

Abstract
Increased arterial stiffness and vascular remodeling precede and are consequences of hypertension. They also contribute to the development and progression of life-threatening cardiovascular diseases. Yet, there are currently no agents specifically aimed at preventing or treating arterial stiffening and remodeling. Previous research indicates that vascular smooth muscle actin polymerization participates in the initial stages of arterial stiffening and remodeling, and that LIM kinase promotes F-actin formation and stabilization via cofilin phosphorylation and consequent inactivation. Herein, we hypothesize that LIM kinase inhibition is able to prevent vasoconstriction- and hypertension-associated arterial stiffening and inward remodeling. We found that small visceral arteries isolated from hypertensive subjects are stiffer and have greater cofilin phosphorylation than those from non-hypertensives. We also show that LIM kinase inhibition prevents arterial stiffening and inward remodeling in isolated human small visceral arteries exposed to prolonged vasoconstriction. Using cultured vascular smooth muscle cells, we determined that LIM kinase inhibition prevents vasoconstrictor agonists from increasing cofilin phosphorylation, F-actin volume and cell cortex stiffness. We further show that localized LIM kinase inhibition prevents arteriolar inward remodeling in hypertensive mice. This indicates that hypertension is associated with increased vascular smooth muscle cofilin phosphorylation, cytoskeletal stress fiber formation and heightened arterial stiffness. Our data further suggest that pharmacological inhibition of LIM kinase prevents vasoconstriction-induced arterial stiffening, in part via reductions in vascular smooth muscle F-actin content and cellular stiffness. Accordingly, LIM kinase inhibition should represent a promising therapeutic means to stop the progression of arterial stiffening and remodeling in hypertension.
Keywords: filamentous actin, cofilin, vascular stiffness, inward remodeling, cytoskeleton
INTRODUCTION
Arterial stiffening and remodeling precede and are consequences of hypertension. They are also strong predictors of cardiovascular disease (CVD) development and associated life-threatening cardiovascular events.1,2 At the level of the resistance vasculature and small arteries, a prominent instigator of arterial stiffening and remodeling is a state of prolonged vasoconstriction. Thus, arterial mechanical and structural changes are commonly associated with conditions that increase the activity of the renin-angiotensin-aldosterone system, stimulate sympathetic outflow and promote endothelial dysfunction.3–7 However, the specific mechanisms responsible for increasing arterial stiffness and remodeling in response to prolonged vasoconstriction or hypertension are only partially known and involve processes of cytoskeletal and extracellular matrix (ECM) structural modifications. A better understanding of these mechanisms should lead to new therapeutic targets to ameliorate CVD.
Although vascular stiffening has long been considered the result of excessive collagen deposition and ECM crosslinking,8,9 recent reports show that the mechanical properties of vascular cells also contribute to increasing the stiffness of blood vessels.7,10–12 In particular, we have shown that the initial stages of vascular stiffening and inward remodeling in small arteries are associated with activation of actin polymerization pathways,13,14 and that inhibition of actin polymerization prevents prolonged vasoconstriction-induced stiffening and remodeling of isolated small arteries.5,13,15 We have also shown that destruction of the actin cytoskeleton is able to enlarge the passive diameter and soften the wall of inwardly remodeled small vessels.15 All this suggests that actin polymerization is a prominent component of the mechanisms that lead to increased arterial stiffness and vascular remodeling.
Multiple vasoconstrictor agonists provoke vascular smooth muscle actin polymerization,16–18 increase vascular stiffness and induce inward remodeling in isolated arteries.14,19–22 Such agonists, which include angiotensin-II (AngII), norepinephrine (NE), endothelin-1 and serotonin, are also well-known activators of the small GTPase, RhoA and its downstream effector, Rho-associated protein kinase (ROCK).23,24 In turn, ROCK has been shown to be partly responsible for reducing the activity of myosin light chain phosphatase and inducing calcium sensitization, a process that sustains vasoconstriction, even as intracellular calcium diminishes over time.25,26 Notably, ROCK is also an activator of LIM Kinase (LIMK),14,27–30 an enzyme that phosphorylates and inactivates cofilin.31 As cofilin’s primary activity is to sever filamentous (F)-actin cytoskeletal stress fibers, cofilin inactivation results in enhanced actin polymerization.32 Indeed, consistent with the notion that actin polymerization is involved in vasoconstriction-induced arterial remodeling, we have previously shown that LIMK inhibition prevents rat isolated small arteries from undergoing inward remodeling following prolonged exposure to vasoconstrictor agonists.14 Herein, we designed a series of experiments to determine the role of LIMK in hypertension and vasoconstriction-induced stiffening and remodeling in human vessels, and ascertain the ability of LIMK inhibition to prevent inward remodeling in an animal model of hypertension. We tested the hypothesis that vascular LIMK activity is greater in hypertensive vs. non-hypertensive subjects and that LIMK inhibition prevents vasoconstriction-induced arterial stiffening and remodeling in human isolated arteries and in an AngII-infusion mouse model of hypertension.
METHODS
Data supporting the findings presented here are available upon reasonable request to the corresponding author. See the online-only Data Supplement for a detailed version of methodologies.
Ethics and approvals
Procedures for the collection and use of human samples conformed to the Declaration of Helsinki and were approved by the University of Missouri Institutional Review Board (IRB#2003033). All subjects provided written informed consent prior to participation. All animal procedures also received prior approval by the University of Missouri Institutional Animal Care and Use Committee and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Analyses in human omental vessels
Small arteries were isolated from omental samples obtained from 43 subjects undergoing elective abdominal surgery at the University of Missouri hospital and used for assessment of vascular function, mechanics and structure as previously described.4,20,33,34 Subject characteristics are described in the Table.
Table.
Demographic and clinical characteristics of the subjects included in the comparison of vascular features under different experimental conditions
| Vascular Experimental Condition | |||
|---|---|---|---|
| Characteristic | Basal | 4-hour Constriction | 18-hour Constriction |
| Number of Subjects | 23 | 8 | 12 |
| Age (years) | 43.7 ± 2.2 | 55.5 ± 5.2 | 40.8 ± 3.1 |
| BMI (Kg/m2) | 48.2 ± 1.2 | 44.6 ± 4.3 | 40.4 ± 3.4 |
| Sex (females/males) | 19/5 | 7/1 | 9/3 |
| Non-Diabetics / Diabetics | 24/0 | 6/2 | 5/7 |
| Non-Hypertensives / Hypertensives | 15/8 | 3/5 | 9/3 |
| Medications | Albuterol, Alprazolam, Amlodipine, Aripiprazole, Aspirin, Atorvastatin, Bifidobacterium, Bimatoprost, Canagliflozin, Carvedilol, Celecoxib, Cetirizine, Cholecalciferol, Chondroitin, Citalopram, Clopidogrel, Cyanocobalamin, Cyclobenzaprine, Diphenhydramine, Enalapril, Ergocalciferol, Flurosemide, Fluticasone, Folic acid, Glargine, Glimepiride, Glucosamine, Hydrochlorothiazide, Ibuprofen, Insulin, Lactobacillus, Lisinopril, Lisonopril, Lispro, Loratadine, Lorazepam, Losartan, Lovastatin, Lutein, Metformin, Methotrexate, Methylsulfonylmethane, Metoprolol, Metronidazole, Neomycin, Nitroglycerin, Omeprazole, Oxycodone, Pantoprazole, Paroxetine, Polyethylene glycol, Propranolol, Psyllium, Ranitidine, Rosuvastatin, Sertraline, Sitagliptin, Spironolactone, Tiotropium, Tramadol, Triamterene, Verapamil, Zonisamide | ||
Analyses in cultured vascular smooth muscle cells
Human coronary artery smooth muscle cells (HCASMCs) were used to assess F-actin content, protein expression and cell cortical stiffness as previously described.14,35,36
Arterial remodeling in a mouse model of hypertension
Transgenic mice (OTO2–10) expressing green fluorescent protein (GFP) guided by the alpha-actin smooth muscle promoter were used to assess arterial remodeling in vivo.37 These mice were developed by Jen-Yue Tsai, backcrossed to C57BL6 and provided to us by Robert Fariss, Senior Investigator at the National Eye Institute. OTO2–10 male mice were implanted minipumps delivering AngII at either a non-pressor dose of 100ng/kg/min (n=4 mice) or a pressor dose of 1000ng/kg/min (n=4 mice) for two weeks. Arterioles at the base of both ears were imaged with a Leica SP5 multiphoton microscope. After imaging, and every fourth day thereafter, one ear was injected with 20μL of a 100μM LIMK inhibitor (LIMKi, 1μM, Calbiochem #435930) solution, while the contralateral ear was injected with the vehicle control. Fourteen days after pump implantation, ears were imaged again. Paired comparisons were made between the arterial diameter 14 days after pump implantation with that before pump implantation to assess vascular remodeling.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism version-8. The Shapiro-Wilk test was used to determine if data were normally distributed. Data not normally distributed were subjected to logarithmic transformation to achieve normality (i.e., cell stiffness), or analyzed using the Mann-Whitney non-parametric test (i.e., vasoconstriction at 4h of exposure to NE+AngII and mouse blood pressure). Analyses included two-tailed T-tests for experiments with two groups, one-way ANOVA for experiments with three groups and two-way ANOVA for factorial designs including that with repeated measures (i.e., pressure myography data). Fisher’s protected least significance difference followed ANOVA for pre-planned multiple comparisons. A value of P<0.05 was considered significant.
RESULTS
Hypertension is associated with increased arterial stiffness and LIMK activity.
Omental arteries from hypertensive subjects, as clinically diagnosed and reported in their medical record, were not different from those of non-hypertensives in their passive diameters across intraluminal pressures from 5 to 120mmHg (Figure 1A). However, they were less distensible and significantly stiffer as determined by their percent increase in diameter, strain-stress relationship, Young’s and incremental moduli of elasticity, and calculated pulse wave velocity (cPWV, Figure 1A). Fluorescence images showed that arteries from hypertensives also had greater levels of cofilin phosphorylation and F-actin content (Figure 1B–C).
Figure 1.
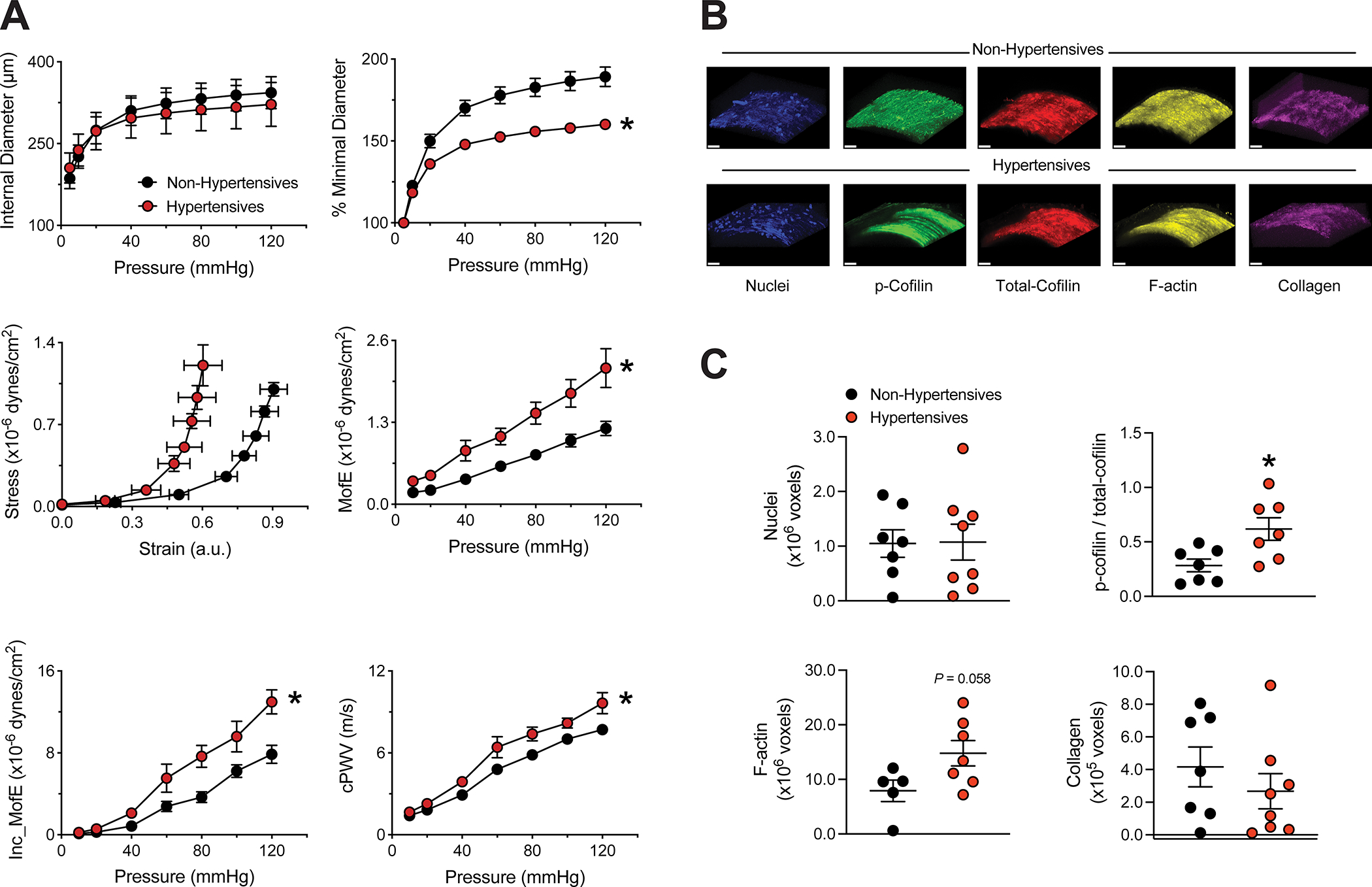
Omental small arteries from hypertensive subjects are stiffer and have greater cofilin phosphorylation and F-actin content than those from non-hypertensives. Small arteries were isolated from omental samples of subjects undergoing elective abdominal surgery and subsequently cannulated in pressure myography systems. A, Pressure-internal diameter curves, pressure-percent minimal diameter curves, strain-stress curves, Young’s moduli of elasticity (MofE), incremental moduli of elasticity (Inc_MofE), and calculated pulse wave velocities (cPWV) of arteries under calcium-free passive conditions obtained from non-hypertensive and hypertensive subjects. B, Representative images of small arteries from non-hypertensive and hypertensive subjects stained for detection of nuclei (blue) p-cofilin (green), total-cofilin (red), F-actin (yellow) and stain-free collagen (magenta); bar=30μm. C, Comparison of nuclei, p-cofilin / total-cofilin, F-actin and collagen content between small arteries from non-hypertensive and hypertensive subjects. Data are expressed as means ± SEM. *P<0.05 vs. non-hypertensives, n=7–15/group.
LIMK inhibition prevents vasoconstriction-induced inward remodeling in human isolated arteries.
To determine if LIMK inhibition prevents vasoconstriction-induced inward remodeling in human vessels, we treated isolated, cannulated and pressurized human omental arteries with AngII+NE for 4h (Figure 2A). Arteries treated or not with LIMKi showed similar levels of constriction in response to AngII+NE throughout the 4-hour exposure to the agonists (Figure 2A–C). In the absence of LIMKi, vessels had a reduction in passive diameter after the 4-hour exposure to the vasoconstrictors. In comparison, vessels treated with LIMKi had increased passive diameters. This differential remodeling response in the absence vs. presence of LIMKi was significant (Figure 2D). The reduction in passive diameter in vessels not treated with LIMKi was observed across all intraluminal pressures tested (Figure 2E) and resulted in a reduced distensibility (strain) of the vessels (Figure 2F) without significant changes in elastic moduli (data not shown).
Figure 2.
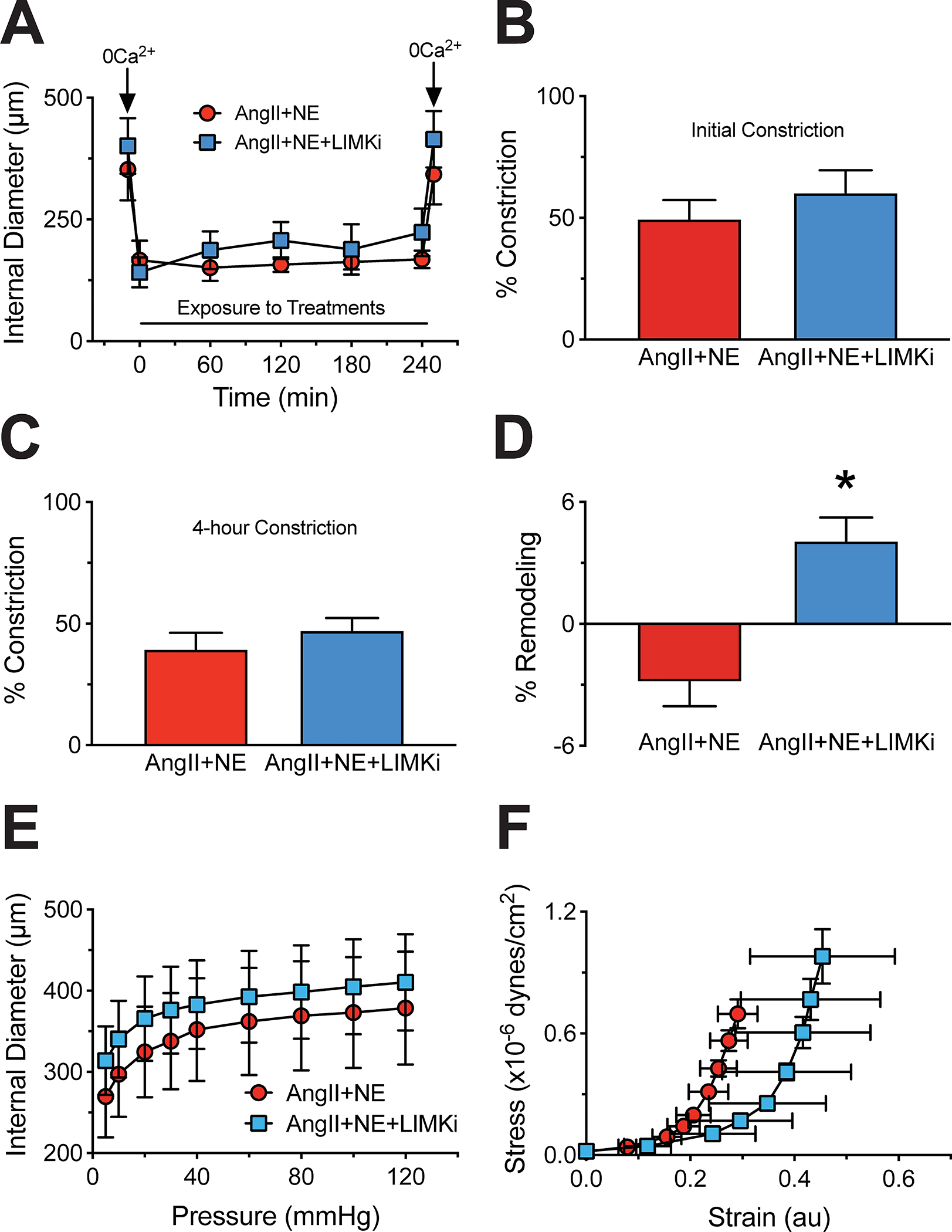
LIM kinase (LIMK) inhibition does not influence human omental small artery vasoconstriction, but prevents inward remodeling following a 4-hour exposure to angiotensin-II (AngII) and norepinephrine (NE). Small arteries were isolated from omental samples of subjects undergoing elective abdominal surgery and subsequently exposed to AngII (100nM) + NE (3μM) in the absence or presence of LIMK inhibition (LIMKi, 1μM) for 4 hours, while cannulated in pressure myography systems. A, Internal diameter during the initial exposure of the arteries to calcium-free conditions; then, while in the presence of the vasoconstrictor agonists without and with LIMKi, and after a second exposure to calcium-free conditions. B, Percent level of constriction upon the initial exposure (5 minutes) to AngII+NE. C, Percent level of constriction after 4 hours of exposure to the vasoconstrictor agonists. D, Change between post- and pre-constriction maximal passive (calcium-free) diameters expressed as percent remodeling following the 4-hour exposure to AngII+NE without and with LIMKi. E, Passive pressure-internal diameter curves following the 4-hour exposure to AngII+NE without and with LIMKi. F, Passive strain-stress curves following the 4-hour exposure to AngII+NE without and with LIMKi. Data are expressed as means ± SEM. *P<0.05 vs. AngII+NE without LIMKi, n=4/group.
In contrast to results from the 4-hour vasoconstriction experiment, arteries exposed to AngII+NE for 18h had reduced distensibility and compliance, and increased stiffness (Figure 3A). Notably, LIMKi prevented prolonged vasoconstriction from inducing those mechanical changes in the arteries (Figure 3A). The prolonged 18-hour exposure to AngII+NE failed to cause a significant increase in cofilin phosphorylation due to a differential response in vessels from hypertensives vs. non-hypertensives. Nonetheless, LIMK inhibition consistently and significantly decreased p-cofilin (Figure 3A). To determine if LIMK inhibition had additional differential effects on arteries from hypertensives vs. non-hypertensives, data were divided on the basis of hypertension diagnosis (Figure 3B). Results show that while LIMKi was effective at reducing the distensibility of vessels in both groups, it reduced the incremental modulus of elasticity only in vessels from hypertensives (Figure 3B). In addition, while LIMKi reduced cofilin phosphorylation in AngII+NE-treated arteries from both hypertensives and non-hypertensives, the effect of LIMKi was greater in hypertensives when compared to control untreated vessels (Figure 3B).
Figure 3.
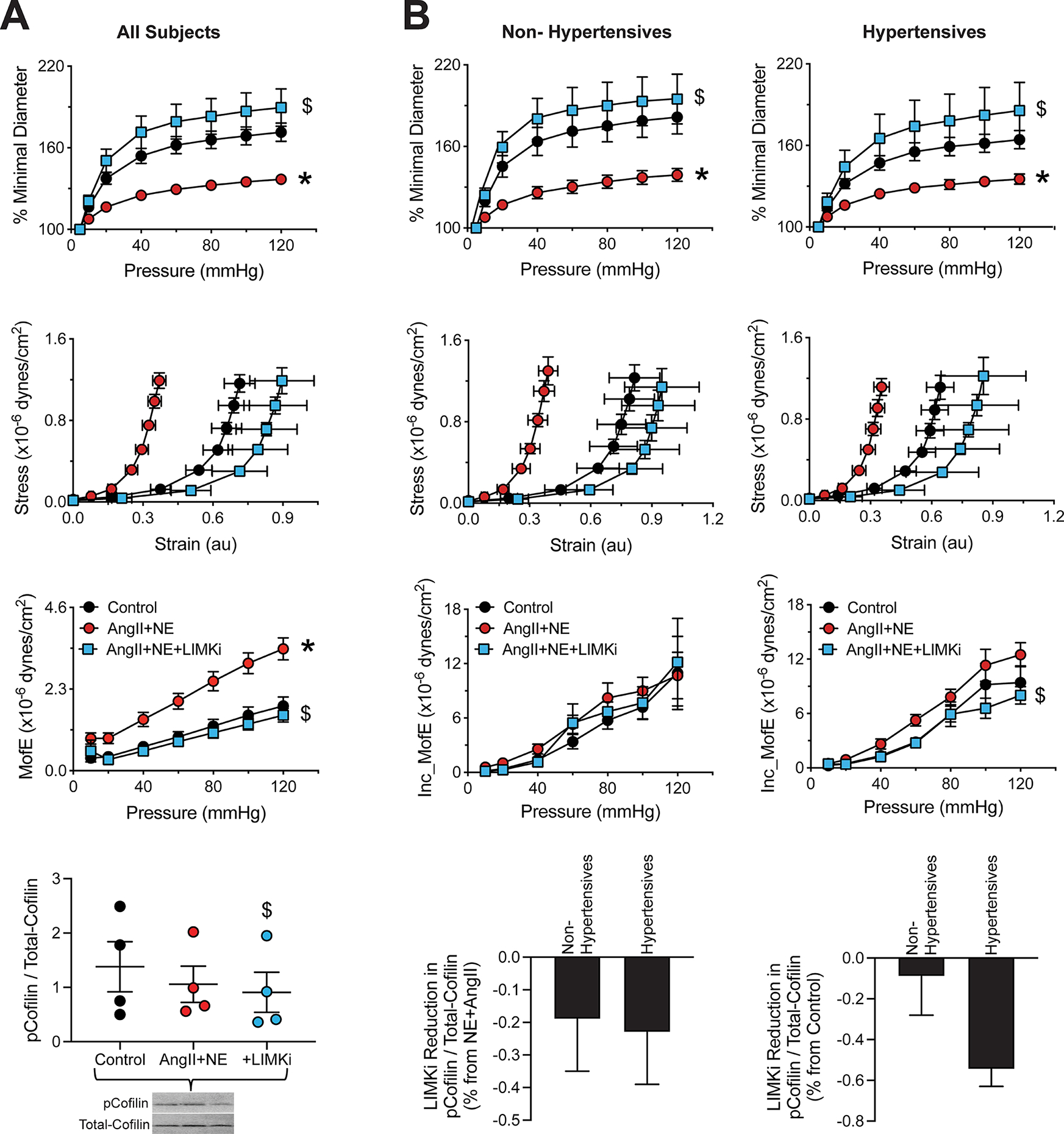
LIM kinase (LIMK) inhibition prevents the arterial stiffening associated with an 18-hour exposure to angiotensin-II (AngII) and norepinephrine (NE), while also decreasing cofilin phosphorylation. Small arteries were isolated from omental samples of subjects undergoing elective abdominal surgery and subsequently exposed to vehicle (control) or AngII (100nM) + NE (3μM) in the absence or presence of LIMK inhibition (LIMKi, 1μM) for 18 hours. A, Passive pressure-percent minimal diameter curves, strain-stress curves, moduli of elasticity, phosphorylated (p)- to total-cofilin ratios and representative blot obtained after the 18-hour incubation treatments. B, Passive pressure-percent minimal diameter curves, strain-stress curves, incremental moduli of elasticity (Inc_MofE) and phosphorylated (p)- to total-cofilin ratios obtained after the 18-hour incubation treatments separated according to the absence or presence of hypertension in the sample’s donor. Data are expressed as means ± SEM. *P<0.05 vs. Control. $P<0.05 vs. AngII+NE, n=4–12 subjects/group.
LIMK inhibition reduces actin stress fibers and cortical stiffness in cultured vascular smooth muscle cells.
We previously showed that a 20-minute exposure to AngII increases actin stress fiber thickness and cortical stiffness in cultured vascular smooth muscle cells obtained from the rat cremaster.38 Here, we first determined that an 18-hour exposure to AngII+NE increased the volume of F-actin in cultured HCASMCs and that LIMK inhibition consistently prevented that cytoskeletal change (Figure 4A–B). Western blots further showed that exposure of HCASMCs to AngII+NE for 18h caused an increase in cofilin phosphorylation, which was prevented by LIMKi (Figure 4C–D). Moreover, LIMK inhibition was able to prevent AngII+NE from increasing cell cortical stiffness as determined by atomic force microscopy (Figure 4E–F).
Figure 4.
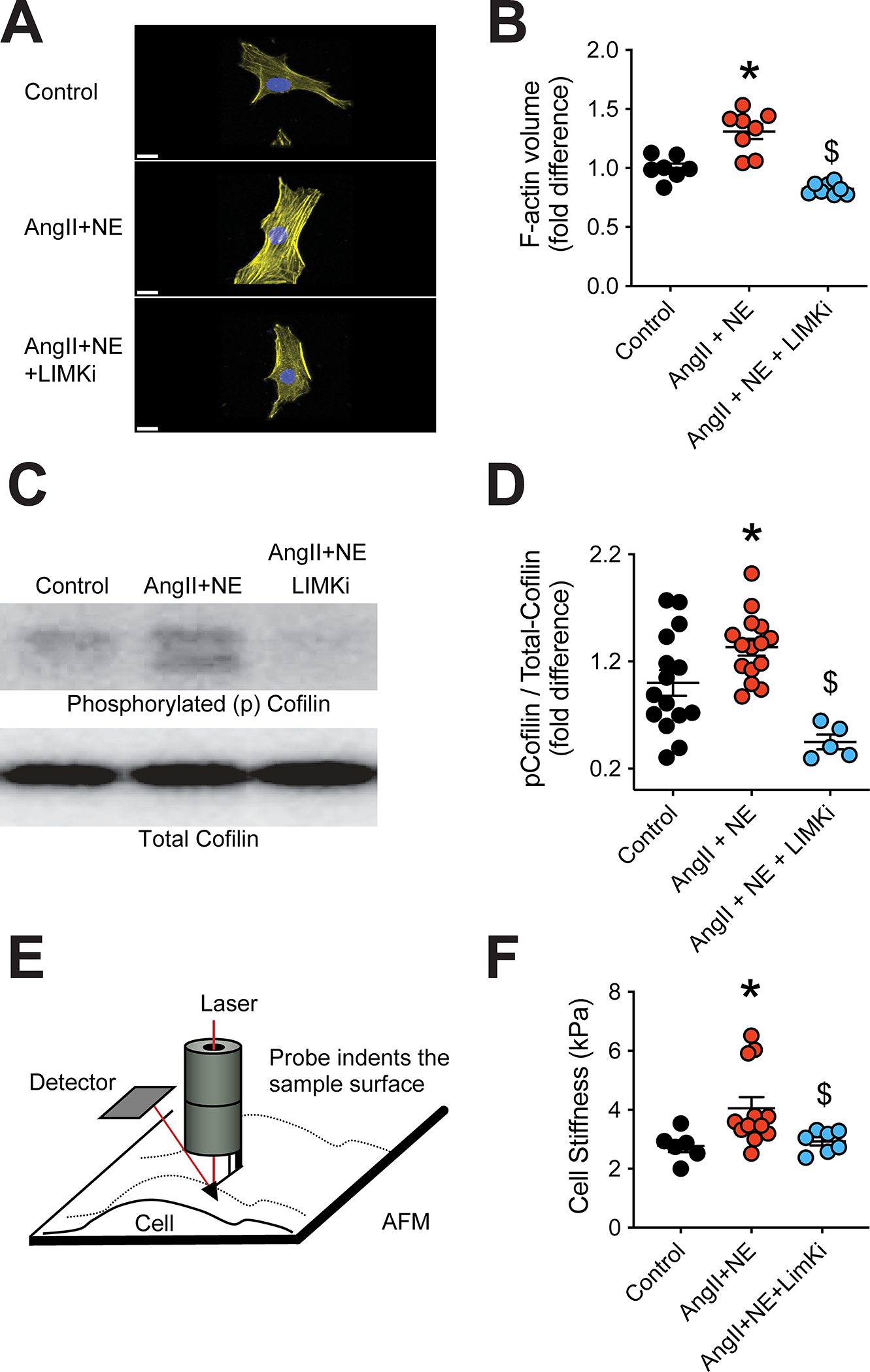
LIM kinase (LIMK) inhibition prevents angiotensin-II (AngII) and norepinephrine (NE) from increasing actin stress fibers and cortical stiffness, while decreasing cofilin phosphorylation in cultured human coronary artery smooth muscle cells (HCASMCs). HCASCMs in culture were exposed to AngII (100nM) + NE (3μM) in the absence or presence of LIMK inhibition (LIMKi, 1μM) for 18 hours. A, Representative confocal microscope images of F-actin (yellow) and nuclei (blue) present in HCASMCs after the 18-hour exposure treatments, bar=15μm. B, Quantification of F-actin volume within HCASMCs after the 18-hour exposure treatments. C, Representative Western blot of phosphorylated (p)- and total-cofilin of HCASMCs after the 18-hour exposure treatments. D, Quantification of p-cofilin to total-cofilin ratios in HCASMCs after the 18-hour exposure treatments. E, Schematic representation of the use of atomic force microscopy for the measurement of cellular cortical stiffness. F, Quantification of cell cortical stiffness of HCASMCs after the 18-hour exposure treatments. Data are expressed as means ± SEM. *P<0.05 vs. Control. $P<0.05 vs. AngII+NE, n=6–15 / treatment group.
LIMK inhibition prevents arteriolar inward remodeling in a mouse model of hypertension.
In order to determine the effects of LIMK inhibition on vascular remodeling in vivo, we used an AngII-infusion mouse model of hypertension. First, we determined that infusion of AngII at a rate of 1000ng/kg/min for 14 days increased mean arterial pressure by 19mmHg compared to mice infused with 100ng/kg/min (Figure 5A). Presence of hypertension was associated with a 16% reduction in arteriolar diameter in the ears treated with vehicle control (Figure 5B–C). The contralateral ears treated with LIMKi showed no change in vascular diameter after 14 days of AngII (1000ng/kg/min) infusion (Figure 5B). In comparison, arterioles in the ears of mice treated with the sub-pressor dose of AngII (100ng/kg/min) showed no changes in diameter, regardless of the presence or absence of LIMKi (Figure 5B).
Figure 5.
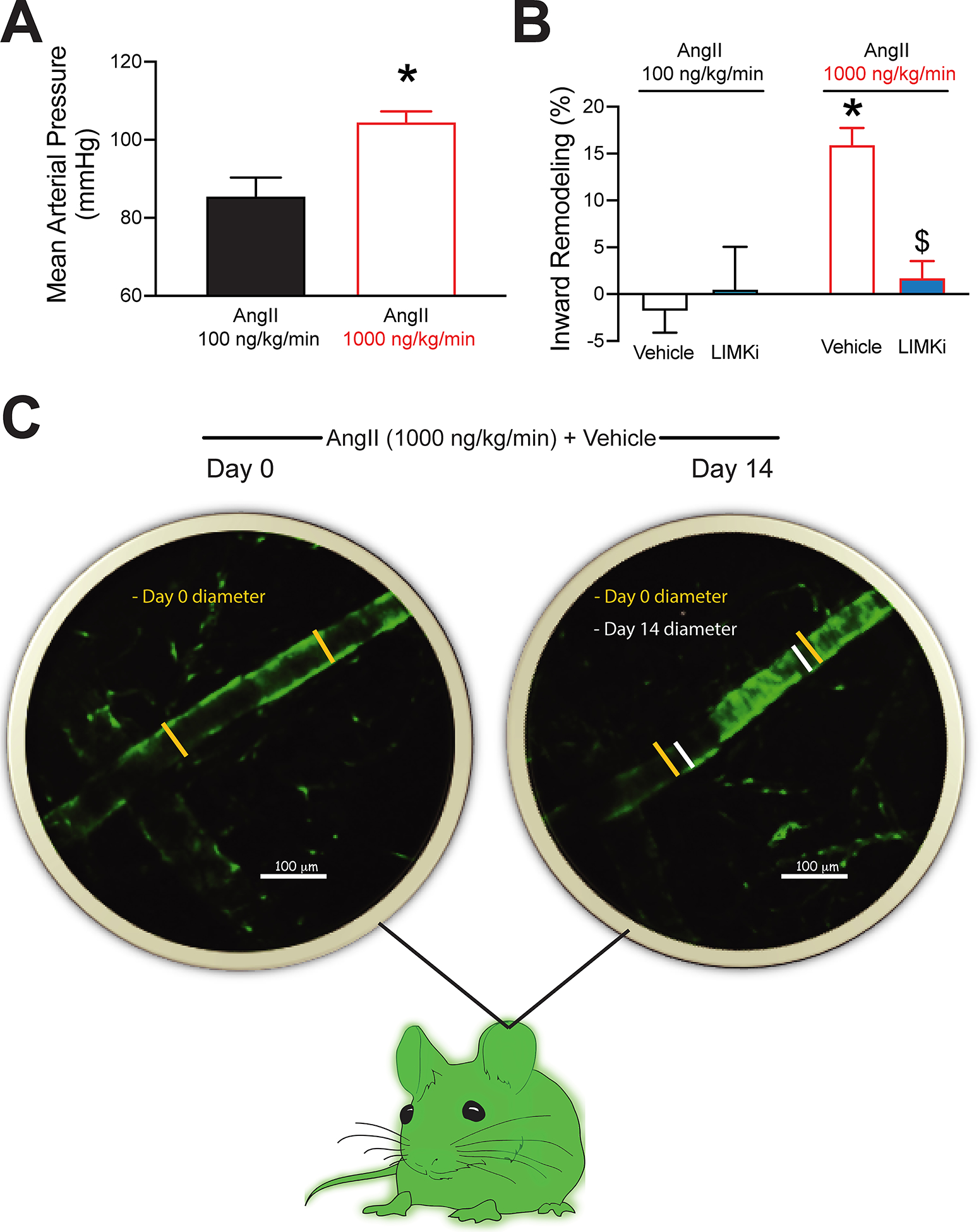
LIM kinase (LIMK) inhibition prevents arteriolar inward remodeling in a mouse model of angiotensin-II (AngII) infusion-dependent hypertension. Mice were infused with AngII (100 or 1000ng/kg/min), while one of their ears was injected subcutaneously with vehicle control and the contralateral with 20μL LIMKi (100μM). A, Carotid-catheterization-acquired mean arterial pressure in mice implanted with minipumps delivering AngII at 100 or 1000ng/kg/min. B, Changes in auricular subcutaneous arteriolar diameter expressed as percent remodeling after 14 days of AngII infusion in the ears treated with vehicle or LIMKi. C, Representative multiphoton intravital images of arterioles (green) within an ear treated with vehicle on days 0 and 14 of AngII (1000ng/kg/min) infusion. Data are expressed as means ± SEM. *P<0.05 vs. AngII (100ng/kg/min). $P<0.05 vs. AngII (1000ng/kg/min) + vehicle, n=4 / treatment group.
DISCUSSION
The primary findings of the present study are that small omental arteries from hypertensive subjects exhibit increased levels of phosphorylated cofilin associated with augmented F-actin stress fiber volume and vascular stiffness. Additional novel findings include the discovery that LIMK inhibition prevents arterial stiffening in omental human arteries exposed to prolonged vasoconstriction as well as arteriolar inward remodeling in hypertensive mice.
Hypertension is associated with adverse remodeling of the vasculature and an increased incidence of life-threatening cardiovascular events.39,40 Prominent among the remodeling processes are the presence of overall stiffening and inward remodeling of small and large arteries.41–44 Furthermore, evidence indicates that vascular remodeling and stiffening precede the development of hypertension,1 and a plethora of studies show that a primary stimulus driving adverse vascular remodeling and stiffening is prolonged vasoconstriction.15,19,22 Previously, we reported that vasoconstriction-induced inward remodeling of isolated rat arterioles is inhibited by preventing the phosphorylation of cofilin via LIMK inhibition.14 These results prompted us to investigate the potential association between LIMK activity, cofilin phosphorylation and arterial remodeling in hypertension. For this purpose, we first isolated human omental arteries from non-hypertensive and hypertensive subjects undergoing elective abdominal surgery and assessed their level of remodeling and cofilin phosphorylation. Results indicate that arteries from hypertensive subjects are less distensible and stiffer than those from non-hypertensives, as determined by their moduli of elasticity and cPWV. The latter is an estimation of stiffness derived from the geometrical and structural properties of isolated arteries under passive conditions using stepwise pressure increments. The physiological relevance of this index remains to be determined; however, we recently showed that it resembles in vivo measurements of aortic PWV,34,45 the gold standard assessment of arterial stiffness. Data also show that arteries from hypertensives have a greater level of cofilin phosphorylation that coincides with measures of arterial stiffness and F-actin content in their medial layer. While it has been shown that diabetic subjects have increased vascular cofilin phosphorylation,46 to the best of our knowledge, this is the first report of an association between cofilin phosphorylation and arterial stiffness in hypertension.
LIMK is a family of two isoenzymes (LIMK-1 and -2) that regulates the conversion of F-actin filaments into G-actin monomers.47–49 The expression of LIMK-1 and -2 varies among different tissue, with the cerebral cortex being a prominent expresser of LIMK-1 and smooth muscle an expresser of LIMK-2.49,50 The effects of LIMK on actin occur via the phosphorylation and consequent inactivation of cofilin, the primary function of which is to sever F-actin fibers.47–49 Consequently, an increased activity of LIMK effectively reduces actin depolymerization. Current research also indicates that LIMK-dependent inactivation of cofilin facilitates cell migration and participates in the progression of different types of cancer.49 Accordingly, LIMK inhibition is considered a potential therapeutic means to treat a number of pathological neoplasias.49 At the vascular level, activation of LIMK is associated with disturbed blood flow in the aortic arch and reduced endothelial-cell barrier function,51 the latter of which is associated with vascular stiffening.52 In comparison, decreased LIMK expression has been associated with the development of thoracic aortic aneurism and aortic coarctation.53 Similarly, defects in the gene responsible for LIMK expression are known to increase the risk for cerebral aneurism formation.54,55 All this suggests that changes in the activity or expression of LIMK associated with vasomotor pathways are able to stiffen or weaken the vascular wall. Indeed, numerous reports indicate that the process of vasoconstriction, in addition to involving actin-myosin cross-bridge cycling and calcium sensitization, also encompasses smooth muscle actin polymerization processes.14,38,56 Moreover, LIMK activation has been shown to occur in vascular smooth muscle cells exposed to vasoconstrictor agonists.57,58 Notably, we previously reported that prolonged vasoconstriction-induced stiffening and inward remodeling of small resistance arteries is associated with rapid vascular smooth muscle cell repositioning within the arterial wall.13,59 All this is consistent with a scenario in which LIMK phosphorylates and inactivates cofilin and allows for F-actin formation, vascular smooth muscle cell stiffening and cell movement.
In accordance with the role that LIMK plays in the vascular wall, we designed a series of experiments to determine if LIMK inhibition prevents prolonged vasoconstriction-induced inward remodeling in isolated human arteries. We chose a combination of AngII and NE as the vasoconstriction stimulus in order to simulate the increased activities of the renin-angiotensin-aldosterone system and sympathetic outflow that occur in hypertension. To block LIMK activity, we chose LIMKi, which is a specific LIMK inhibitor with limited effects on other kinases.48 Our results show that LIMK inhibition not only prevented the reduction in passive diameter associated with a 4-hour exposure of the arteries to AngII+NE, but caused an increase in passive diameter, so that the percent remodeling in arteries exposed only to the vasoconstrictors was significantly different from that of those exposed to the vasoconstrictors in combination with LIMKi. The fact that LIMK inhibition did not prevent AngII+NE from inducing or maintaining vasoconstriction indicates that the effects of LIMKi observed on vascular remodeling were not associated with a reduced capacity of the arteries to constrict. However, we found that the vasoconstriction response to AngII+NE was diminished when isolated mouse arteries were pre-incubated with LIMKi at a concentration above 1μM (data not shown). Those data were obtained in preparation for applying LIMKi in isolated human omental arteries and in order to use a concentration of the inhibitor not affecting vasoconstriction. Accordingly, it awaits to be determined whether sufficient actin polymerization capabilities remained to allow for vasoconstriction, but not for remodeling in the vessels treated with 1μM LIMKi; or if only a specific pool of actin needed for remodeling and not for vasoconstriction was targeted by the inhibition of cofilin occurring in response to 1μM LIMKi exposure.
We also performed a series of experiments in which we exposed human arteries to AngII+NE for 18 hours in the absence or presence of LIMKi in order for the vasoconstrictors to induce a robust stiffening of the arteries and determine the effects of LIMK inhibition in this process. Results indicate that LIMK inhibition prevented vascular stiffening as arteries treated with AngII+NE+LIMKi had slightly increased distensibility compared to those treated with vehicle and significantly greater than those treated only with the vasoconstrictors. Accordingly, LIMK inhibition also preserved arterial moduli of elasticity, as well as cross-sectional compliance, both of which were significantly affected by the prolonged exposure to AngII+NE. As expected, LIMK inhibition significantly reduced the ratio of phosphorylated to total cofilin, but AngII+NE alone failed to increase this ratio. We reason that the latter occurred because cofilin is already highly phosphorylated in vessels from hypertensive subjects as indicated in our initial test performed in human omental arteries. Therefore, we decided to separate the data of the 18-hour incubation experiment into groups of vessels from non-hypertensive and hypertensive subjects. Results from this analysis indicate that while LIMK inhibition prevented arterial stiffening in arteries from both non-hypertensives and hypertensives, the reduction in stiffness was greater in the arteries from hypertensives as determined by their level of distensibility or strain. Consequently, LIMK inhibition was only able to successfully reduce the incremental modulus of elasticity in hypertensive vessels. Moreover, while LIMK inhibition reduced the level of cofilin phosphorylation induced by AngII+NE similarly in arteries from both non-hypertensive and hypertensive subjects, it reduced cofilin phosphorylation to less that 50% of control only in hypertensive arteries. No levels of statistical significance could be determined in the comparisons of cofilin phosphorylation between arteries from non-hypertensives and hypertensives due to the fact that in order to obtain adequate amounts of protein for Western blot analysis, three arteries (each artery from a different subject) were pooled into a single homogenate and only two samples per subject group were available to perform the analyses.
We have previously shown that the inward remodeling associated with prolonged (4h) vasoconstriction in isolated rat arterioles depends on actin cytoskeletal structures, as disruption of F-actin stress fibers increases the passive diameter of remodeled arterioles to levels similar to those observed before the induction of vasoconstriction.15 In order to determine that similar actin cytoskeletal structures form in human vascular smooth muscle cells, we exposed HCASMCs to AngII+NE for 18 hours and show that exposure to these vasoconstrictors increases F-actin stress fiber volume and the ratio of phosphorylated to total cofilin. We also showed before that rat arteriolar smooth muscle cells stiffen in response to a short exposure to AngII.38 Here we demonstrate that comparable stiffening occurs in human vascular smooth muscle cells exposed to AngII+NE for 18 hours and that LIMK inhibition limits cofilin phosphorylation, as well as the formation of F-actin stress fibers and the increase in stiffness associated with exposure to the vasoconstrictors.
A primary goal of our research is to elucidate novel approaches aimed at preventing and/or treating arterial stiffening and remodeling. Therefore, in order to test the potential capacity of LIMK inhibition to prevent arterial remodeling in hypertension, we treated arterioles within the ear of normotensive and hypertensive mice with either vehicle control or LIMKi. Results from these experiments indicate that LIMK inhibition blocks the arteriolar inward remodeling process associated with AngII-induced hypertension in mice. Given that LIMKi does not affect the vasoconstriction capacity of human omental or mouse mesenteric arteries,14 these findings collectively suggest that LIMK inhibition can ameliorate arterial stiffening without affecting arterial vasoconstrictive function.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What is New?
Small arteries of hypertensive subjects have increased cofilin phosphorylation in conjunction with increased stiffness and greater fibrillar actin content.
LIM kinase (LIMK) inhibition prevents vasoconstriction-induced stiffening and inward remodeling, as well as cofilin phosphorylation in human isolated small arteries.
LIMK inhibition averts vasoconstrictor-induced F-actin formation, cofilin phosphorylation and cortical stiffening in cultured human vascular smooth muscle cells.
LIMK inhibition blocks arteriolar inward remodeling in a mouse model hypertension.
What is Relevant?
Arterial stiffening and inward remodeling precede and contribute to the development and progression of hypertension and cardiovascular disease. We provide evidence that LIMK inhibition blocks their development, which indicates that downstream actin polymerization is one of the mechanisms that promote vasoconstriction- and hypertension-associated arterial stiffening and vascular remodeling.
Summary
Herein we show that vasoconstriction- and hypertension-induced arterial stiffening and remodeling are associated with increased cofilin phosphorylation and F-actin stress fiber formation in vascular smooth muscle cells. We also demonstrate that inhibition of LIMK, the enzyme responsible for cofilin phosphorylation, prevents vasoconstriction and hypertension-associated arterial stiffening and remodeling. Therefore, LIMK inhibition should be considered as a potential means to treat vascular disease in hypertension.
Perspectives.
Although the mechanisms responsible for the processes of arterial stiffening and remodeling in hypertension have not been fully elucidated, cumulative evidence indicates that vascular smooth muscle cytoskeletal stress fiber formation participates in vasoconstriction- and hypertension-associated arterial stiffening and remodeling. Herein we show that inhibition of LIMK, the primary enzyme responsible for the phosphorylation of cofilin, prevents stiffening and remodeling in isolated human small arteries and cultured vascular smooth muscle cells exposed to vasoconstrictor agonists. Moreover, we show that LIMK inhibition prevents arteriolar inward remodeling in a mouse model of hypertension. As the inactivation of cofilin via its phosphorylation promotes actin polymerization, our results suggest that inhibition of vascular smooth muscle cell cytoskeletal fiber formation prevents vasoconstriction- and hypertension-associated arterial stiffening and remodeling. Based on these results, we propose that pharmacological inhibition of LIMK should be considered as a potential strategy to stop the progression of arterial stiffening and remodeling in hypertension.
ACKNOWLEDGMENTS
We would like to thank all the subjects who provided omental tissue for the study.
SOURCES OF FUNDING
This work was supported in part by the National Institutes of Health R01 HL088105 (LM-L), R01 HL137769 (JP), and R01 HL142770 (CM-A). This work was also supported by the use of resources and facilities at the Harry S. Truman Memorial Veterans’ Hospital in Columbia, MO.
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Weisbrod RM, Shiang T, Al Sayah L, Fry JL, Bajpai S, Reinhart-King CA, Lob HE, Santhanam L, Mitchell G, Cohen RA, Seta F. Arterial stiffening precedes systolic hypertension in diet-induced obesity. Hypertension. 2013;62(6):1105–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palombo C, Kozakova M. Arterial stiffness, atherosclerosis and cardiovascular risk: Pathophysiologic mechanisms and emerging clinical indications. Vascular pharmacology. 2016;77:1–7. [DOI] [PubMed] [Google Scholar]
- 3.Xiong XQ, Chen WW, Zhu GQ. Adipose afferent reflex: sympathetic activation and obesity hypertension. Acta Physiol (Oxf). 2014;210(3):468–478. [DOI] [PubMed] [Google Scholar]
- 4.Bender SB, Castorena-Gonzalez JA, Garro M, Reyes-Aldasoro CC, Sowers JR, DeMarco VG, Martinez-Lemus LA. Regional variation in arterial stiffening and dysfunction in Western diet-induced obesity. Am J Physiol Heart Circ Physiol. 2015;309(4):H574–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castorena-Gonzalez JA, Staiculescu MC, Foote C, Martinez-Lemus LA. Mechanisms of the inward remodeling process in resistance vessels: is the actin cytoskeleton involved? Microcirculation. 2014;21(3):219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davel AP, Jaffe IZ, Tostes RC, Jaisser F, Belin de Chantemele EJ. New roles of aldosterone and mineralocorticoid receptors in cardiovascular disease: translational and sex-specific effects. Am J Physiol Heart Circ Physiol. 2018;315(4):H989–H999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aroor AR, Habibi J, Nistala R, Ramirez-Perez FI, Martinez-Lemus LA, Jaffe IZ, Sowers JR, Jia G, Whaley-Connell A. Diet-Induced Obesity Promotes Kidney Endothelial Stiffening and Fibrosis Dependent on the Endothelial Mineralocorticoid Receptor. Hypertension. 2019;73(4):849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayashi K, Hirayama E. Age-related changes of wall composition and collagen cross-linking in the rat carotid artery - In relation with arterial mechanics. Journal of the mechanical behavior of biomedical materials. 2017;65:881–889. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, Zhao X, Vatner DE, McNulty T, Bishop S, Sun Z, Shen YT, Chen L, Meininger GA, Vatner SF. Extracellular Matrix Disarray as a Mechanism for Greater Abdominal Versus Thoracic Aortic Stiffness With Aging in Primates. Arterioscler Thromb Vasc Biol. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sehgel NL, Sun Z, Hong Z, Hunter WC, Hill MA, Vatner DE, Vatner SF, Meininger GA. Augmented vascular smooth muscle cell stiffness and adhesion when hypertension is superimposed on aging. Hypertension. 2015;65(2):370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sehgel NL, Zhu Y, Sun Z, Trzeciakowski JP, Hong Z, Hunter WC, Vatner DE, Meininger GA, Vatner SF. Increased vascular smooth muscle cell stiffness: a novel mechanism for aortic stiffness in hypertension. Am J Physiol Heart Circ Physiol. 2013;305(9):H1281–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez-Lemus LA, Aroor AR, Ramirez-Perez FI, Jia G, Habibi J, DeMarco VG, Barron B, Whaley-Connell A, Nistala R, Sowers JR. Amiloride Improves Endothelial Function and Reduces Vascular Stiffness in Female Mice Fed a Western Diet. Front Physiol 2017;8:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castorena-Gonzalez JA, Staiculescu MC, Foote CA, Polo-Parada L, Martinez-Lemus LA. The obligatory role of the actin cytoskeleton on inward remodeling induced by dithiothreitol activation of endogenous transglutaminase in isolated arterioles. Am J Physiol Heart Circ Physiol. 2014;306(4):H485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foote CA, Castorena-Gonzalez JA, Staiculescu MC, Clifford PS, Hill MA, Meininger GA, Martinez-Lemus LA. Brief serotonin exposure initiates arteriolar inward remodeling processes in vivo that involve transglutaminase activation and actin cytoskeleton reorganization. Am J Physiol Heart Circ Physiol. 2016;310(2):H188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Staiculescu MC, Galinanes EL, Zhao G, Ulloa U, Jin M, Beig MI, Meininger GA, Martinez-Lemus LA. Prolonged vasoconstriction of resistance arteries involves vascular smooth muscle actin polymerization leading to inward remodelling. Cardiovasc Res. 2013;98(3):428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang DD, Anfinogenova Y. Physiologic Properties and Regulation of the Actin Cytoskeleton in Vascular Smooth Muscle. Journal of cardiovascular pharmacology and therapeutics. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brozovich FV, Nicholson CJ, Degen CV, Gao YZ, Aggarwal M, Morgan KG. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol Rev 2016;68(2):476–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerthoffer WT. Actin cytoskeletal dynamics in smooth muscle contraction. Can J Physiol Pharmacol. 2005;83(10):851–856. [DOI] [PubMed] [Google Scholar]
- 19.Martinez-Lemus LA, Zhao G, Galinanes EL, Boone M. Inward remodeling of resistance arteries requires reactive oxygen species-dependent activation of matrix metalloproteinases. Am J Physiol Heart Circ Physiol. 2011;300(6):H2005–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez-Lemus LA. Persistent Agonist-Induced Vasoconstriction Is Not Required for Angiotensin II to Mediate Inward Remodeling of Isolated Arterioles with Myogenic Tone. J Vasc Res. 2008;45(3):211–221. [DOI] [PubMed] [Google Scholar]
- 21.Bakker EN, Buus CL, VanBavel E, Mulvany MJ. Activation of resistance arteries with endothelin-1: from vasoconstriction to functional adaptation and remodeling. J Vasc Res. 2004;41(2):174–182. [DOI] [PubMed] [Google Scholar]
- 22.Bakker EN, van der Meulen ET, van den Berg BM, Everts V, Spaan JA, VanBavel E. Inward remodeling follows chronic vasoconstriction in isolated resistance arteries. J Vasc Res. 2002;39(1):12–20. [DOI] [PubMed] [Google Scholar]
- 23.Calo LA, Davis PA, Rossi GP. Understanding the mechanisms of angiotensin II signaling involved in hypertension and its long-term sequelae: insights from Bartter’s and Gitelman’s syndromes, human models of endogenous angiotensin II signaling antagonism. J Hypertens. 2014;32(11):2109–2119; discussion 2119. [DOI] [PubMed] [Google Scholar]
- 24.Nuno DW, England SK, Lamping KG. RhoA localization with caveolin-1 regulates vascular contractions to serotonin. Am J Physiol Regul Integr Comp Physiol. 2012;303(9):R959–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83(4):1325–1358. [DOI] [PubMed] [Google Scholar]
- 26.Puetz S, Lubomirov LT, Pfitzer G. Regulation of smooth muscle contraction by small GTPases. Physiology (Bethesda). 2009;24:342–356. [DOI] [PubMed] [Google Scholar]
- 27.Chen YL, Ren Y, Xu W, Rosa RH Jr., Kuo L, Hein TW. Constriction of Retinal Venules to Endothelin-1: Obligatory Roles of ETA Receptors, Extracellular Calcium Entry, and Rho Kinase. Invest Ophthalmol Vis Sci. 2018;59(12):5167–5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bagnato A, Rosano L. Endothelin-1 receptor drives invadopodia: Exploiting how beta-arrestin-1 guides the way. Small GTPases. 2018;9(5):394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai YP, Bongalon S, Mutafova-Yambolieva VN, Yamboliev IA. Distinct effects of contraction agonists on the phosphorylation state of cofilin in pulmonary artery smooth muscle. Adv Pharmacol Sci. 2008;2008:362741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yi R, Xiao-Ping G, Hui L. Atorvastatin prevents angiotensin II-induced high permeability of human arterial endothelial cell monolayers via ROCK signaling pathway. Biochem Biophys Res Commun. 2015;459(1):94–99. [DOI] [PubMed] [Google Scholar]
- 31.Bernard O Lim kinases, regulators of actin dynamics. The international journal of biochemistry & cell biology. 2007;39(6):1071–1076. [DOI] [PubMed] [Google Scholar]
- 32.Ostrowska Z, Moraczewska J. Cofilin - a protein controlling dynamics of actin filaments. Postepy Hig Med Dosw (Online). 2017;71(0):339–351. [DOI] [PubMed] [Google Scholar]
- 33.Padilla J, Ramirez-Perez FI, Habibi J, Bostick B, Aroor AR, Hayden MR, Jia G, Garro M, DeMarco VG, Manrique C, Booth FW, Martinez-Lemus LA, Sowers JR. Regular Exercise Reduces Endothelial Cortical Stiffness in Western Diet-Fed Female Mice. Hypertension. 2016;68(5):1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aroor AR, Das NA, Carpenter AJ, Habibi J, Jia G, Ramirez-Perez FI, Martinez-Lemus L, Manrique-Acevedo CM, Hayden MR, Duta C, Nistala R, Mayoux E, Padilla J, Chandrasekar B, DeMarco VG. Glycemic control by the SGLT2 inhibitor empagliflozin decreases aortic stiffness, renal resistivity index and kidney injury. Cardiovasc Diabetol. 2018;17(1):108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hussain T, Hong M, Osmond PJ, Izzo JL. Protective effects of amiloride monotherapy on blood pressure, hemodynamics, and arterial stiffness in glucocorticoid-remediable aldosteronism (GRA). Journal of the American Society of Hypertension. 8(4):e137–e138. [Google Scholar]
- 36.Jia G, Habibi J, Aroor AR, Martinez-Lemus LA, DeMarco VG, Ramirez-Perez FI, Sun Z, Hayden MR, Meininger GA, Mueller KB, Jaffe IZ, Sowers JR. Endothelial Mineralocorticoid Receptor Mediates Diet-Induced Aortic Stiffness in Females. Circ Res. 2016;118(6):935–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yokota T, Kawakami Y, Nagai Y, Ma JX, Tsai JY, Kincade PW, Sato S. Bone marrow lacks a transplantable progenitor for smooth muscle type alpha-actin-expressing cells. Stem Cells. 2006;24(1):13–22. [DOI] [PubMed] [Google Scholar]
- 38.Hong Z, Sun Z, Li M, Li Z, Bunyak F, Ersoy I, Trzeciakowski JP, Staiculescu MC, Jin M, Martinez-Lemus L, Hill MA, Palaniappan K, Meininger GA. Vasoactive agonists exert dynamic and coordinated effects on vascular smooth muscle cell elasticity, cytoskeletal remodelling and adhesion. J Physiol. 2014;592(Pt 6):1249–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niiranen TJ, Kalesan B, Hamburg NM, Benjamin EJ, Mitchell GF, Vasan RS. Relative Contributions of Arterial Stiffness and Hypertension to Cardiovascular Disease: The Framingham Heart Study. J Am Heart Assoc. 2016;5(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mancusi C, Losi MA, Izzo R, Canciello G, Carlino MV, Albano G, De Luca N, Trimarco B, de Simone G. Higher pulse pressure and risk for cardiovascular events in patients with essential hypertension: The Campania Salute Network. Eur J Prev Cardiol. 2018;25(3):235–243. [DOI] [PubMed] [Google Scholar]
- 41.Diaz-Otero JM, Fisher C, Downs K, Moss ME, Jaffe IZ, Jackson WF, Dorrance AM. Endothelial Mineralocorticoid Receptor Mediates Parenchymal Arteriole and Posterior Cerebral Artery Remodeling During Angiotensin II-Induced Hypertension. Hypertension. 2017;70(6):1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rizzoni D, Porteri E, Boari GE, De Ciuceis C, Sleiman I, Muiesan ML, Castellano M, Miclini M, Agabiti-Rosei E. Prognostic significance of small-artery structure in hypertension. Circulation. 2003;108(18):2230–2235. [DOI] [PubMed] [Google Scholar]
- 43.Jia G, Aroor AR, Martinez-Lemus LA, Sowers JR. Potential Role of Antihypertensive Medications in Preventing Excessive Arterial Stiffening. Curr Hypertens Rep 2018;20(9):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Safar ME. Arterial stiffness as a risk factor for clinical hypertension. Nature reviews. Cardiology 2017. [DOI] [PubMed] [Google Scholar]
- 45.Xiong Y, Aroor AR, Ramirez-Perez FI, Jia G, Habibi J, Manrique-Acevedo C, Lastra G, Chen D, DeMarco VG, Martinez-Lemus LA, Hill MA, Jaisser F, Sowers JR, Whaley-Connell A. Western diet induces renal artery endothelial stiffening that is dependent on the epithelial Na(+) channel. Am J Physiol Renal Physiol. 2020;318(5):F1220–F1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hien TT, Turczynska KM, Dahan D, Ekman M, Grossi M, Sjogren J, Nilsson J, Braun T, Boettger T, Garcia-Vaz E, Stenkula K, Sward K, Gomez MF, Albinsson S. Elevated Glucose Levels Promote Contractile and Cytoskeletal Gene Expression in Vascular Smooth Muscle via Rho/Protein Kinase C and Actin Polymerization. J Biol Chem. 2016;291(7):3552–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manetti F LIM kinases are attractive targets with many macromolecular partners and only a few small molecule regulators. Medicinal research reviews. 2012;32(5):968–998. [DOI] [PubMed] [Google Scholar]
- 48.Manetti F Recent advances in the rational design and development of LIM kinase inhibitors are not enough to enter clinical trials. Eur J Med Chem. 2018;155:445–458. [DOI] [PubMed] [Google Scholar]
- 49.Lee MH, Kundu JK, Chae JI, Shim JH. Targeting ROCK/LIMK/cofilin signaling pathway in cancer. Arch Pharm Res. 2019;42(6):481–491. [DOI] [PubMed] [Google Scholar]
- 50.Scott RW, Olson MF. LIM kinases: function, regulation and association with human disease. J Mol Med (Berl). 2007;85(6):555–568. [DOI] [PubMed] [Google Scholar]
- 51.Miyazaki T, Honda K, Ohata H. m-Calpain antagonizes RhoA overactivation and endothelial barrier dysfunction under disturbed shear conditions. Cardiovasc Res. 2010;85(3):530–541. [DOI] [PubMed] [Google Scholar]
- 52.Huynh J, Nishimura N, Rana K, Peloquin JM, Califano JP, Montague CR, King MR, Schaffer CB, Reinhart-King CA. Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Science translational medicine. 2011;3(112):112ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tian L, Liao MF, Zhang L, Lu QS, Jing ZP. A study of the expression and interaction of Destrin, cofilin, and LIMK in Debakey I type thoracic aortic dissection tissue. Scand J Clin Lab Invest. 2010;70(7):523–528. [DOI] [PubMed] [Google Scholar]
- 54.Akagawa H, Tajima A, Sakamoto Y, Krischek B, Yoneyama T, Kasuya H, Onda H, Hori T, Kubota M, Machida T, Saeki N, Hata A, Hashiguchi K, Kimura E, Kim CJ, Yang TK, Lee JY, Kimm K, Inoue I. A haplotype spanning two genes, ELN and LIMK1, decreases their transcripts and confers susceptibility to intracranial aneurysms. Human molecular genetics. 2006;15(10):1722–1734. [DOI] [PubMed] [Google Scholar]
- 55.Low SK, Zembutsu H, Takahashi A, Kamatani N, Cha PC, Hosono N, Kubo M, Matsuda K, Nakamura Y. Impact of LIMK1, MMP2 and TNF-alpha variations for intracranial aneurysm in Japanese population. J Hum Genet. 2011;56(3):211–216. [DOI] [PubMed] [Google Scholar]
- 56.Kim HR, Gallant C, Leavis PC, Gunst SJ, Morgan KG. Cytoskeletal remodeling in differentiated vascular smooth muscle is actin isoform dependent and stimulus dependent. Am J Physiol Cell Physiol. 2008;295(3):C768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fediuk J, Gutsol A, Nolette N, Dakshinamurti S. Thromboxane-induced actin polymerization in hypoxic pulmonary artery is independent of Rho. Am J Physiol Lung Cell Mol Physiol. 2012;302(1):L13–26. [DOI] [PubMed] [Google Scholar]
- 58.Du H, Wang X, Wu J, Qian Q. Phenylephrine induces elevated RhoA activation and smooth muscle alpha-actin expression in Pkd2+/− vascular smooth muscle cells. Hypertens Res. 2010;33(1):37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martinez-Lemus LA, Hill MA, Bolz SS, Pohl U, Meininger GA. Acute mechanoadaptation of vascular smooth muscle cells in response to continuous arteriolar vasoconstriction: implications for functional remodeling. Faseb J. 2004;18(6):708–710. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.