珠蛋白<0.125 g/L,IgG 14.6 g/L,补体C3 0.40 g/L,补体C4 0.02 g/L;直接Coombs试验IgG+C3阳性(IgG效价1∶256,C3效价1∶512),冷凝集试验阳性(效价1∶1 024);CD4+/CD8+淋巴细胞比值1.33,CD19+淋巴细胞比例为5.8%。伊布替尼单药治疗起始剂量为280 mg/d(口服)。治疗第2周HGB升至75 g/L,淋巴细胞绝对值4.55×109/L。治疗第3周加量为420 mg/d。治疗第5周EPO水平(30.93 IU/L)及血清铁饱和度(14%)降低,给予EPO 10 000 U隔日1次皮下注射联合口服多糖铁复合物(150 mg,每日2次)治疗,2周后停用。伊布替尼治疗第9周减量为280 mg/d。目前随访16周,患者持续达完全缓解伴代偿性溶血状态(CRi)标准[HGB≥110 g/L(女性)或HGB≥120 g/L(男性),溶血相关实验室指标改善]。治疗期间未发生3~4级不良反应,1~2级不良反应包括粒细胞减少、淋巴细胞增多及高尿酸血症,均缓解。
例2,女,7岁,“乏力、浓茶色尿7个月”于2019年10月22日入我院。2019年3月起病,4月当地医院查HGB 40 g/L,Ret比例60%,WBC、PLT正常,直接Coombs阳性,诊断AIHA。首次治疗给予甲泼尼龙500 mg/d联合丙种球蛋白12 g/d冲击治疗3 d,后甲泼尼龙40 mg/d维持治疗有效,甲泼尼龙减量至16 mg/d后溶血复发。2019年9月予大剂量甲泼尼龙(1 000 mg/d)冲击治疗3 d,联合利妥昔单抗100 mg每周1次×4周,10月再次给予丙种球蛋白15 g/d连续3 d治疗,无效后来我院。入院HGB 66 g/L,Ret比例32.72%、绝对值589×109/L;直接Coombs试验IgG阳性,冷凝集素试验阳性(效价1∶64,积分40)。淋巴瘤、病毒感染均排除,风湿免疫抗体除抗Ro52(±)、抗线粒体M2(±)外均阴性。诊断为复发/难治原发性AIHA(温冷双抗体型)。给予环磷酰胺10 mg/kg每周1次联合环孢素A 100 mg/d,持续4周无效。征得患者与家属知情同意并签署知情同意书后,于12月2日开始口服伊布替尼单药治疗。治疗前血常规:WBC 3.08×109/L,PLT 248×109/L,HGB 65 g/L,Ret绝对值738.9×109/L,淋巴细胞绝对值1.00×109/L;生化:总胆红素51.0 µmol/L,LDH 255 U/L;结合珠蛋白<0.125 g/L,IgG 6.04 g/L,补体C3 0.73 g/L,补体C4 0.25 g/L;直接Coombs试验IgG阳性(效价1∶128),冷凝集试验阳性(效价1∶64);CD4+/CD8+淋巴细胞比值1.81,CD19+淋巴细胞比例为0。予伊布替尼280 mg/d口服治疗,在基线时最后一次输注红细胞2 U,HGB达65 g/L 2周后稳定在90 g/L,4周时达95 g/L,因此判定在2周时获得部分缓解(PR)(HGB≥100 g/L或较基线值增长≥20 g/L,且脱离输血);第6周HGB达到118 g/L,并维持疗效至末次随访(第12周),达到CRi标准。不良反应包括3~4级白细胞减少、粒细胞减少,1~2级恶心、高尿酸血症、低钾血症。
随访期间2例患者HGB变化见图1。2例患者均在治疗第2周脱离输血依赖、HGB增长超过基线值20 g/L,达到PR。例1在第10周HGB达到113 g/L,并维持疗效至末次随访第16周,符合CRi标准;例2在第6周HGB达到118 g/L,并维持疗效至末次随访第12周,达到CRi标准。截至末次随访,2例患者均维持有效反应分别达14和12周,其中CRi疗效均维持6周。溶血实验室指标改善状况如图2所示,例2 Ret绝对值基线水平较高,治疗后呈持续下降,但即使HGB完全恢复正常,网织红细胞仍然有代偿性增高(图2A)。治疗后总胆红素水平呈明显下降(例2)和稳定下降(例1),但尚未到正常水平(图2B)。与Ret和总胆红素不同,治疗后乳酸脱氢酶水平迅速降到正常水平(图2C),与这2例患者主要是血管外溶血为主有关。监测患者免疫功能状况评估显示(表1),免疫球蛋白、补体C3、C4含量没有降低,外周血CD19+B淋巴细胞比例,CD4+、CD8+T细胞比例均无异常变化。文献报道,伊布替尼治疗慢性淋巴细胞白血病会有短暂的、一过性淋巴细胞增多[12]–[13],我们在例1中的确看到治疗第2周开始淋巴细胞数增高,第4周达高峰,8周以后开始下降,16周恢复到基线值。2例患者伊布替尼治疗中ANC未发生持续性下降。
图1. 2例复发/难治原发性自身免疫性溶血性贫血患者伊布替尼治疗后HGB改善情况.
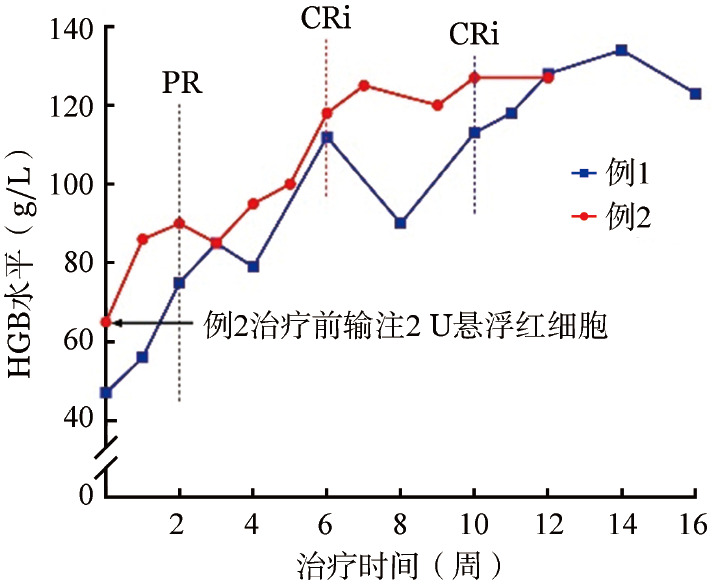
PR:部分缓解;CRi:完全缓解伴代偿性溶血状态
图2. 2例复发/难治原发性自身免疫性溶血性贫血患者伊布替尼治疗后溶血实验室指标改善情况.

表1. 2例复发/难治原发性AIHA患者伊布替尼治疗不同时间免疫功能指标变化.
| 指标 | 例1 |
例2 |
|||||||
| 基线 | 2周 | 4周 | 8周 | 12周 | 16周 | 基线 | 4周 | 9周 | |
| IgG(g/L) | 14.6 | 12.7 | 12.3 | 14.1 | - | - | 6.04 | 6.49 | 5.99 |
| C3(g/L) | 0.4 | 0.47 | 0.47 | 0.53 | - | - | 0.73 | 0.71 | 0.66 |
| C4(g/L) | 0.02 | 0.02 | 0.03 | 0.05 | - | - | 0.25 | 0.20 | 0.22 |
| CD19+ B淋巴细胞比例(%) | 5.8 | 6.0 | 4.1 | 4.6 | - | - | 0.0 | 0.3 | 0.1 |
| CD4+ T淋巴细胞比例(%) | 42.4 | 44.6 | 41.4 | 42.2 | - | - | 56.5 | 50.4 | 46.0 |
| CD8+ T淋巴细胞比例(%) | 32.0 | 39.0 | 32.1 | 28.0 | - | - | 31.2 | 25.9 | 36.4 |
| 淋巴细胞绝对值(×109/L) | 2.41 | 4.55 | 7.41 | 5.42 | 4.88 | 3.20 | 1.00 | 0.82 | 1.00 |
| 中性粒细胞绝对值(×109/L) | 3.86 | 1.41 | 3.01 | 3.38 | 2.97 | 2.65 | 1.50 | 1.48 | 3.48 |
注:AIHA:自身免疫性溶血性贫血;-:未检测
讨论
复发/难治AIHA尚无国际国内统一诊断标准,我们综合文献[2]-[5],[18]拟定复发/难治AIHA诊断标准:①糖皮质激素1.0~2.0 mg/kg治疗3周无效者;②糖皮质激素治疗依赖者(每日维持量≥15 mg泼尼松);③2种或2种以上治疗措施(糖皮质激素、利妥昔单抗、脾切除术)无效或复发者;④2种或2种以上免疫抑制药物联合治疗HGB仍然<110 g/L者;⑤需要3种或3种以上免疫抑制药物联合治疗方能维持HGB>110 g/L者。
鉴于糖皮质激素3周内高达80%以上的有效率,糖皮质激素治疗依赖者极容易反复溶血发作,长期使用不良反应大,多篇文献将糖皮质激素治疗无效或治疗依赖者归于复发/难治[4],[7],[18]。因为考虑到糖皮质激素长期使用的严重不良反应,多数临床诊治共识不建议长期应用糖皮质激素,认为糖皮质激素治疗3周无效、治疗依赖、治疗后首次复发时即应该启动二线治疗措施[2]–[5],[20]。早期文献[2],[5],[21]将利妥昔单抗及脾切除作为二线治疗,但是2019年国际专家共识[1],[4]已将利妥昔单抗作为唯一二线治疗药物,也可以针对性选择重度溶血贫血患者、合并症较多的老年患者作为一线选择用药,与糖皮质激素联合。脾切除术与其他免疫抑制剂如硫唑嘌呤、环孢素A、吗替麦考酚酯作为三线治疗。2个随机对照临床研究也证实了利妥昔单抗联合糖皮质激素虽然不提高3~6个月总有效率,但是可以明显提高完全缓解率及1~3年无复发生存率(70%~80%),而单用糖皮质激素1年无复发生存率仅50%[22]–[23]。文献报道复发/难治AIHA发生的危险因素可能与发病时贫血严重程度,非温抗体型AIHA(温冷双抗体型或冷抗体型),伴有血小板减少,利妥昔单抗延迟使用有关[24]。复发/难治AIHA患者接受目前的三四线治疗药物疗效不能很好维持,且不良反应较大[25]–[26],寻找新的高效且不良反应较低的治疗药物将为AIHA整个治疗路径带来新的治疗前景,患者群体有较大的获益。
分子靶向治疗血液肿瘤的优势在于良好的疗效和耐受性,针对惰性小B淋巴瘤的BTK抑制剂已经作为初诊老年患者及复发/难治患者的有效药物[11]–[13]。文献[14]–[17]报道5例继发于慢性淋巴细胞白血病、套细胞淋巴瘤的AIHA患者经伊布替尼治疗后溶血获得缓解。本研究结果显示减低剂量伊布替尼治疗短期内即可明显控制溶血,脱离输血依赖,获得了有效反应,第6、10周血常规指标正常,溶血指标持续改善,疗效维持至治疗第12、16周均未复发。既往报道的5例中4例为伊布替尼联合糖皮质激素治疗,药物剂量为420~560 mg/d,而我们报道的2例无原发疾病,单用伊布替尼治疗,减低剂量也可以获得满意疗效,可能与无原发淋巴瘤病因有关。我们报道的2例和之前报道的5例在起效时间上比较一致,一般在8周内获得完全缓解(表2)。安全性评价数据显示安全性良好,对机体免疫功能没有抑制作用。
表2. 伊布替尼治疗7例复发/难治AIHA患者临床资料汇总.
| 病例来源 | 性别 | 年龄(岁) | 原发病 | HGB(g/L) |
治疗方案 | 疗效 | 起效时间(周) | |
| 基线 | 治疗有效 | |||||||
| Manda等[14] | 男 | 70 | CLL伴17p− | 76 | 115 | 伊布替尼420 mg/d联合糖皮质激素 | CR | 3 |
| St Bernard等[15] | 男 | 70 | CLL伴13q− | 85 | 120 | 伊布替尼420 mg/d联合糖皮质激素 | CR | 16 |
| St Bernard等[15] | 女 | 70 | CLL伴13q− | 60 | 125 | 伊布替尼420 mg/d单药 | CR | 8 |
| Cavazzini等[16] | 男 | 62 | CLL伴17p− | 63 | 105 | 伊布替尼420 mg/d联合糖皮质激素 | PR | 5 |
| Barot等[17] | 女 | 75 | MCL | 92 | 110 | 伊布替尼560 mg/d联合糖皮质激素 | CR | 7 |
| 例1 | 女 | 17 | − | 47 | 113 | 伊布替尼280 mg/d单药(第3~8周加量为420 mg/d) | PR/CRi | 2/10 |
| 例2 | 女 | 7 | − | 65(输血后) | 118 | 伊布替尼280 mg/d单药 | PR/CRi | 2/6 |
注:AIHA:自身免疫性溶血性贫血;CLL:慢性淋巴细胞白血病;MCL:套细胞淋巴瘤;−:不适用;CR:完全缓解;PR:部分缓解;CRi:完全缓解伴代偿性溶血状态
综合以上数据,我们认为BTK抑制剂有望成为复发/难治原发性AIHA新的二线治疗药物选择。本报道尚属于探索性研究,仍然需要在规范的临床试验中使用BTK抑制剂治疗复发/难治AIHA进一步验证其疗效及安全性。
Funding Statement
基金项目:国家自然科学基金(81670120);中国医学科学院医学与健康科技创新工程项目(2017-I2M-3-018);天津市科学技术委员会重大疾病防治科技重大专项(18ZXDBSY00070)
Fund program:National Natural Science Foundation of China(81670120); CAMS Initiative Fund for Medical Sciences(2017-I2M-3-018); Tianjin Municipal Science and Technology Commission Major Project(18ZXDBSY00070)
References
- 1.Brodsky RA. Warm Autoimmune Hemolytic Anemia[J] N Engl J Med. 2019;381(7):647–654. doi: 10.1056/NEJMcp1900554. [DOI] [PubMed] [Google Scholar]
- 2.Hill A, Hill QA. Autoimmune hemolytic anemia[J] Hematology Am Soc Hematol Educ Program. 2018;2018(1):382–389. doi: 10.1182/asheducation-2018.1.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barcellini W, Fattizzo B, Zaninoni A. Current and emerging treatment options for autoimmune hemolytic anemia[J] Expert Rev Clin Immunol. 2018;14(10):857–872. doi: 10.1080/1744666X.2018.1521722. [DOI] [PubMed] [Google Scholar]
- 4.Jäger U, Barcellini W, Broome CM, et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: Recommendations from the First International Consensus Meeting[J] Blood Rev. 2019:100648. doi: 10.1016/j.blre.2019.100648. [DOI] [PubMed] [Google Scholar]
- 5.Hill QA, Stamps R, Massey E, et al. The diagnosis and management of primary autoimmune haemolytic anaemia[J] Br J Haematol. 2017;176(3):395–411. doi: 10.1111/bjh.14478. [DOI] [PubMed] [Google Scholar]
- 6.Lechner K, Jäger U. How I treat autoimmune hemolytic anemias in adults[J] Blood. 2010;116(11):1831–1838. doi: 10.1182/blood-2010-03-259325. [DOI] [PubMed] [Google Scholar]
- 7.Reynaud Q, Durieu I, Dutertre M, et al. Efficacy and safety of rituximab in auto-immune hemolytic anemia: A meta-analysis of 21 studies[J] Autoimmun Rev. 2015;14(4):304–313. doi: 10.1016/j.autrev.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 8.Barcellini W, Fattizzo B, Zaninoni A, et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients[J] Blood. 2014;124(19):2930–2936. doi: 10.1182/blood-2014-06-583021. [DOI] [PubMed] [Google Scholar]
- 9.Roumier M, Loustau V, Guillaud C, et al. Characteristics and outcome of warm autoimmune hemolytic anemia in adults: New insights based on a single-center experience with 60 patients[J] Am J Hematol. 2014;89(9):E150–155. doi: 10.1002/ajh.23767. [DOI] [PubMed] [Google Scholar]
- 10.Dierickx D, Kentos A, Delannoy A. The role of rituximab in adults with warm antibody autoimmune hemolytic anemia[J] Blood. 2015;125(21):3223–3229. doi: 10.1182/blood-2015-01-588392. [DOI] [PubMed] [Google Scholar]
- 11.Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia[J] N Engl J Med. 2013;369(1):32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang X, Qiu L, Jin J, et al. Ibrutinib versus rituximab in relapsed or refractory chronic lymphocytic leukemia or small lymphocytic lymphoma: a randomized, open-label phase 3 study[J] Cancer Med. 2018;7(4):1043–1055. doi: 10.1002/cam4.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shanafelt TD, Wang XV, Kay NE, et al. Ibrutinib-rituximab or chemoimmunotherapy for chronic lymphocytic leukemia[J] N Engl J Med. 2019;381(5):432–443. doi: 10.1056/NEJMoa1817073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manda S, Dunbar N, Marx-Wood CR, et al. Ibrutinib is an effective treatment of autoimmune haemolytic anaemia in chronic lymphocytic leukaemia[J] Br J Haematol. 2015;170(5):734–736. doi: 10.1111/bjh.13328. [DOI] [PubMed] [Google Scholar]
- 15.St Bernard R, Hsia CC. Safe utilization of ibrutinib with or without steroids in chronic lymphocytic leukemia patients with autoimmune hemolytic anemia[J] Ann Hematol. 2015;94(12):2077–2079. doi: 10.1007/s00277-015-2487-8. [DOI] [PubMed] [Google Scholar]
- 16.Cavazzini F, Lista E, Quaglia FM, et al. Response to ibrutinib of refractory life-threatening autoimmune hemolytic anemia occurring in a relapsed chronic lymphocytic leukemia patient with 17p deletion[J] Leuk Lymphoma. 2016;57(11):2685–2688. doi: 10.3109/10428194.2016.1154955. [DOI] [PubMed] [Google Scholar]
- 17.Barot SV, Lee SS, Patel BJ, et al. Ibrutinib is Effective in Refractory Type II Cryoglobulinemia[J] Clin Lymphoma Myeloma Leuk. 2019;19(12):e629–629e632. doi: 10.1016/j.clml.2019.07.442. [DOI] [PubMed] [Google Scholar]
- 18.范 斯斌, 王 志军, 毛 强, et al. 复发/难治自身免疫性溶血性贫血患者脾切除术疗效分析[J] 中华血液学杂志. 2019;40(2):132–136. doi: 10.3760/cma.j.issn.0253-2727.2019.02.007. [DOI] [Google Scholar]
- 19.中华医学会血液学分会红细胞疾病(贫血)学组. 自身免疫性溶血性贫血诊断与治疗中国专家共识(2017年版)[J] 中华血液学杂志. 2017;38(4):265–267. doi: 10.3760/cma.j.issn.0253-2727.2017.04.001. [DOI] [Google Scholar]
- 20.Sys J, Provan D, Schauwvlieghe A, et al. The role of splenectomy in autoimmune hematological disorders: Outdated or still worth considering?[J] Blood Rev. 2017;31(3):159–172. doi: 10.1016/j.blre.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Zanella A, Barcellini W. Treatment of autoimmune hemolytic anemias[J] Haematologica. 2014;99(10):1547–1554. doi: 10.3324/haematol.2014.114561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia[J] Br J Haematol. 2013;163(3):393–399. doi: 10.1111/bjh.12541. [DOI] [PubMed] [Google Scholar]
- 23.Michel M, Terriou L, Roudot-Thoraval F, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study)[J] Am J Hematol. 2017;92(1):23–27. doi: 10.1002/ajh.24570. [DOI] [PubMed] [Google Scholar]
- 24.Barcellini W, Zaninoni A, Fattizzo B, et al. Predictors of refractoriness to therapy and healthcare resource utilization in 378 patients with primary autoimmune hemolytic anemia from eight Italian reference centers[J] Am J Hematol. 2018;93(9):E243–243E246. doi: 10.1002/ajh.25212. [DOI] [PubMed] [Google Scholar]
- 25.Piatek CI, Bocian H, Algaze S, et al. A Retrospective Study of the Combination of Rituximab, Cyclophosphamide and Dexamethasone for the Treatment of Relapsed/Refractory Warm Antibody Autoimmune Hemolytic Anemia[J] Acta Haematol. 2019:1–6. doi: 10.1159/000501538. [DOI] [PubMed] [Google Scholar]
- 26.Ratnasingam S, Walker PA, Tran H, et al. Bortezomib-based antibody depletion for refractory autoimmune hematological diseases[J] Blood Adv. 2016;1(1):31–35. doi: 10.1182/bloodadvances.2016001412. [DOI] [PMC free article] [PubMed] [Google Scholar]