

Abstract
We compared therapeutic properties of natural and engineered chemokine (C-X-C motif) receptor 4 (CXCR4) agonists in a rat acute respiratory distress syndrome (ARDS) model utilizing the PaO2/FiO2-ratio as a clinically relevant primary outcome criterion. Ventilated rats underwent unilateral lung ischemia from t = 0–70 min plus hemorrhage to a mean arterial blood pressure (MAP) of 30 mmHg from t = 40–70 min, followed by reperfusion/fluid resuscitation until t = 300 min. Natural CXCR4 agonists (CXCL12, ubiquitin) and engineered CXCL12 variants (CXCL121, CXCL22, CXCL12K27A/R41A/R47A, CXCL12 (3–68)) were administered within 5 min of fluid resuscitation. Animals treated with vehicle or CXCL12 (3–68) reached criteria for mild and moderate ARDS between t = 90–120 min and t = 120–180 min, respectively, and remained in moderate ARDS until t = 300 min. Ubiquitin, CXCL12, CXCL121 and CXCL122 prevented ARDS development. Potencies of CXCL12/CXCL121/CXCL122 were higher than the potency of ubiquitin. CXCL12K27A/R41A/R47A was inefficacious. CXCL121 > CXCL12 stabilized MAP and reduced fluid requirements. CXCR4 agonists at doses that preserved lung function reduced histological injury of the post-ischemic lung and reduced mortality from 55 to 9%. Our findings suggest that CXCR4 protein agonists prevent development of ARDS and reduce mortality in a rat model, and that development of new engineered protein therapeutics with improved pharmacological properties for ARDS is possible.
Subject terms: Drug discovery, Physiology, Diseases, Medical research, Molecular medicine
Introduction
Acute respiratory distress syndrome (ARDS) remains a major contributor to morbidity and mortality in trauma patients. Although the incidence of ARDS in trauma patients has decreased in the last decade, mortality from ARDS has increased1. The treatment of ARDS is currently limited to supportive therapy and lung-protective ventilation strategies. Drugs that attenuate development and progression of ARDS are not available, but highly desirable for their potential to improve outcomes.
Several lines of evidence suggest that natural and synthetic agonists of chemokine (C-X-C motif) receptor 4 (CXCR4) protect lung endothelial barrier function and have lung protective properties in animal models2–11. We showed previously that treatment with the non-cognate CXCR4 agonist ubiquitin protects lung endothelial barrier function during resuscitation from hemorrhagic shock and prevents development of ARDS in pre-clinical lung ischemia–reperfusion injury, endotoxemia and polytrauma models2,5,6,10. The therapeutic potential of the cognate CXCR4 agonist chemokine (C-X-C motif) ligand 12 (CXCL12, stromal cell-derived factor 1α) during development of ARDS, however, is unknown. Furthermore, we recently reported that various engineered CXCL12 variants, which possess biochemical and pharmacological characteristics distinct from wild-type CXCL12, protect human lung endothelial barrier function in vitro similar to wild-type CXCL129. These engineered CXCL12 variants have not been tested in an in vivo lung injury model.
The aim of the present study was to test and compare efficacy and potency of ubiquitin, CXCL12 and engineered CXCL12 variants to prevent ARDS development in a rat model.
Thoracic injury and hypotension on admission have been identified as important risk factors for the development of trauma-induced ARDS in patients1,12–14. Thus, in the present study, we employed a rat model that mimics clinical predictors of trauma-induced ARDS development by combining a thoracic injury component, i.e. thoracotomy and subsequent lung ischemia–reperfusion injury, with systemic hypotension during the hemorrhagic shock period. This animal model was designed to suffice key criteria of the Berlin definition of ARDS15, i.e. to result in a ratio of arterial oxygen partial pressure to fractional inspired oxygen (P/F) < 300 mmHg under positive end-expiratory pressure ≥ 5 cmH2O. Our findings show that treatment with CXCR4 agonists during the early fluid resuscitation period prevents development of lung ischemia–reperfusion injury and hemorrhage induced ARDS in rats and suggest that the development of engineered CXCR4 agonists with improved therapeutic efficacy is possible.
Results
Ubiquitin treatment prevents development of ARDS
In the first series of experiments animals were treated with vehicle or ubiquitin (0.14 and 0.7 μmol/kg). There were no statistically significant differences in any of the measured physiological parameters between groups at baseline (Fig. 1). Hemorrhage volumes to achieve the target MAP of 40 mmHg during the shock period were comparable among the groups (Fig. 1A). Two of 6 (2/6), 2/3 and 3/5 animals after treatment with vehicle, 0.14 and 0.7 μmol/kg ubiquitin, respectively, survived until t = 300 min. MAP, resuscitation fluid requirements and hematocrit values were comparable between ubiquitin and vehicle treated animals, despite some statistically significant differences in MAP and fluid requirements during short time periods (Fig. 1B–D). Vehicle treated animals fulfilled criteria for mild ARDS between t = 90–120 min (P/F < 300 mmHg) and for moderate ARDS (P/F < 200 mmHg) at t = 180 min. The average P/F of vehicle treated animals at the end of the observation period was 110 ± 33 mmHg (Fig. 1E). While animals treated with 0.14 μmol/kg ubiquitin were indistinguishable from vehicle-treated animals, treatment with 0.7 μmol/kg ubiquitin prevented development of ARDS (Fig. 1E).
Figure 1.

Administration of the non-cognate CXCR4 agonist ubiquitin prevents development of ARDS. Animals were treated with vehicle (n = 6), 0.7 (n = 3) or 0.14 (n = 5) μmol/kg ubiquitin. Arrows indicate the time point of drug administration. LI + HEM: Lung ischemia and hemorrhage period. Data are mean ± SE. *p < 0.05 for 0.7 μmol/kg ubiquitin vs. vehicle. (A) Hemorrhage volumes (mL/kg) during LI + HEM. (B) MAP: Mean arterial blood pressure (mmHg). (C) RES: Resuscitation fluid requirements (mL/kg). (D) Hct: Hematocrit (%). (E) PaO2/FiO2 (mmHg).
CXCL12 treatment prevents development of ARDS
We then performed the second series of experiments, in which animals were treated with various doses of CXCL12 (0.07, 0.14 and 0.7 μmol/kg). The inactive N-terminal truncated CXCL12 (3–68) was used as a specific control protein. Animals in all groups were indistinguishable at baseline (Fig. 2). Hemorrhage volumes were comparable among the groups (Fig. 2A). One of 3 animals treated with CXCL12 (3–68) survived until t = 300 min. One of 3 (1/3), 5/5 and 3/3 animals after treatment with 0.07, 0.14 and 0.7 μmol/kg CXCL12, respectively, survived until t = 300 min. Despite similar hematocrit values, MAPs were higher and fluid requirements lower in animals treated with 0.7 μmol/kg and 0.14 μmol/kg CXCL12, as compared with animals treated with 0.7 μmol/kg CXCL12 (3–68) and 0.07 μmol/kg CXCL12 (Fig. 2B–D). Similar to vehicle treated animals in series 1, P/F in animals treated with 0.7 μmol/kg CXCL12 (3–68) and 0.07 μmol/kg CXCL12 progressively decreased and animals fulfilled criteria for moderate to severe ARDS at t = 300 min (Fig. 2E). Treatment with 0.7 μmol/kg and 0.14 μmol/kg CXCL12 prevented development of ARDS (P/F > 300 mmHg at all time points, Fig. 2E).
Figure 2.

Administration of the cognate CXCR4 agonist CXCL12 prevents development of ARDS. Animals were treated with 0.7 μmol/kg inactive N-terminal truncated CXCL12 (3–68) (control, n = 3) or with 0.7 (n = 3), 0.14 (n = 5) or 0.07 (n = 3) μmol/kg CXCL12. Arrows indicate the time point of drug administration. LI + HEM: Lung ischemia and hemorrhage period. Data are mean ± SE. #p < 0.05 for 0.7 μmol/kg CXCL12 vs. CXCL12 (3–68). *p < 0.05 for 0.14 μmol/kg CXCL12 vs. CXCL12 (3–68). (A) Hemorrhage volumes (mL/kg) during LI + HEM. (B) MAP: Mean arterial blood pressure (mmHg). (C) RES: Resuscitation fluid requirements (mL/kg). (D) Hct: Hematocrit (%). (E) PaO2/FiO2 (mmHg).
Comparison of CXCL12 with engineered CXCL12 variants for prevention of ARDS
In the third series of experiments, we selected a dose of 0.14 μmol/kg as the lowest effective dose of CXCL12 from series 2 to compare the effects of wild-type CXCL12 with vehicle and the engineered CXCL12 variants. As in series 1 and 2, animals in all groups were indistinguishable at baseline (Fig. 3) and hemorrhage volumes to achieve the target MAP during the shock period were comparable among the groups (Fig. 3A). Three of 5 (3/5) vehicle treated animals and 4/4, 3/3, 3/4 and 2/3 animals treated with CXCL12, CXCL121, CXCL122 and CXCL12 K27A/R41A/R47A, respectively, survived until t = 300 min. Despite indistinguishable hematocrit values among groups, MAPs were higher and fluid requirements lower in animals treated with CXCL12 and with the CXCL12 variants, as compared with vehicle treated animals (Fig. 3B–D). Fluid requirements after CXCL121 treatment were significantly lower than after treatment with CXCL12 (Fig. 3C). P/F in animals treated with vehicle fulfilled criteria for mild ARDS at t = 120 min and for moderate ARDS from t = 180 min until t = 300 min (Fig. 3E). P/F in animals treated with CXCL12 K27A/R41A/R47A were indistinguishable from vehicle-treated animals. The mean P/F of animals treated with CXCL12 fulfilled the criteria of mild ARDS not until t = 300 min. Animals treated with CXCL121 and CXCL122 did not fulfill criteria for ARDS at any time point (Fig. 3E).
Figure 3.

Administration of engineered CXCL121 and CXCL122 prevent development of ARDS. Animals were treated with vehicle (n = 5) or with 0.14 μmol/kg of CXCL12 (n = 4), CXCL121 (n = 3), CXCL122 (n = 4) or CXCL12 K27A/R41A/R47A (n = 3). Arrows indicate the time point of drug administration. LI + HEM: Lung ischemia and hemorrhage period. Data are mean ± SE. #p < 0.05 for vehicle vs. CXCL12. *p < 0.05 for vehicle vs. CXCL121. %p < 0.05 for vehicle vs. CXCL122. &p < 0.05 for vehicle vs. CXCL12 K27A/R41A/R47A. $p < 0.05 for CXCL12 vs. CXCL121. (A) Hemorrhage volumes (mL/kg) during LI + HEM. (B) MAP: Mean arterial blood pressure (mmHg). (C) RES: Resuscitation fluid requirements (mL/kg). (D) Hct: Hematocrit (%). (E) PaO2/FiO2 (mmHg).
Treatment with CXCR4 agonists reduces mortality
Figure 4 shows the cumulative survival curves for vehicle treated animals, animals treated with inefficacious doses of the CXCR4 agonists and for animals treated with the CXCR4 agonists at doses that prevented ARDS from series 1–3. In vehicle-treated animals and animals treated with CXCR4 agonists at doses that did not attenuate ARDS development mortality was 55% and median survival time was 240 min (p > 0.05). In animals treated with CXCR4 agonists that prevented ARDS development mortality was 9% and median survival time was > 300 min (p = 0.0023 vs. vehicle-treated animals).
Figure 4.
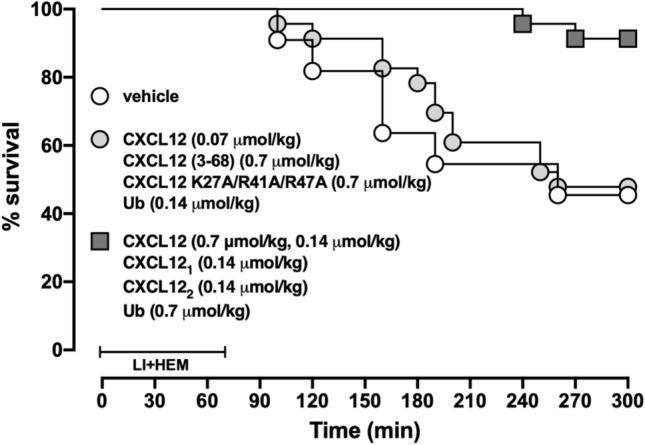
Administration of CXCR4 agonists reduce mortality from ARDS. Cumulative survival curves from animals treated with vehicle (open circles, n = 11), CXCR4 agonists administered at doses that did not prevent ARDS (grey circles, n = 12) and CXCR4 agonists administered at doses that prevented ARDS (grey squares, n = 23, p = 0.0023 vs. vehicle and CXCR4 agonists at doses that did not prevent ARDS).
Treatment with CXCR4 agonists reduces histological lung injury
Next, we assessed whether the effects of the CXCR4 agonists on lung function are accompanied by alterations in lung histomorphology. Representative images from H&E stained lung slices of the uninjured (left) lungs are shown in Fig. 5A. There were no obvious differences in the histomorphology of the uninjured lungs among groups. LIS in the groups in which lungs from at least three animals could be obtained at t = 300 min were not significantly different among groups (p > 0.05 vs. vehicle for all; Fig. 5B). Figure 6A shows representative images from H&E stained lung slices of the injured (right) lungs. In vehicle-treated animals and animals treated with CXCL12 (3–68) and CXCL12K27A/R41A/R47A, lung histology showed alveolar congestion, hemorrhage, interstitial edema, increased alveolar wall thickness and leukocyte infiltration, which were obviously reduced with CXCL12, CXCL121, CXCL122 and ubiquitin treatment (Fig. 6A). Accordingly, LIS were significantly reduced in animals after treatment with CXCR4 agonists at doses that prevented ARDS development, as compared with vehicle-treated animals (Fig. 6B). Furthermore, LIS after treatment with ubiquitin, CXCL121 and CXCL122 were lower than after treatment with wild-type CXCL12 (Fig. 6B).
Figure 5.

Histology of the uninjured lungs after treatment with vehicle and CXCR4 agonists. (A) Representative images from H&E stained lung sections from animals after treatment with vehicle and the various CXCR4 agonists. (B) Lung injury scores (LIS) from treatment groups in which lungs from at least 3 animals could be harvested. Data are median ± interquartile ranges. Vehicle: n = 4. 0.7 μmol/kg CXCL12: n = 3. 0.7 μmol/kg ubiquitin: n = 3. 0.14 μmol/kg CXCL12: n = 5. 0.14 μmol/kg CXCL121: n = 3. 0.14 μmol/kg CXCL122: n = 3. There were no statistically significant differences among groups.
Figure 6.

Administration of CXCR4 agonists reduce histological lung injury in the injured lung. (A). Representative images from H&E stained lung sections from animals after treatment with vehicle and the various CXCR4 agonists. (B) Lung injury scores (LIS) from treatment groups in which lungs from at least 3 animals could be harvested. Data are median ± interquartile ranges. Vehicle: n = 4. 0.7 μmol/kg CXCL12: n = 3. 0.7 μmol/kg ubiquitin: n = 3. 0.14 μmol/kg CXCL12: n = 5. 0.14 μmol/kg CXCL121: n = 3. 0.14 μmol/kg CXCL122: n = 3. *p < 0.05 vs. vehicle. #p < 0.05 vs. 0.7 μmol/kg CXCL12.
Discussion
In the present study, we evaluated the pharmacodynamic properties of various CXCR4 protein agonists in a rat model of lung ischemia–reperfusion injury and hemorrhage induced ARDS. There are several new findings from the present study. First, wild-type CXCL12 and the engineered variants CXCL121 and CXCL122 show higher potency than ubiquitin to prevent development of ARDS. Second, administration of CXCR4 agonists at doses that prevent ARDS development reduce mortality. Third, the engineered variant CXCL121 shows the most favorable pharmacodynamics among the tested CXCR4 agonists.
We showed previously that in rats ventilated with FiO2 of 1.0, the hemorrhage protocol alone does not lead to impairment of lung function during crystalloid resuscitation within the time period studied16. Therefore, the thoracic injury component, i.e. thoracotomy and lung ischemia–reperfusion injury, appears to be the primary cause of ARDS in the present study. Furthermore, several model-specific procedures, such as use of isoflurane as anesthetic, continuous ventilation of the animals with FiO2 of 1.0 or the strict crystalloid resuscitation regimen, may constitute confounding variables that influence cardiovascular and pulmonary function17–19. Thus, it is obvious that our model does not fully resemble all aspects of the etiology and treatment of severely injured patients who develop ARDS. Nevertheless, and as documented by the indistinguishable physiological parameters that we measured in three different sets of control animals, the rat model that we utilized in the present study was able to induce ARDS after lung injury and hemorrhage in a highly reproducible manner.
Besides the natural CXCR4 agonists CXCL12 and ubiquitin, we selected the engineered variants CXCL121, CXCL122 and CXCL12 K27A/R41A/R47A for testing in the present study. These CXCL12 variants were chosen based on their efficacy and potency to protect lung endothelial cell barrier function in vitro, and based on their distinct physicochemical properties, such as resistance to proteolytic cleavage and heparan sulfate proteoglycan binding affinity, which could result in altered functional outcomes after administration of the proteins in vivo9,20,21.
The effects of the non-cognate CXCR4 agonist ubiquitin on ARDS development in the present study are consistent with our previous observations2,5,6,10. Although a synthetic CXCL12 analogue and wild-type CXCL12 have been reported to attenuate lipopolysaccharide and oleate induced lung injury, their effects on lung function, i.e. oxygenation, have not been evaluated in these previous studies7,22. The findings from the present study demonstrate that wild-type CXCL12, CXCL121 and CXCL122 are equally efficacious but more potent than ubiquitin to preserve lung function in our ARDS model. These observations are in agreement with the previously described detrimental effects of the CXCR4 antagonist AMD3100 on lung function after isolated lung ischemia–reperfusion injury, and with the lower affinity of ubiquitin for CXCR4, when compared with the affinity of CXCL125,23. The observation that CXCL12 (3–68) did not affect ARDS development is consistent with the loss of function of N-terminal truncated CXCL12 in other assay systems9,24,25 and documents specificity of the effects of CXCL12, CXCL121 and CXCL122.
Although we observed previously that the CXCR4 agonist activity of CXCL12 K27A/R41A/R47A is approximately 30-fold lower than of wild-type CXCL12, this triple mutant protected in vitro lung endothelial barrier function with the same potency as CXCL12 and CXCL1219. While the absence of therapeutic efficacy of CXCL12 K27A/R41A/R47A in the present study can be explained by its reduced potency to activate CXCR4, these findings also suggest that the reduced heparan sulfate binding affinity does not compensate for the reduced potency to activate CXCR4 in vivo.
In contrast to CXCL12 and the engineered CXCL12 variants, ubiquitin binding to CXCR4 does not induce β-arrestin recruitment to recombinant CXCR4 and ubiquitin does not bind to atypical chemokine receptor 3 (ACKR3)9,23,26–28. Because CXCL12 K27A/R41A/R47A activates ACKR3 with high potency similar to wild-type CXCL129, our findings imply that activation of ACKR3 does not contribute to the lung protective properties of CXCL12 and the CXCL12 variants. Therefore, our observations support the concept that natural and engineered CXCR4 agonists could be used as lung protective therapeutics for ARDS. Based on the significant effects of the CXCR4 agonists on survival in the present study, such a treatment may have the potential to reduce mortality from ARDS. The majority of animals who died, however, had P/F ratios above 100 mmHg and hematocrit values were indistinguishable among groups. While these observations make hypoxemia as well as fluid overload with subsequent right heart failure as causes of mortality unlikely, circulatory failure during fluid resuscitation could explain mortality in our model. These findings resemble the clinical observations that refractory hypoxemia is a rare cause of death in ARDS patients and that mortality is predominantly caused by multiple organ failure, with cardiovascular and acute renal failure being the most common organ failures29.
It is known that CXCL12 exists at a physiological pH in a monomer–dimer equilibrium30. While monomeric CXCL12 exists at low concentrations, dimerization occurs at high concentrations and is influenced by pH, phosphate, sulfate or heparin20,31. Although wild type CXCL12, CXCL121 and CXCL122 showed comparable CXCR4 and ACKR3 activities in Presto-Tango β-arrestin recruitment assays, distinct pharmacokinetic and pharmacodynamic properties have been observed in various other signaling and cell function assays in vitro, in an isolated rat heart ischemia–reperfusion injury model and in a melanoma lung metastasis model21,32,33. The direct side-by-side comparison of wild-type CXCL12, CXCL121 and CXCL122 in the present study suggests that CXCL121 is the protein therapeutic with the most favorable pharmacodynamic properties when effects on lung function, lung histomorphology and hemodynamics are considered comprehensively.
While the present study was not designed to address the molecular mechanisms by which CXCR4 activation prevents ARDS development, several lines of evidence suggest that CXCR4 activation protects endothelial barrier function, and regulates vascular smooth muscle function and blood pressure through heteromerization with α1-adrenergic receptors9,11,34–37. Thus, direct protective effects of CXCR4 agonists on lung endothelial barrier function as well as improvement of cardiovascular function with subsequently reduced fluid resuscitation requirements upon CXCR4 activation likely contributed to the beneficial effects that we observed in the present study. CXCR4 and ACKR3, however, are known to hetero-oligomerize with each other and with several other G protein coupled receptors, which likely affects their signaling properties depending on the available repertoire of interacting receptor partners34,35,38–40. Furthermore, the natural CXCR4 agonists CXCL12 and ubiquitin are proteolytically processed in the systemic circulation, leading to their inactivation or the generation of CXCR4 antagonists, which will result in altered ligand binding kinetics at the receptor and modulate subsequent signaling events24,41,42. These complex mechanisms likely correspond to the observations that signaling and functions of CXCR4 and its agonists depend on the cellular context and the specific pathophysiological environment34,38. Thus, context-dependent consequences of CXCR4 activation with CXCL12 may explain that CXCL12 administration desensitized phenylephrine-mediated vasoconstriction of isolated arteries and reduced blood pressure in a model of isolated hemorrhagic shock in spontaneously breathing rats, whereas CXCL12 sensitized the phenylephrine induced blood pressure response in normal rats and stabilized hemodynamics in the present ARDS model34,37.
In conclusion, our findings suggest that the class of CXCR4 protein agonists prevent development of ARDS and reduce mortality in rats after lung ischemia–reperfusion injury and hemorrhage when administered during the early resuscitation period. The observation that engineered CXCL121 outperformed ubiquitin and wild-type CXCL12 in its overall pharmacodynamic properties implies that the development of new engineered protein therapeutics with improved pharmacological properties for ARDS is possible. While the context-dependency of CXCL12 mediated effects restricts translational potential, engineered protein therapeutics may overcome this limitation. Although our study demonstrates that administration of CXCR4 agonists prevents ARDS development when administered within a few minutes upon initiation of crystalloid resuscitation, the therapeutic window for such an intervention as well as the possible therapeutic potential to improve lung function and to reduce lung injury in animals that already fulfill criteria for ARDS remain to be determined in the future. The findings from the present study provide justification for such translational pre-clinical studies.
Methods
Proteins
Ubiquitin, CXCL12, the engineered constitutively monomeric (CXCL121) and dimeric (CXCL122) CXCL12 variants, N-terminal truncated CXCL12 (3–68), a posttranslational modification that occurs in vivo after cleavage of CXCL12 by CD26/dipeptidyl peptidase 4 and inactivates the chemokine, and the triple mutant CXCL12 K27A/R41A/R47A, which shows significantly reduced heparan sulfate proteoglycan binding properties, were as described9.
ARDS model
All procedures were performed according to National Institutes of Health Guidelines for Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee and the Animal Care and Use Review Office of the U.S. Army Medical Research and Materiel Command. Male Sprague–Dawley rats (300–350 g) were purchased from Harlan (Indianapolis, IN, USA). Anesthetized (isoflurane inhalation) animals were oro-tracheally intubated with a 16-gauge EXEL disposable safelet angiocatheter (EXELINT International, Los Angeles, CA, USA) and mechanically ventilated with a SomnoSuite small animal anesthesia system (Kent Scientific Corporation, Torrington, CT, USA). Animals were ventilated with a pressure-controlled ventilator mode with an initial positive end expiratory pressure (PEEP) of 2 mmHg, a fraction of inspired oxygen (FiO2) of 1.0 and anesthetized with isoflurane at 2.5%. Tidal volumes were set based on the weight of the animals then titrated to maintain normal PaCO2 (35–45 mmHg). The femoral artery was then cannulated with 24-gauge BD angiocath shielded IV catheters (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) to allow for monitoring of arterial blood pressure and blood withdrawal, and the femoral vein was cannulated with 1.5-french tubing for fluid and drug administration. Animals underwent a right lateral thoracotomy and a suture was placed around the hilum of the right lung. The suture was then tied around the hilum of the right lung, occluding the pulmonary artery, vein, and right main stem bronchus (t = 0 min). After 40 min of lung ischemia animals were hemorrhaged to a mean arterial blood pressure (MAP) of 40 mmHg until t = 70 min. At t = 70 min the suture was removed, animals were ventilated with FiO2 1.0, PEEP 5 mmHg and resuscitated to a MAP of 60 mmHg with normal saline (NS). After initial bolus injections of NS to achieve the MAP target, NS infusion was limited to maximal infusion rates (2 mL/min until t = 75 min; 1 mL/min from t = 75–300 min) irrespective of the MAP target to prevent acute fluid overload. A sigh breath was administered every 15 breaths for the first 5 min following resuscitation and then every 90 breaths until t = 300 min to fully expand the post-ischemic lung. Hemodynamics were continuously monitored with the surgivet invasive blood pressure monitor (Med-Electronics, Beltsville, MD, USA) and blood pressures values were recorded every 5 min throughout the experiment. Arterial blood gases and routine laboratory parameters were determined in regular intervals throughout the experiment. At t = 300 min, animals were euthanized (5% isoflurane, bilateral pneumothorax, arterial exsanguination). Both lungs were then harvested for the analysis of lung histopathology. All experiments were performed randomized and blinded. All drugs were administered in 0.5 mL NS (= vehicle) within 5 min of fluid resuscitation. In series 1, animals received vehicle (n = 6), 0.7 μmol/kg ubiquitin (n = 3) or 0.14 μmol/kg ubiquitin (n = 5). In series 2, animals were treated with 0.7 (n = 3), 0.14 (n = 5) or 0.07 (n = 3) μmol/kg CXCL12 or with CXCL12 (3–68) (0.7 μmol/kg, n = 3). In series 3 animals were treated with vehicle (n = 5) or with 0.14 μmol/kg of CXCL12 (n = 4) or the engineered variants CXCL121 (n = 3), CXCL122 (n = 4) and CXCL12 K27A/R41A/R47A (n = 3). The dosing of the CXCR4 agonists was selected based on our previous observations in other animal models5,10,43.
Arterial blood gases and routine laboratory parameters
Arterial blood gases, electrolytes, creatinine, lactate, hematocrit and hemoglobin were analyzed using the Element point of care veterinary blood gas, electrolyte and critical care analyzer (Cuattro Veterinary USA, Loveland, CO, USA). Complete blood counts were analyzed using the Hematrue hematology analyzer (Cuattro Veterinary USA, Loveland, CO, USA).
Histopathology
For histomorphological examination, lung specimens were placed in formalin fixative, embedded in paraffin wax, sliced into 5 µm sections and stained with hematoxylin and eosin (H&E). From each lung specimen, 3 slides were prepared. The slides were examined under a light microscope by 4 investigators who were blinded as to the identity of the specimens. Histopathology was assessed using a previously described lung injury score (LIS)5,44. In brief, each investigator rendered a score of 0 (no damage) to 4 (maximal damage) based on the following criteria: (1) alveolar congestion, (2) presence of hemorrhage, (3) interstitial edema, (4) alveolar wall thickness, (5) infiltration of polymorphonuclear neutrophils (PMN) in the vessel wall and (6) infiltration of PMN in the alveoli. For each criterion the median of the scores from each investigator was used as the final score for each criterion and the sum of the scores for all six criteria calculated for each animal.
Data analyses and statistics
Data are presented as mean ± standard error (SE) or median with interquartile range (25th/75th percentile). Data were analyzed by 1-way ANOVA or 2-way ANOVA with Dunnett's multiple comparisons test, as appropriate. Survival curves were analyzed using the log-rank test. Data analyses were calculated with the GraphPad Prism program (GraphPad Software). A two-tailed p < 0.05 was considered significant.
Conference presentation
Parts of this research have been presented at the 42nd Conference on Shock, Coronado, CA, 2019 (Shock, 51(S1):36, 2019).
Acknowledgements
This work was supported by the National Institute of General Medical Sciences (Awards R01GM107495 and T32GM008750) and by the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program under Award No. W81XWH-15-1-0262. The content is solely the responsibility of the authors.
Author contributions
F.S.B., X.L., G.A.E., A.J.D., X.G. performed experiments. F.S.B., X.L., G.A.E., A.J.D., X.G. and M.M. analyzed data. B.F.V. provided reagents. M.M. wrote the manuscript. All authors reviewed and commented on the manuscript and approved the final version.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
Competing interests
BFV and MM are inventors on a patent application for the use of CXCL121 filed by Medical College of Wisconsin. BFV is an inventor on US Patents No. 9,346,871, 8,524,670, 7,923,016 and 9,908,923 relating to the composition and methods of use of CXCL121 and CXCL122. BFV and MM have not received income related to the patents or patent application. The remaining authors have disclosed that they do not have any conflicts of interest.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Tignanelli CJ, Hemmila MR, Rogers MAM, Raghavendran K. Nationwide cohort study of independent risk factors for acute respiratory distress syndrome after trauma. Trauma Surg. Acute Care Open. 2019;4:e000249. doi: 10.1136/tsaco-2018-000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Majetschak M, et al. Effects of exogenous ubiquitin in lethal endotoxemia. Surgery. 2004;135:536–543. doi: 10.1016/j.surg.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Earle SA, Proctor KG, Patel MB, Majetschak M. Ubiquitin reduces fluid shifts after traumatic brain injury. Surgery. 2005;138:431–438. doi: 10.1016/j.surg.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Covarrubias L, Manning EW, 3rd, Sorell LT, Pham SM, Majetschak M. Ubiquitin enhances the Th2 cytokine response and attenuates ischemia-reperfusion injury in the lung. Crit. Care Med. 2008;36:979–982. doi: 10.1097/CCM.0B013E318164E417. [DOI] [PubMed] [Google Scholar]
- 5.Nassoiy SP, Babu FS, LaPorte HM, Majetschak M. Pharmacological modulation of C-X-C motif chemokine receptor 4 influences development of acute respiratory distress syndrome after lung ischaemia-reperfusion injury. Clin. Exp. Pharmacol. Physiol. 2018;45:16–26. doi: 10.1111/1440-1681.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baker TA, et al. Effects of exogenous ubiquitin in a polytrauma model with blunt chest trauma. Crit. Care Med. 2012;40:2376–2384. doi: 10.1097/CCM.0b013e3182514ed9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo C, et al. A stromal cell-derived factor 1alpha analogue improves endothelial cell function in lipopolysaccharide-induced acute respiratory distress syndrome. Mol. Med. 2016;22:115–123. doi: 10.2119/molmed.2015.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo W, et al. Stromal cell-derived factor-1alpha attenuates oleate-induced acute lung injury in rabbits. Biochem. Biophys. Res. Commun. 2014;452:191–196. doi: 10.1016/j.bbrc.2014.07.033. [DOI] [PubMed] [Google Scholar]
- 9.Cheng YH, Eby JM, LaPorte HM, Volkman BF, Majetschak M. Effects of cognate, non-cognate and synthetic CXCR4 and ACKR3 ligands on human lung endothelial cell barrier function. PLoS ONE. 2017;12:e0187949. doi: 10.1371/journal.pone.0187949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Babu FS, LaPorte HM, Nassoiy SP, Majetschak M. Chemokine (C-X-C motif) receptor 4 regulates lung endothelial barrier permeability during resuscitation from hemorrhagic shock. Physiol. Res. 2019;68:675–679. doi: 10.33549/physiolres.934105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi K, et al. Stromal cell-derived factor-1alpha/C-X-C chemokine receptor type 4 axis promotes endothelial cell barrier integrity via phosphoinositide 3-kinase and Rac1 activation. Arterioscler. Thromb. Vasc. Biol. 2014;34:1716–1722. doi: 10.1161/ATVBAHA.114.303890. [DOI] [PubMed] [Google Scholar]
- 12.Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin. Respir. Crit. Care Med. 2006;27:337–349. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- 13.Miller PR, Croce MA, Kilgo PD, Scott J, Fabian TC. Acute respiratory distress syndrome in blunt trauma: identification of independent risk factors. Am. Surg. 2002;68:845–850. [PubMed] [Google Scholar]
- 14.Pierrakos C, Karanikolas M, Scolletta S, Karamouzos V, Velissaris D. Acute respiratory distress syndrome: pathophysiology and therapeutic options. J. Clin. Med. Res. 2012;4:7–16. doi: 10.4021/jocmr761w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Force ADT, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 16.Nassoiy SP, Babu FS, LaPorte HM, Byron KL, Majetschak M. Effects of the Kv7 voltage-activated potassium channel inhibitor linopirdine in rat models of haemorrhagic shock. Clin. Exp. Pharmacol. Physiol. 2018 doi: 10.1111/1440-1681.12958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Gara B, Talmor D. Lung protective properties of the volatile anesthetics. Intensive Care Med. 2016;42:1487–1489. doi: 10.1007/s00134-016-4429-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brugniaux JV, et al. Highs and lows of hyperoxia: physiological, performance, and clinical aspects. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018;315:R1–R27. doi: 10.1152/ajpregu.00165.2017. [DOI] [PubMed] [Google Scholar]
- 19.Kornblith LZ, et al. Predictors of postinjury acute respiratory distress syndrome: lung injury persists in the era of hemostatic resuscitation. J. Trauma Acute Care Surg. 2019;87:371–378. doi: 10.1097/TA.0000000000002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ziarek JJ, et al. Heparin oligosaccharides inhibit chemokine (CXC motif) ligand 12 (CXCL12) cardioprotection by binding orthogonal to the dimerization interface, promoting oligomerization, and competing with the chemokine (CXC motif) receptor 4 (CXCR4) N terminus. J. Biol. Chem. 2013;288:737–746. doi: 10.1074/jbc.M112.394064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takekoshi T, Ziarek JJ, Volkman BF, Hwang ST. A locked, dimeric CXCL12 variant effectively inhibits pulmonary metastasis of CXCR4-expressing melanoma cells due to enhanced serum stability. Mol. Cancer. Ther. 2012;11:2516–2525. doi: 10.1158/1535-7163.MCT-12-0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guan S, et al. Combined treatment with a CXCL12 analogue and antibiotics improves survival and neutrophil recruitment and function in murine sepsis. Immunology. 2014 doi: 10.1111/imm.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saini V, Marchese A, Majetschak M. CXC chemokine receptor 4 is a cell surface receptor for extracellular ubiquitin. J. Biol. Chem. 2010;285:15566–15576. doi: 10.1074/jbc.M110.103408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janssens R, et al. Truncation of CXCL12 by CD26 reduces its CXC chemokine receptor 4- and atypical chemokine receptor 3-dependent activity on endothelial cells and lymphocytes. Biochem. Pharmacol. 2017;132:92–101. doi: 10.1016/j.bcp.2017.03.009. [DOI] [PubMed] [Google Scholar]
- 25.Proost P, et al. Processing by CD26/dipeptidyl-peptidase IV reduces the chemotactic and anti-HIV-1 activity of stromal-cell-derived factor-1alpha. FEBS Lett. 1998;432:73–76. doi: 10.1016/s0014-5793(98)00830-8. [DOI] [PubMed] [Google Scholar]
- 26.Saini V, et al. The CXC chemokine receptor 4 ligands ubiquitin and stromal cell-derived factor-1alpha function through distinct receptor interactions. J. Biol. Chem. 2011;286:33466–33477. doi: 10.1074/jbc.M111.233742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eby JM, et al. Functional and structural consequences of chemokine (C-X-C motif) receptor 4 activation with cognate and non-cognate agonists. Mol. Cell. Biochem. 2017;434:143–151. doi: 10.1007/s11010-017-3044-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saini V, Marchese A, Tang WJ, Majetschak M. Structural determinants of ubiquitin-CXC chemokine receptor 4 interaction. J. Biol. Chem. 2011;286:44145–44152. doi: 10.1074/jbc.M111.298505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pierrakos C, Vincent JL. The changing pattern of acute respiratory distress syndrome over time: a comparison of two periods. Eur. Respir. J. 2012;40:589–595. doi: 10.1183/09031936.00130511. [DOI] [PubMed] [Google Scholar]
- 30.Holmes WD, Consler TG, Dallas WS, Rocque WJ, Willard DH. Solution studies of recombinant human stromal-cell-derived factor-1. Protein Exp. Purif. 2001;21:367–377. doi: 10.1006/prep.2001.1402. [DOI] [PubMed] [Google Scholar]
- 31.Veldkamp CT, Peterson FC, Pelzek AJ, Volkman BF. The monomer-dimer equilibrium of stromal cell-derived factor-1 (CXCL 12) is altered by pH, phosphate, sulfate, and heparin. Protein. Sci. 2005;14:1071–1081. doi: 10.1110/ps.041219505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drury LJ, et al. Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc. Natl. Acad. Sci. USA. 2011;108:17655–17660. doi: 10.1073/pnas.1101133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veldkamp CT, et al. Monomeric structure of the cardioprotective chemokine SDF-1/CXCL12. Protein Sci. 2009;18:1359–1369. doi: 10.1002/pro.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tripathi A, et al. Heteromerization of chemokine (C-X-C motif) receptor 4 with alpha1A/B-adrenergic receptors controls alpha1-adrenergic receptor function. Proc. Natl. Acad. Sci. USA. 2015;112:E1659–1668. doi: 10.1073/pnas.1417564112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Albee LJ, et al. Alpha1-adrenergic receptors function within hetero-oligomeric complexes with atypical chemokine receptor 3 and chemokine (C-X-C motif) receptor 4 in vascular smooth muscle cells. J. Am. Heart Assoc. 2017 doi: 10.1161/JAHA.117.006575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans AE, et al. New insights into mechanisms and functions of chemokine (C-X-C Motif) receptor 4 heteromerization in vascular smooth muscle. Int. J. Mol. Sci. 2016 doi: 10.3390/ijms17060971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bach HH, et al. Chemokine (C-X-C motif) receptor 4 and atypical chemokine receptor 3 regulate vascular alpha(1)-adrenergic receptor function. Mol. Med. 2014;20:435–447. doi: 10.2119/molmed.2014.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heuninck J, et al. Context-dependent signaling of CXC chemokine receptor 4 and atypical chemokine receptor 3. Mol. Pharmacol. 2019;96:778–793. doi: 10.1124/mol.118.115477. [DOI] [PubMed] [Google Scholar]
- 39.Gomes I, et al. G protein-coupled receptor heteromers. Annu. Rev. Pharmacol. Toxicol. 2016;56:403–425. doi: 10.1146/annurev-pharmtox-011613-135952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Albee LJ, et al. Identification and functional characterization of arginine vasopressin receptor 1A: atypical chemokine receptor 3 heteromers in vascular smooth muscle. Open Biol. 2018 doi: 10.1098/rsob.170207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tripathi A, et al. Modulation of the CXC chemokine receptor 4 agonist activity of ubiquitin through C-terminal protein modification. Biochemistry. 2013;52:4184–4192. doi: 10.1021/bi400254f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jackson EK, Mi E, Ritov VB, Gillespie DG. Extracellular ubiquitin(1–76) and ubiquitin(1–74) regulate cardiac fibroblast proliferation. Hypertension. 2018;72:909–917. doi: 10.1161/HYPERTENSIONAHA.118.11666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bach HH, Wong YM, LaPorte HM, Gamelli RL, Majetschak M. Pharmacological targeting of chemokine (C-X-C motif) receptor 4 in porcine polytrauma and hemorrhage models. J. Trauma Acute Care Surg. 2016;80:102–110. doi: 10.1097/TA.0000000000000865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manning EW, 3rd, et al. Proteasome peptidase activities parallel histomorphological and functional consequences of ischemia-reperfusion injury in the lung. Exp. Lung. Res. 2009;35:284–295. doi: 10.1080/01902140802668823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during the current study are available from the corresponding author on reasonable request.