

Abstract
This work describes the design and implementation of an automated device for catalytic materials testing by direct modifications to a gas chromatograph (GC). The setup can be operated as a plug-flow isothermal reactor and enables the control of relevant parameters such as reaction temperature and reactant partial pressures directly from the GC. High-quality kinetic data (including reaction rates, product distributions, and activation barriers) can be obtained at almost one-tenth of the fabrication cost of analogous commercial setups. With these key benefits including automation, low cost, and limited experimental equipment instrumentation, this implementation is intended as a high-throughput catalyst screening reactor that can be readily utilized by materials synthesis researchers to assess the catalytic properties of their synthesized structures in vapor-phase chemistries.
Keywords: micro-flow reactor, automated kinetic measurements, reactive gas chromatography, alcohol dehydration, automated analysis, high-throughput experimentation, packed bed reactors
Graphical Abstract

Highlights
-
•
Fabrication to convert a gas chromatograph to an automated micro-flow reactor
-
•
Setup operation possible under plug-flow hydrodynamics and isothermal conditions
-
•
Quality and reliability of kinetic data investigated and found satisfactory
Progress and Potential
Automation to reduce research labor is an important area in reaction engineering, and reactors capable of operating without manual intervention are sought for rapid catalyst testing. The emergence of the COVID-19 pandemic provides further impetus to a transition away from labor-intensive material testing techniques to new automated approaches without compromising on data quality, and at costs viable for academic laboratories. Here, we convert common analytical equipment employed in catalysis laboratories, namely a gas chromatograph (GC), into a low-cost packed bed flow reactor that can be operated isothermally. The quality of catalytic data is validated by comparisons with prior reports on more traditional reactors. Ultimately, this user-friendly implementation only requires limited additional instrumentation and cost to GC operation and puts a standardized high-throughput reactor into the hands of materials synthesis researchers for screening their synthesized catalysts.
Through simple hardware and software modifications, it is possible to convert a typical gas chromatograph into an automated packed bed reactor capable of accurate kinetic measurements in vapor-phase catalytic chemistries. Such automation can potentially aid the collection and processing of a large amount of high-quality catalytic data in a manner that does not require constant human presence, allowing laboratories to run efficiently under strict work-place restrictions due to the COVID-19 pandemic.
Introduction
The rapid onset of coronavirus disease 2019 (COVID-19), a dangerous infection spreadable by people in close contact, has transformed the nature of materials and chemical research.1 , 2 While the past two decades have produced a wave of new catalyst materials including hierarchical zeolites,3 metal-organic frameworks,4 single-atom surfaces,5 intermetallic structures,6 and other low-dimensional materials,7 testing these catalysts to determine structure-performance relationships requires researchers to work in close proximity. Such in-person catalytic performance evaluation tends to be a multi-step process whereby the traditional approaches have involved qualitative screening techniques8, 9, 10, 11, 12 in search for the “best” catalyst for the envisioned application, followed by kinetic interrogations of the shortlisted catalysts to establish reactivity, selectivity, and stability trends and subsequent detailed mechanistic inquiries into reaction pathways.9 , 12 To continue this effective general approach to assessing materials performance, laboratory operations must transition to more automated methods requiring only minimal manual intervention.
The challenge of transitioning laboratories to more robotic operations arises from the complexity of catalytic materials testing. In particular, kinetic studies frequently utilize differential reactors to measure either initial or steady-state rates of reaction over a wide range of operating conditions. For reactions occurring on gas-solid interfaces, such measurements are typically performed on packed bed flow reactors (PBRs),12, 13, 14, 15, 16, 17 which remain the workhorse of any heterogeneous catalysis laboratory. Importantly, laboratory-scale cost-effective PBRs are often custom-fabricated and require constant human monitoring owing to multiple independent process control elements, and separate, often non-coupled, reaction and separation/quantification components.9 , 12 The manual interventions on this equipment typically involves operations such as switching valves and/or precisely altering reaction conditions at determined intervals. Furthermore, data analysis from common analytical instruments on these PBRs (such as GC units) remains largely manual. Together, these factors lead to cumbersome experimentation and analysis, especially in scenarios requiring kinetic measurements for many catalysts or assessing catalyst stability on-stream for prolonged periods.
The ability to perform high-fidelity kinetic studies with minimal human oversight would greatly reduce the labor and costs associated with catalyst development and discovery, and recent research efforts reflect this interest.18 Flow reactors19, 20, 21 as well as control algorithms22, 23, 24, 25 are increasingly integrated with online analytical tools to collect and analyze kinetic data without user supervision. However, these experimental systems are almost always fabricated for a specific targeted application, and important heat and mass transfer considerations are often not explicitly reported, dissuading other research groups to invest time and resources to fabricate them. One straightforward implementation of low-cost automated micro-reactors with broad applicability among previously reported approaches is the pulsed-flow technique, which involves dosing a pulse of reactant over a catalytic material to study product distribution trends. This approach is readily integrated within a commercial GC,26, 27, 28, 29, 30, 31, 32 but the transient nature of the technique limits kinetic parameter estimation. In addition, the product distributions under such transient conditions may vary from corresponding steady-state values.16 Extending this approach to modify existing GCs, enabling reliable but largely automated kinetics measurements in a continuous flow method while also facilitating safe laboratory operation, has been the focus of this report.
Here, the automated vapor-phase continuous micro-flow reactor is described as integrated within a GC unit capable of measuring the reaction kinetics of vapor-phase catalytic chemistries with volatile feeds (either gases or vaporizable liquids). The design and implementation of the setup is presented in the Methods section. This is followed by detailed investigation of heat and mass transfer characteristics under cold-flow as well as reaction conditions by a recently developed online toolbox (GradientCheck) reported by the Ribeiro group.33 Thereafter, kinetic parameters (namely turnover rates, apparent activation barriers, and product distributions) for acid-catalyzed vapor-phase dehydration of three alcohols (ethanol, 2-propanol, and 1-butanol) and a cyclic ether (2-methyltetrahydrofuran [2-MTHF]) on a solid acid catalyst (HZSM-5, Si/Al 140) are measured and compared with previously reported values under similar conditions. To address the labor-intensive nature of chromatogram data handling, we developed a chromatogram analysis tool in-house and integrated it into the experimentation workflow for automatic GC peak detection and peak area quantification.
The fabricated setup and the reported data analysis tool are primarily intended for materials researchers to obtain kinetic data of vapor-phase chemistries with minimal user supervision and safe working conditions. The provided information along with the detailed analysis of the performance of the device enables simple implementation in any laboratory that is testing catalytic materials. Extensive supporting information provides detailed parts lists and instructions for implementation with both hardware and software modifications (Section S.1 of Supplemental Experimental Procedures and Figures S1–S4). Additionally, the provided analysis means that immediate transition of a laboratory to safer, more robotic catalyst materials testing can be conducted with the justification that resulting data achieve the quality obtained using conventional methods.
Results and Discussion
Reactor Temperature Distribution: Unreactive Conditions
Laboratory-scale fixed-bed reactors conventionally involve heat transfer across a temperature-controlled wall, and gradients between the bulk phase in the reactor and the wall might lead to non-isothermal conditions inside the reactor. Similarly, a GC inlet is heated by a cartridge heater, and the controlled temperature is that of the inner inlet wall. To gauge the differences between the reactor set temperature and the actual temperature of the catalyst bed, we conducted independent measurements at different temperature set points as a function of axial positions inside the liner, the results of which are shown in Figure 1 . Notably, the axial region in the range 0.15 < h/H < 0.35 (h is the height for packing the bed; H is total height of the liner) showed <1% difference between measured and set temperature under non-reacting conditions. Furthermore, the deviation in temperature from the set point outside this region was significantly higher (>10%) at higher temperatures (>473 K). These results highlight that the axial location of the catalyst bed is sensitively linked to the temperature control attainable on this setup.
Figure 1.

The Deviation in Set and Actual Temperature as a Function of the Non-dimensionalized Height of the Splitless Inlet Liner Used to Hold the Catalyst Bed
It is important to note that these measurements only probe the temperature distribution of the reactor under non-reacting conditions. Temperature gradients across the bed as well as within catalyst particles may also develop under reacting conditions, depending on reaction thermodynamics. As expected, highly endo-/exothermic reactions are more likely to result in axial temperature gradients.34 We have evaluated the temperature distribution of the catalyst bed under the reaction conditions for the probe reactions, and the results are discussed in Reaction Kinetics Measurements.
Residence Time Distributions
Axial mass dispersion in PBRs can limit the accurate determination of kinetic parameters. This manifests in the form of channeling and dead volumes, leading to back-mixing and non-plug-flow hydrodynamics.35 The extent of dispersion is described by, and inversely proportional to, the Peclet number (Equation 6), and packed beds can only be assumed to be free from axial dispersion effects for Pe > 100.35 The extent of axial dispersion is inversely proportional to the bed length, and residence time distributions (RTD) experiments were therefore performed on the reactor packed with quartz wool alone mimicking the limit .36
Figure 2 depicts the experimental RTDs obtained at different gas flow rates. The obtained experimental data were fit to the RTD distribution of an axially dispersed plug-flow reactor model with open boundary conditions (Equation 2). Using the average residence times (Equation 3), experimental variances (Equation 4) were then compared with the corresponding values derived from the model (Equation 5) to calculate Peclet numbers (Equation 6).
| (Equation 2) |
| (Equation 3) |
| (Equation 4) |
| (Equation 5) |
| (Equation 6) |
Figure 2.

The Residence Time Distribution Obtained from the Inlet Liner Packed with Deactivated Quartz Wool at Four Different Carrier Gas Flow Rates
The experiments were performed with ethanol pulses and the inlet was maintained at 523 K. Different symbols (circles, triangles, and squares) represent replicate runs under identical conditions. Corresponding Peclet numbers given by Equation 6 are also indicated. Errors indicate the 95% confidence interval (CI) on the Peclet numbers resulting from independent experimental runs.
Gas flow rates ≤25 sccm led to Peclet numbers in the range ~50–90, indicating non-negligible degree of back-mixing under these flow rates. However, the RTD curve at a slightly higher flow rate (40 sccm) exhibited significantly lower variance, leading to Pe ~ 280. It is therefore possible to operate the setup with plug-flow hydrodynamics at moderately low flow rates (>40 sccm).
Reaction Kinetics Measurements
Any new reactor needs to be characterized and benchmarked against traditional designs; this can be accomplished by measuring and comparing reaction kinetics for relatively simple and well understood probe chemistries. To this extent, we utilized C2-C4 alcohol dehydration, and the dehydra-decyclization of a five-membered saturated ether, 2-MTHF. Alcohol dehydration on Brønsted acid sites (BAS) proceeds by two mechanisms: a unimolecular pathway to the corresponding olefin and a bimolecular pathway to a di-alkyl ether (Scheme 1 ).37, 38, 39, 40 Alternatively, 2-MTHF can either undergo dehydra-decyclization to linear pentadienes, namely 1,3-pentadiene and 1,4-pentadiene, or fragment to butenes and formaldehyde by a competing retro-Prins condensation pathway (Scheme 1). Other typical side products are propene and large aromatics (C6+ fraction).41
Scheme 1.

Known Reaction Pathways for the Brønsted Acid Catalyzed Dehydration of Ethanol, 2-Propanol, 1-Butanol, and Dehydra-Decyclization of 2-Methyltetrahydrofuran
In the case of alcohol dehydration, the kinetic preference to unimolecular/bimolecular dehydration pathways was found to be temperature dependent. Increasing temperature consistently led to unimolecular dehydration being favored over the bimolecular pathway for all three alcohols. Taking ethanol as a representative case, this observation is highlighted in Figure 3 A. While di-ethyl ether was the only observed dehydration product at low temperatures (≤413 K), ethylene selectivities exhibited a monotonic rise with increase in temperature for the range investigated (388–483 K). Corresponding product distributions from previous reports are also shown for the same chemistry in Figure 3A, and direct comparisons reveal good agreement between the product distributions obtained on the micro-flow reactor setup and previously reported values. It has been widely reported that the 10-membered ring channels in the MFI framework (for ZSM-5) enthalpically stabilize the bulkier bimolecular pathway transition state for light alcohol (C1-C4) dehydration at low temperatures due to tighter pore confinement.37 , 38 , 40 , 42 Increasing temperature increases the contribution of entropy, thereby favoring looser transition state fits and leading to higher unimolecular product selectivities at higher temperatures. Consistent with this discussion, the same behavior was indeed observed for the other two alcohols (2-propanol and 1-butanol) employed in this study, and the results can be found in Table S3 and Section S.4 of Supplemental Experimental Procedures.
Figure 3.

Product Selectivity Comparisons between Traditional PBRs and Reported Micro-flow Reactor Setup
(A) The product distribution obtained from ethanol dehydration at different reaction temperatures compared with previously reported values on HZSM-5. The conversions are indicated on the chart in red; the values for Phillips and Datta43 were not explicitly reported but differential. WHSV for the data from the micro-flow reactor is 7.35 g EtOH/g cat./h. Error bars in data represent a 95% CI on multiple injections from the same experimental run to measure steady-state product selectivities.
(B) The product distribution obtained from 2-MTHF dehydra-decyclization at different reaction temperatures compared with measurements carried out on HZSM-5 (Zeolyst CBV8014, Si/Al 40) under similar experimental conditions on a traditional packed bed reactor (PBR) as described by Li et al.44 The conversions are indicated on the chart in red. WHSV for the data from the micro-flow reactor is 5.5 g 2-MTHF/g cat./h, while WHSV for the data from the PBR is in the range 3.2–7.5 g 2-MTHF/g cat./h. Carbon balances for both sets of data are within ±7%.
For 2-MTHF dehydra-decyclization, linear pentadienes were found to be the dominant product with selectivities of ~70%–75% under low conversions (2.9%–8.0%) in the temperature range investigated (483–523 K) (Figure 3B). The obtained product distributions were again found to be in reasonable agreement with the values obtained from the traditional PBR setup as well as previously reported values under similar reaction conditions.45
The Arrhenius dependence of proton-normalized rates of the most dominant reaction products for all the test reactions are plotted in Figure 4 . The corresponding values of site-time yields (STYs) previously reported under similar experimental conditions are also plotted to directly compare them against the values obtained from the micro-flow reactor setup. The STYs for all the probe reactions obtained from our setup are typically within a factor of ~2× the corresponding values reported in literature. The calculation of STYs inherently has considerable errors associated with it, in part due to the different measurement methods for estimating the BAS count. Furthermore, the absolute value of STYs depends on the experimental variables such as the space velocities and consequently the conversions at which the product formation rates are calculated, and hence it is safe to conclude that the micro-flow reactor reported here is capable of accurate STY measurements provided the conversions are near-differential (<15%). The apparent activation barriers extracted from the Arrhenius plots measured in this work are listed in Table 1 along with previously reported values. Considering the apparent nature of our measurements (which are likely a convolution of intrinsic kinetics and thermodynamics), the agreement with previously reported values is also reasonable, especially given that some of these referenced values are intrinsic zero-order rate constants, and are expected to be higher than apparent (nearly first-order) rate constants due to adsorption enthalpy contributions built into the apparent values.
Figure 4.

Rates and Activation Barrier Comparisons between Traditional PBRs and Reported Micro-flow Reactor Setup
Site-time yields of major products plotted as a function of inverse temperature for (left to right) (A) ethanol, (B) 2-propanol, (C) 1-butanol, and (D) 2-MTHF (reaction conditions: preactant ~ 25 torr, He flow rate = 60 sccm, WHSVs in the range 5.0–7.6 g reactant/g cat./h; all conversions kept below 15%). Error bars in (A) to (C) represent the 95% CI on multiple injections from the same experimental run to measure steady-state rates; error bars in (D) represent 95% CI on replicate independent measurements on fresh/recalcined catalyst beds. The references indicated in the insets38,40,44, [45, 46, 47, 48, 49, 50 are also listed in the last column of Table 1.
Table 1.
Apparent Activation Energies for the Major Dehydration Product of Ethanol, 2-Propanol, 1-Butanol, and 2-MTHF under reaction conditions measured on HZSM-5 (Si/Al 140) compared with previously reported values
| Reactant | Product(s) | Apparent Activation Barrier [kcal/mol] |
|
|---|---|---|---|
| This Work | Literature | ||
| Ethanol | di-ethyl ether | 19.7 ± 1.5 | 21.9,47 23.2,40 24.538 |
| 2-Propanol | propene | 34.5 ± 1.9 | 34.9 ± 1.646 |
| 1-Butanol | di-butyl ether | 17.6 ± 0.7 | 18 ± 2,51 21.0 ± 0.844 |
| 2-MTHF | (1,3 + 1,4)-pentadienes | 17.4 ± 1.9 | 21.5 ± 1.5a, 17.7 ± 1.745b |
These measurements were performed on a traditional packed bed reactor as described in Li et al.44
These measurements were performed on amorphous silica-alumina (13.7 wt % alumina content).
Another application of a laboratory-scale PBR is the estimation of rate orders by varying reactant partial pressures. We highlight the applicability of the micro-flow reactor setup by considering ethanol dehydration at low temperatures (388–409 K); under these conditions, di-ethyl ether (DEE) is the only reaction product. As expected, DEE production rates were sensitive to the ethanol partial pressures in the low partial pressure regime (<10 torr) (Figure 5 ). Overall, the order of the reaction decreased and approached a zero-order behavior indicative of a fully saturated surface. Notably, the absolute DEE STY values as well as ethanol partial pressure dependence showed reasonably good agreement with values previously reported under similar reaction conditions on ZSM-5 by Chiang and Bhan.38 Along with all the applications highlighted so far, long-term stability analysis of a catalyst can also be performed on this setup, provided that the reaction conditions ensure complete bed utilization.52 An example case for a test chemistry suffering from deactivation (2-MTHF dehydra-decyclization) carried out on HZSM-5 is included Section S5 of Supplemental Experimental Procedures, Table S3, and Figure S10 to illustrate the efficacy of this setup to perform automated long-term stability investigations.
Figure 5.

Di-Ethyl Ether Site-Time Yields Measured as a Function of Ethanol Partial Pressure on HZSM-5 (Si/Al 140) at 388 K, 398 K, and 409 K, Respectively, on the Micro-Flow Reactor Setup
The error bars represent a 95% CI in the steady-state rate measurements on one catalyst bed (reaction conditions: WHSV in the range 0.81–28.6 g EtOH/g cat/h, carrier gas flow rate = 60 sccm, all conversions were kept below 1.2%). The reported data are compared with experimental and modeled data from Chiang and Bhan38 where the rates were reported at conversions <1.5% on HZSM-5 (Si/Al 42.5).
Calculations to probe the temperature distribution of the reactor under reaction conditions were carried out by (1) the measurement of axial bed temperature gradients, combined with (2) calculation of inter-, and intra-particle temperature gradients (Section S.4 of Supplemental Experimental Procedures). For the reported kinetics collected under strictly differential conversions, both external and internal particle gradients were found to be negligible (Section S.4 of Supplemental Experimental Procedures). At the highest temperature of kinetic measurements, the axial temperature change across the catalyst bed were estimated to be ~0.6 K, ~ −2.4 K, ~0.7 K, and ~−0.4 K for dehydration of ethanol, 2-propanol, 1-butanol, and 2-MTHF, respectively (see Section S.4 of Supplemental Experimental Procedures). With the exception of 2.4 K, these temperature drops are all within the resolution limits of a thermocouple and indicate the nearly isothermal operation of the reactor, at least under these low conversion conditions.
These results, taken together, highlight that the product distributions, absolute values of STYs along with their partial pressure dependencies, and the apparent activation barriers measured using this setup are under nearly isothermal reaction conditions, and hence are in good agreement with previous studies that reported on more traditional PBRs. Comparable reactor performance underpins the utility of this automated setup to obtain kinetic parameters.
Data Automation
The developed chromatogram analysis tool allowed parsing of the raw (.ch) files generated from the flame ionization detector (FID) through the analysis pipeline as described in Section S.6 of Supplemental Experimental Procedures to detect chromatogram peaks and calculate peak areas. The peak areas calculated with this tool were then compared with manually calculated peak areas to investigate its reliability in data analysis. Data of the analytes from the four probe reactions resulted in a total of >200 data points, spanning more than two orders of magnitude, and the results are presented in the parity plot shown in Figure 6 . There was generally good agreement between the manually calculated areas and the corresponding values predicted by the tool irrespective of compound identity, although the values for manual integration were found to be scaled up by a fixed factor. The presented results indicate that the chromatography analysis tool developed can be effectively utilized to analyze raw data files generated by this automated reactor system.
Figure 6.

Parity Plot Comparing the Peak Areas of Different Compounds Calculated by the Chromatography Analysis Tool vis-a-vis Manually Calculated Values
Inset shows a magnified view of the data corresponding to high area values (10–35 pA.min) on a linear scale.
Benefits and Limitations of the Reported Micro-Flow Reactor Setup
As highlighted through the earlier discussions, the micro-flow reactor affords automated operation without requiring constant human intervention. In addition, the fabrication of the setup is fairly straightforward and requires minimal added instrumentation over and above typical GC operation. Many catalysis laboratories across the world are indeed equipped with fully automated state-of-the-art setups operating in a 24/7 mode, but the key benefit of using the reported approach rather than these high-end commercial reactor systems lies in the significant cost savings in implementing this approach. As shown in Figure 7 , depending on the desired level of instrumentation (e.g., new/refurbished GC or the presence/absence of online detectors), the cost benefit of fabricating the micro-flow reactor setup can range anywhere from ~2- to 10-fold over these commercial systems, which amount to cost savings of US$20,000–80,000. Therefore, this approach can potentially enable significantly more kinetic data per unit cost without compromising its quality. With the ease of operation, relatively simple fabrication, and low cost, we view this approach as a tool for standardized experimental reaction systems that can be readily utilized by materials synthesis researchers.
Figure 7.

Fabrication Cost Comparison between Traditional Lab-Scale Automated PBRs and Reported Micro-flow Reactor Setup
Comparison of the fabrication cost for the reported micro-flow reactor under different considered scenarios (e.g., choice of gas chromatograph, presence/absence of an online detector and a methanizer [quantitative carbon detector]) compared with typical prices of commercial low-end automated lab-scale flow reactors. The error bar represents standard error from three different commercial automated setup quotes.
One can reasonably argue that only one catalyst can be tested at any given time, limiting the reactor throughput, while also requiring manual intervention for catalyst changeover. However, a typical mid-size heterogeneous catalysis laboratory in the United States employs anywhere between two to ten GC units, some of which can be converted to these automated micro-flow reactors given the minimal additional fabrication costs (Figure 7). Therefore, the possible shortcomings arising from the testing capability being limited to one catalyst at any given time can be solved by added investment enabling operation of multiple units in parallel. With the reduced-workforce regulations whereby staggered work hours in laboratories is going to be the norm for the foreseeable future, these low-cost automated setups offer another key advantage: to the extent that a laboratory has multiple such units, one researcher can operate ~2–5 systems (potentially being used on different research projects/chemistries/researchers) at a given time depending on familiarity and know-how, meaning that multiple researchers do not need to be present for “their” systems to operate in 24/7 mode.
What about the gas cylinder changeovers and setting up feed-stream dosing (which are not controlled by a GC)? With the increasing global shortage of helium, researchers are moving to low-cost alternatives such as hydrogen for carrier gases for their gas chromatography needs,53 which can be generated in-house using hydrogen generators. Even in cases where helium gas cylinders are used, it is possible to set up helium cylinder multi-packs in conjunction with manifolds for such equipment, enabling continuous operation for weeks without requiring replacement. Setting up liquid feed dosing using syringe pump/HPLC pumps remains manual but is usually quick; as pointed out before, for applications only requiring an initial estimation of product distributions, a significantly simpler version of this setup as a transient pulsed micro-reactor from our earlier works26 , 27 can be utilized, which would barely require any manual intervention in setting up feeds, as the dosing is through the GC autosampler itself. Similarly, for chemistries utilizing light gases as feeds (e.g., methane activation, carbon dioxide valorization), it is possible to set up individual lines with separate mass flow controllers (MFCs), and such systems can run practically indefinitely for a given catalyst bed.
The ease of fabrication does lead to some limitations of using the proposed system, one of which is the feasible conditions of catalyst temperature control. While a small range of h/H ratios ensures minimal deviation temperature, one should be mindful that temperature deviations can be as high as >10%, even without considering the reaction thermodynamics, if the catalyst bed packing extends outside the range of 0.15 < h/H < 0.35. This criterion limits the total mass of catalyst that can be packed in this setup to ~30–45 mg, meaning that highly exothermic reactions requiring bed dilutions may not be suited for this setup. Furthermore, the maximum operating temperature for the setup is limited by the maximum allowable temperature of the GC inlet (673 K, Agilent 7890). Therefore, in situ catalyst pre-treatments requiring higher temperatures cannot be achieved. Lastly, the unit is not intended to be used for multi-phase mixtures; if liquids with low vapor pressures are used, separate heat tracing of individual transfer lines may be required, which compromises the simplicity of fabricating the setup.
Conclusions
We provide a detailed design and implementation strategy of an automated micro-flow reactor integrated within a typical GC unit, enabling kinetic data acquisition without manual intervention for vapor-phase chemistries carried out at near-ambient pressures. Temperature control inside the reactor depends on the axial position of the catalyst bed, where deviations are negligible in the region 0.15 < h/H < 0.35. Residence time distribution analysis reveals near plug-flow hydrodynamics at relatively small gas flow rates (>40 sccm). A combination of four Brønsted acid catalyzed reactions further highlight the absence of heat and mass transport limitations under strictly differential conversions (≤5%), and the axial temperature change remains <2% for the investigated chemistries. Furthermore, typical kinetic parameters are in reasonable agreement with previous reports. Lastly, a chromatogram analysis tool developed in-house integrated with the experimental workflow enables the handling of raw chromatography files for automated evaluation of peak areas. The setup has significant cost savings compared with commercial automated lab-scale reactors, and coupled with its automated implementation affords an opportunity to carry out catalytic evaluation of materials in a manner that is both reliable and consistent with reduced-workforce guidelines in place due to the COVID-19 pandemic.
Experimental Procedures
Resource Availability
Lead Contact
Paul J. Dauenhauer (hauer@umn.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The source code of the automated chromatography analysis tool is available on GitHub.54 All the raw data files (including the .JSON files used for heat/mass transfer calculations on GradientCheck) can be found on the UMN data repository.55 Any further input for fabricating the setup is available from the corresponding author upon reasonable request.
Methods
This section discusses a detailed design and implementation of the reactor, independent temperature measurements to investigate axial variations under non-reaction conditions, and residence time distribution studies within the micro-flow reactor. The methodology used in the apparent kinetic measurements for all the example reactions is also discussed. Detailed heat and mass transfer characteristics specific to each of the probe reactions are discussed in Section S.4 of Supplemental Experimental Procedures.
Design and Implementation
The following modifications were performed on an Agilent 7890 GC unit to convert it to the micro-flow reactor (Scheme 2 ). The automatic liquid sampler (ALS) was uninstalled, and the front inlet of the GC was installed as an auxiliary heater. The flow control to this front inlet (housing the catalyst bed) was achieved using upstream referenced MFCs (Vici Valco Model 100) in conjunction with an auxiliary electronic pressure controller (EPC; Agilent G3452) (gas delivery details can be found in Section S.1 of Supplemental Experimental Procedures and Figure S4). Given the upstream reference configuration of the MFC, a fixed and repeatable gas flow rate could be delivered for a particular supply pressure set by the EPC. Gas flow rates delivered by the MFC were therefore calibrated as a function of the auxiliary EPC pressure set point, which could be directly controlled through the GC (Figure S4).
Scheme 2.
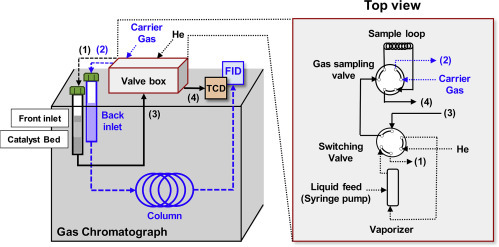
A Schematic for the Vapor-Phase Micro-Flow Reactor Integrated within a Gas Chromatograph
The instrumentation of the setup consists solely of a typical gas chromatograph, where the front inlet is used as a packed bed reactor. The gas flows are routed to either bypass or contact the catalyst bed by using a combination of two six-port valves housed within a heated valve box unit, and a thermal conductivity detector (TCD) is used for online reactor effluent monitoring. The back inlet is utilized for effluent quantification and consequent rate measurements through periodic gas-sampling followed by separation by a GC column and quantification through a flame ionization detector (FID).
A splitless inlet liner (Agilent, 5190-2293) was utilized as the reactor tube (internal diameter 4 mm) (Figure S2). The split vent for the front inlet was blocked using a capping nut (Swagelok SS-200-P); doing so forced gas flows exiting the reactor through one single outlet (Figures S1 and S3). The back inlet was used for chromatography, where analyte separation was performed by an HP-PLOTQ column (Agilent, 19091P-QO4) connected to a quantitative carbon detector (QCD; Polyarc)36 in conjunction with an FID.
Two six-port valves (Vici Valco, DC6UWE) (shown in Scheme 2; identified as V-1 and V-2 in Scheme 3 ) housed within a heated valve box (Agilent G1581A) were used to route gas flows and perform gas-phase injections on the back inlet utilized for chromatography. Valve V-1 was installed as a gas-sampling valve; V-2 was installed as a switching valve (Scheme 3 and Figure S1). A pressure gauge upstream of the reactor was used to monitor the pressure drops across the catalyst bed by comparing with a reference pressure measured in absence of a catalyst bed (indicative of pressure drop due to process tubing and fittings). A vaporization section was housed within the heated valve box, and consisted of a ¼″ tube (316 SS) filled with deactivated quartz chips (SiO2, 4-20 mesh, Sigma-Aldrich) to facilitate static mixing and effective vaporization of the injected liquid (Figure S1). A thermal conductivity detector (TCD) placed in-line with the flow exiting the reactor stream was used as an online detector (Scheme 3).
Scheme 3.

Methodology for Vapor-Phase Micro-Flow Reactor Integrated within a Gas Chromatograph
Catalyst samples are loaded into the front inlet liner of a gas chromatograph and reaction kinetics are studied by operating the setup in three configurations. (i) In situ catalyst pre-treatment mode (A. Configuration 1; no feed): catalysts are calcined in situ in flowing air at 673 K and then cooled down to reaction temperature and the carrier gas is switched from Air to He. (ii) Reactant dosing in bypass mode (A. Configuration 1; feed on): a syringe pump delivers a steady flow of liquid reactant to the vaporizer, bypassing the reactor, and is routed directly to the online detector (TCD) until a stable signal is observed. and (iii) Reactant introduction and product quantification mode (combination of Configurations 2 and 3 [B and C]): the vaporized reactant stream is contacted with the catalyst bed in configuration 2, followed by periodic gas sampling (configuration 3) leading to the separation and quantification of products through QCD/FID.
Both front and back inlets were insulated with fiberglass insulation sheet (McMaster) to ensure that bed temperatures were close to the inlet wall temperature controlled by the auxiliary heater; this was later confirmed by secondary measurements on an empty bed (discussed in Reactor Temperature Distribution: Unreactive Conditions).
Scheme 3 depicts the overall experimental methodology for performing kinetic measurements using the setup in three different configurations discussed below. The different valve positions to operate the setup for these specific configurations are also discussed and listed in Table 2 .
-
(a)
Catalyst pre-treatment mode (configuration 1; no reactant feed). Catalyst powders were pressed and sieved into aggregates of 500–1,000 μm and placed between deactivated quartz wool (Restek 24324) to keep the catalyst bed in place. Typically, ~25–30 mg of catalyst was used for the kinetic measurements. Catalyst masses were restricted to keep the bed located in the normalized height h/H range of 0.15–0.35, where differences between actual and GC indicated temperature were found to be negligible; this is discussed in more detail in Reactor Temperature Distribution: Unreactive Conditions. Following the placement of the reactor in the front inlet, catalysts were pre-treated in air (99.997%, Minneapolis Oxygen) at 673 K for 5 h at a ramp rate of 3 K min−1 (Scheme 3A). After pre-treatment, the catalyst was cooled down to reaction temperature and the gas supply was switched to He (99.995%, Matheson). The catalyst was kept at reaction temperature and purged with He for at least 30 min prior to reactant introduction.
-
(b)
Reactor bypass mode (configuration 1; reactant feed on). In the same configuration, liquid reactants were then pumped into the vaporization section through a 1/16″ PEEK capillary line (0.01″ internal diameter) using a syringe pump (74,905-04, Cole Parmer) and swept by the He stream exiting the reactor. In this way, the reactant stream bypassed the reactor by placing the vaporization section downstream of the reactor (configuration 1, Scheme 3A). The valve box temperature was maintained at 473 K to ensure that all species were retained in the vapor phase. The vaporized reactant stream was swept through the vaporization section to V-2 (Scheme 3B) and routed to the TCD after filling the sampling loop located on V-1. Prior to introducing the reactant stream to the reactor, a steady reactant stream was ensured by observing a stable TCD signal for at least 15 min. Periodic gas-sampling injections in this bypass configuration allowed for the total carbon quantification to gauge carbon balances during the reaction.
-
(c)
Reactant introduction and product quantification mode (combination of configurations 2 and 3). Once a stable vapor stream of reactant was established, V-2 was switched to direct the flow of He through the vaporizer section first (i.e., after the V-2 switch, the vaporizer becomes upstream of the reactor). The vaporized stream of reactants thus passed through the reactor and contacted the catalyst bed (configuration 2, Scheme 3B). Effluent from the reactor continued to fill the sample loop attached to V-1 and subsequently passed through the online TCD. Quantitative analysis of the reactor effluent was achieved by periodically switching the gas-sampling valve (V-1) at the start of every chromatography analysis (configuration 3, Scheme 3C). This effectively allowed for the quantitative transfer of information from the reactive front inlet portion of the GC to the analytical chromatography back inlet portion. He carrier gas carried the contents of the sampling loop through the GC column to the QCD/FID for separation and quantification, and V-1 switched back after the sample injection was complete (0.5 min).
Table 2.
Possible Valve Positions and the Corresponding Configurations as Highlighted in Scheme 3
| Valve Position |
Gas | Function | Configuration | |
|---|---|---|---|---|
| V1 | V2 | |||
| off | off | air | calcination | 1 |
| off | off | He | bypass | 1 |
| off | on | He | reaction | 2 |
| on | on | He | reaction-sample injection | 3 |
Configured this way, all kinetic measurements were performed in downflow mode. Notably, the setup could be operated in either of the three configurations without any manual intervention. This ease of operation was due to the integration of all control elements within the GC circuitry and software without the use of any programmable logic controllers and/or customized LabView programs. These benefits are specifically listed below (see Section S1.2 of Supplemental Experimental Procedures and Figures S5–S8 for details on method development in ChemStation v.8.2.1 to allow control of all reactor parameters directly from method files).
-
(i)
Reaction temperatures could be varied by adjusting the front inlet temperature (installed as an auxiliary heater) in the corresponding method files.
-
(ii)
V-2 switching from bypass-to-reactor mode, as well as V-1 injection to the back inlet for product separation/quantification, was also controlled directly through ChemStation method files.
-
(iii)
Gas flows were set by calibrating the MFCs on auxiliary EPC (set in method files) with the final flow rates at the TCD outlet.
We note that the methodology described here is for the specific application reported and can be easily tuned to other applications. For example, the choice of online detector (TCD in this work) is up to the user, and even the absence of an online detector and a methanizer (a QCD, POLYARC in this work) does not compromise the performance of the setup. Secondary detectors such as a mass spectrometer can be added to augment the quantitative information with real-time product identification. Similarly, for the reactant delivery, if reactants need to be introduced only for a short period of time (<5–10 min), an ALS can be used for a continuous pumping of reactants and can essentially act as a syringe pump. Indeed, if preliminary product distributions on a variety of different materials is the only intended application, a much simpler version of this setup with pulsed dosing of reactants followed immediately by separation and detection of products can be utilized, as highlighted by our earlier works.26 , 27 While we use syringe pumps for liquid delivery, gaseous reactants can also be dosed using (a) separate gas line(s) equipped with MFC(s). Total reaction pressures in excess of atmospheric levels can be achieved by using back-pressure regulator, and we have been able to achieve pressures as high as ~100 psi with this simple modification, possibly allowing for moderately high-pressure chemistries to be probed (e.g., hydrodeoxygenation). In short, there are many conceivable modifications using hardware that can be readily integrated with the GC depending on the specific application, rendering this approach versatile.
Temperature Variations under Non-reacting Conditions
Temperature measurements were obtained on the front liner packed with deactivated quartz wool by inserting a 1/16″ thermocouple (Omega). This “empty reactor” was maintained at a particular set temperature as indicated by the GC for at least 20 min prior to every measurement, and the measurements were repeated in at least ten different axial positions along the length of the liner.
Hydrodynamic Behavior under Non-reacting Conditions
RTDs were measured to establish the range of flow rates under which flow in the setup was sufficiently plug-flow. All residence time studies were conducted by connecting the reactor effluent directly to the inlet of the TCD so as to minimize the volume between the point of injection and detection. The reactor inlet was packed with deactivated quartz wool and maintained at 523 K; pulses of liquid ethanol were used as tracer. Liquid injections of ethanol pulses were manually performed with a 0.5-μL syringe (Agilent). Tracer identity is unlikely to change the hydrodynamic behavior under non-reacting conditions,56 and the results obtained with ethanol are assumed to be broadly applicable. Typical injection volumes were 0.4 μL, and the experiments were conducted at low carrier flow rates (13–40 sccm) to capture the transition from axially dispersed to a non-dispersed regime.
Catalytic Evaluation
Comparison of the turnover rates of reactions involves the calculation of STYs (Equation 1). For Brønsted acid catalyzed chemistries, this calculation requires normalizing mass-based rates of reaction by the total Brønsted acid site density (BAS). The BAS (N H+) of the catalyst (HZSM-5) was measured by the quantification of butenes resulting from the Hofmann elimination of tert-butylamine using reactive gas chromatography methodology described elsewhere,46 and the results are discussed in the Section S.2 of Supplemental Experimental Procedures and Figure S9. These values (NH+) were used in the calculation of STYs (Equation 1) in all probe reactions.
| (Equation 1) |
All kinetic experiments were performed under near-differential conversions (<~15%) for a given feed by adjusting the weight-hourly space velocities (WHSVs). The reactions were investigated at ~25 torr partial pressure of reactant. Alcohol dehydration reactions were carried out in the temperature range 388–483 K, whereas 2-MTHF dehydra-decyclization was conducted in the temperature range 463–543 K. Due to an abundant literature on alcohol dehydration on solid acids, kinetic data obtained from the micro-flow reactor were compared with previously reported values. For the case of 2-MTHF dehydra-decyclization, however, the kinetic parameters obtained on the micro-flow reactor were directly compared with measurements performed on a traditional PBR as described in our earlier work.44 Minimal catalyst deactivation was observed after initial transients for alcohol dehydration kinetics under the investigated conditions, and the rates reported here are steady-state values. Replicate experiments were carried out using randomized reactor temperature sequencing to minimize any systematic errors. Significant catalyst deactivation was observed during 2-MTHF dehydra-decyclization, and the methods to correct for deactivation to report initial rates are described in detail in Section S.3 of Supplemental Experimental Procedures.
All carbon balances closed to within ±10%. Pressure drops across the reactor were maintained below 13%–16% of total pressure for all kinetic experiments. Error bars represent the 95% confidence interval on at least three independent measurements unless otherwise stated.
Data Automation
The data analysis tool reported in this work is capable of processing data files (.ch files) from the Agilent Gas Chromatograph with ChemStation as well as OpenLab CDS ChemStation software. We note that these are the most commonly used GC and chromatography analysis software packages. The source code for this tool is available on GitHub,54 and the details of raw data parsing, peak detection, and peak area calculation and other analysis pipeline steps, along with illustration of each step, can be found in Section S.6 of Supplemental Experimental Procedures and Figures S11 and S12.
Materials
Ethanol (200 proof, ≥99.5%, Sigma-Aldrich), 2-propanol (≥99.5%, Sigma-Aldrich), 1-butanol (≥99.5%, Sigma-Aldrich), and 2-MTHF (≥98%, stabilized with butylated hydroxytoluene, TCI Chemicals) were used without further purification. Ammonium form of a high-silica ZSM-5 (Si/Al 140) (Zeolyst CBV28014) was calcined ex situ under air flow (~40 sccm) (zero grade, Minneapolis Oxygen) at 823 K for 10 h at a ramp rate of 3 K min−1 (Lindberg Blue M tubular furnace). An exhaustive list of all major instrumentation parts as well as a manual guide for both hardware and software modifications on an existing GC to fabricate the setup is included in Section S.1 of Supplemental Experimental Procedures and Table S1.
Acknowledgments
We acknowledge financial support of the Catalysis Center for Energy Innovation, a US Department of Energy – Energy Frontier Research Center under Grant DE-SC0001004.
Author Contributions
Conceptualization, O.A.A. and P.J.D; Methodology, G.K., H.B., M.A.A., O.A.A., A.C., Y.P., C.L., and D.M.; Investigation, G.K. and H.B.; Writing – Original Draft, G.K.; Writing – Review & Editing, G.K., D.M., M.A.A., O.A.A., P.J.D., and M.T.; Funding Acquisition, M.T. and P.J.D.; Supervision, P.J.D., O.A.A., and M.T.
Declaration of Interests
The authors declare no competing interests.
Published: July 10, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.matt.2020.06.025.
Supplemental Information
References
- 1.Bao Y., Leader G., Bossion A. Snapshots of life—early career materials scientists managing in the midst of a pandemic. Chem. Mater. 2020 doi: 10.1021/acs.chemmater.0c01624. [DOI] [PubMed] [Google Scholar]
- 2.Degnan T. How will the COVID-19 pandemic affect the catalysis community? Focus Catal. 2020 doi: 10.1016/j.focat.2020.04.001. [DOI] [Google Scholar]
- 3.Hartmann M. Hierarchical zeolites: a proven strategy to combine shape selectivity with efficient mass transport. Angew. Chem. Int. Ed. 2004;43:5880–5882. doi: 10.1002/anie.200460644. [DOI] [PubMed] [Google Scholar]
- 4.Chughtai A.H., Ahmad N., Younus H.A., Laypkov A., Verpoort F. Metal-organic frameworks: versatile heterogeneous catalysts for efficient catalytic organic transformations. Chem. Soc. Rev. 2015;44:6804–6849. doi: 10.1039/c4cs00395k. [DOI] [PubMed] [Google Scholar]
- 5.Wang A., Li J., Zhang T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018;2:65–81. [Google Scholar]
- 6.Furukawa S., Komatsu T. Intermetallic compounds: promising inorganic materials for well-structured and electronically modified reaction environments for efficient catalysis. ACS Catal. 2017;7:735–765. [Google Scholar]
- 7.Voiry D., Shin H.S., Loh K.P., Chhowalla M. Low-dimensional catalysts for hydrogen evolution and CO2 reduction. Nat. Chem. Rev. 2018;2 doi: 10.1038/s41570-017-0105. [DOI] [Google Scholar]
- 8.Holzwarth A., Schmidt H.W., Maier W.F. Detection of catalytic activity in combinatorial libraries of heterogeneous catalysts by IR thermography. Angew. Chem. Int. Ed. 1998;37:2644–2647. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2644::AID-ANIE2644>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 9.Cong P., Doolen R.D., Fan Q., Giaquinta D.M., Guan S., Mcfarland E.W., Poojary D.M., Self K., Turner H.W., Weinberg W.H. High-throughput synthesis and screening of combinatorial heterogeneous catalyst libraries. Angew. Chem. 1999;4:483–488. doi: 10.1002/(SICI)1521-3773(19990215)38:4<483::AID-ANIE483>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 10.Hendershot R.J., Snively C.M., Lauterbach J. High-throughput heterogeneous catalytic science. Chemistry. 2005;11:806–814. doi: 10.1002/chem.200400613. [DOI] [PubMed] [Google Scholar]
- 11.Van Veen A.C., Farrusseng D., Rebeilleau M., Decamp T., Holzwarth A., Schuurman Y., Mirodatos C. Acceleration in catalyst development by fast transient kinetic investigation. J. Catal. 2003;216:135–143. [Google Scholar]
- 12.Corma A., Serra J.M. Heterogeneous combinatorial catalysis applied to oil refining, petrochemistry and fine chemistry. Catal. Today. 2005;107–108:3–11. [Google Scholar]
- 13.Huybrechts W., Mijoin J., Jacobs P.A., Martens J.A. Development of a fixed-bed continuous-flow high-throughput reactor for long-chain n-alkane hydroconversion. Appl. Catal. A Gen. 2003;243:1–13. [Google Scholar]
- 14.Hendershot R.J., Lasko S.S., Fellmann M.F., Oskarsdottir G., Nicholas Delgass W., Snively C.M., Lauterbach J. A novel reactor system for high throughput catalyst testing under realistic conditions. Appl. Catal. A Gen. 2003;254:107–120. [Google Scholar]
- 15.Hahndorf I., Buyevskaya O., Langpape M., Grubert G., Kolf S., Guillon E., Baerns M. Experimental equipment for high-throughput synthesis and testing of catalytic materials. Chem. Eng. J. 2002;89:119–125. [Google Scholar]
- 16.Pérez-Ramírez J., Berger R.J., Mul G., Kapteijn F., Moulijn J.A. Six-flow reactor technology a review on fast catalyst screening and kinetic studies. Catal. Today. 2000;60:93–109. [Google Scholar]
- 17.Moulijn J.A., Pérez-Ramírez J., Berger R.J., Hamminga G., Mul G., Kapteijn F. High-throughput experimentation in catalyst testing and in kinetic studies for heterogeneous catalysis. Catal. Today. 2003;81:457–471. [Google Scholar]
- 18.Parrott A.J., Bourne R.A., Akien G.R., Irvine D.J., Poliakoff M. Self-optimizing continuous reactions in supercritical carbon dioxide. Angew. Chem. Int. Ed. 2011;50:3788–3792. doi: 10.1002/anie.201100412. [DOI] [PubMed] [Google Scholar]
- 19.Waldron C., Pankajakshan A., Quaglio M., Cao E., Galvanin F., Gavriilidis A. An autonomous microreactor platform for the rapid identification of kinetic models. React. Chem. Eng. 2019;4:1623–1636. [Google Scholar]
- 20.Perera D., Tucker J.W., Brahmbhatt S., Helal C.J., Chong A., Farrell W., Richardson P., Sach N.W. A platform for automated nanomole- scale reaction screening and micromole-scale synthesis in flow. Science. 2018;359:429–434. doi: 10.1126/science.aap9112. [DOI] [PubMed] [Google Scholar]
- 21.Holmes N., Akien G.R., Blacker A.J., Woodward R.L., Meadows R.E., Bourne R.A. Self-optimisation of the final stage in the synthesis of EGFR kinase inhibitor AZD9291 using an automated flow reactor. React. Chem. Eng. 2016;1:366–371. [Google Scholar]
- 22.Cherkasov N., Bai Y., Expósito A.J., Rebrov E.V. OpenFlowChem-a platform for quick, robust and flexible automation and self-optimisation of flow chemistry. React. Chem. Eng. 2018;3:769–780. [Google Scholar]
- 23.Reizman B.J., Jensen K.F. An automated continuous-flow platform for the estimation of multistep reaction kinetics. Org. Process. Res. Dev. 2012;16:1770–1782. [Google Scholar]
- 24.Watanabe R., Komatsu T., Sakamoto S., Uranoc Y., Noji H. High-throughput single-molecule bioassay using micro-reactor arrays with a concentration gradient of target molecules. Lab Chip. 2018;18:2849–2853. doi: 10.1039/c8lc00535d. [DOI] [PubMed] [Google Scholar]
- 25.https://github.com/richardingham/octopus
- 26.Abdelrahman O.A., Park D.S., Vinter K.P., Spanjers C.S., Ren L., Cho H.J., Vlachos D.G., Fan W., Tsapatsis M., Dauenhauer P.J. Biomass-derived butadiene by dehydra-decyclization of tetrahydrofuran. ACS Sustain. Chem. Eng. 2017;5:3732–3736. [Google Scholar]
- 27.Abdelrahman O.A., Park D.S., Vinter K.P., Spanjers C.S., Ren L., Cho H.J., Zhang K., Fan W., Tsapatsis M., Dauenhauer P.J. Renewable isoprene by sequential hydrogenation of itaconic acid and dehydra-decyclization of 3-methyl-tetrahydrofuran. ACS Catal. 2017;7:1428–1431. [Google Scholar]
- 28.Sica A.M., Valles E.M., Gigola C.E. Kinetic data from a pulse microcatalytic reactor-hydrogenation of benzene on a nickel catalyst. J. Catal. 1978;51:115–125. [Google Scholar]
- 29.Deshpande A., Krishnaswamy S., Ponnani K. Pulsed Micro-reactor: an alternative to estimating kinetic parameters of non-catalytic gas–solid reactions. Chem. Eng. Res. Des. 2017;117:382–393. [Google Scholar]
- 30.Wang H., Zhao P., Jiang B., Liu W. A pulse reactor-gas chromatographic investigation of the kinetics of catalytic carbon monoxide hydrogenation. J. Mol. Catal. 1992;71:357–364. [Google Scholar]
- 31.Bett J.A.S., Hall K.W. The microcatalytic technique applied to a zero order reaction: the dehydration of 2-butanol over hydroxyapatite catalysts. J. Catal. 1968;10:105–113. [Google Scholar]
- 32.Verma A. Kaliaguine Estimation of rate coefficients from pulsed microcatalytic reactors: oxidation of ethylene over silver catalyst. J. Catal. 1973;30:430–437. [Google Scholar]
- 33.Hickman D.A., Degenstein J.C., Ribeiro F.H. Fundamental principles of laboratory fixed bed reactor design. Curr. Opin. Chem. Eng. 2016;13:1–9. [Google Scholar]
- 34.Froment G.F., Bischoff K.B. Wiley; 1974. Chemical Reaction Engineering. [DOI] [Google Scholar]
- 35.Levenspiel O. Third Edition. Wiley; 1998. Chemical Reaction Engineering. [Google Scholar]
- 36.Maduskar S., Teixeira A.R., Paulsen A.D., Krumm C., Mountziaris, Fan W., Dauenhauer P.J. Quantitative carbon detector (QCD) for calibration-free, high-resolution characterization of complex mixtures. Lab Chip. 2015;15:440–447. doi: 10.1039/c4lc01180e. [DOI] [PubMed] [Google Scholar]
- 37.Gounder R., Iglesia E. The roles of entropy and enthalpy in stabilizing ion-pairs at transition states in zeolite acid catalysis. Acc. Chem. Res. 2012;45:229–238. doi: 10.1021/ar200138n. [DOI] [PubMed] [Google Scholar]
- 38.Chiang H., Bhan A. Catalytic consequences of hydroxyl group location on the rate and mechanism of parallel dehydration reactions of ethanol over acidic zeolites. J. Catal. 2010;271:251–261. [Google Scholar]
- 39.Williams C., Makarova M.A., Malysheva L.V., Paukshtis E.A., Zamaraev K.I., Thomas J.M. Mechanistic studies of the catalytic dehydration of isobutyl alcohol on NaH-ZSM-5. J. Chem. Soc. Faraday Trans. 1990;86:3473–3485. [Google Scholar]
- 40.Liu D., Bhan A., Tsapatsis M., Al Hashimi S. Catalytic behavior of Brønsted acid sites in MWW and MFI zeolites with dual meso- and microporosity. ACS Catal. 2011;1:7–17. [Google Scholar]
- 41.Kumar G., Liu D., Xu D., Tsapatsis M., Dauenhauer P.J. Dehydra-decyclization of 2-methyltetrahydrofuran to pentadienes on boron- containing zeolites. Green. Chem. 2020 doi: 10.1039/D0GC00136H. [DOI] [PubMed] [Google Scholar]
- 42.Gounder R., Iglesia E. The catalytic diversity of zeolites: confinement and solvation effects within voids of molecular dimensions. Chem. Commun. 2013;49:3491. doi: 10.1039/c3cc40731d. [DOI] [PubMed] [Google Scholar]
- 43.Phillips C.B., Datta R. Production of ethylene from hydrous ethanol on H-ZSM-5 under mild conditions. Ind. Eng. Chem. Res. 1997;36:4466–4475. [Google Scholar]
- 44.Li S., Abdelrahman O.A., Kumar G., Tsapatsis M., Vlachos D., Caratzoulas S., Dauenhauer P.J. Dehydra-decyclization of tetrahydrofuran on H-ZSM5: mechanisms, pathways, and role of transition state entropy. ACS Catal. 2019;9:10279–10293. [Google Scholar]
- Kumbhalkar M.D., Buchanan J.S., Huber G.W., Dumesic J.A. Ring opening of biomass-derived cyclic ethers to dienes over silica/alumina. ACS Catal. 2017;8:5248–5256. [Google Scholar]
- 46.Abdelrahman O., Vinter K.P., Ren L., Xu D., Gorte R.J., Tsapatsis M., Dauenhauer P.J. Simple quantification of zeolite acid site density by reactive gas chromatography. Catal. Sci. Technol. 2017;7:3831–3841. [Google Scholar]
- 47.Alexopoulos K., John M., Van Der Borght K., Galvita V., Reyniers M.F., Marin G.B. DFT-based microkinetic modeling of ethanol dehydration in H-ZSM-5. J. Catal. 2016;339:173–185. [Google Scholar]
- 48.Moser W.R., Thompson R.W., Chiang C.C., Tong H. Silicon-rich H-ZSM-5 catalyzed conversion of aqueous ethanol to ethylene. J. Catal. 1989;117:19–32. [Google Scholar]
- 49.John M., Alexopoulos K., Reyniers M.F., Marin G.B. First-principles kinetic study on the effect of the zeolite framework on 1-butanol dehydration. ACS Catal. 2016;6:4081–4094. [Google Scholar]
- 50.John M., Alexopoulos K., Reyniers M., Marin G.B. Reaction path analysis for 1-butanol dehydration in H-ZSM-5 zeolite: ab initio and microkinetic modeling. J. Catal. 2015;330:28–45. [Google Scholar]
- 51.Makarova M.A., Paukshtis E.A., Thomas J.M., Williams C., Zamaraev K.I. Dehydration of n-butanol on zeolite H-ZSM-5 and amorphous aluminosilicate: detailed mechanistic study and the effect of pore confinement. J. Catal. 1994;149:36–51. [Google Scholar]
- 52.Scott S.L. A matter of life(time) and death. ACS Catal. 2018;8:8597–8599. [Google Scholar]
- 53.Reisch M.S. Coping with the helium shortage. C&EN. 2013;91:18–19. https://cen.acs.org/articles/91/i5/Coping-Helium-Shortage.html [Google Scholar]
- 54.https://github.com/dauenhauer-umn/micro-flow-reactor-paper
- 55.Kumar G., Bossert H., McDonald D., Chatzidimitriou A., Ardagh A.M., Pang Y., Lee C.S., Tsapatsis M., Abdelrahman O.A., Dauenhauer P. Supporting data for "Catalysis-in-a-Box: robotic screening of catalytic materials in the times of COVID-19 and beyond. 2020. http://hdl.handle.net/11299/213839 [DOI] [PMC free article] [PubMed]
- 56.Nauman E.B. Residence time theory. Ind. Eng. Chem. Res. 2008;47:3752–3766. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source code of the automated chromatography analysis tool is available on GitHub.54 All the raw data files (including the .JSON files used for heat/mass transfer calculations on GradientCheck) can be found on the UMN data repository.55 Any further input for fabricating the setup is available from the corresponding author upon reasonable request.