

Abstract
The redesign of a macromolecular binding interface and corresponding alteration of recognition specificity is a challenging endeavor that remains recalcitrant to computational approaches. This is particularly true for the redesign of DNA binding specificity, which is highly dependent upon bending, hydrogen bonds, electrostatic contacts, and the presence of solvent and counterions throughout the molecular interface. Thus, redesign of protein-DNA binding specificity generally requires iterative rounds of amino acid randomization coupled to selections. Here we describe the importance of scaffold thermostability for protein engineering, coupled with a strategy that exploits the protein’s specificity profile, to redesign the specificity of a pair of meganucleases towards three separate genomic targets. We determine and describe a series of changes in protein sequence, stability, structure and activity that accumulate during the engineering process, culminating in fully retargeted endonucleases.
Graphical Abstract
eTOC Blurb
This paper describes an approach for protein engineering to recognize new DNA targets. The authors demonstrate that counteracting the destabilizing effect of increasing numbers of mutations, by computationally ‘pre-stabilizing’ the protein scaffold, improves the recovery of active engineered proteins and robust activity and function.
Protein engineering has produced notable results and successes ranging from computational design of novel protein folds and assemblages (Huang et al., 2016), to the creation of ligand-binding proteins (Stoddard, 2016; Yang and Lai, 2017). This includes the design of a novel protein fold with ligand-binding activity (Dou et al., 2018) and alteration of protein-protein binding specificity (Berger et al., 2016; Procko et al., 2014) or protein-DNA binding specificity (Thyme and Baker, 2014; Thyme et al., 2014; Thyme et al., 2009). Much of this recent success is attributable to improved computational strategies for sampling protein conformational space and sequences (Dou et al., 2018), and accurate calculation of energetic terms and forces (Alford et al., 2017).
While computational approaches have become increasingly reliable for the design of protein folds and assemblages (Huang et al., 2016), the design of molecular binding function usually requires additional rounds of mutagenesis and selection (Stoddard, 2016). For example, the creation of a protein that binds and activates a small fluorescent molecule required screening panels of protein mutants followed by selection for optimal activity (Dou et al., 2018). Similar efforts have also illustrated that changes to the designed protein’s sequence during engineering alter protein conformation and behavior in a manner that improves function, but is difficult to predict computationally (Bick et al., 2017; Day et al., 2018; Dou et al., 2017; Smith et al., 2016; Tinberg et al., 2013).
Approaches that rely upon rounds of mutagenesis and selection can produce compromised stability and solution behavior as the protein scaffold endures increasing mutational loads. Whereas natural selection continuously enforces an appropriate balance of stability versus functional capability (as illustrated by Ashenberg et al., 2013; Gong et al., 2013), laboratory screens and/or selection experiments are less successful at maintaining protein stability.
Attempts in the laboratory to generate altered DNA recognition specificity are illustrative of these challenges (Bogdanove et al., 2018). This is due to the size, complexity and flexibility of the protein-DNA interface and the important role of hydrogen-bonds, electrostatic interactions, solvent molecules and counterions in recognition and binding (Joyce et al., 2015; Thyme and Baker, 2014). For example, the proteins described in this study (LAGLIDADG homing endonucleases, also termed ‘meganucleases’) recognize 22 basepair DNA target via interactions involving up to 50 residues and a mechanism that involves significant DNA bending, water-mediated contacts, and cross-talk between neighboring residues and basepairs (Stoddard, 2014; Werther et al., 2017). While computational approaches have exhibited some success for small-scale redesign of such systems (Ashworth et al., 2010; Thyme et al., 2014; Thyme et al., 2009), substantial retargeting of this type of DNA binding protein and similar systems such as site-specific recombinases (Karpinski et al., 2016) require extensive manual intervention and screening as described above. Nonetheless, such efforts have produced many redesigned meganucleases and recombinases that are now employed as highly active gene targeting reagents (Bogdanove et al., 2018).
In this study, we describe protein engineering using two different meganucleases that result in the alteration of their DNA recognition specificity towards three different genomic targets. We use an approach that accounts for their natural specificity profiles and demonstrate that protein scaffolds that are sufficiently stabilized deliver superior performance during selections for altered specificity. We also illustrate how the stepwise accumulation of mutations on a protein scaffold is accompanied by reductions in stability and changes in structure and activity.
Results
Engineering I-SmaMI to disrupt a human regulatory sequence.
We initiated this project by engineering the I-SmaMI meganuclease (a naturally evolved thermostable LAGLIDADG homing endonuclease encoded in a mobile group I intron within the mitochondrial genome of the fungus Sordaria macrospora (Shen et al., 2016) to recognize and cleave a genomic target corresponding to a putative GATA transcription factor target site found in the β-globin locus on human chromosome 11 (Orkin and Bauer, 2019). The new recognition site differs at 11 out of 22 basepair positions relative to the enzyme’s original DNA target, with the altered positions being localized to the 5’ and 3’ ends of the target (Figure 1a). The rationale for choosing this target site (aside from its potential role in regulation of globin synthesis) corresponds to an established feature of these proteins’ specificity: the center of the target site is largely recognized via indirect readout of DNA bending and conformation, resulting in sequence preferences across those basepairs that are difficult to alter (Lambert et al., 2016).
Figure 1: Engineering a naturally occurring thermostable meganuclease scaffold.
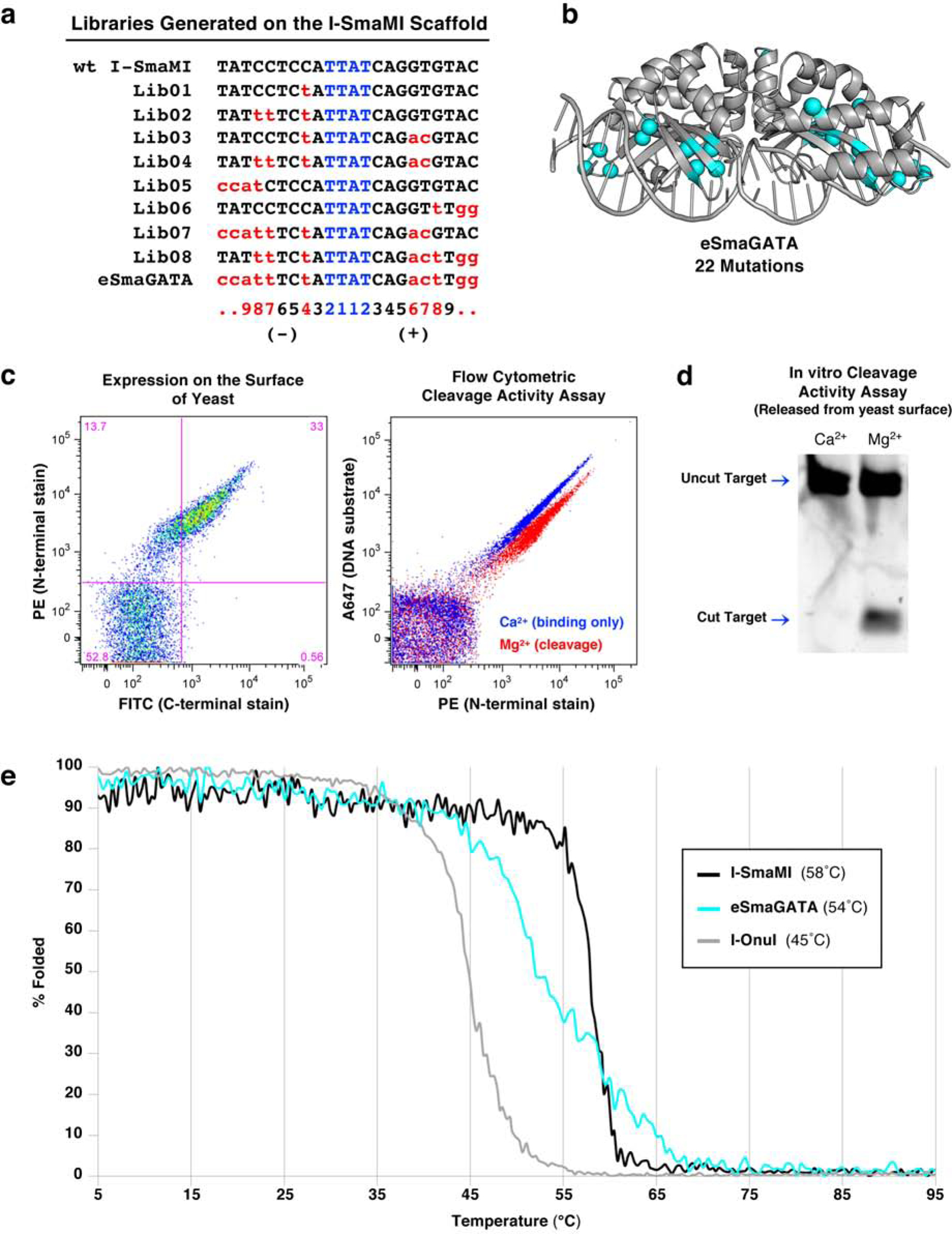
(a) List of target sites for the series of libraries generated on the I-SmaMI meganuclease scaffold and screened for cleavage of the desired hGATA sequence. The numbering of positions in the 22 basepair target is indicated below the target sequences; the 5’ half-site is numbered from −11 to −1; the 3’ half-site is numbered from +1 to +11. (b) Locations of 22 mutations introduced across the I-SmaMI scaffold to create the hGATA-targeting meganuclease (eSmaGATA) are illustrated with cyan spheres on the alpha carbons of the mutated sidechains. (c) Expression and activity of the re-engineered eSmaGATA meganuclease on the surface of yeast using flow cytometry (see Methods and Supplementary Figure S1 for a detailed description of the approach). A drop in the A647 signal from the tethered DNA substrate indicates cleavage of the DNA substrate by the meganuclease. (d) In vitro analysis of DNA cleavage activity with eSmaGATA meganuclease released from the surface of yeast and no physical tethering of the DNA substrate. (e) Circular dichroism (CD) thermal denaturation for the analysis of protein thermostability. The original wild type I-SmaMI meganuclease (black) unfolds at 58°C, while the final re-engineered eSmaGATA protein (cyan) shows a reduced melting temperature of 54°C. The melting curve for the less thermostable meganuclease I-OnuI is shown for comparison (gray), with an even lower melting temperature of 45°C.
Consequently, we used an engineering strategy in which we identified a DNA sequence in the target gene that is largely matched at those central basepairs, and then incorporated changes to the protein that altered specificity towards the 5’ and 3’ ends of the target. The individual ‘stages’ of altered DNA recognition leading to the final desired target are shown in Figure 1a. The approach for generating and screening libraries of meganuclease variants for altered specificity has been previously described (Baxter et al., 2013; Baxter et al., 2014) and is also illustrated in Supplementary Figure S1 and described fully in ‘STAR Methods’. The sequences of the engineered endonuclease variants described in this study and alignments of those sequences with their original wildtype scaffolds, are provided in Supplementary Information, including Supplementary Table S1.
The final engineered variant of I-SmaMI (termed ‘I-SmaMI-e-hGATA’ according to naming convention (Roberts et al., 2003) and referred to hereafter as ‘eSmaGATA’; see Table 1 and Table S1 for formal and abbreviated names of the constructs in this study) contains 22 amino acid substitutions distributed across its DNA-binding surface (Figure 1b and Figure S2a). The mutated residues are located in clusters corresponding to positions that are proximal to the altered DNA basepairs in the new DNA target sequence. The enzyme is expressed strongly on the surface of yeast and displays efficient cleavage of the desired human DNA target in a flow cytometric cleavage assay (Figure 1c). Release of the enzyme from the surface and subsequent in vitro cleavage assays confirms cleavage activity in solution (Figure 1d). The final enzyme construct was then subcloned into a bacterial expression vector, overexpressed and purified as an untagged recombinant protein, and its cleavage activity measured more quantitatively (Figure S2b). The specificity of the final engineered enzyme was assessed using the flow cytometric cleavage assay against a panel of mismatched DNA substrates that each contain a single basepair substitution away from the desired target (Figure S2c).
TABLE 1.
Crystallographic Data and Refinements
| Structure | I-OnuI | I-OnuI-e-Therm | I-OnuI-e-ThermE178D | I-OnuI-e-Therm-bCtxA | I-OnuI-e-Therm-hChr11v1 | IOnuI-e-Therm-hChr11v2 | I-OnuI-e-Therm-hChr11v3 |
|---|---|---|---|---|---|---|---|
| (shortened name in manuscript) | eOnuTherm | eOnuTherm-E178D | eOnuTherm-bCtxA |
eOnuTherm-hChr11v1 (‘Intermediate #1’) |
eOnuTherm-hChr11v2 (‘Intermediate #2’) |
eOnuTherm-hChr11v3 (‘Final’) |
|
| PDB Code | 3QQY | 6UVW | 6UWG | 6UW0 | 6UWH | 6UWJ | 6UWK |
| Data Collection | |||||||
| Space group | P 21 21 21 | P 21 21 21 | P 21 21 21 | P 21 21 21 | P 21 21 21 | P 21 21 21 | P 21 21 21 |
| Cell dimensions | |||||||
| a, b, c (Å) | 37.95, 73.93, 166.93 | 75.43, 82.26, 167.40 | 39.32, 73.52, 163.53 | 75.25, 79.68, 168.94 | 38.89, 72.99, 163.75 | 38.29, 62.59, 157.40 | 38.37, 62.64, 160.20 |
| α, β, γ (°) | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 |
| Resolution (Å) | 44.46 – 2.40 | 50.00 – 2.55 | 50.00 – 2.22 | 50.00 – 2.70 | 50.00 – 2.30 | 50.00 – 1.85 | 50.00 – 2.53 |
| Rmerge | 0.080 (0.140) | 0.073 (0.377) | 0.057 (0.341) | 0.075 (0.416) | 0.058 (0.428) | 0.035 (0.162) | 0.071 (0.335) |
| I/σI | 18.6 (9.62) | 32.6 (4.17) | 30.09 (4.08) | 34.00 (4.63) | 30.32 (4.32) | 51.34 (10.53) | 32.34 (5.75) |
| Completeness (%) | 96.8 (91.0) | 98.6 (90.0) | 99.5 (95.8) | 98.7 (90.5) | 97.4 (99.3) | 99.7 (96.7) | 99.0 (91.7) |
| Redundancy | 6.7 (4.8) | 11.9 (8.5) | 6.5 (5.0) | 11.4 (9.1) | 8.6 (7.2) | 11.3 (7.3) | 11.2 (8.7) |
| CC 1/2 | 0.955 | 0.909 | 1.003 | 0.908 | 0.997 | 0.990 | |
| Refinement | |||||||
| No. Reflections | 18568 | 34193 | 24196 | 27508 | 20908 | 33033 | 13362 |
| Rwork (Rfree) | 0.189 (0.240) | 0.223 (0.261) | 0.193 (0241) | 0.223 (0.281) | 0.204 (0.250) | 0.191 (0.234) | 0.233 (0.288) |
| No. Complex in ASU | 1 | 2 | 1 | 2 | 1 | 1 | 1 |
| No. Atoms | |||||||
| Protein | 2419 | 4522 | 2338 | 4396 | 2280 | 2283 | 2273 |
| DNA | 1060 | 2200 | 1060 | 2204 | 1060 | 1025 | 1022 |
| Ca2+ | 0 | 5 | 4 | 5 | 4 | 4 | 8 |
| Mg2+ | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Water | 73 | 23 | 88 | 10 | 36 | 139 | 31 |
| Ethylene glycol | 0 | 0 | 30 | 30 | 10 | 0 | 10 |
| B-factor | 30.56 | 51.30 | 35.80 | 52.50 | 40.54 | 27.44 | 37.22 |
| RMS deviations | |||||||
| Bond lengths (Å) | 0.018 | 0.005 | 0.006 | 0.008 | 0.006 | 0.010 | 0.002 |
| Bond angles (°) | 2.217 | 0.743 | 0.799 | 0.968 | 0.785 | 1.136 | 0.469 |
| Ramachandran | |||||||
| Preferred (%) | 90.40 | 94.70 | 97.30 | 93.13 | 95.89 | 96.56 | 96.23 |
| Allowed (%) | 8.50 | 5.13 | 2.70 | 6.53 | 3.77 | 3.09 | 3.42 |
| Outliers (%) | 1.10 | 0.17 | 0.00 | 0.34 | 0.34 | 0.34 | 0.34 |
The thermal stabilities of the wildtype I-SmaMI scaffold and the final engineered enzyme were measured using circular dichroism (CD) spectroscopy (Figure 1e). Whereas the wildtype enzyme displays a highly cooperative unfolding transition at 58°C, the final engineered construct displays a less cooperative unfolding transition with an approximate midpoint (‘Tm’) value of 54°C. Thus, the point mutations that alter the enzyme’s recognition specificity also resulted in measurable (albeit tolerable) reductions in the enzyme’s thermal stability.
Thermostabilization of the I-OnuI engineering scaffold.
The results from the engineering of I-SmaMI described above indicated that our campaign of protein mutagenesis and selections for altered specificity resulted in reduced protein stability, which was tolerated due to the protein’s initial thermostability. This led us to hypothesize that (1) similar efforts using a protein scaffold with a lower initial stability would be more prone to failure due to reduced tolerance for destabilization, and (2) that structure-based stabilization of such an enzyme scaffold might increase the ‘engineerability’ of the enzyme.
We therefore engineered the I-OnuI meganuclease (a mesophilic LAGLIDADG homing endonuclease encoded in a mobile group I intron within the mitochondrial genome of the fungus Ophiostoma novo-ulmi (Sethuraman et al., 2009; Takeuchi et al., 2011)) for improved thermostability. Because the wildtype I-OnuI enzyme displays a lower thermal denaturation midpoint (45°C) and less cooperative unfolding than I-SmaMI (Figure 1e), we initiated the project by conducting a structure-based computational analysis of the wildtype I-OnuI enzyme to identify amino acid substitutions that increase the stability of the wildtype enzyme.
To conduct this analysis, we used the PROSS (‘Protein One Stop Shop’) server (Goldenzweig et al., 2016), which combines a phylogenetic analysis (to identify potential sites of mutation that would restore the sequence of the enzyme back towards an evolutionary ‘consensus’) with computational predictions of the effect of such mutations on the protein’s folding energetics. This approach (which relies on a Rosetta-based algorithm to predict protein folding energetics) thereby reduces the likelihood of incorporating deleterious mutations. Residues within the DNA-contacting surface or the enzyme’s active sites were excluded from possible mutation.
The initial enzyme variants spanned 6 constructs with increasing numbers of potential stabilizing mutations (ranging from 9 to 26 amino acid substitutions) (Figure S3a). Each construct was expressed and purified as recombinant protein. Examination of the thermal stability of each purified construct indicated increased thermal denaturation midpoints relative to the wildtype I-OnuI enzyme (ranging from 51°C to well over 60°C) with varying degrees of cooperativity associated with the thermal unfolding transition (Figure S3b).
We eventually selected construct ‘P4’ (termed ‘eOnuTherm’) as a starting scaffold for engineering and for comparison with the wildtype enzyme during the engineering process. This construct harbored 15 individual amino acid substitutions that were largely located across the surface of the protein and acted to redistribute charged side chains (8 substitutions either introduced, eliminated or reversed side chain charge), while maintaining the overall charge of the protein and also leaving its hydrophobic core unchanged (Figure 2a,b). It displayed a significantly increased Tm (56°C) and a similar degree of unfolding cooperativity as the wildtype enzyme (Figure 2c). It displayed cleavage activity that was comparable to the wildtype enzyme (Figure 2d). Its structure bound to DNA (solved at 2.6 Å resolution, PDB code 6UVW; Table 1) demonstrated that the overall structure was closely superimposable on that of the wildtype enzyme (superimposed α-carbon rmsd = 0.44 Å).
Figure 2. Generation of a thermostable I-OnuI meganuclease engineering scaffold (I-OnuI-e-Therm’ or ‘eOnuTherm’).
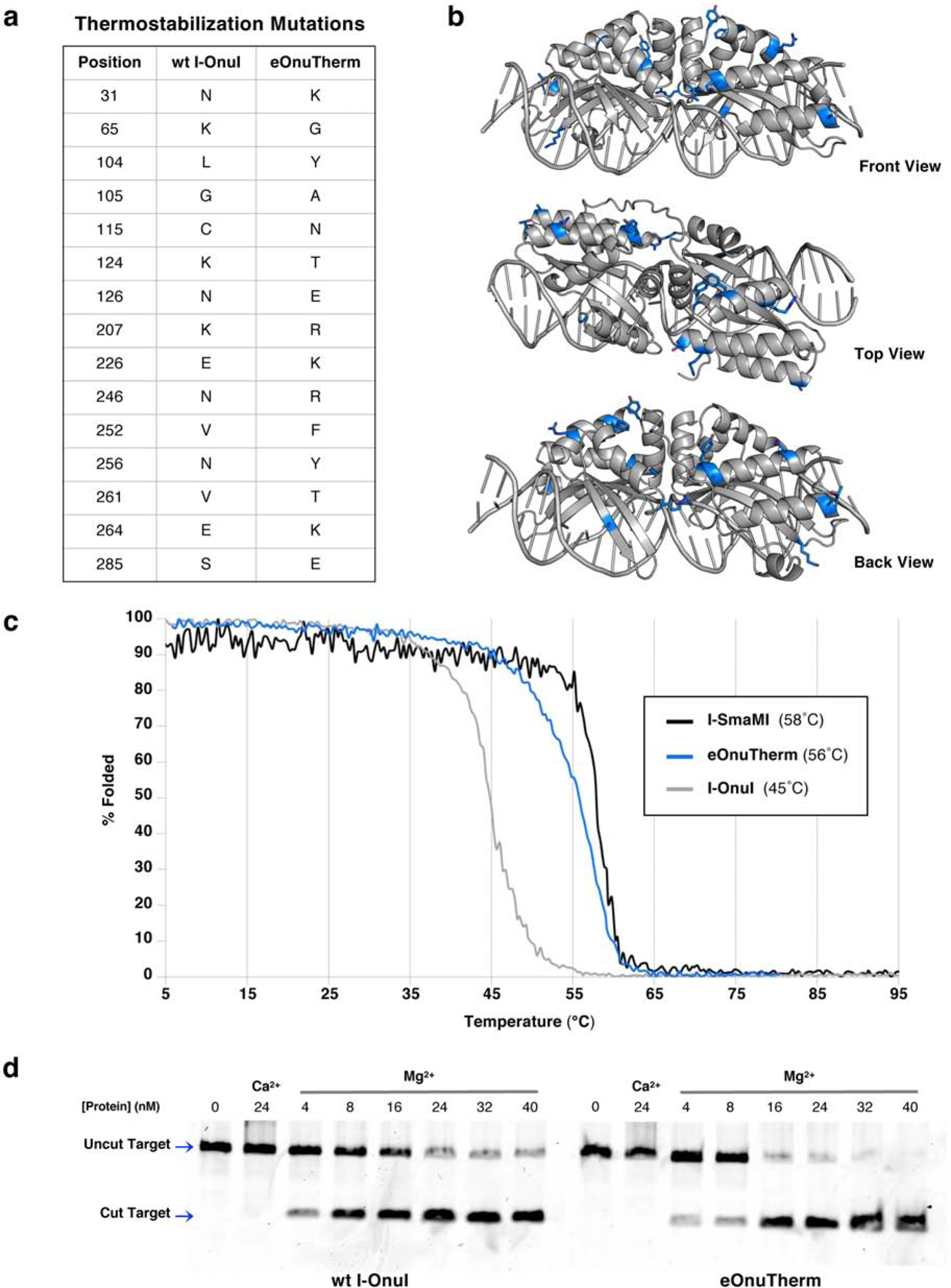
(a) A total of fifteen mutations were made to the wild type I-OnuI meganuclease protein to create the thermostabilized eOnuTherm engineering scaffold. (b) The positions of the mutations introduced to create the eOnuTherm protein are highlighted blue on the eOnuTherm crystal structure (PDB ID 6UVW), with sidechains represented as sticks. The structure is shown in various orientations (front, top, and back views) to illustrate the distribution of mutations across the surface of the entire protein. (c) Circular dichroism (CD) thermal denaturation for the analysis of protein thermostability. The original wild type I-OnuI meganuclease (gray) unfolds at 45°C, while the thermostabilized eOnuTherm protein (blue) shows a noticeably improved melting temperature of 56°C. The melting curve for the naturally evolved thermostable meganuclease I-SmaMI is shown for comparison (black), with a melting temperature of 58°C. (d) Verification of DNA cleavage activity after thermostabilization. Both the wild type I-OnuI (left gel) and thermostabilized eOnuTherm (right gel) enzymes were purified as recombinant protein and tested for cleavage of their DNA target sequence. 40nM fluorescent DNA target substrate was incubated with increasing concentrations of enzyme and 10mM divalent (Ca2+ for no cleavage and Mg2+ to allow cleavage) at 37°C for 30 minutes. Samples of the digest reactions were separated on an acrylamide gel, and fluorescent DNA cleavage products were visualized with a Typhoon imager. Cleavage activity is observed at the lowest enzyme concentration for both the wild type and thermostabilized proteins.
Engineering I-OnuI to disrupt a bacterial toxin gene.
We next conducted parallel engineering campaigns, using wildtype I-OnuI and eOnuTherm as the starting scaffolds, to produce meganucleases that recognize and cleave a highly conserved sequence gene encoding the Vibrio cholerae toxin A-subunit (Figure 3a), for potential use in a gene-drive application. This target differs from the original target site of the I-OnuI enzyme at 8 basepair positions out of 22, with all mismatches again outside the central four positions in the target site. Similar to the strategy employed for I-SmaMI, we selected for altered basepair recognition starting at positions nearest to the center of the target site and then incorporated additional changes in the protein that altered specificity further towards the 5’ and 3’ ends of the target. The individual steps of altered DNA recognition (corresponding to six sequential selection steps and corresponding enzyme libraries) are shown in Figure 3a, right.
Figure 3: Comparison of engineering on the I-OnuI vs. the thermostabilized eOnuTherm meganuclease scaffolds.
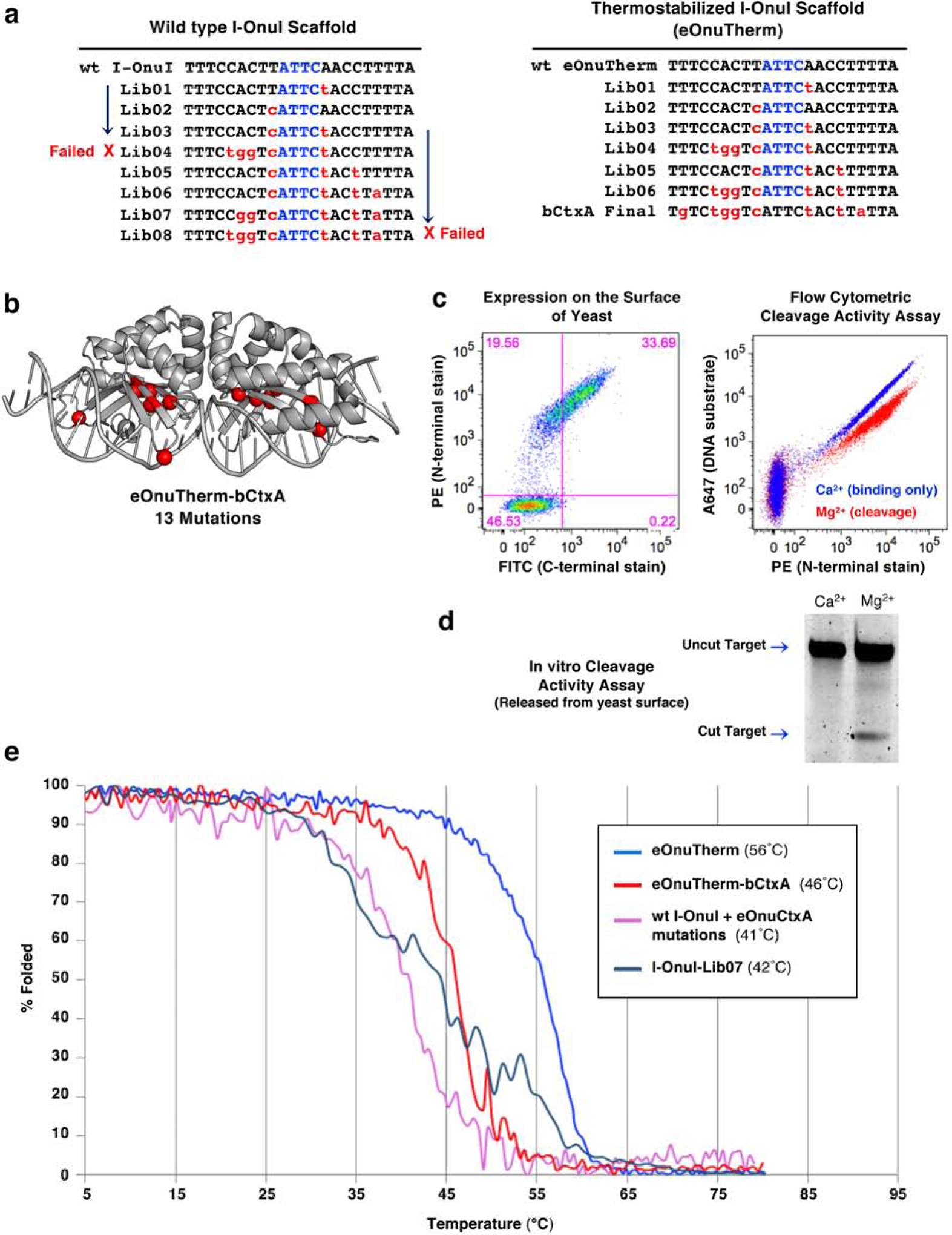
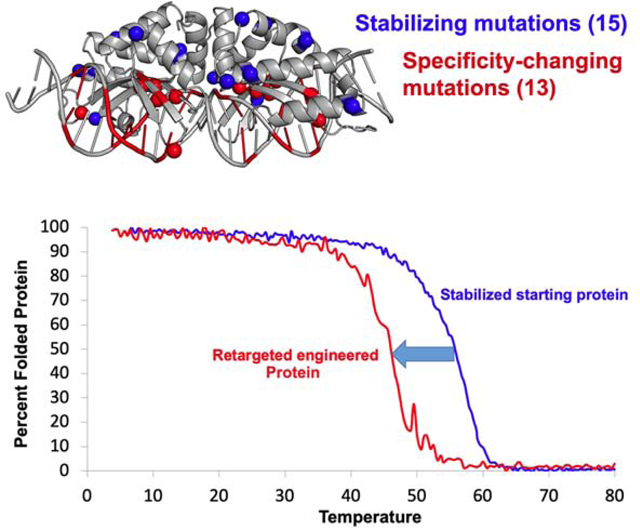
(a) List of target sites for the series of libraries designed, generated, and screened on both the I-OnuI and eOnuTherm protein scaffolds toward the desired bCtxA sequence. Library 04 (Lib04) produced no active variants on the I-OnuI scaffold (marked “Failed” with a red X) but was successful on the eOnuTherm scaffold. Additional libraries were screened on the I-OnuI scaffold (toward changes on the right half-site of the DNA target), but selections failed again at Library 08 (Lib08, also marked “Failed”). In contrast, the thermostabilized eOnuTherm scaffold was successfully re-engineered to target the bCtxA DNA sequence after the step-wise generation of seven libraries, with no selection steps failing to produce active variants. (b) Locations of 13 mutations introduced across the eOnuTherm protein to create the bCtxA-targeting meganuclease (eOnuTherm-bCtxA) are illustrated with red spheres on the alpha carbons of the mutated sidechains. (c) Expression and activity of the re-engineered eOnuTherm-bCtxA meganuclease on the surface of yeast using flow cytometry, as previously described in Figure 1, ‘Methods’ and Supplementary Figure S1. (d) In vitro analysis of DNA cleavage activity with eOnuTherm-bCtxA meganuclease released from the surface of yeast and no physical tethering of the DNA substrate. (e) Circular dichroism (CD) thermal denaturation for the analysis of protein thermostability. The original eOnuTherm meganuclease (blue) unfolds at 56°C, while the final re-engineered eOnuTherm-bCtxA protein (red) shows a reduced melting temperature of 46°C. The best-performing active clone from Lib07 on the I-OnuI scaffold (slate) melts at an even lower temperature of 42°C. The thirteen mutations required to generate the final eOnuTherm-bCtxA-targeting enzyme were grafted onto the wild type I-OnuI scaffold and resulted in a protein with a Tm value of 41°C (pink), which represents a 4° reduction relative to wild-type I-OnuI (Figure 1e, 2c).
The outcome of the engineering, and corresponding behavior of enzymes at each stage of the selection process, differed significantly for the wildtype I-OnuI enzyme as compared to the thermostabilized construct (Figure 3a). Selections with the wildtype I-OnuI scaffold failed to produce active variants at the stage of Library 04 (designed to alter specificity at the −5, −6, −7 positions of the target site) and at Library 08 (designed to alter specificity at the −7 position) (Figure 3a, left).
In contrast, the eOnuTherm construct progressed readily through the workflow and yielded populations of strongly displayed, active clones at every stage of the engineering process (Figure 3a, right). The final construct (‘eOnuTherm-bCtxA’) harbored 13 additional amino acid substitutions beyond those already incorporated to increase its stability (Figures 3b, S4a) and were again distributed throughout its DNA-binding surface to either side of the center of the DNA target. The final construct displayed strong expression on the yeast surface and DNA cleavage activity both as a surface-displayed construct (Figure 3c) and after release from the yeast surface with no tethering of the DNA substrate (Figure 3d). In vitro cleavage assays with purified eOnuTherm-bCtxA enzyme (Figure S4b) validated the activity of the final construct. The specificity profile of surface displayed meganuclease was also determined (Figure S4c).
The final enzyme construct displays a near wild-type rate of cleavage combined with reduced binding affinity towards its cognate target site (as illustrated by the fact that significant cleavage requires high nanomolar concentrations of enzyme in those assays). Since this engineered enzyme (like all others in this study) is destined for application and use in the context of a ‘megaTAL’ gene targeting reagent, the reduction in affinity for the uncoupled meganuclease is not problematic (but rather, is actually a desirable feature that improves the overall fidelity of the final construct).
The thermostability of the final engineered eOnuTherm-bCtxA enzyme was determined via a thermal denaturation analysis using CD spectroscopy as before (Figure 3e) and was found to be significantly lower (Tm = 46°C) than the initial eOnuTherm scaffold (Tm = 56°C). This once again demonstrates that the introduction of point mutations throughout the DNA interaction surface to alter its specificity was accomplished at the cost of a significant reduction in stability, which was tolerated due to thermostabilization of the initial protein scaffold.
To further examine this conclusion, we also examined the stability of (1) a selected variant derived from the wildtype scaffold that failed to advance through selections beyond ‘Library 07’ (Figure 3a, left) and (2) a construct in which the thirteen DNA-contacting mutations selected for the ‘eOnuTherm-CtxA’ enzyme were transferred back to the wildtype I-OnuI scaffold. As expected, both constructs displayed significantly reduced thermal stabilities of 42°C and 41°C, respectively (Figure 3e).
Visualizing the process of altering DNA recognition specificity at the cost of reduced stability.
Having demonstrated the importance of exploiting elevated protein stability for extensive selections leading to altered protein function, we wished to determine if our prior results would be observed a second time if we engineered the same eOnuTherm enzyme scaffold to recognize a different genomic target. We also aimed to more systematically visualize the accumulation of structural changes, in a step-by-step manner, that lead to altered function and reduced stability (and to also determine if that loss in stability occurs in an additive manner that parallels the accumulation of amino acid substitutions from sequential selection steps).
We therefore returned to the eOnuTherm enzyme scaffold and engineered its DNA recognition specificity towards an intergenic target site in human chromosome 11, as part of a larger project to study site-specific trans gene insertion events in human cells. The genomic target contains 5 basepair substitutions relative to the enzyme’s natural DNA target (Figure 4a). The engineered enzyme (‘eOnuTherm-hChr11v3’) contained 13 amino acid substitutions (Figure 4b, S5a), distributed across the enzyme’s DNA binding surface to either side of the center of the target site. Included in those 13 mutations was a mutation involving a metal-ion binding residue in the enzyme’s active site (E178D) that had been previously observed to improve enzyme activity and tolerance of DNA basepair substitutions near the center of the enzyme’s target (McMurrough et al., 2018; Takeuchi et al., 2011).
Figure 4: Targeting a DNA sequence on human chromosome 11 with the thermostabilized eOnuTherm enzyme scaffold.
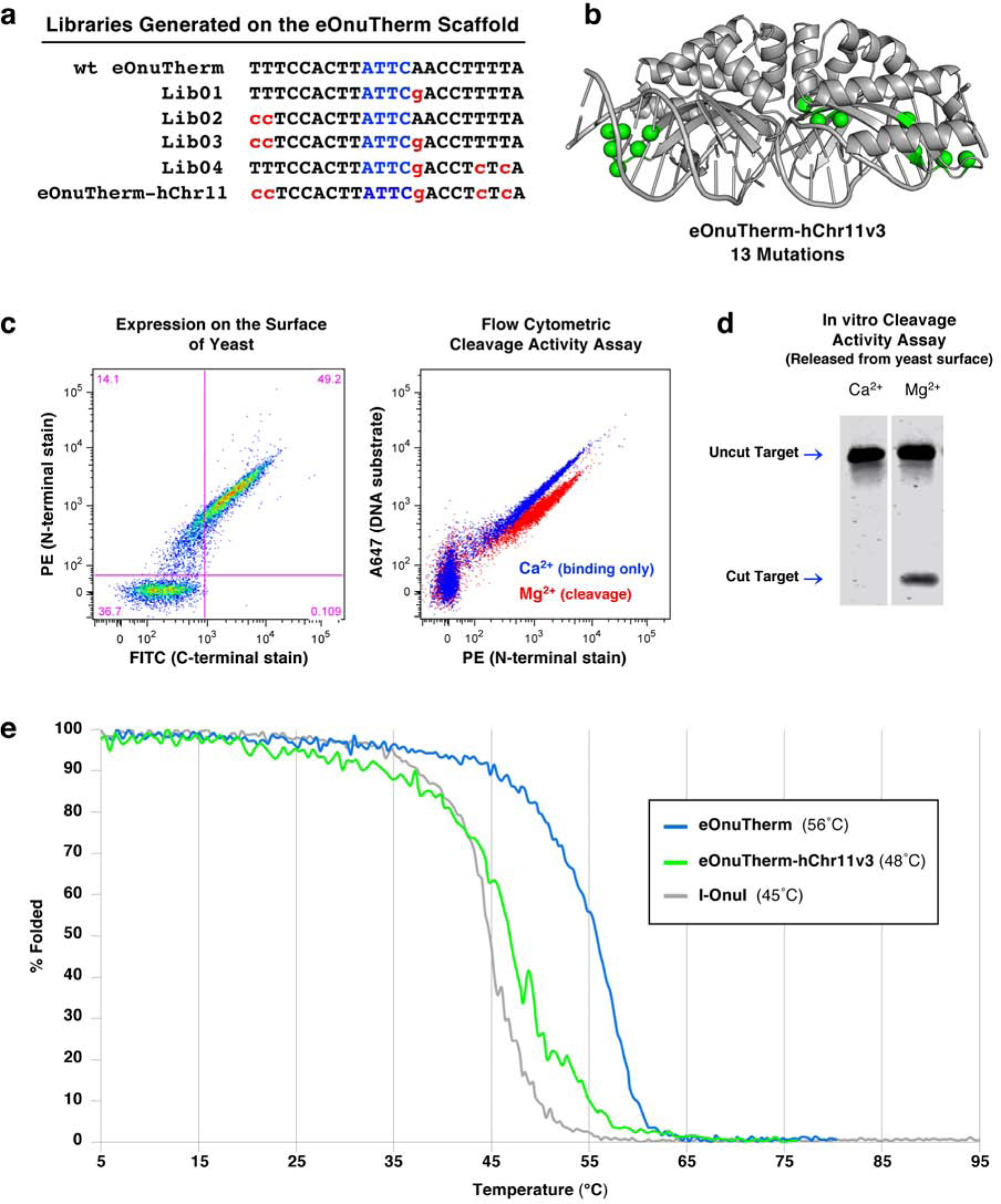
(a) List of target sites for the series of libraries designed, generated, and screened on the eOnuTherm protein scaffold toward the desired hChr11 sequence. (b) The locations of 13 mutations introduced across the eOnuTherm protein to create the hChr11-targeting meganuclease (eOnuTherm-hChr11) are illustrated with green spheres on the alpha carbons of the mutated sidechains. (c) Expression and activity of the re-engineered eSmaGATA meganuclease on the surface of yeast using flow cytometry, as previously described in Figure 1, ‘Methods’ and Supplementary Figure S1. (d) In vitro analysis of DNA cleavage activity with eOnuTherm-hChr11 meganuclease released from the surface of yeast and no physical tethering of the DNA substrate. (e) Circular dichroism (CD) thermal denaturation for the analysis of protein thermostability. The original thermostabilized eOnuTherm meganuclease (blue) unfolds at 56°C, while the final re-engineered eOnuTherm-hChr11 protein (green) shows a reduced melting temperature of 48°C. The eOnuTherm-hChr11 enzyme built on the thermostabilized eOnuTherm scaffold is still more stable than the wild type I-OnuI meganuclease (gray, 45°C melting temperature).
The final eOnuTherm-hChr11v3 meganuclease displayed strong expression on the surface of yeast, as well as robust DNA cleavage activity both as a surface-displayed construct with tethered DNA substrate (Figure 4c) and after release from the yeast surface with no tethering of the DNA substrate (Figure 4d). In vitro cleavage assays with purified recombinant eOnuTherm-hChr11v3 (Figure S5b) and specificity profile determination with yeast surface expressed enzyme (Figure S5c) validated the activity of the final engineered construct.
We then decided to evaluate the behavior of the enzyme scaffold at individual stages of the engineering process (Figure 5). This analysis included two intermediate constructs along with the final engineered enzyme, each harboring increasing number of mutations (Figures 5a, S6a). We measured their relative cleavage activity (using both surface-displayed enzyme and surface-released enzyme) and thermal stability and determined their DNA-bound structures. One question was whether the decreased thermal stability observed in prior engineered constructs was the product of incremental drops in stability at sequential selection stages or the product of a more abrupt loss of stability during the final engineering steps.
Figure 5. Intermediates along the eOnuTherm-hChr11 engineering pathway.
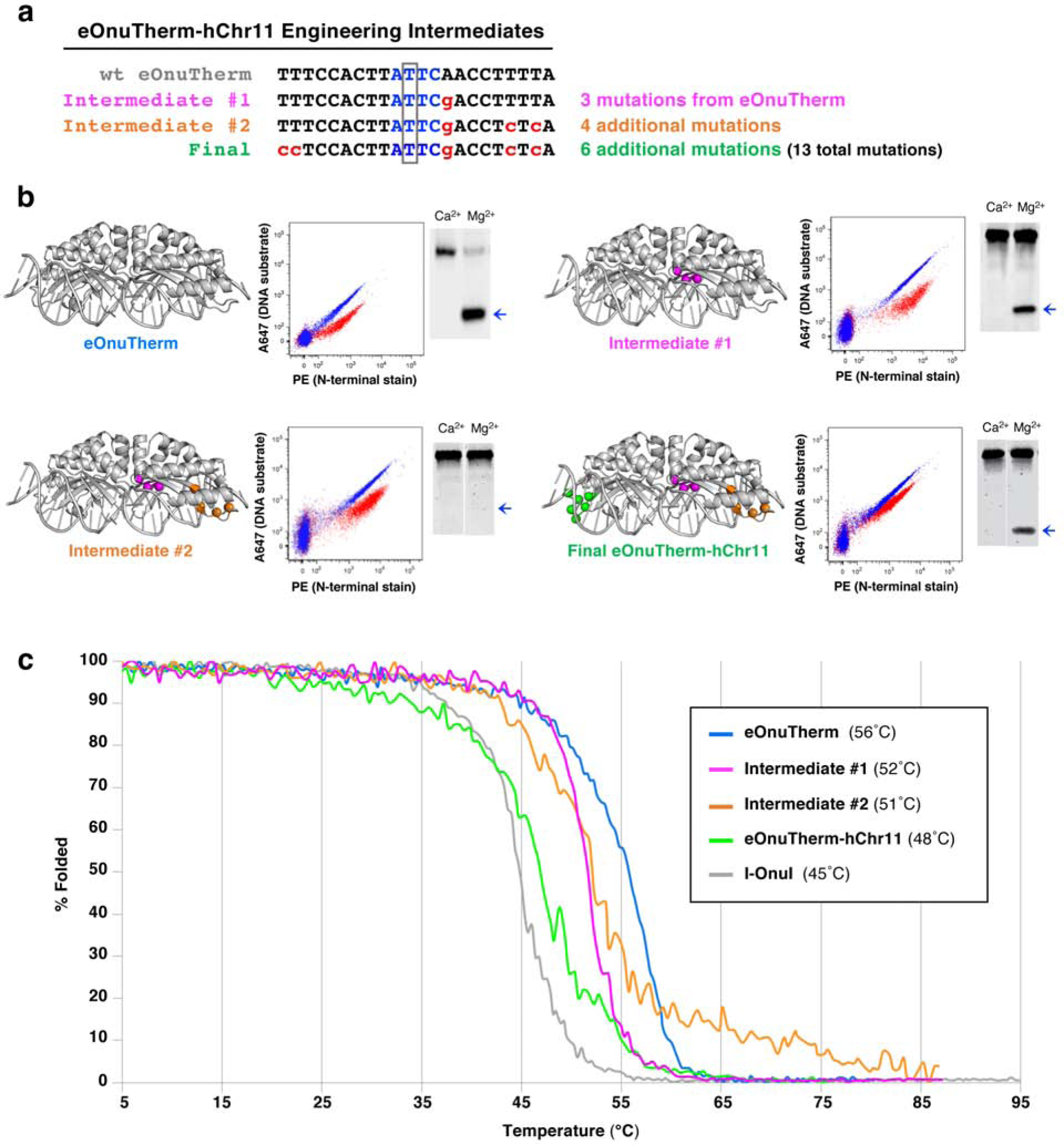
(a) Intermediates were isolated for further characterization at two different stages during the process of engineering eOnuTherm towards the hChr11 target. Intermediate #1 (eOnuTherm-hChr11v1) was the best performing variant isolated from Library 01 (designed for altered specificity at the +3 position of the target site, see Figure 4a). Only 3 mutations were introduced to the eOnuTherm scaffold at this stage. Intermediate #2 (eOnuTherm-hChr11v2) was isolated from Library 04, designed for altered specificity at positions +3, +8, and +10 of the target site (see Figure 4a). An additional 4 mutations were introduced on top of those from intermediate #1. The final engineered meganuclease against the full hChr11 target site included 6 additional mutations along with those from intermediate #2, for a total of 13 mutations. (b) The locations of mutations introduced to the eOnuTherm scaffold at various stages of the engineering pathway are highlighted with colored spheres. The colors of the spheres indicate the stage at which each mutation was made (Intermediate #1: magenta, Intermediate #2: orange, Final engineered enzyme: green). Each meganuclease variant was expressed on the surface of yeast and tested for cleavage activity against its corresponding DNA target using the tethered flow cytometric cleavage assay and the complementary surface-released in vitro cleavage assay. Results from the two activity assays are presented next to the corresponding structures. For the in vitro assay, the bands representing cleaved DNA are designated with a blue arrow. (c) Circular dichroism (CD) thermal denaturation for the analysis of protein thermostability. The original thermostabilized eOnuTherm meganuclease (blue) unfolds at 56°C, and we observe decreased melting temperatures for each subsequent stage of engineering. Intermediate #1 unfolds at 52°C (magenta), intermediate #2 at 51°C (orange), and the final engineered meganuclease unfolds at 48°C (green). The melting curve for the wild type I-OnuI meganuclease is shown for comparison (gray), with the lowest melting temperature at 45°C.
The progression of cleavage activity (Figure 5b) through the engineering process indicates that substrate cleavage efficiency was consistently maintained on the yeast surface (in a reaction where the DNA is physically tethered in proximity to the enzyme, such that reductions in affinity have relatively modest effects on cleavage). In contrast, activity when the enzyme and DNA were both free in solution was more variable. This pattern of behavior indicated variation from one step to the next in the enzyme’s overall DNA binding affinity, leading to an intermediate construct where efficient cleavage on the yeast surface is matched by inefficient cleavage in solution (Figure 5b; Intermediate #2 middle panel). The re-acquisition of cleavage in solution for the final construct (Figure 5b; right panels) likely corresponds to re-establishment of tighter DNA binding affinity in the final stage of engineering.
We further examined the variation in activity for sequentially selected constructs by examining their relative binding affinity towards cognate and fully noncognate DNA target sequences, using surface-displayed enzymes incubated with increasing concentrations of dsDNA targets presented in trans. As illustrated in Supplementary Figure S6b, sequential rounds of selection are accompanied by a reduction in cognate DNA binding affinity for the intermediate constructs, followed by an increase in relative affinity for the final construct (most pronounced at lower DNA concentrations). At the same time, each of the engineered constructs displays a small increase in non-specific binding of an unrelated (noncognate) DNA target sequence.
The sequential stages of selection that produced incremental changes in DNA specificity and activity (Figure 5a,b) were accompanied by incremental (but non-uniform) decreases in the enzymes’ measured thermal denaturation midpoints (Tm values) and corresponding stability (Figure 5c). From an initial Tm value of 56°C for the starting eOnuTherm scaffold, the first stage of selection resulted in a 4° reduction in Tm, while actually increasing the cooperativity of enzyme unfolding. The second stage of selection reduced the enzyme’s Tm by only one additional degree, but at the cost of decreased cooperativity of unfolding. Finally, the introduction of a final six additional amino acid substitutions (that altered specificity at two positions at the 5’ end of the target site) decreased the enzyme’s Tm by an additional 3°C (to its final value of 48°C).
In addition to the crystal structure of the starting ‘eOnuTherm’ protein scaffold, we determined the DNA-bound structures of the two intermediate constructs that were sequentially isolated during the engineering process (Intermediate #1 and Intermediate #2), a variant of the starting scaffold containing a point mutation that was found to improve recovery of active constructs (eOnuTherm-E178D) and the final engineered ‘eOnuTherm-hChr11v3’ enzyme (Tables 1, 2, Figures 6, 7 and S7). The structures demonstrated several features that accompanied the functional and behavioral changes in the enzyme described above:
Table 2.
Protein α-carbon rmsd values
| eOnuTherm | E178D | Intermediate #1 | Intermediate #2 | Final | |
|---|---|---|---|---|---|
| E178D | +T203S,D236E | +I86S, S188K, S190T, Q197K | +N32Y, N33S, K34Q, S35T, S36R, S40L | ||
| eOnuTherm | - | 0.57 Å | 0.52 Å / (−4°)* | 0.47 Å / (−5°)* | 0.43 Å / (−8°)* |
| E178D | --- | 0.20 Å | 0.29 Å | 0.40 Å | |
| Intermediate #1 | --- | 0.23 Å | 0.38 Å | ||
| Intermediate #2 | --- | 0.28 Å | |||
| Final | --- | ||||
Values in parentheses are the decrease in denaturation midpoint temperatures (Tm values) relative to the ‘eOnuTherm’ scaffold.
Figure 6. Structural analyses of sequential constructs from the protein engineering workflow.
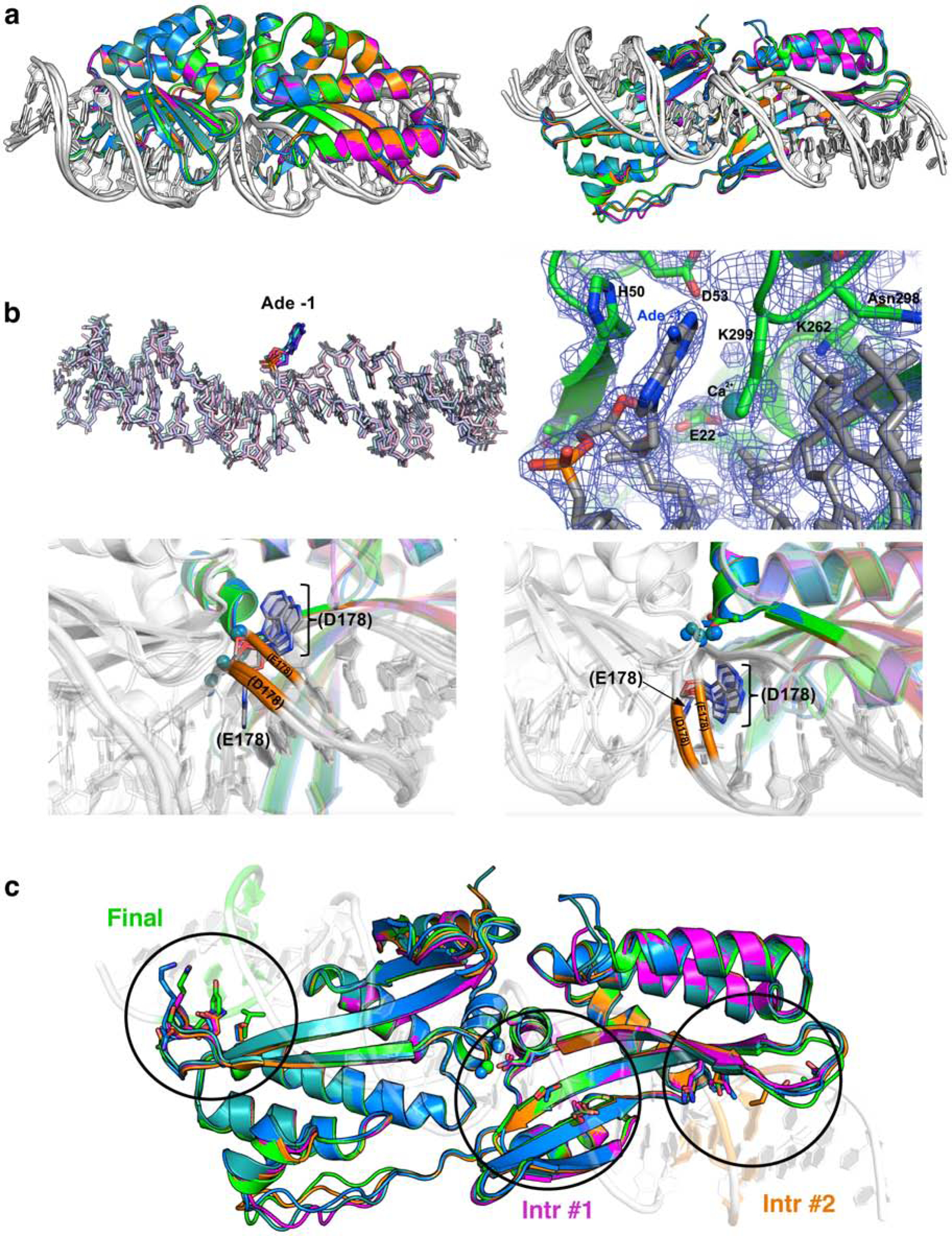
(a) Superposition of the initial scaffold (‘eOnuTherm’; blue), a variant harboring an E178D active site mutation (dark teal), ‘Intermediate 1’ (magenta) and ‘Intermediate 2’ (orange) and the final construct (‘eOnuTherm-hChr11v3’; green). Pairwise protein RMSD values for superimposed structures and the reductions in Tm values (relative to the initial scaffold) are reported in Table 2. (b) Conformation and electron density map indicating the extrusion (‘flipping’) of the adenine base at position −1 in every construct containing the E178D mutation. The lower panels in (b) illustrate the corresponding motion of the DNA phosphodiester backbone at and near the flipped base. (c) Location of clusters of amino acid randomization and eventual mutations corresponding to the three sequential steps of selection for endonuclease variants with altered specificity at corresponding DNA basepair positions.
Figure 7. Alterations to protein-DNA contacts during the course of sequential selection steps.
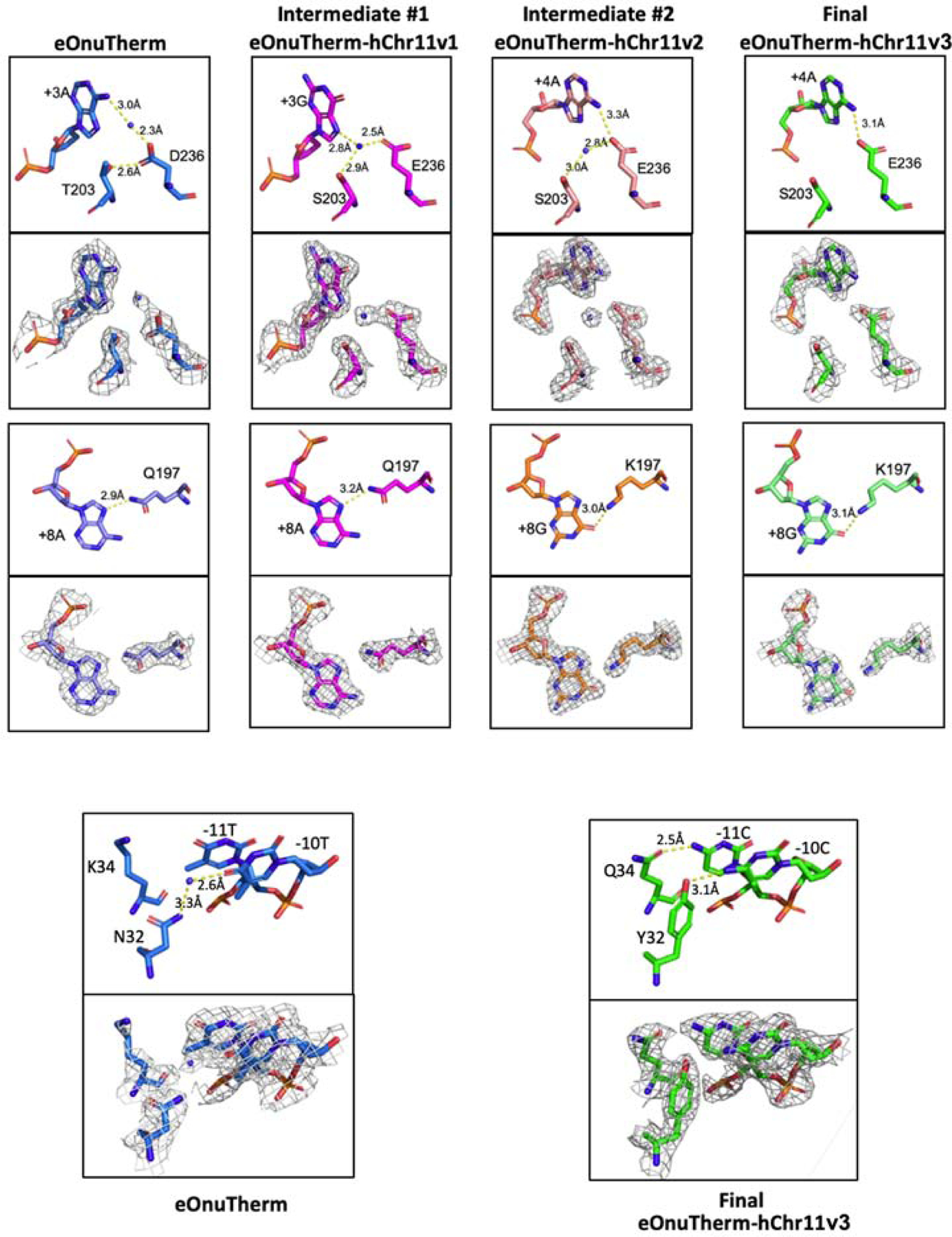
Also see Figure 5 and Figure 6 and the text. The density shown in each panel is displayed at a 1σ contour level.
1. The accumulated mutations impart relatively small changes to the protein conformation; all pairwise superpositions of the protein coordinates yield rmsd values of less than 0.6 Å (Figure 6a and Table 2). The majority of the small changes in conformation that contribute to those values appear to largely be derived from the early incorporation of the E178D mutation (which was incorporated to improve recovery of subsequent constructs that need to accommodate the initial A:T to G:C basepair substitution near the center of the target site).
2. The E178D mutation (that improves recovery of active constructs during the selection workflow, and was previously identified as increasing enzyme activity (Takeuchi et al., 2011, McMurrough et al., 2018)) appears to cause an adenine base at the center of the target site to flip out of the DNA duplex (Figure 6b, Figure S7; the flipped base is located on the bottom strand opposite the boxed thymine in Figure 5a). This motion is accompanied by a corresponding ~2 Å movement of the phosphodiester backbone at that nucleotide position which widens the major groove near the center of the DNA target. While the mechanistic basis for enhanced cleavage due to this mutation and conformational alteration is not obvious, we have previously described how small structural changes in the vicinity of the enzyme active site can dramatically influence catalytic behavior of the enzyme (Lambert et al., 2016).
As expected, the engineering workflow introduces a series of discrete changes to protein-DNA hydrogen bond contacts that contribute to altered specificity. The first and perhaps most notable change not only demonstrates the concept that such mutations act to define new basepair specificities, but also demonstrates that the conformations and contacts made by newly introduced amino acids (the identities of which are fixed during later selections) can change further as additional amino acid substitutions are incorporated (Figure 7, upper panels). The amino acid substitutions in question are a pair of mutations at neighboring residues T203 and D236 that originally interact with one another and with adenine +3 (along with the participation of a water molecule). After mutation to S203 and E236 in the “Intermediate #1” complex, they form a new water-mediated interaction network with the altered guanine base at position +3. However, additional changes made to the protein at the distant end of the DNA target for “Intermediate #2” complex (Figure 6c) are accompanied by further alteration of the contacts made by T203 and E236 to the DNA, eventually resulting in a single contact between a carboxylate oxygen on the E236 side chain to the extracyclic amine group on the adenine base at the +4 position.
Additional changes to protein-DNA contacts (Figure 7, lower panels) that were selected during the engineering process and subsequently visualized crystallographically are straightforward, involving (for Intermediate #2) a change in basepair identity (+8 A:T to +8 G:C) and corresponding change in an amino acid (Q197K) and protein-side chain contact; and (during the final selection step) changes to neighboring residues (N32Y and K34Q) that generate new and additional contacts to two adjoining, altered basepairs at positions −10 and −11.
Discussion
We have demonstrated the relationship between the accumulation of function-altering point mutations onto a protein scaffold during extensive protein selection protocols and the corresponding effects of those mutations on both activity and on protein stability. An additional observation, derived by systematically examining structures of engineering intermediates, is the extent to which clusters of amino acid substitutions at distant locations within a molecular interface can impart long-range structural effects on one another. This led us to conclude that such protein engineering projects (in which individual selections are conducted simultaneously and their individual outputs subsequently combined in a ‘modular’ style of engineering) may suffer from (1) reductions in protein stability below a threshold for sufficient activity, and (2) from unexpected co-dependencies between clusters of amino acids distant from one another. Thus, for this and many similar types of protein engineering projects (specifically, those that involve alteration of molecular binding specificity), we believe that attention to scaffold stabilization, followed by iterative, sequential strategies of selection experiments (that fix early mutations while gradually building towards the final desired construct) are generally advisable.
A second point that this study also illustrates is the importance of a detailed fundamental understanding of the basis for recognition for a given system, particularly when the features of recognition vary significantly across the expanse of the molecular complex. In this example, a fundamental aspect of the behavior of LAGLIDADG DNA binding proteins that must be accounted for is the difference between determinants of specificity near the center of the site (which is dictated largely by the need for a precise DNA bend for high affinity binding and subsequent cleavage, which in turn constrains the sequence of the DNA in the vicinity of the bend) versus the determinants of specificity at the more distal ends of the DNA target (which are more directly dictated by steric and chemical complementarity between protein sequence and conformation relative to the DNA sequences).
Adding an additional layers of complexity to the engineering process are the observations that (1) for many LAGLIDADG protein-DNA complexes, overall binding affinity is dominated by formation of contacts across one-half of the protein-DNA interface (Thyme et al., 2009); (2) that distal mutations can unexpectedly alter specificity elsewhere in the protein-DNA interface (Laforet et al. (2019); and (3) that a frequently observed amino acid substitution in the LAGLIDADG protein family (corresponding to E178D in I-OnuI) that increases tolerance for (and activity against) target sites with altered central basepairs (McMurrough et al., 2018; Takeuchi et al., 2011) is a key element in the engineering process.
As a result of these studies and observations, it has been possible to arrive at an engineering strategy for I-OnuI that involves three elements: (1) early incorporation of an enabling catalytic residue mutation, (2) thermostabilization of the protein scaffold to increase yields of active constructs throughout the selection process and (3) exploitation of an approach (in this particular instance, an ‘inside-out’ order of iterative selection steps) that accommodates the specificity profile and recognition behavior of the enzyme. Elements (2) and (3) of this strategy would translate to engineering other LAGLIDADG homing endonucleases.
Finally, an unexpected observation from this study, that may also serve as a cautionary tale for investigators conducting extensive, selection-based protein engineering campaigns, is that the accumulation of mutations that are almost entirely localized to the protein surface can significantly affect protein stability and thereby impact the recovery (and subsequent function) of active constructs. While it is widely understood that surface-exposed residues can and do contribute to protein folding and stability (Kumar et al., 2000), we had expected that mutations at the DNA interface, derived through selections that maintained enzymatic function (albeit towards an altered target) would display negligible effects on stability.
The concept that thermostable protein scaffolds offer considerable advantages for protein engineering is not novel to this study by any means. A recent review described studies over the past few years that collectively demonstrate that “engineers must carefully balance the mutation-mediated creation and/or optimization of function with the destabilizing effect of those mutations…. protein stability is positively correlated with “evolvability” or the ability to support mutations which bestow new functionality on the protein” (Finch and Kim, 2018). Our study contributes to the growing appreciation of this point through the systematic examination of the manner in which protein destabilization accompanies functional selection and engineering.
The field of protein engineering is rapidly improving its ability to create new types of protein functions, including new or altered forms of ligand recognition. This expansion can be attributed to the development of improved computational algorithms that generate de novo models of protein folds and interactions, and to increasingly robust methods to generate and screen complex protein libraries. Management of complex libraries has benefitted from more powerful protein expression systems (both in cellulo and cell-free), the combination of surface display and flow cytometric-based selections, and the exploitation of high-throughput sequencing approaches that can accurately determine protein amino acid and/or DNA basepair co-variation and context dependence that influence the course of protein selection experiments.
The capabilities of protein engineering are also further enhanced by the rapid accumulation of genomic sequence information. Our own engineering efforts for the systems described above have been aided significantly by the identification of considerable numbers of homologous LAGLIDADG homing endonucleases, which in turn has facilitated improved understanding and prediction of their structure-function relationships.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact.
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Barry L. Stoddard PhD (bstoddar@fredhutch.org).
Materials availability.
There are no restrictions on materials or information from this study. The sequences of all engineered meganucleases described in the text are provided in Supplementary Table S1. All expression plasmids corresponding to engineered meganucleases (for yeast surface display and for generation of recombinant protein from E. coli) are immediately available upon request.
Data and code availability.
This study did not utilize novel code or generate/analyze computational datasets, other than crystallographic structures. All crystallographic data, refined coordinates and validation reports for structures solved in this study (PDB ID codes 6UVW, 6UWG, 6UWO, 6UWH, 6UWJ and 6UWK) are publicly available at www.rcsb.org. Details for each structure can also be found in Table 1.
Experimental Model and Subject Details.
All recombinant proteins were expressed from the pET21d vector in E.coli strain BL21-CodonPlus (DE3)-RIL or BL21 (DE3) pLysS in Luria Broth (LB) culture media. All individual yeast constructs and engineering libraries were expressed from the pETCON yeast surface expression vector with Saccharomyces cerevisiae strain EBY100 in rich growth media or selective growth media. Further details for culture, growth, and induction conditions are described in the Method Details section.
METHOD DETAILS
Nomenclature and Accession Numbers.
Engineered variants of the meganucleases in this work were named using established conventions (Roberts et al., 2003). X-ray diffraction data and coordinates for new crystal structures have been deposited in the RCSB database and are publicly available. See Table 1 and Supplementary Table S1 for a list of formal and abbreviated names and sequences of the constructs described in this study as well as the corresponding PDB IDs for all crystal structures.
Yeast Expression Constructs.
All yeast constructs and engineering libraries were cloned into the pETCON yeast surface display vector (Addgene #41522).
Yeast Growth Media and Plates
2xYPAD rich growth media
2xYPAD rich growth media was used at a “2x” concentration (i.e. not diluted prior to use) for nonselective growth of EBY100 Saccharomyces cerevisiae yeast (ATCC). Recipe for 990 mL of media: 20 g Bacto Yeast Extract (ThermoFisher), 40 g Bacto Peptone (Fisher Scientific), 40 g D-glucose (Fisher Scientific), and 100 mg Adenine hemisulfate (Sigma-Aldrich) (prepared as a 50x stock, filter sterilized, stored in the fridge, and added to the liquid media at 1x concentration before use). The media was brought to a pH of 6.0 and autoclaved for sterilization. After cooling, 10 mL of 100x pen/strep solution (ThermoFisher) and 500 μL of kanamycin (50 mg/mL) (Fisher Scientific) was added.
Selective culture (SC) media
Selective culture (SC) media (SC -Ura -Trp) was prepared along with separate 10x stock solutions of the following sugars: 20% w/v of glucose, 20% w/v raffinose (Fisher Scientific) + 1% w/v glucose, and 20% w/v galactose (Fisher Scientific). The desired sugar source was added to the SC media prior to use. Recipe for 890 mL of SC media (100 mL volume reserved for 10x stock of sugar solution and 10 mL of antibiotic solution): 6.7 yeast nitrogen base (Sunrise Science Products), 5 g casamino acids (Fisher Scientific), and 2.13 g MES (Fisher Scientific), brought to a pH of 5.25 and autoclaved. After cooling, 10 mL of 100x pen/strep solution and 500 μL of kanamycin (50 mg/mL) was added.
Selective culture (SC) media + glucose plates
Selective yeast plates were made from 445 mL SC media with 18 grams of bacteriological agar (Fisher Scientific) added prior to autoclaving. After cooling, 50 mL pre-warmed 20% glucose, 5 mL 100x pen/strep solution and 250 μL of kanamycin (50 mg/mL) were added before pouring into plates.
Transformation of Individual Yeast Constructs.
Yeast expression constructs were transformed into EBY100 Saccharomyces cerevisiae (ATCC) using the lithium acetate method. Full details sufficient to reproduce this method are provided here, and the original method can be consulted at (Gietz and Schiestl, 2007).
Small aliquots of frozen competent EBY100 yeast cells were used for the transformation of individual plasmid-encoded constructs (see below for the procedure scaled up for use with libraries). The reaction mixture added to a 50 μL aliquot of yeast cells contained the following components: 50 μL boiled/snap-cooled salmon sperm DNA (2 mg/mL) (Sigma-Aldrich), 260 μL 50% (w/v) PEG 3350 (Hampton Research), 36 μL 1M lithium acetate, and 14 μL sterile water.
The cells/reaction mixture was portioned out in 25–35 μL aliquots (depending on the number of constructs being transformed) into 1.5 mL microcentrifuge tubes. Lastly, 300 ng of plasmid DNA was added to each transformation reaction. The transformations were incubated in a 42°C water bath for 40 minutes, mixing gently every 7 minutes. After incubation, the reaction was stopped by adding 1 mL sterile water to the tube, spinning down the cells, and resuspending the cell pellet in 500 μL selective culture (SC) media + 2% w/v glucose. 20–50 μL of the resuspended pellet was plated onto selective media plates. The plates were incubated at 30°C for approximately 3 days before colonies had grown large enough. Individual yeast colonies were transferred to growth in 1.5 – 2.0 mL SC media + 2% w/v glucose in 15 mL polypropylene culture tubes and incubated overnight at 30°C with shaking. These cultures could then be stored in the fridge or moved on to induction of surface expression.
Yeast Growth and Induction of Surface Expression.
Single colonies were grown in 1.5–2.0 mL of SC media + 2% glucose at 30°C with shaking overnight. The next day, 36 million cells from the glucose cultures were transferred to 1.5 mL of SC + 2% raffinose + 0.1% glucose media and grown at 30°C with shaking until reaching a density of 80–100 million cells/mL (6–7 hr). Finally, 30 million cells were washed with water and transferred to 1.5 mL of SC + 2% galactose media for overnight induction on the benchtop at room temperature (14–16 hr).
For larger scale cultures, overnight SC + 2% glucose cultures were used to start SC + 2% raffinose + 1% glucose cultures at a density of 24 million cells/mL. Raffinose cultures were grown at 30°C with shaking until reaching a density of 80–100 million cells/mL. The desired number of yeast were washed with water and transferred to SC + 2% galactose media at a density of 20 million cells/mL and left on the benchtop overnight for room-temperature induction of surface expression.
Analysis of enzyme expression and activity on the yeast surface using flow cytometry.
Full details sufficient to reproduce these assays are provided here; the original methodological descriptions can also be consulted at (Baxter et al., 2013; Baxter et al., 2014; Jarjour et al., 2009).
Enzyme expression on the yeast surface.
The pETCON yeast surface expression vector adds an N-terminal hemagglutinin (HA) epitope tag and a C-terminal Myc epitope tag to the expressed protein (Figure S1a). Successful surface expression was confirmed by staining the C-terminal Myc epitope tag with an anti-Myc fluorescein isothiocyanate (FITC) antibody (ICL).
The N-terminus was stained with an anti-HA-biotin antibody (BioLegend) which was then conjugated to a fluorescent streptavidin-phycoerythrin (SAV-PE) reagent (BD Biosciences). In a 96-well v-bottom plate, 2 million induced yeast were incubated in a 50 μL volume with yeast staining buffer (YSB): 180 mM KCl, 10 mM NaCl, 0.2% BSA, 0.1% galactose, and 10 mM HEPES (pH 7.5), plus 1:250 anti-HA-biotin (considered a 250x stock) and 1:100 anti-Myc-FITC (considered a 100x stock) for 45 minutes at 4°C. Cells were washed with YSB and then incubated in a 50 μL volume with YSB and 5nM SAV-PE at 4°C for 30 minutes. Stained cells were washed with YSB and run on a FACSymphony flow cytometer (BD Biosciences). Observation of the both the PE and FITC signals indicates full-length expression of the desired protein on the surface of the yeast.
Generation of Labeled DNA Target Substrates.
Double-stranded oligonucleotide target site substrates were generated by PCR using Platinum Taq High Fidelity DNA polymerase (Invitrogen) with biotin- and AlexaFluor647-labeled primers (see Supplementary Table S2). A 54-base pair single-stranded oligo served as the template for the PCR reaction, designed to contain the 22-base pair target site of the meganuclease flanked by two primer sequences. The PCR reaction was performed in 40 μL volumes with final component concentrations of 1x reaction buffer (supplied with enzyme), 3.25 mM MgSO4, 0.2 mM dNTPs, 0.55 μM of each primer, 2 nM template oligo, and 0.2 μL Platinum Taq High Fidelity polymerase enzyme. Thermalcycling conditions: 92°C × 1 min, 50 cycles of (90°C × 15 sec, 55°C × 15 sec, 68°C × 30 sec), 68°C × 5 min, 40 cycles of (68°C × 30 sec, decreasing by 1°C every cycle), hold at 4°C. Contaminating single-stranded template and/or primers were removed with a 6-hr Exonuclease I (New England Biolabs) digest at 37°C (0.2 μL ExoI in 3 μL total volume of water added to each 40 μL reaction) followed by purification on a G-100 Sephadex (GE Healthcare) column in a filter plate. The final labeled substrates were analyzed for purity on a 12% acrylamide gel with tris-borate-EDTA (TBE) buffer and quantitated via absorbance at 260nm (A260).
Tethered Flow Cytometric DNA Cleavage Assay.
In a 96-well V-bottom plate (for analysis of individual constructs), 3 million yeast with surface-expressed meganuclease were washed with yeast staining buffer (YSB): 180 mM KCl, 10 mM NaCl, 0.2% BSA, 0.1% galactose, and 10 mM HEPES, pH 7.5. The cells were incubated in a 50 μL volume primary stain containing 1:250 anti-HA-biotin antibody and 1:100 anti-Myc-FITC antibody for 45 min at 4°C. During the incubation, 40 nM (or 80nM for I-SmaMI constructs) purified A647/biotin-labeled DNA target substrate was conjugated to 5 nM streptavidin-phycoerythrin SAV-PE in YSB supplemented with an additional 400 mM KCl (DNA was added to the mixture last and mixed immediately, then incubated at room temperature for 10 minutes before moving to an ice bucket until ready for use). Primary-stained cells were washed once with YSB and again with YSB + 400 mM KCl and resuspended in 50 μL secondary stain with the pre-conjugated SAV-PE + DNA target substrate. This step resulted in the formation of a physical biotin-streptavidin tethering of the DNA substrate to the N-terminus of the surface-expressed enzyme (Figure S1b). The high-salt staining buffer (YSB + an additional 400 mM KCl) was used in the presence of DNA to discourage undesired binding of substrates tethered to neighboring cells. With the tethered DNA target substrate in place, the cells were washed with oligo cleavage buffer (OCB): 250 mM KCl, 10 mM NaCl, 5 mM K-glutamate, 0.05% BSA and 10 mM HEPES, pH 7.4 and divided equally into two separate samples. The duplicate samples were incubated in either OCB + 5 mM CaCl2 (supports DNA binding without cleavage) or OCB + 5 mM MgCl2 (supports DNA cleavage) for 20–45 minutes at 37°C. After final washing in YSB, the yeast cells were run on a FACSymphony flow cytometer (BD Biosciences), and the data were analyzed with FlowJo™ software (https://www.flowjo.com). Graphs of A647 signal (DNA substrate, y-axis) vs. PE signal (N-term of the protein, x-axis) were plotted, and overlapping of the Ca2+ and Mg2+ samples allowed for visualization of a drop in A647 signal when cleavage of the DNA substrate had occurred (Figure S1c).
This assay was scaled up as necessary for working with yeast libraries, with incubations performed at a density of 20 million induced cells in a 200 μL volume (up to 100 million cells in a 1000 μL volume) using 1.5 mL microcentrifuge tubes. All other buffer conditions, dilutions of antibody reagents, and concentrations of DNA target substrates remained the same.
Flow Cytometric DNA Binding Assay.
In a V-bottom 96-well plate, 1.5 million induced yeast cells were washed with oligo cleavage buffer (OCB): 150 mM KCl, 10 mM NaCl, 5 mM K-glutamate, 0.05% BSA, and 10 mM HEPES, pH 7.4, supplemented with 5 mM CaCl2 (to allow binding of DNA while preventing cleavage activity) and stained with 1:100 anti-Myc-FITC antibody (to tag the C-terminus of the surface-expressed meganuclease). The induced yeast were incubated for 1.5 hours at 4°C in a 50 μL volume containing OCB with 5 mM CaCl2 and an increasing concentration of A647-labeled DNA target substrate (0, 0.1, 0.25, 0.5, 1, 2.5, 5, 10, 20 nM). Each enzyme was assayed for binding against its cognate DNA target sequence and an unrelated non-cognate DNA target of the same length (sequence ACTTCAGATGTAGACTGTCAGT). After incubation, cells were washed with OCB + 5 mM CaCl2 and run on a FACSymphony flow cytometer (BD Biosciences). The presence of A647 signal indicated binding of the labeled DNA target substrates. Data was quantified with FlowJo™ software by measuring the mean A647 signal in the upper-right quadrant of each flow cytometric plot (cells with both FITC signal representing full-length expression of the surface-expressed meganuclease and A647 signal representing labeled DNA target substrate bound by the enzyme). We observed some “noise” in the baseline/zero DNA conditions (0nM) due to sample carryover from the robotic sample loader on the instrument (despite maximum volume of buffer rinse between samples). As a result, all flow cytometric plots were examined by eye, and quantification for conditions with this type of sample carryover or other significant noise were manually adjusted.
Flow Cytometric DNA Cleavage Specificity Profiles.
Labeled DNA target substrates were generated with each position of the 22bp meganuclease target sequence systematically substituted with each of the three alternative bases. This created a set of 66 unique “one-off” target substrates. The tethered flow cytometric cleavage assay (described above) was performed in 96-well plate format for each of the 66 one-off DNA target substrates. Cleavage was allowed to occur for 30 minutes. The resulting cleavage shift (drop in A647 signal) was quantified by calculating the ratio of median A647 signal from the Ca2+/Mg2+ conditions for each one-off target. The ratios were arranged in bar-graph format relative to cleavage of the wildtype base at each position.
Surface-Released In Vitro DNA Cleavage Assay.
A second DNA cleavage assay was performed to complement the tethered flow cytometric cleavage assay, using enzyme released from the surface of yeast and the same labeled DNA target substrates free in solution. The presence of dithiothreitol (DTT) disrupts the AGA1-AGA2 disulfide bond holding the expressed enzyme on the yeast surface. The physical tethering of the DNA target substrate to the N-terminus of the surface-expressed meganuclease in the tethered flow cytometric assay can mask deficiencies in DNA binding. In the absence of tethered substrate, the enzyme must be able to both bind and cleave the DNA target substrate for activity to be observed. For this assay, 5 million induced yeast cells were washed with oligo cleavage buffer (OCB) and incubated in OCB with 20 nM labeled DNA target substrate, 10 mM DTT, and 5 mM divalent metal (either CaCl2 or MgCl2) for 30 min at 37°C. The digest reactions were stopped by moving to an ice bucket, and a colorless Ficoll loading dye was added to each reaction (6x Ficoll loading dye: 18% w/v Ficoll-400 (Sigma-Aldrich) in 6x tris-borate-EDTA buffer). The yeast cells were pelleted by centrifugation, and samples of the supernatant were separated by electrophoresis on a 12% acrylamide gel with tris-borate-EDTA (TBE) buffer for 1 hour at 120V. Cleavage products were visualized on an Amersham Typhoon 5 Biomolecular Imager (GE Healthcare).
Meganuclease Engineering Library Design.
Meganuclease engineering libraries were designed using an “inside-out” strategy, in which specificity changes were made in an iterative manner starting at the center of the meganuclease target site (nearest the active sites) and moving progressively outward. The positions of sidechain variation were chosen based on the crystal structure of the engineering scaffold (wt I-SmaMI, wt I-OnuI, or eOnuTherm). A maximum of six amino acid positions were designated for complete variation (using an NNS codon, including all 20 amino acid possibilities plus a STOP codon) in any one library. This would produce a theoretical library size of 21 × 21 × 21 × 21 × 21 × 21 = 85.8 million variants. A cluster of six sidechain positions is typically sufficient to cover a “window” of 2–3 base pairs of the target sequence. This allows for alteration of direct sidechain contacts to individual bases as well as the network of nearby interactions typically found within the DNA recognition surface of a meganuclease enzyme. Larger numbers of sidechains positions could be varied at a time using degenerate codons defining more limited variation. Libraries with limited sidechain variation were designed based on the sequences of unique active variants isolated from previous library stages. Sidechain identities were locked down whenever sequence variation converged to a single amino acid at any given position.
Generation of Meganuclease Library Inserts.
Yeast libraries were generated by exploiting the natural proficiency of yeast to perform homologous recombination. During the transformation procedure (described in the following section), a mixture containing open pETCON vector and insert DNA is added to the reaction. Both pieces of DNA are taken up into the yeast during the heat shock, and intact pETCON plasmid is formed through homologous recombination once inside the cells. The insert DNA is designed with ~100-base pair overlaps between the insert DNA and the open vector. For libraries in this study, the insert coding for the meganuclease scaffold was generated using assembly PCR, a method which produces a longer DNA sequence from a collection of shorter overlapping top-strand and bottom-strand oligos. The positions of sidechain variation (degenerate codons like “NNS”) were introduced on longer oligonucleotide ultramers (IDT). A sample set of ultramers for generating a library on the wild type I-OnuI engineering scaffold is provided in Supplementary Table S2. In these sample ultramers, positions of DNA-contacting sidechains are highlighted to show where degenerate codons could be introduced (the DNA sequence for the wild type I-OnuI sequence is shown in the table).
The insert assembly procedure was performed using two sequential PCR reactions with Accuprime Pfx DNA polymerase (Invitrogen). The primary reaction was performed in a 50 μL volume with the supplied reaction buffer, 1 μL of an assembly oligo mix as template (a mixture of all assembly oligos and ultramers at a final concentration of 1 μM each), and 0.5 μL Accuprime Pfx polymerase. A secondary PCR reaction with a total volume of 100 μL included the supplied reaction buffer, 10 μL of the primary reaction as template (no purification), 3 μL each of two primers situated just inside the outermost ends of the full-length insert, and 1 μL of Accuprime Pfx polymerase. Thermalcycling conditions were set according to the manufacturer’s specifications. The full volume of the secondary PCR reaction was loaded onto a 1% agarose gel and purified via gel extraction. Following isolation of the full-length PCR product, the gel-purified insert was amplified according to manufacturer’s specifications with Platinum Taq High Fidelity DNA polymerase (Invitrogen) and the same two primers from the secondary assembly reaction. Multiple 50-uL reactions were pooled to reach a total volume of 340 μL when combined with a volume containing 20 μg of purified open pETCON library vector (multiplied by the total number of library transformations being performed for each single library). The pooled amplified insert DNA was added directly to the transformation reaction with no purification necessary.
Transformation of Yeast Libraries.
Transformations of yeast libraries were performed using the same lithium acetate transformation method described above at a larger scale. Multiple large-scale transformation reactions were performed for each library, depending on the theoretical size of the library and the number of unique transformants desired. Instead of using frozen competent yeast, fresh EBY100 cells were cultured on the day of library transformation. The night before, a 25 mL overnight culture of 2xYPAD rich growth media was inoculated with 2 μL of frozen competent EBY100 yeast cells. In the morning, the overnight culture was used to start one or more 100 mL 2xYPAD cultures (one 100-mL culture provides enough cells for approximately 3 library transformation reactions). The EBY100 cells were grown in 1-L baffled flasks at 30°C with shaking until reaching a density of 80–100 million/mL (~2.5–3.5 hours) and then moved to an ice bucket. Culture media containing 2500 million cells was measured out for each library transformation reaction and spun down in 50-mL conical tubes. The cells were washed once with sterile water, spun down, and resuspended in the following library transformation mixture: 2.4 mL 50% PEG 3350, 360 μL 1M lithium acetate, 500 μL boiled/snap-cooled salmon sperm DNA (2 mg/mL), and a total volume of 340 μL containing 20 μg purified open library vector and the remaining volume filled with amplified library insert DNA (no purification necessary). The transformation reactions were transferred to 15-mL polypropylene cultures tubes and incubated in a 42°C water bath for 35 minutes, mixing gently by swirling the tubes every 7 minutes. After incubation, the cells + transformation reaction mixtures were added to 25 mL of a 50:50 mix of 20% w/v glucose and SC media for washing and recovery. The cells were spun down, and the pellets were resuspended in 100 mL of selective culture media + 2% w/v glucose in 1-L baffled flasks. A 100 μL sample of each large culture was diluted in 900 μL sterile water, and 10 μL of this dilution was plated onto a selective media plate. The plates were incubated at 30°C until the colonies were large enough to count (for quantification of achieved library size).
FACS Analysis and Sorting of Meganuclease Libraries.
Transformed yeast libraries were cultured and induced for surface expression. A scaled-up version of the tethered flow cytometric cleavage assay (described above) was performed with 25–100 million induced yeast in 250–1000 μL volume (depending on the size of the library being sorted). Five million stained and DNA substrate-tethered cells were incubated in oligo cleavage buffer with CaCl2 for use as a no-cleavage control (for the purpose of drawing sort gates), while the remaining cells were incubated in oligo cleavage buffer containing MgCl2 to allow for cleavage to occur. Two subsequent rounds of sorting were performed, with a 45-minute digest for the first sort and a 30-minute digest for the second sort. The yeast cells were run over a FACS Aria II cell sorter instrument (BD Biosciences), with sort gates chosen to collect only those cells demonstrating a downward shift in A647 fluorescence (indicating cleavage of the tethered DNA substrate). Cells were sorted into 2xYPAD media for recovery and growth, and then transferred to selective media. The second sorts were performed in both normal oligo cleavage buffer (OCB) as well as OCB + 150mM KCl for additional selective pressure to collect variants with stronger DNA binding affinity. Also, gates for the second sorts were drawn to collect only those cells with the largest A647 shifts. The cells from the second sorts were grown overnight in 2xYPAD media and plasmid DNA was isolated from the yeast. The plasmid DNA was transformed into chemically competent bacteria and individual colonies were sequenced to determine the identities of the enzyme variants isolated. Bacterial cultures were grown for each unique sequence, plasmids were isolated, and the plasmid DNA was transformed back into yeast. From there, each unique clone was expressed on the surface of yeast and verified for cleavage activity in both the tethered flow cytometric cleavage assay and the complementary non-tethered cleavage assay (described above). Sequences of the clones with the best activity in both cleavage assays were used to guide the next round of library design.
Protein Expression and Purification.
Wild type and engineered constructs were each subcloned into commercially available, T7-inducible pET21d vector (EMD Millipore). Expression of tagless protein was performed in Escherichia coli BL21-CodonPlus (DE3)-RIL competent cells (Agilent) for all constructs except eOnuTherm-hChr11v1 which required BL21 (DE3) pLysS competent cells (EMD Millipore) to prevent cell toxicity during growth. Both cell types were cultured in Luria Broth (LB) media (RPI) with 100 μg/mL ampicillin (Fisher Scientific) added for selective growth. Each 1-L LB + Amp culture was inoculated with 10 mL overnights and grown at 37°C with shaking until reaching an OD600 of 0.6–0.8. Subsequent induction with 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) (Teknova) was followed by overnight shaking at 16°C, pelleting of the cells, and storage at −20°C.
Resuspension of cell pellets in 25 mM Tris-HCl pH 7.5, 200 mM NaCl, 5% glycerol, 50 μM phenylmethylsulfonyl fluoride (PMSF) (Sigma-Aldrich), and 0.01 U/μL benzonase nuclease (Sigma-Aldrich) was followed by sonication and centrifugation. Clarified supernatant was filtered using a pore size of 0.45 μm and loaded onto a 5 mL Heparin HP HiTrap column (GE Life Sciences). A linear salt gradient (from 25 mM Tris-HCl pH 7.5, 200 mM NaCl, 5% glycerol to 25 mM Tris-HCl pH 7.5, 1M NaCl, 5% glycerol) was used for elution.
The tagless protein was concentrated to about 5 mg/mL and further purified using a Superdex HiLoad prep grade 16/60 size exclusion column (GE Life Sciences) equilibrated in 25 mM Tris-HCl pH 7.5, 200 mM NaCl, and 5% glycerol.
Cleavage Assays with Purified Recombinant Enzyme.
Purified recombinant meganuclease protein was incubated at 37°C for 15–30 minutes with 40 nM A647-labeled DNA target substrates in the presence of 5 mM divalent (CaCl2 or MgCl2) in oligo cleavage buffer at pH 7.4 or 7.9. A colorless Ficoll loading dye was added, and digest products were separated by electrophoresis on a 12% acrylamide gel for 1 hour at 120V. Cleavage products were visualized on an Amersham Typhoon 5 Biomolecular Imager (GE Healthcare).
Thermal Denaturation and CD Spectroscopy.
Plane polarized light was measured with a J-800 CD spectrometer (JASCO). Each purified meganuclease was scanned in a 1 mm path length cuvette at 210 nm wavelength across a range of temperatures from 5°C – 95°C. Melting temperatures (Tm) were calculated for each meganuclease using denaturation analysis in Spectra Manager II software (JASCO).
Protein Crystallography.
Each meganuclease-DNA complex was formed in the presence of 10 mM CaCl2 with double-stranded DNA. Meganucleases were concentrated to 200 – 400 μM and mixed with DNA target oligo to achieve a protein:DNA molar ratio of 1:1.5. Each cocrystallization DNA duplex included the 22-base pair target sequence flanked by random bases with a singlebase 5’ overhang on each strand. Crystals were grown at room temperature using hanging drop vapor diffusion in 24-well trays. A 20% ethylene glycol, 80% reservoir solution was used as a cryoprotectant prior to freezing in liquid nitrogen. Data collection was performed with either an in-house Rigaku Micromax 007HF rotating anode generator using a Saturn 944+ CCD area detector or the Advanced Light Source synchrotron beamline 5.0.1 at Lawrence Berkeley National Laboratory with a Pilatus 6M detector. The sequences of crystallization oligos, reservoir conditions, and X-ray sources for each crystal structure are listed below:
eOnuTherm
Crystallized in 100 mM HEPES pH 8.5, 200 mM ammonium sulfate, 28% PEG 3350 in the presence of duplexed 27-nucleotide DNA strands with single base 5’ overhangs.
Top strand DNA oligo: 5’- GGGTTTCCACTTATTCAACCTTTTAGG −3’
Bottom strand DNA oligo: 5’- CCCTAAAAGGTTGAATAAGTGGAAACC −3’
Data collection: ALS beamline 5.0.1, Pilatus 6M detector
eOnuTherm-E178D
Crystallized in 200 mM calcium acetate, 100 mM sodium acetate pH 5.5, 100 mM sodium acetate pH 6.0, 35% PEG 400 in the presence of duplexed 27 nucleotide DNA strands with single base 5’ overhangs:
Top strand DNA oligo: 5’- GGGTTTCCACTTATTCAACCTTTTAGG −3’
Bottom strand DNA oligo: 5’- CCCTAAAAGGTTGAATAAGTGGAAACC −3’
Data collection: In-house rotating anode, Saturn 944+ CCD area detector
eOnuTherm-bCtxA
Crystallized in 100 mM HEPES pH 8.5, 200 mM ammonium sulfate, 30% PEG 3350 in the presence of duplexed 27 nucleotide DNA strands with single base 5’ overhangs:
Top strand DNA oligo: 5’- GGGTGTCTGGTCATTCTACTTATTAGG −3’
Bottom strand DNA oligo: 5’- CCCTAATAAGTAGAATGACCAGACACC −3’
Data collection: ALS beamline 5.0.1, Pilatus 6M detector
eOnuTherm-hChr11v1 (Intermediate #1)
Crystallized in 100 mM sodium acetate pH 6.0, 200 mM calcium acetate, 35% PEG 400 in the presence of duplexed 26 nucleotide DNA strands with single base 5’ overhangs:
Top strand DNA oligo: 5’- GGGTTTCCACTTATTCGACCTTTTAG −3’
Bottom strand DNA oligo: 5’- CCTAAAAGGTCGAATAAGTGGAAACC −3’
Data collection: In-house rotating anode, Saturn 944+ CCD area detector
eOnuTherm-hChr11v2 (Intermediate #2)
Crystallized in 100mM sodium acetate pH 6.0, 200mM calcium acetate, 35% PEG 400 in the presence of duplexed 27 nucleotide DNA strands with single base 5’ overhangs:
Top strand DNA oligo: 5’- GGGTTTCCACTTATTCGACCTCTCAGG-3’
Bottom strand DNA oligo: 5’- CCCTGAGAGGTCGAATAAGTGGAAACC −3’
Data collection: ALS beamline 5.0.1, Pilatus 6M detector
eOnuTherm-hChr11v3
Crystallized in 100 mM sodium acetate pH 6.0, 200 mM calcium acetate, 35% PEG 400 in the presence of duplexed 27 nucleotide DNA strands with single base 5’ overhangs:
Top strand DNA oligo: 5’- GGGCCTCCACTTATTCGACCTCTCAGG −3’
Bottom strand DNA oligo: 5’- CCCTGAGAGGTCGAATAAGTGGAGGCC −3’
Data collection: In-house rotating anode, Saturn 944+ CCD area detector
The HKL2000 software suite (Otwinowski and Minor, 1997) was used for crystallographic data processing. Molecular replacement was performed with PHENIX (Liebschner et al. 2019) using the I-OnuI structure as a search model. COOT molecular graphics software (Emsley et al., 2010) and PHENIX were used for model building and refinement of structures. Structural images were generated using the PyMOL molecular visualization program (Schrödinger, LLC). Crystallographic statistics for each meganuclease-DNA complex are listed in Table 1.
QUANTIFICATION AND STATISTICAL ANALYSIS
The data shown in all Figures in the main text (Figures 1 through 6) are shown in their entirety (surface display and flow cytometric assays of protein expression and DNA cleavage; cleavage of DNA by enzyme liberated from yeast surface, and analyses of thermostability using circular dichroism spectroscopy) and do not require or utilize statistical analyses.
The data shown in Supplementary Figures S2 through S7 are also shown in their entirety, including cleavage of DNA targets with recombinant purified enzymes, additional protein denaturation (thermostability) measurements via CD spectroscopy, and specificity profiles.
For meganuclease specificity profiling (Supplementary Figures S2, S4 and S5), the experiments were each performed at least twice using independently induced yeast cultures. For each construct and corresponding figure, a single replicate is shown (because the absolute magnitude of surface expression levels varies between individual yeast cultures, thereby confounding the calculation of average values).
For target site binding assays (Supplementary Figure S6), all assays were performed in triplicate; error bars represent standard deviation from the mean at each DNA concentration.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Biotin anti-HA.11 Epitope Tag, Clone 16B12 Lot # B293295 |
BioLegend | Cat# 901505 RRID: AB_2565023 |
| anti-C-Myc Antibody (Chicken) - FITC Conjugated Lot #48 | ICL | Cat# CMYC-45F |
| PE Streptavidin (SAV-PE) Lot #9049827 | BD Biosciences | Cat# BDB554061 |
| Bacterial and Virus Strains | ||
| BL21-Codon Plus (DE3)-RIL Escherichia coli competent cells | Agilent | Cat# 230245 |
| Novagen BL21(DE3) pLysS Escherichia coli competent cells | EMD Millipore | Cat# 69451 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bacto™ Yeast Extract | ThermoFisher | Cat# 212750 |
| Gibco™ Bacto™ Peptone | Fisher Scientific | Cat# DF0118-17-0 |
| alpha-D(+)-Glucose, 99+%, anhydrous, Acros Organics™ | Fisher Scientific | Cat# AC170080010 |
| Adenine hemisulfate salt | Sigma-Aldrich | Cat# A3159 |
| Gibco™ Penicillin-Streptomycin (10,000 U/mL) | ThermoFisher | Cat# 15140122 |
| Kanamycin Sulfate (White Powder), Fisher BioReagents | Fisher Scientific | Cat# BP906-5 |
| MP Biomedicals™ D-(+)- Raffinose pentahydrate (powder) | Fisher Scientific | Cat# MP114061522 |
| D(+)-Galactose, 99+%, Acros Organics™ | Fisher Scientific | Cat#AC150615000 |
| YNB + Nitrogen Powder | Sunrise Science Products | Cat# 1501-500 |
| Casamino acids | Fisher Scientific | Cat# J851-1KG |
| MES (Fine White Crystals), Fisher BioReagents | Fisher Scientific | Cat# BP300-100 |
| Thermo Scientific Bacteriological Agar, Ultrapure | Fisher Scientific | Cat# AAJ10906P2 |
| Deoxyribonucleic acid sodium salt from salmon testes | Sigma-Aldrich | Cat# D1626-1G |
| 50% w/v Polyethylene glycol 3,350 Monodisperse | Hampton Research | Cat# HR2-527 |
| Platinum™ Taq DNA Polymerase High Fidelity | Invitrogen | Cat#11304029 |
| Exonuclease I (E.coli) | New England Biolabs | Cat# M0293S |
| Ficoll PM 400 | Sigma-Aldrich | Cat# F4375 |
| AccuPrime™ Pfx DNA Polymerase | Invitrogen | Cat# 12344024 |
| Luria Broth (LB) 1 Liter buffered capsules | RPI | Cat# L24045 |
| Ampicillin sodium salt (crystalline powder) | Fisher Scientific | Cat# BP1760 |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | Teknova | Cat# I3326 |
| Phenylmethylsulfonyl fluoride (PMSF) | Sigma-Aldrich | Cat# P7626 |
| Benzonase Nuclease | Sigma-Aldrich | Cat# E1014-25KU |
| Critical Commercial Assays | ||
| Deposited Data | ||
| I-OnuI structure | Takeuchi et al., 2011 | PDB: 3QQY |
| I-OnuI-e-Therm (eOnuTherm) structure | This paper | PDB: 6UVW |
| I-OnuI-e-ThermE178D (eOnuTherm-E178D) structure | This paper | PDB: 6UWG |
| I-OnuI-e-Therm-bCtxA (eOnuTherm-bCtxA) structure | This paper | PDB: 6UW0 |
| I-OnuI-e-Therm-hChr11v1 (Intermediate #1) structure | This paper | PDB: 6UWH |
| I-OnuI-e-Therm-hChr11v2 (Intermediate #2) structure | This paper | PDB: 6UWJ |
| I-OnuI-e-Therm-hChr11v3 (eOnuTherm-hChr11) structure | This paper | PDB: 6UWK |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Saccharomyces cerevisiae (Strain: EBY100) | ATCC | Cat# MYA-4941 |
| Oligonucleotides | ||
| See Table S2 for oligonucleotides used in this study | Integrated DNA Technologies (IDT) | N/A |
| Recombinant DNA | ||
| pETCON yeast surface display vector | Baxter, S.K. et al., 2014 | Addgene Cat# 41522 |
| Novagen pET21d bacterial protein expression vector | EMD Millipore | Cat# 69743-3 |
| Software and Algorithms | ||
| PROSS: the Protein Repair One-Stop Shop | Goldenzweig, A. et al., 2016 | https://pross.weizmann.ac.il/step/pross-terms/ |
| FlowJo™ Software for Mac. Version 9.9.6. | Ashland, OR: Becton, Dickinson and Company; 2019. | https://www.flowjo.com |
| Spectra Manager II Version 2.10.1.1 | JASCO | https://www.jasco.de/Spectroscopy/Software |
| HKL-2000 v715 for Mac | HKL Research, Inc | https://hkl-xray.com/download-instructions-hkl-2000 |
| Phenix Software Suite Version 1.17-3644 | Liebschner, D. et al., 2019 | https://www.phenix-online.org |
| COOT Crystallographic Object-Oriented Toolkit Version 0.8.9.2 |
Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PyMOL Molecular Graphics System Version 2.3.0 | Schrödinger, LLC | https://pymol.org |
| Other | ||
| illustra™ Sephadex™ G-100 DNA grade | GE Healthcare | Cat# 17057402 |
| HiTrap® Heparin High Performance column | GE Healthcare | Cat# 17-0407-03 |
| HiLoad® 16/600 Superdex® 75 prep grade column | GE Healthcare | Cat# 28-9893-33 |
Highlights.
Redesign of molecular recognition specificity, such as DNA binding, is difficult.
Rounds of mutation and selection during protein engineering destabilize the scaffold.
Stabilization of the protein scaffold prior to engineering increases success.
These points are illustrated via a series of three protein engineering experiments.
Acknowledgements
DG was supported by the grant “Mobilność Plus IV” agreement 1288/MOB/IV/2015/0, provided by the Ministry of Science and Higher Education of Poland. BLS and investigators in his laboratory (JH, AL, RW) were supported by the NIH (R01 GM105691), Bluebird Bio Inc. and the Fred Hutchinson Cancer Research Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
BLS is a listed co-inventor on IP patent filings (US13/702,972; WO2011156430A2) describing methods for the production of engineered meganucleases, for which he has received royalties on licensing revenue from the Hutchinson Center. His lab has been funded for this work by Bluebird Bio, Inc., which has licensed IP related to these studies.
REFERENCES
- Alford RF, Leaver-Fay A, Jeliazkov JR, O’Meara MJ, DiMaio FP, Park H, Shapovalov MV, Renfrew PD, Mulligan VK, Kappel K, et al. (2017). The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J Chem Theory Comput 13, 3031–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashenberg O, Gong LI, and Bloom JD (2013). Mutational effects on stability are largely conserved during protein evolution. Proceedings of the National Academy of Sciences of the United States of America 110, 21071–21076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth J, Taylor GK, Havranek JJ, Quadri SA, Stoddard BL, and Baker D (2010). Computational reprogramming of homing endonuclease specificity at multiple adjacent base pairs. Nucleic Acids Res 38, 5601–5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter SK, Lambert AR, Scharenberg AM, and Jarjour J (2013). Flow cytometric assays for interrogating LAGLIDADG homing endonuclease DNA-binding and cleavage properties. Methods in molecular biology (Clifton, N J) 978, 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter SK, Scharenberg AM, and Lambert AR (2014). Engineering and flow-cytometric analysis of chimeric LAGLIDADG homing endonucleases from homologous I-OnuI-family enzymes. Methods in molecular biology (Clifton, N J) 1123, 191–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger S, Procko E, Margineantu D, Lee EF, Shen BW, Zelter A, Silva D-A, Chawla K, Herold MJ, Garnier J-M, et al. (2016). Computationally designed high specificity inhibitors delineate the roles of BCL2 family proteins in cancer. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bick MJ, Greisen PJ, Morey KJ, Antunes MS, La D, Sankaran B, Reymond L, Johnsson K, Medford JI, and Baker D (2017). Computational design of environmental sensors for the potent opioid fentanyl. Elife 6 pii: e28909 (doi: 10.7554/eLife.28909). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanove AJ, Bohm A, Miller JC, Morgan RD, and Stoddard BL (2018). Engineering altered protein-DNA recognition specificity. Nucleic Acids Res 46, 4845–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day AL, Greisen P, Doyle L, Schena A, Stella N, Johnsson K, Baker D, and Stoddard BL (2018). Unintended specificity of an engineered ligand-binding protein facilitated by unpredicted plasticity of the protein fold. Protein Eng Des Sel 31, 375 – 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou J, Doyle L, Jr Greisen P, Schena A, Park H, Johnsson K, Stoddard BL, and Baker D (2017). Sampling and energy evaluation challenges in ligand binding protein design. Protein Sci 26, 2426–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou J, Vorobieva AA, Sheffler W, Doyle LA, Park H, Bick MJ, Mao B, Foight GW, Lee MY, Gagnon LA, et al. (2018). De novo design of a fluorescence-activating b-barrel. Nature 561, 485 – 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott W, and Cowtan K Acta Cryst. (2010) Features and Development of Coot. Acta Cryst Section D-Biological Crystallography 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch AJ, and Kim JR (2018). Thermophilic proteins as versatile scaffolds for protein engineering. Microorganisms 6, 97 ( 10.3390/microorganisms6040097). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, and Schiestl RH (2007). High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2, 31 – 34. [DOI] [PubMed] [Google Scholar]
- Goldenzweig A, Goldsmith M, Hill SE, Gertman O, Laurino P, Ashani Y, Dym O, Unger T, Albeck S, Prilusky J, et al. (2016). Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol Cell 63 (2), 337 – 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong LI, Suchard MA, and Bloom JD (2013). Stability-mediated epistasis constrains the evolution of an influenza protein. Elife 2, e00631 ( 10.7554/eLife.00631). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P-S, Boyken SE, and Baker D (2016). The coming of age of de novo protein design. Nature 537, 320–327. [DOI] [PubMed] [Google Scholar]
- Jarjour J, West-Foyle H, Certo MT, Hubert CG, Doyle L, Getz MM, Stoddard BL, and Scharenberg AM (2009). High-resolution profiling of homing endonuclease binding and catalytic specificity using yeast surface display. Nucleic Acids Res 37, 6871–6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce AP, Zhang C, Bradley P, and Havranek JJ (2015). Structure-based modeling of protein: DNA specificity. Brief Funct Genomics 14, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpinski J, Hauber I, Chemnitz J, Schafer C, Paszkowski-Rogacz M, Chakraborty D, Beschorner N, Hofmann-Sieber H, Lange UC, Grundhoff A, et al. (2016). Directed evolution of a recombinase that excises the provirus of most HIV-1 primary isolates with high specificity. Nat Biotechnol 34, 401–409. [DOI] [PubMed] [Google Scholar]
- Kumar S, Tsai C-J, and Nussinov R (2000). Factors enhancing protein thermostability. Protein Eng Des Sel 13, 179 – 191. [DOI] [PubMed] [Google Scholar]
- Laforet M, McMurrough TA, Vu M, Brown CM, Zhang K, Junop MS, Gloor GB and Edgell DR (2019) Modifying a covarying protein-DNA interaction changes substrate preference of a site-specific endonuclease. Nucleic Acids Research 47 (20), 10830 – 10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AR, Hallinan JP, Shen BW, Chik JK, Bolduc JM, Kulshina N, Robins LI, Kaiser BK, Jarjour J, Havens K, et al. (2016). Indirect DNA Sequence Recognition and Its Impact on Nuclease Cleavage Activity. Structure 24, 862–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung L-W, Jain S, McCoy AJ, Moriarty NW, Oeffner RD, Poon BK, Prisant MG, Read RJ, Richardson JS, Richardson DC, Sammito MD, Sobolev OV, Stockwell DH, Terwilliger TC, Urzhumtsev AG, Videau LL, Williams CJ, and Adams PD (2019) Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix Acta Cryst. D75, 861–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurrough TA, Brown CM, Zhang K, Hausner G, Junop MS, Gloor GB, and Edgell DR (2018). Active site residue identity regulates cleavage preference of LAGLIDADG homing endonucleases. Nucleic Acids Res 46, 11990–12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH, and Bauer DE (2019). Emerging genetic therapy for sickle celll disease. Ann Rev Med. 70, 257 – 271. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode In Methods Enzymol, Carter CW, and Sweet RM, eds. (New York: Academic Press; ), pp. 307 – 326. [DOI] [PubMed] [Google Scholar]
- Procko E, Berguig G, Shen B, Song Y, Frayo S, Convertine A, Margineantu D, Booth G, Correia B, Cheng Y, et al. (2014). A computationally designed inhibitor of an Epstein-Barr viral Bcl-2 protein induces apoptosis in infected cells. Cell 157, 1644 – 1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PyMOL Molecular Graphics System, Version 2.3.0 Schrödinger, LLC. [Google Scholar]
- Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, Blumenthal RM, Degtyarev SK, Dryden DTF, Dybvig K, et al. (2003). A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res 31, 1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethuraman J, Majer A, Friedrich NC, Edgell DR, and Hausner G (2009). Genes within genes: multiple LAGLIDADG homing endonucleases target the ribosomal protein S3 gene encoded within an rnl group I intron of Ophiostoma and related taxa. Mol Biol Evol 26, 2299–2315. [DOI] [PubMed] [Google Scholar]
- Shen BW, Lambert A, Walker BC, Stoddard BL, and Kaiser BK (2016). The Structural Basis of Asymmetry in DNA Binding and Cleavage as Exhibited by the I-SmaMI LAGLIDADG Meganuclease. J. Mol. Biol 428, 206 – 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RD, Damm-Ganamet KL, Dunbar JB Jr., Ahmed A, Chinnaswamy K, Delproposto JE, Kubish GM, Tinberg CE, Khare SD, Dou J, et al. (2016). CSAR Benchmark Exercise 2013: Evaluation of Results from a Combined Computational Protein Design, Docking, and Scoring/Ranking Challenge. J Chem Inf Model 56, 1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddard BL (2014). Homing endonucleases from mobile group I introns: discovery to genome engineering. Mobile DNA 5, 7 – 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddard BL (2016). Design and Creation of Ligand-Binding Proteins In Computational Design of Ligand Binding Proteins, Stoddard BL, ed. (New York: Humana Press; ), pp. v – ix. [Google Scholar]
- Takeuchi R, Lambert AR, Mak AN, Jacoby K, Dickson RJ, Gloor GB, Scharenberg AM, Edgell DR, and Stoddard BL (2011). Tapping natural reservoirs of homing endonucleases for targeted gene modification. Proceedings of the National Academy of Sciences of the United States of America 108, 13077–13082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyme S, and Baker D (2014). Redesigning the specificity of protein-DNA interactions with Rosetta. Methods in molecular biology (Clifton, N J) 1123, 265–282. [DOI] [PubMed] [Google Scholar]
- Thyme SB, Boissel SJS, Arshiya Quadri S, Nolan T, Baker DA, Park RU, Kusak L, Ashworth J, and Baker D (2014). Reprogramming homing endonuclease specificity through computational design and directed evolution. Nucleic Acids Res 42, 2564–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyme SB, Jarjour J, Takeuchi R, Havranek JJ, Ashworth J, Scharenberg AM, Stoddard BL, and Baker D (2009). Exploitation of binding energy for catalysis and design. Nature 461, 1300–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinberg CE, Khare SD, Dou J, Doyle L, Nelson JW, Schena A, Jankowski W, Kalodimos CG, Johnsson K, Stoddard BL, et al. (2013). Computational design of ligand-binding proteins with high affinity and selectivity. Nature 501, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werther RA, Hallinan JP, Lambert AR, Havens K, Jarjour J, Galizi R, Windbichler N, Crisanti A, Nolan T, and Stoddard BL (2017). Crystallographic analyses illustrate exceptional plasticity and efficient recoding of meganuclease target specificity. Nucleic Acids Res 45, 8621 – 3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, and Lai L (2017). Computational design of ligand-binding proteins. Current Opinion in Structural Biology 45, 67 – 73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not utilize novel code or generate/analyze computational datasets, other than crystallographic structures. All crystallographic data, refined coordinates and validation reports for structures solved in this study (PDB ID codes 6UVW, 6UWG, 6UWO, 6UWH, 6UWJ and 6UWK) are publicly available at www.rcsb.org. Details for each structure can also be found in Table 1.